
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic covalent chemistry of imine polymers at the liquid/solid interface investigated by scanning tunneling microscopy†
Yanxia Yuac,
Jianbin Lin*b and
Shengbin Lei *ac
*ac
aTianjin Key Laboratory of Molecular Optoelectronic Science, Department of Chemistry, School of Science & Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin University, Tianjin, 300072, P. R. China. E-mail: shengbin.lei@tju.edu.cn
bDepartment of Chemistry, College of Chemistry and Chemical Engineering, MOE Key Laboratory of Analytical Sciences, Xiamen University, Xiamen, 361005, P. R. China. E-mail: jb.lin@xmu.edu.cn
cKey Laboratory of Microsystems and Microstructures Manufacturing, Ministry of Education, Harbin Institute of Technology, Harbin, 150080, P. R. China
First published on 14th February 2017
Abstract
How the constitutional dynamics of a dynamic covalent library (DCL) adapts to the presence of a surface is of fundamental importance for the design of adaptive materials. In this work, we show that amine exchange can be achieved at the liquid/solid interface within one-dimensional and two-dimensional imine polymers, which were characterized on the submolecular scale by scanning tunneling microscopy (STM). The potential of a surface to influence the thermodynamic equilibrium in a mixture of constitutionally interrelated building blocks with different molar ratio was explored. Our investigation demonstrates that the substrate and molar ratio of precursors markedly rearrange the composition of the DCC libraries.
Introduction
Recently, dynamic covalent chemistry (DCC), which is based on reversible chemical reactions where the covalent bond has the ability to be formed and broken reversibly under equilibrium control, has received much attention in synthetic and material chemistry.1–3 The constitutional dynamics allow for adaptation through component exchange and selection in response to physical stimuli, chemical effectors, or environmental effects, thus allows fast screening of bioactive substances from a so-called dynamic covalent library (DCL), and also opens the way towards adaptive chemistry and dynamic materials.4 Significantly, DCC has a lot of features in common with supramolecular chemistry, both involve bonding processes that are reversible and the formation of products occurs under thermodynamic control. These common characteristics result not only in the possibility of carrying out synthesis under thermodynamic control, but also in making it possible for the products to undergo error-checking and correction, a process which ensures the eradication of thermodynamically unstable products.1 Besides, DCC has the additional advantage of constructing robust molecules on account of the formation of covalent bonds rather than fragile supramolecules resulting from noncovalent bonding interactions.1,2 The imine bond, formed via the reversible condensation between an amine and an aldehyde, displays many ideal dynamic covalent bond characteristics.5–7In a recent publication, P. Samorì et al. reported for the first time a submolecularly resolved STM mapping of the surface-mediated reversible exchange process of bisimines that occur at the liquid/solid interface.8 This work opens new avenues for the understanding of the parameters affecting the thermodynamic and kinetic features of covalent dynamic processes at the liquid/solid interface. J. Plas et al. address both the solution composition and surface composition of a DCL using a combination of STM and NMR.9 These investigations mainly focus on the DCC of small imine molecules or imine oligomers. Although the reversible exchange of components in polymers has been investigated in solutions over 10 years ago,5 where an external input such as ZnII ions is required to promote component exchange in polyimine dynamers,10 dynamic adaptation of imine polymers to a surface that occur at the liquid/solid interface have not previously been reported. In the DCL containing imine polymers the effects of surface in accelerating the reaction, fractionation of polymers with different degree of polymerization,11 and its van der Waals epitaxy effect in templating the growth and assembling of imine polymers will all play important roles during the adaptation process.
Kunitake and our group recently have reported the successful in situ synthesis of long-range ordered one-dimensional (1D)/two-dimensional (2D) imine polymer at the liquid/solid interface at room temperature, either in aqueous or organic media.12–15 Comparing with sample preparation in solution,8,16,17 at the gas/solid interface18–23 or under UHV conditions,24–30 the liquid/solid interface has a number of advantages: (1) the dynamic exchange between matters on the surface and in the supernatant liquid phase promotes self-healing of defects during the polymer growth; (2) the solvation effect helps to lower the energy barrier for matter exchange, promote the formation of well-defined covalent architectures, (3) the concentration and van der Waals epitaxy effects of the surface accelerate the reaction speed and promote the formation of long-range ordered structures.31
Herein, we present a submolecular resolution STM investigation of the reversible diamine exchange process within 1D and 2D Schiff-base polymers that occur at the octanoic acid/highly oriented pyrolitic graphite (HOPG) interface. Freshly cleaved HOPG is an ideal substrate because it is hydrophobic, atomically flat, and relatively chemical inert.32 The van der Waals epitaxy effect can promote the formation of ordered 1D/2D nanostructures.18–23 The compounds utilized in this study are illustrated in Scheme 1: benzene-1,3,5-tricarbaldehyde (1), 2,5-bis(octyloxy)terephthalaldehyde (2), benzidine (3), 9,10-bis(p-aminophenylethynyl)anthracene (4), and 4,4′′-diamino-p-terphenyl (5). Among numerous aldehydes and diamines, aldehyde 1–2 and diamine 3–5 were chosen because of the formation of well-ordered 1D polymer (1DP) or 2D polymers (2DP) at the liquid/solid interface.14,15 Moreover, diamines 3, 4 and 5 show different adsorption capacity on the surface. We focused our attention on the adaptation of such polymer containing DCL to the presence of a surface, as well as on the covalent dynamic process of amine exchange at the liquid/solid interface within these polymers.
