
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect synthesis of phenol by novel [FeFe]-hydrogenase model complexes as catalysts of benzene hydroxylation with H2O2†
Xia Zhangabc,
Tianyong Zhang*abc,
Bin Li *abc,
Guanghui Zhanga,
Li Haia,
Xiaoyuan Maa and
Wubin Wua
*abc,
Guanghui Zhanga,
Li Haia,
Xiaoyuan Maa and
Wubin Wua
aTianjin Key Laboratory of Applied Catalysis Science and Technology, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300354, China. E-mail: tyzhang@tju.edu.cn; libin@tju.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering, Tianjin 300354, China
cTianjin Engineering Research Center of Functional Fine Chemicals, Tianjin 300354, China
First published on 13th January 2017
Abstract
Three new [FeFe]-hydrogenase model complexes, μ-(SCH(CH2CH3)CH2S)–Fe2(CO)6 (complex 1), μ-(SCH(CH2CH3)CH2S)–Fe2(CO)5PCy3 (complex 2) and μ-(SCH(CH2CH3)CH2S)–Fe2(CO)5PPh3 (complex 3) were prepared. The structures of complexes 1–3 were characterized by FT-IR, UV-vis, 1H, 13C, 31P NMR spectra and single-crystal analyses. The electron density of these model complexes was studied by IR spectra, UV spectra and electrochemical analysis and evaluated against their respective catalytic performances. The CV (cyclic voltammetry) study of complex 2 showed a less positive oxidation event at 0.6 V and a more negative reduction event at −1.94 V, which is in accordance with the enlargement of electron density at diiron centers when CO were substituted by better electron donor ligands. Of all these three complexes, complex 2 exhibited the best catalytic activity, with a yield of phenol of up to 24.6% and selectivity up to 92%, which is consistent with its higher electron density of the Fe–Fe bond. This study revealed the correlations between the electron density of the catalytic site of catalysts and their performance in catalytic hydroxylation of benzene. Based on these experimental results, a catalytic oxidation mechanism via an Fe2+–μ-O–Fe2+ intermediate as oxygen transfer reagent has been proposed.
Introduction
Being an important industrial feedstock used in production of fibres, resins, antioxidants, plastics, agrochemicals and medicines, phenol is in high demand.1 The current method for industrial scale production of phenol is mostly based on the cumene process, which produces a large amount of acetone as the by-product. Since the market demand for phenol is much higher than for acetone, a method of direct oxidation of benzene to phenol is expected to bring better economic benefits.2–4 The direct catalytic hydroxylation of benzene to phenol under mild conditions has attracted great interest and has been extensively investigated due to the reduced expense of the base materials. Some new catalysts have already been tried in this reaction, like iron complexes, TS-1 and Py4PMo11V.5–8 However, the yield of phenol by the direct hydroxylation of benzene has been found to be lower than produced through the traditional method. The hydroxylation process can not avoid the excessive oxidation and subsequent formation of by-products derived from phenol thereby leading to an overall decline in desirable yield.9 Therefore, the seeking for novel catalysts with high selectivity and efficiency for the direct catalytic hydroxylation of benzene to phenols under mild circumstances has been an ongoing process and a challenge.10,11Among diversified catalysts, iron-based catalysts attracted great research interest for many years due to the convenience, low cost, environmental amity and low toxicity of iron. As reported in the previous studies,12–14 iron-based catalysts usually have high selectivity and efficiency in a series of chemically challenging oxidative processes. Especially, iron-based biomimetic catalysts with low barriers to suitable redox states for selective oxidations, have been studied. These catalysts mimic the active site of natural oxygenase enzymes and serve as high-performance catalysts for selective hydrocarbon oxidation.11,15–17
[FeFe]-hydrogenases are also iron-based bio-catalysts which are much more efficient in the evolution and oxidation of H2 under mild conditions.17–24 However, oxygenation is found to occur at the dithiolato-sulfur or at the Fe–Fe bond site of the [FeFe]-hydrogenase active site model complexes.25,26 This oxygenation may lead to S-oxygenated products which having relatively stable O atom. The other route is the formation of Fe-based oxidative addition products of Fe2+–μ-O–Fe2+. Although this species are demonstrated thermodynamically favourable, they are not isolated to date. According to the reaction kinetics research on [FeFe]-hydrogenase active site model complexes, the diiron Fe+–Fe+ subsite is thermodynamically favored to be oxidized to the active Fe2+–μ-O–Fe2+ species.25,27 The iron-based highly active Fe2+–μ-O–Fe2+ species as the crucial intermediates during oxygenation could be used for transfer of oxygen in the biomimetic hydrocarbon hydroxylation.28–31 This is due to the fact that the [FeFe]-hydrogenases model complexes have an active site that remains unconstrained towards a suitable redox state. This also implies high activity towards the oxygenation reactions. Our group's previous research showed the Fe–Fe bond based oxygenated μ-O complex has the ability of transferring oxygen atom to the aromatic substrates. Supporting DFT calculations and the experimental catalytic activity towards hydroxylation tend to confirm the mechanism.32 Although the selectivity for phenol is high, the yield is not ideal yet. Chen's group showed the benzene conversions by the catalytic system can be improved by adding PPh3 analogues rather than using the catalysts alone.33 Thus, it is essential to prepare some new [FeFe]-hydrogenase model compound with a PPh3 analogue ligand aimed at enhancing the yield of catalytic hydroxylation of benzene.