 | ||
| Scheme 1 Chemical structure of aldehyde and amine monomers used in this work: 1, benzene-1,3,5-tricarbaldehyde; 2, 2,5-bis(octyloxy)terephthalaldehyde; 3, benzidine; 4, 9,10-bis(p-aminophenylethynyl)anthracene; 5, 4,4′′-diamino-p-terphenyl. | ||
Results and discussion
In order to explore the adsorption capacity of monomers on graphite, self-assembly of the precursors were first investigated using STM at the octanoic acid/HOPG interface. While as expected, monomers 1, 3 and 5 did not show any clue of monolayer formation on HOPG surface due to their weak intermolecular and molecule–substrate interactions. Self-assembly of 4 only forms ordered domains with tens of nanometers in size, no long-range ordered monolayer was formed. This suggests that the supramolecular motif is mainly stabilized by relatively strong molecule–substrate interactions. High-resolution image (Fig. S1b†) shows that molecule 4 forms a zigzag lamellae structure via weak hydrogen bonding between amino groups. The self-assembly of 2 on HOPG surface leads to ordered crystalline monolayer that consists of lamellar architectures with co-adsorption of octanoic acid, which is in agreement with that reported in our previous work.14Synthesis of 1D and 2D polymers at the octanoic acid/HOPG interface
Schiff-base coupling of 2 and 3 (with concentration of both monomers c2 = c3 = 1.6 × 10−4 mol L−1) was previously reported by our group.14 Here, at lower concentration (c2 = c3 = 1.6 × 10−5 mol L−1), again 1DP2+3 is also observed, the assembling structure is identical to that with higher concentration, as shown in Fig. 1b. The repeating period of 1DP2+3 chain measures 1.7 ± 0.1 nm, which agrees well with that reported in the previous studies.14 Co-condensation of 2 with 4 (c2 = c4 = 1.1 × 10−5 mol L−1) at the octanoic acid/HOPG interface at room temperature also results in highly ordered parallel-packed adlayers of linear polymer arrays several minutes after deposition, almost covering the entire surface, as shown in Fig. 1d. Along the polymer backbone of 1DP2+4, the repeating period is 2.6 ± 0.1 nm, in good agreement with those expected from the chemical structure, confirming the covalent bond formation. More detailed structural information can be obtained from the high-resolution STM images in Fig. 1e and f. Different from the case of 1DP2+3, in which the interchain distance (1.7 ± 0.1 nm) is determined by the interdigitation of the alkoxy side chains, the interchain distance of 1DP2+4 (1.1 ± 0.1 nm) is apparently smaller than that expected with alkoxy chain interdigitation (1.8 nm). Thus we assume that these octyloxy groups are floated in solution. This hypothesis is confirmed by the observation of identical structure obtained by co-condensation of terephthaldicarboxaldehyde with 4, as shown in Fig. S2.† The aligned wider bright spots along the polymer chains typically appeared in the high resolution STM images (Fig. 1f and S2b†) can be attributed to the anthracenyl groups of the 4 unit, which contributes to stabilize the assembly of 1DP2+4 by relatively strong molecule–molecule van der Waals interaction. While for 1DP2+3, the octyloxy side chains can be clearly revealed on the surface, which are fully extended and interdigitated with that from the neighbouring 1DP2+3 chains (Fig. 1c). Therefore, the assembling structures of 1DP2+3 and 1DP2+4 can be easily distinguished on the surface. | ||
| Fig. 1 (a) Schematic diagram of the synthesis of 1D imine polymers at the liquid/solid interface at room temperature. (b and c) STM images of 1DP2+3. The lower panel of (c) shows the structural model of 1DP2+3. (d) Large-scale STM image of 1DP2+4; (e) and (f) are enlargements of the regions as marked in (d) showing the domain boundaries and defects of 1DP2+4 (marked by the green and red arrows); (g) tentative structural model of 1DP2+4. Imaging conditions: (b) Iset = 39 pA, Vbias = 0.52 V; (c) Iset = 21 pA, Vbias = 0.52 V; (d–f) Iset = 30 pA, Vbias = 0.90 V. | ||
Within the 1DP2+4 assembly, besides the typical domain boundaries between differently orientated domains, some other characteristic domain boundaries can also be observed, as shown in Fig. 1e, which are resulted from incomplete reaction due to space confinement and lattice mismatch. As marked by the green arrows at such boundaries, only one of the amino groups of 4 is reacted with aldehyde. Due to the mismatch of the position and the limited space, further reaction with aldehyde 2 is impossible. In addition, defects in the crystalline domain were also observed (see the red arrows in Fig. 1f), which are caused by the absence of aldehyde 2, or diamine 4, or a combination of both units.
Similarly, co-condensation of 1 with 3 or 4 at the octanoic acid/HOPG interface at room temperature was also investigated by STM, both results in formation of well-defined 2DPs by delicate control of monomer concentration and molar ratio.14 Fig. 2 shows the typical images of both 2DPs. The experimental lattice parameters of 2DP1+3 and 2DP1+4 are a = b = 3.1 ± 0.2 nm, α = 60 ± 2°, and a = b = 4.4 ± 0.2 nm, α = 60 ± 2°, respectively, which are in good agreement with the chemical structure models and confirm the covalent bond formation.14 In the 2D patterns, the six sides of the hexagonal ring are ascribed to the backbone of diamines. In the high resolution STM image (Fig. 2e) the anthracenyl groups on the sides of 2DP1+4 are clearly visible with higher contrast and larger width, while 2DP1+3 appears with more homogeneous contrast. Thus the difference in lattice parameters and contrast of the diamine backbone helps to distinguish both 2DPs when coexist on the surface.
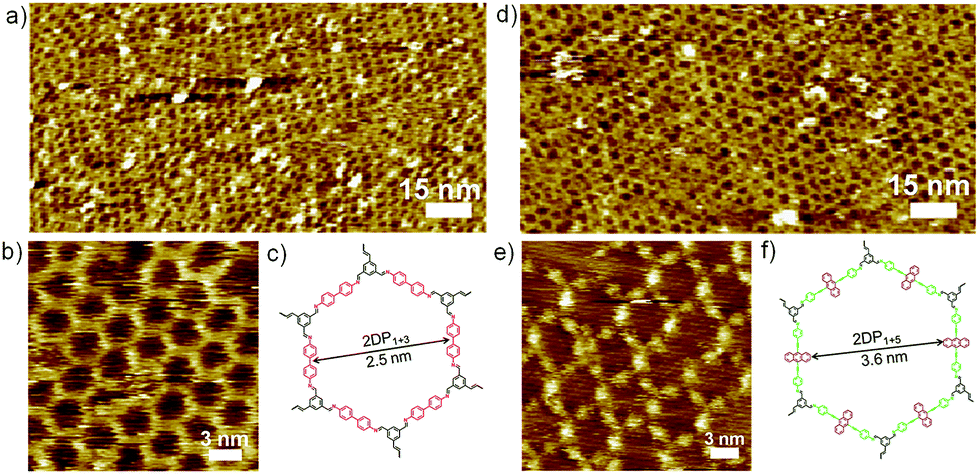 | ||
| Fig. 2 Large-scale and high resolution STM images of 2DP1+3 (a and b) and 2DP1+4 (d and e) formed at the octanoic acid/HOPG interface, concentration of amine: (a and b) 9.3 × 10−6 mol L−1, (d and e) 5.3 × 10−6 mol L−1. (c and f) Scheme illustrating the structure of 2DPs. Imaging conditions: (a) Iset = 67 pA, Vbias = 0.56 V, (b) Iset = 28 pA, Vbias = 0.86 V, (d) Iset = 33 pA, Vbias = 0.66 V, (e) Iset = 19 pA, Vbias = 0.68 V. | ||
Constitutional dynamic process of three-component libraries at the octanoic acid/HOPG interface
Before investigating the amine exchange process of the polymers at the liquid/solid interface, we first studied the adaptation of a three-component dynamic library constitutes one dialdehyde or trialdehyde and two diamines towards the HOPG surface. By applying a droplet of mixture of 2, 3 and 4 with the molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5:1.5 on HOPG surface, crystalline structure that mainly consists of 1DP2+4 architectures was observed, as shown in Fig. 3b and S3a.† Fig. 3c shows the high resolution image of crystalline domain of 1DP2+4. Seldomly, polymer chains consist of both 3 and 4, denoted as 1DP2+3+4 hereafter, can be observed with this molar ratio of monomers. The crystalline domains of 1DP2+4 cover a proportion of ca. 80.0% of the surface on average, and the other ca. 20.0% by defects and 1DP2+3+4 chains. This observation suggests that 1DP2+4 is the thermodynamically most stable product at the liquid/solid interface, thus confirming that the affinity of the building blocks to the surface plays an important role in the selective formation of polymers at the octanoic acid/HOPG interface. In contrast to the well-ordered structure at the octanoic acid/HOPG interface, 1D polymers consist randomly arranged 3 and 4 are expected in solution due to the possible diffusion of the precursors in all directions. However, at the liquid/solid interface the affinity of the precursors and the assembling properties of the 1DPs help the selection and self-sorting of 1DP2+4 as the primary product. When mixing 2, 3 and 4 in a 3:2:1 ratio, the surface coverage of 1DP2+4 crystalline domains was reduced to ca. 20%. About 80% of the surface was covered by 1DP2+3+4. Fig. 3d shows the representative STM image. This statistical analysis indicated that the substrate influences the distribution of reaction products, and the assembling on the surface induces self-sorting of 1DP2+4. The yellow arrows in Fig. 3d highlighted some 1DP2+3+4 chains, connecting two crystalline domains of 1DP2+4, and the interchain distance (1.8 ± 0.2 nm) is larger than the distance (1.1 nm) between 1DP2+4 chains in the crystalline domain, indicating that the octyloxy side chains in 1DP2+3+4 are adsorbed on the surface.
:1.5:1.5 on HOPG surface, crystalline structure that mainly consists of 1DP2+4 architectures was observed, as shown in Fig. 3b and S3a.† Fig. 3c shows the high resolution image of crystalline domain of 1DP2+4. Seldomly, polymer chains consist of both 3 and 4, denoted as 1DP2+3+4 hereafter, can be observed with this molar ratio of monomers. The crystalline domains of 1DP2+4 cover a proportion of ca. 80.0% of the surface on average, and the other ca. 20.0% by defects and 1DP2+3+4 chains. This observation suggests that 1DP2+4 is the thermodynamically most stable product at the liquid/solid interface, thus confirming that the affinity of the building blocks to the surface plays an important role in the selective formation of polymers at the octanoic acid/HOPG interface. In contrast to the well-ordered structure at the octanoic acid/HOPG interface, 1D polymers consist randomly arranged 3 and 4 are expected in solution due to the possible diffusion of the precursors in all directions. However, at the liquid/solid interface the affinity of the precursors and the assembling properties of the 1DPs help the selection and self-sorting of 1DP2+4 as the primary product. When mixing 2, 3 and 4 in a 3:2:1 ratio, the surface coverage of 1DP2+4 crystalline domains was reduced to ca. 20%. About 80% of the surface was covered by 1DP2+3+4. Fig. 3d shows the representative STM image. This statistical analysis indicated that the substrate influences the distribution of reaction products, and the assembling on the surface induces self-sorting of 1DP2+4. The yellow arrows in Fig. 3d highlighted some 1DP2+3+4 chains, connecting two crystalline domains of 1DP2+4, and the interchain distance (1.8 ± 0.2 nm) is larger than the distance (1.1 nm) between 1DP2+4 chains in the crystalline domain, indicating that the octyloxy side chains in 1DP2+3+4 are adsorbed on the surface.
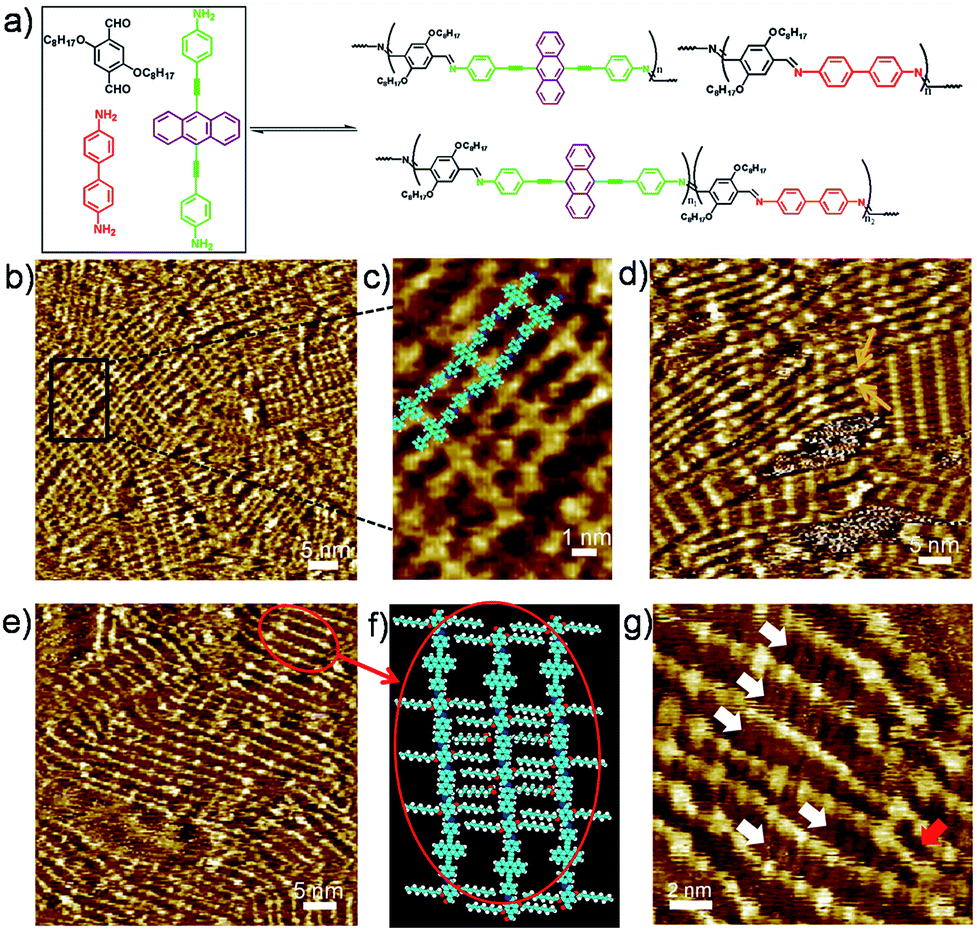 | ||
| Fig. 3 (a) Schematic illustration of the possible products from a three-component library at the liquid/solid interface. STM images of the monolayer formed by a mixture of 2, 3 and 4 in octanoic acid with molar ratio 3:1.5:1.5 (b and c), 3:2:1 (d), 3:2.5:0.5 (e and g). (f) shows a molecular packing model of 1DP2+3+4 corresponding to the area marked by the red box in (e). Imaging conditions: (b and c) Iset = 39 pA, Vbias = 0.60 V; (d) Iset = 24 pA, Vbias = 0.60 V; (e) Iset = 26 pA, Vbias = 0.52 V; (g) Iset = 24 pA, Vbias = 0.50 V. | ||
When the molar ratio of 2, 3 and 4 was adjusted to 3:2.5:0.5, the areas occupied by the crystalline domains of 1DP2+4 were further decreased to ca. 5%, indicating the increase of 1DP2+3+4 domain (Fig. 3e). In our previous work, we have demonstrated the important role of octyloxy side chains of 2 in defining the inter-chain distance and interaction of 1DP2+3.14 In the current case, the average inter-chain distance of 1DP2+3+4 (1.8 ± 0.2 nm) matches well with that of 1DP2+3 (1.7 ± 0.1 nm). It is worth noting that the interchain distance of 1DP2+3+4 is not uniform and the backbone of 1DP2+3+4 is not aligned as straight as 1DP2+3 or 1DP2+4 (Fig. 3e). Typical high-resolution STM image clearly shows that along the polymer backbone, in some domain as marked by the white arrows in Fig. 3g, the adsorbed octyloxy chains are visible, however, in other domains as marked by the red arrow in Fig. 3g, the alkyl chains are not visible, which indicates it is not adsorbed on the surface. We consider this as an indication of inter-chain interaction dominated by side chain interdigitation or van der Waals interaction between the backbones, which is determined by the relative abundance of diamine 3 or 4.
The above experiments have clearly demonstrated that the molar ratio and adsorption ability of building blocks plays a vital role in the selectivity of the final products at the liquid/solid interface. The surface coverage of 1DP2+4/1DP2+3+4 domain can be adjusted by tuning the molar ratio of 3 and 4 gradually. Typical high-resolution STM images (Fig. 3d and e) clearly revealed that the backbone of 3 and 4 distribute randomly along the backbone of 1DP2+3+4. To have more insight into the reaction mechanism and role of the surface we investigated the reactions in solution by 1H-NMR. To do so, the monomers were dissolved in CDCl3 and then mixing with 1:1 molar ratio (concentration of amine: c3 = 2.3 × 10−3 mol L−1, c4 = 1.9 × 10−3 mol L−1). Right after mixing, 1H-NMR indicates that the condensation between 2 and 4 is neglectable, while in case of 2 and 3, 1H-NMR indicates about ∼37% aldehyde groups have transformed into imine (Fig. S8 & S9†). After six days, the reaction yields increase to ∼13% and 60% for the case of 2 with 4 and 2 with 3, respectively. These results confirms that the Schiff base condensation between these monomers can happen under room temperature, even without the presence of acidic catalysis. If catalysis amount of trifluoroacetic acid (TFA) was added to mimic the acidity of octanoic acid, the solution immediately turns cloudy indicating precipitation of reaction products.
In analogy to the study of the DCC of 1DPs, we also investigated the selectivity of the surface toward a three-component library in the case of 2DPs. STM observation after deposition of a 2:3:3 or 4:3:3 mixture of 1, 3 and 4 in octanoic acid at room temperature reveals exclusive formation of 2DP1+4 at the interface, no sign of presence of 3 was revealed (Fig. S4a and b†). When molar ratio of 1, 3 and 4 was adjusted to 4:5:1, STM revealed the coexistence of both 2DP1+3 and 2DP1+4, and the 2DPs tends to phase separation rather than intermixing, again highlight the self-sorting of reaction products at the interface (Fig. S4c†). This observation further confirms the significance of the preferential adsorption of building blocks in the product selection from a DCC library in the presence of a surface.
Amine exchange at the octanoic acid/HOPG interface
To explore fully the influence of surface, and to gain more insight into the effect of preferential adsorption of building blocks and reaction products on HOPG surface, in situ diamine exchange process of 1DPs or 2DPs were performed. To this end, a drop of equimolar solution of 3 or 4 was applied on top of a pre-existing monolayer of 1DP2+4 or 1DP2+3. In case of exchange of 4 by 3, after 120 minutes, diamine exchange was not observed at the interface. Conversely, when an equimolar solution of 4 was deposited on top of the 1DP2+3 monolayers (Fig. 4b), the equilibrium of the diamine exchange process can be shifted towards the formation of 1DP2+4. After about 170 minutes, coexistence of 1DP2+3 and 1DP2+4 lamellae domains was observed, as indicated in Fig. 4c. In view of the sites occupied by 1DP2+4, we can conclude that the exchange reaction takes place more easily at the domain boundaries of 1DP2+3. However, once the exchange reaction occurs in the middle of a 1DP2+3 polymer chain, that is, one 3 unit is replaced by a 4 unit, due to the mismatch in backbone length, a structural defect in the 2D domain can be created, as indicated by the red arrow in Fig. 4c. Fig. 4d shows the tentative molecular model for the defect formation during exchange of 3 with 4 in the middle of a 1DP2+3 chain. Since the monomer 4 (1.92 nm) is larger than 3 (1.01 nm), removal of a trimer (2.62 nm) is necessary in order to insert a 4 in the middle of a 1DP2+3 chain, and at the same time break one 1DP2+3 chain into two disconnected chains. Apparently, on the surface due to the surface confinement, diamine exchange is more difficult to happen in the middle in comparison with that at the end of a polymer chain, since it needs to break two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds simultaneously. While this problem does not exist in the solution. Our STM observations consist well with this deduction. The difference in diamine exchange of 4 by 3 from 3 by 4 can be rationalized by the difference in their affinity to the surface. Such difference has already been demonstrated by the selection of substrate towards a three-component DCC library in the previous part.
N bonds simultaneously. While this problem does not exist in the solution. Our STM observations consist well with this deduction. The difference in diamine exchange of 4 by 3 from 3 by 4 can be rationalized by the difference in their affinity to the surface. Such difference has already been demonstrated by the selection of substrate towards a three-component DCC library in the previous part.
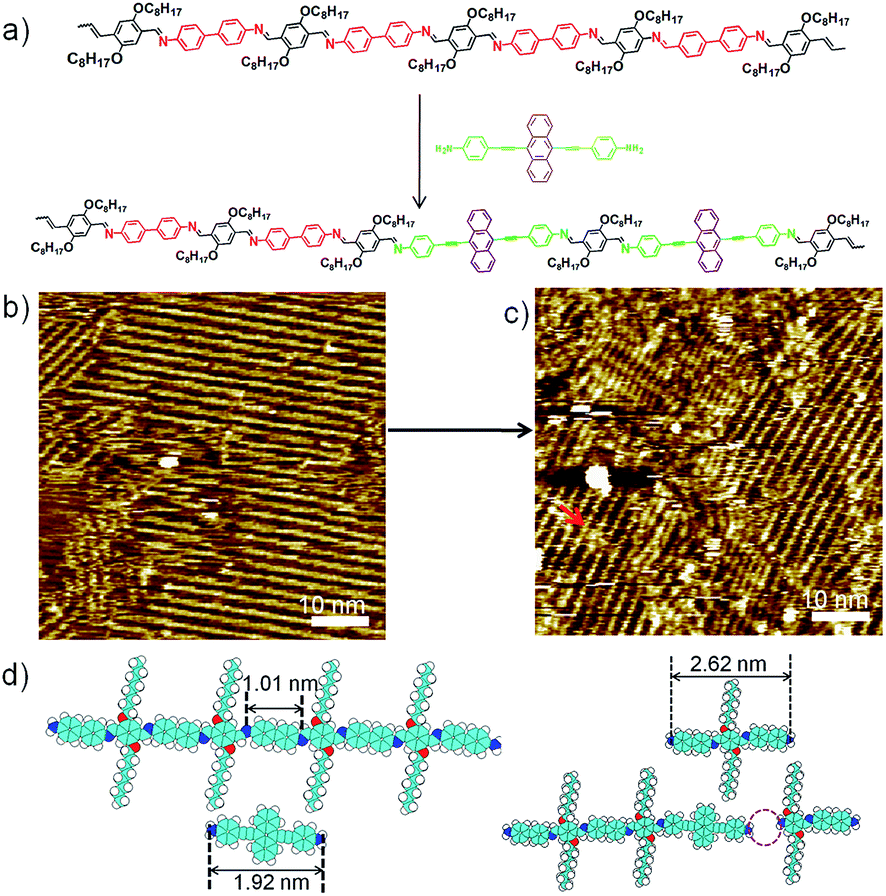 | ||
| Fig. 4 (a) Schematic illustration of the amine exchange reaction of 3 by 4 take place at the liquid/solid interface. Representative STM images show the coexistence of 1DP2+3 and 1DP2+4 (c) on the in situ addition of 4 on top of a pre-existing monolayer of 1DP2+3 (b). (d) A schematic illustration for the defect formation when 3 is replaced by 4. Imaging conditions: (b) Iset = 33 pA, Vbias = 0.52 V; (c) Iset = 18 pA, Vbias = 0.66 V. | ||
However, even in the case of exchange of 3 by 4, after about 780 minutes, the exchange reaction was not complete at room temperature (Fig. S5†). 1DP2+3 still occupies about 34% of the surface. Thus the final structure we got at room temperature from amine exchange is a surface covered by phase separated domains of 1DP2+3, 1DP2+4 and some disordered assembly of 1DP2+3+4. Only after annealing the sample at 100 °C for 30 minutes, the equilibrium of the diamine exchange process can be fully shifted toward the formation of 1DP2+4, 1DP2+3 domains completely disappeared as indicated in Fig. S6.† This means that although the formation of 1DP2+4 at the interface is energetically more favourable, the energy barrier is too high at room temperature, the exchange reaction can only be completed by increasing the temperature to accelerate the exchange process of diamines.
To gain more detailed insight into the DCC process of polymers, we then extended our study to the in situ diamine exchange of the 2DPs at the liquid/solid interface. For this goal, an equimolar solution of 3/4 was drop-casted on top of a pre-existing monolayer of 2DP1+4/2DP1+3. As in the case of 1DPs, the diamine exchange in 2DP1+4 does not happen even we wait for a long time. Instead a large portion of the cavities of the 2DP1+4 network appears filled after application of 3 (Fig. S7†). The bright features in the cavities are most probably imine oligomers formed by 1 and 3. Conversely, when a droplet of solution containing equimolar of 4 was applied on a pre-existing 2DP1+3, after 78 minutes, the surface was covered dominantly by 2DP1+4, indicating complete replacement of 3 by 4 (Fig. 5a). Noticeably, most of the cavities of the 2DP1+4 network formed in this case appear filled with some bright objects, possibly imine oligomers. The different behaviour toward amine exchange in 2DP1+3 and 2DP1+4 can be rationalized by the difference in adsorption energy of both diamines: monomer 4 has stronger molecule–substrate interaction than 3 due to the larger size and extended π-conjugation, thus show higher affinity to the surface.
 | ||
| Fig. 5 Representative STM images show the different results of amine exchange processes in the case of 2DPs. (a and b) STM and schematic illustration of amine exchange process of 3 by 4 from a pre-existing monolayer of 2DP1+3, respectively. 2DP1+4 was observed exclusively after 78 minutes. (c) On the in situ addition of 4 on top of a pre-existing monolayer of 2DP1+5, imine oligomers was incorporated into the cavities of 2DP1+5 network. (d) shows the schematic diagram of formation of host–guest structure. Imaging conditions: (a) Iset = 33 pA, Vbias = 0.60 V; Iset = 44 pA, Vbias = 0.60 V; (c) Iset = 46 pA, Vbias = 0.66 V; Iset = 39 pA, Vbias = 0.90 V. | ||
However, we discovered that the difference in adsorption energy of the diamines is not the only important factor affecting the amine exchange process. Though diamine 4 is expected to have higher affinity toward the surface than monomer 5, when a droplet of equimolar solution of 4 was drop-casted on top of a pre-existing monolayer of 2DP1+5, surprisingly, after 382 minutes, diamine exchange was not observed. Instead, molecule 4 was incorporated into the 2DP1+5 networks, as shown in Fig. 5c. This difference in behaviour is attributed to the difference in pore size of the 2DP1+5 (3.3 nm) from 2DP1+3 (2.5 nm). Since both pores are large enough to accommodate diamine 4 (1.92 nm), we suppose the feature trapped in the cavities are imine oligomers, most probably dimers containing one 4 and one 1, rather than intact monomer 4. The accommodation of these oligomers in the cavity prevents the amine exchange, and in turn helps to stabilize the 2DP1+5.
Finally, we designed a strategy that allows us to construct 2DP hetero-structures on the surface. Schematic illustration on the formation of 2D hetero-structures is outlined in Fig. 6a. In the experiment, first a submonolayer of 2DP1+3 was prepared by applying a 2.5 μL mixture solution of 1 with 3 (1 and 3 in the ratio 2:3, concentration of 3: c3 = 9.3 × 10−6 mol L−1) in octanoic acid on HOPG surface (Fig. 6b). STM revealed that 2DP1+3 covers about 68% of the surface at this concentration. Then a drop of same volume mixture solution of 1 with 4 (1 and 4 in the ratio 2:3, concentration of 4: c4 = 5.3 × 10−6 mol L−1) in octanoic acid was applied on top of this pre-existing submonolayer of 2DP1+3. 2DP1+4 forms at the uncovered area, leads to formation of a hetero two-dimensional polymer, as shown in Fig. 6c. This structure is distinctively different from those formed from simultaneous deposition of a 4:3:3 mixture of 1, 3, 4 in octanoic acid (concentration of 3 and 4: c3 = c4 = 3.4 × 10−6 mol L−1), which leads to a pure 2DP1+4 monolayer as shown in Fig. S6a† due to the higher affinity of 4 towards the surface. Therefore, the 2DP hetero-structures can only be constructed using this stepwise method at the liquid/solid interface. High resolution STM image (Fig. 6c, inset) indicates that the hetero-structure is covalently connected. The two-dimensional network exhibits enough flexibility to accommodate the lattice distortion at the domain edges. Interestingly, with this strategy the formation of 2DP1+4 occurred mainly at the uncovered areas and the orderliness of 2DP1+3 was not disturbed. The surface coverage of 2DP1+3 was 56% in the hetero structure, indicating the hetero structure is formed mainly through formation of 2DP1+4 in the uncovered area instead of replacement of 2DP1+3 by 2DP1+4. This result implies that the Schiff base coupling of 4 with 1 is favourable over amine exchange process at this condition.
 | ||
| Fig. 6 (a) A schematic diagram of the formation of 2DP hetero-structure on the surface. STM images of the submonolayer of 2DP1+3 (b) and in situ synthesized hetero-structure of 2DP1+3/2D1+4 (c) at the octanoic acid/HOPG interface. Imaging conditions: (b) Iset = 28 pA, Vbias = 0.68 V; (c) and inset Iset = 27 pA, Vbias = 0.52 V. | ||
Conclusions
In summary, we have systematically investigated the effect of substrate surface on the selectivity toward specific products from a multi-component dynamic covalent library, as well as the transimination in 1D and 2D imine polymers at the liquid/solid interface. Our STM results successfully achieved evidence for the effect of surface on the reversible dynamic covalent processes of 1D/2D polymers. The self-assembly of 1DPs and the competitive adsorption of monomers affect thermodynamic and kinetic features of covalent dynamic process at the liquid/solid interface and leads to constituent selection and formation of specific copolymers. Furthermore, a covalently connected hetero-structure of 2DPs has been successfully constructed using a stepwise deposition method at the liquid/solid interface.From a conceptual point of view, these results express a synergistic adaptive behaviour: the self-assembling properties of the products and the competitive adsorption of monomer and products on the HOPG surface drives a constitutional evolution of the dynamic mixture toward specific 1D and 2D imine polymers and leads to the yield of reaction products that is essentially different from that can be obtained in solution. This adaptability potentially offers the development of “smart” materials which can respond to substrate through the use of dynamic covalent chemistry.
Materials and methods
All of the chemicals, except 4, were purchased from J&K and used without further purification. The monomer 4 was synthesized with reported method.33To ensure solubilization of all compounds, precursors were first dissolved in dimethyl sulfoxide (1 mg g−1) and diluted with octanoic acid to obtain the desired concentration. For STM characterization, the monomers were mixed with desired molar ratios prior deposition onto the surface. A mixed solution (5 μL) was then directly applied to a freshly cleaved HOPG surface at room temperature. For annealing, the sample was positioned in a preheated oven at 100 °C for ∼30 min. The sample was taken out of the oven and cooled to room temperature, then characterized by scanning tunneling microscopy (STM).
STM measurements were performed by using an Agilent 5100 scanning probe microscope with mechanically formed Pt/Ir (80/20) tips under ambient conditions. All images were taken in the constant current mode. The calibration of STM images was carried out by using an atomic resolution HOPG lattice. The chemical structure models were built with HyperChem software.
Acknowledgements
This work was supported by the National Science Foundation of China (21572157, 21373070, 21302159).References
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- J. M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed.
- M. E. Belowicha and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- M. Ciaccia, R. Cacciapaglia, P. Mencarelli, L. Mandolini and S. D. Stefano, Chem. Sci., 2013, 4, 2253–2261 RSC.
- W. Dai, F. Shao, J. Szczerbiński, R. McCaffrey, R. Zenobi, Y. Jin, A. D. Schlüter and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 213–217 CrossRef CAS PubMed.
- A. Ciesielski, M. El Garah, S. Haar, P. Kovaříček, J. M. Lehn and P. Samorì, Nat. Chem., 2014, 6, 1017–1023 CrossRef CAS PubMed.
- J. Plas, D. Waghray, J. Adisoejoso, O. Ivasenko, W. Dehaen and S. De Feyter, Chem. Commun., 2015, 51, 16338–16341 RSC.
- N. Giuseppone and J. M. Lehn, J. Am. Chem. Soc., 2004, 126, 11448–11449 CrossRef CAS PubMed.
- P. Samori, N. Severin, K. Müllen and J. P. Rabe, Adv. Mater., 2000, 12, 579–582 CrossRef CAS.
- R. Tanoue, R. Higuchi, K. Ikebe, S. Uemura, N. Kimizuka, A. Z. Stieg, J. K. Gimzewski and M. Kunitake, Langmuir, 2012, 28, 13844–13851 CrossRef CAS PubMed.
- R. Tanoue, R. Higuchi, N. Enoki, Y. Miyasato, S. Uemura, N. Kimizuka, A. Z. Stieg, J. K. Gimzewski and M. Kunitake, ACS Nano, 2011, 5, 3923–3929 CrossRef CAS PubMed.
- Y. X. Yu, J. Sun and S. B. Lei, J. Phys. Chem. C, 2015, 119, 16777–16784 CAS.
- Y. X. Yu, J. B. Lin, Y. Wang, Q. D. Zeng and S. B. Lei, Chem. Commun., 2016, 52, 6609–6612 RSC.
- A. P. Cote, A. I Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- A. L. Korich and P. M. Iovine, Dalton Trans., 2010, 39, 1423–1431 RSC.
- X. H. Liu, C. Z. Guan, S. Y. Ding, W. Wang, H. J. Yan, D. Wang and L. J. Wan, J. Am. Chem. Soc., 2013, 135, 10470–10474 CrossRef CAS PubMed.
- L. R. Xu, X. Zhou, Y. Y. Yu, W. Q. Tian, J. Ma and S. B. Lei, ACS Nano, 2013, 7, 8066–8073 CrossRef CAS PubMed.
- X. H. Liu, Y. P. Mo, J. Y. Yue, Q. N. Zheng, H. J. Yan, D. Wang and L. J. Wan, Small, 2014, 10, 4934–4939 CrossRef CAS PubMed.
- N. A. A. Zwaneveld, R. Pawlak, M. Abel, D. Catalin, D. Gigmes, D. Bertin and L. Porte, J. Am. Chem. Soc., 2008, 130, 6678–6679 CrossRef CAS PubMed.
- C. Z. Guan, D. Wang and L. J. Wan, Chem. Commun., 2012, 48, 2943–2945 RSC.
- J. F. Dienstmaier, D. D. Medina, M. Dogru, P. Knochel, T. Bein, W. M. Heckl and M. Lackinger, ACS Nano, 2012, 6, 7234–7242 CrossRef CAS PubMed.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed.
- M. Bieri, M. T. Nguyen, O. Gröning, J. M. Cai, M. Treier, K. Aït-Mansour, P. Ruffieux, C. A. Pignedoli, D. Passerone, M. Kastler, K. Müllen and R. Fasel, J. Am. Chem. Soc., 2010, 132, 16669–16676 CrossRef CAS PubMed.
- Q. T. Fan, C. C. Wang, Y. Han, J. F. Zhu, J. Kuttner, G. Hilt and J. M. Gottfried, ACS Nano, 2014, 8, 709–718 CrossRef CAS PubMed.
- S. Weigelt, C. Busse, C. Bombis, M. M. Knudsen, K. V. Gothelf, E. Lægsgaard, F. Besenbacher and T. R. Linderoth, Angew. Chem., Int. Ed., 2008, 47, 4406–4410 CrossRef CAS PubMed.
- N. A. A. Zwaneveld, R. Pawlak, M. Abel, D. Catalin, D. Gigmes, D. Bertin and L. Porte, J. Am. Chem. Soc., 2008, 130, 6678–6679 CrossRef CAS PubMed.
- A. Wiengarten, K. Seufert, W. Auwärter, D. Ecija, K. Diller, F. Allegretti, F. Bischoff, S. Fischer, D. A. Duncan, A. C. Papageorgiou, F. Klappenberger, R. G. Acres, T. H. Ngo and J. V. Barth, J. Am. Chem. Soc., 2014, 136, 9346–9354 CrossRef CAS PubMed.
- B. Yang, J. Björk, H. P. Lin, X. Q. Zhang, H. M. Zhang, Y. Y. Li, J. Fan, Q. Li and L. F. Chi, J. Am. Chem. Soc., 2015, 137, 4904–4907 CrossRef CAS PubMed.
- P. Samorí, K. Müllen and J. P. Rabe, Adv. Mater., 2004, 16, 1761–1765 CrossRef.
- R. Higuchi, R. Tanoue, N. Enoki, Y. Miyasato, K. Sakaguchi, S. Uemura, N. Kimizuka and M. Kunitake, Chem. Commun., 2012, 48, 3103–3105 RSC.
- M. Imoto, M. Takeda, A. Tamaki, H. Taniguchi and K. Mizuno, Res. Chem. Intermed., 2009, 35, 957–964 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27395e |
| This journal is © The Royal Society of Chemistry 2017 |