Herein, a new dinuclear [2Fe2S] mimic complex μ-(SCH(CH2CH3)CH2S)–Fe2(CO)6 (1) was obtained by reaction of Fe(CO)5 with excess 1,2-butanedithiol. For studying the impact of the ligand at the active site of [FeFe]-hydrogenase model compound, the complexes namely μ-(SCH(CH2CH3)CH2S)–Fe2(CO)5L (2: L = PCy3, tricyclohexyl phosphine; 3: L = PPh3, triphenyl phosphine) have been prepared by controllable CO substitution of 1 with PCy3 and PPh3, as shown in Scheme 1. The structural elucidation of these model compounds featured the use of UV-vis, FT-IR, 1H, 13C, 31P NMR spectra and single-crystal analyses. For investigated the correlations between the catalytic reactivity and the electronic features of the catalysts, their electrochemical properties, FT-IR, UV-vis and their catalytic performance have been discussed. The results indicate that complex 2 has the greatest electron density and also the best catalytic reactivity. This finding suggests that the catalytic reactivity of the catalysts could be improved by enhancing the electron density of active sites in the catalysts.
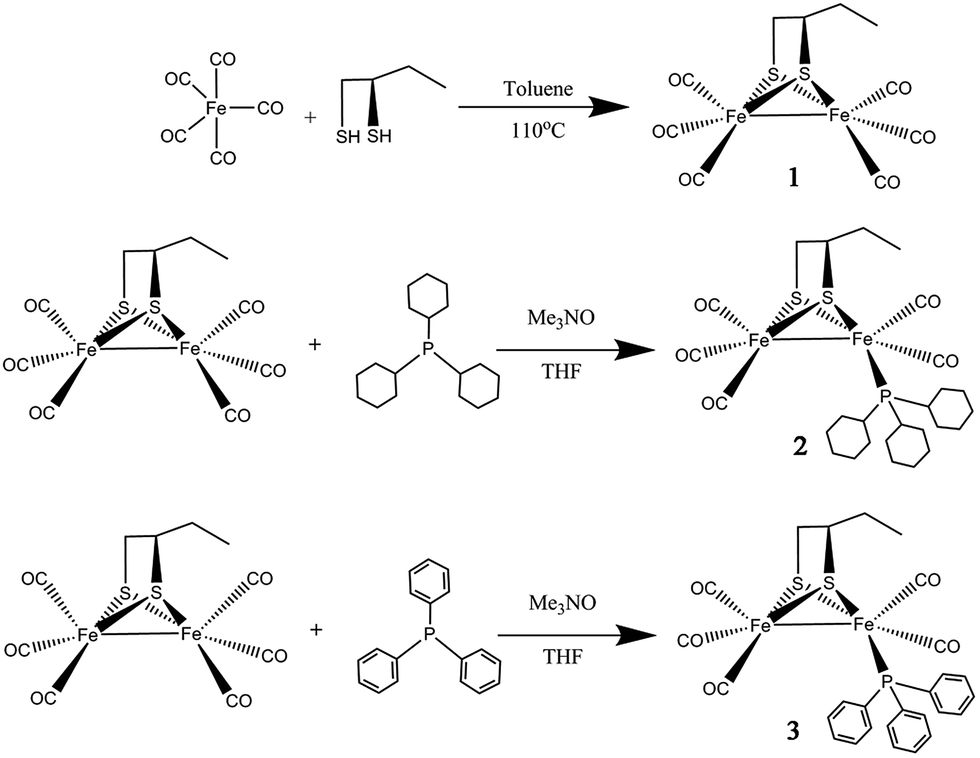 | ||
| Scheme 1 Synthetic route of complexes 1–3. | ||
Results and discussion
Synthesis and spectroscopic characterization of complexes 1–3
The reaction of Fe(CO)5 with 1,2-butanedithiol in refluxing toluene, yielded the anticipated μ-(SCH(CH2CH3)CH2S)–Fe2(CO)6 (1). Mono-substituted complexes μ-(SCH(CH2CH3)CH2S)–Fe2(CO)5L (2: L = PCy3, tricyclohexyl phosphine; 3: L = PPh3, triphenyl phosphine) were obtained by treating 1 with PCy3 and PPh3 respectively, along with Me3NO in THF at ambient temperature with good yields through sufficient stirring. The 1H NMR spectra of complexes 1–3 consist of the expected signals for the 1,2-butanedithiol bridges and the phosphine ligands. In the 13C NMR spectrum, complexes 1–3 show peaks at δ = 208.82, 210.86 and 210.82 ppm, respectively. These peaks can be safely assigned to the Fe–CO carbon atom. The 31P NMR spectrum of 2 and 3 was display a singlet at δ = 70.89 and 63.95 ppm, respectively. The observations from the elemental analysis in all products agree well with the assumed compositions.The IR spectra for the compounds 1–3 obtained in the hexane solution show distinct peaks between 2100 and 1900 cm−1, characteristic of stretching vibrational modes of terminal metal carbonyls.34 As a result of the electron-donating phosphine ligands,35 the IR spectra of complexes 2–3 are shifted by νCO = 30 cm−1 and νCO = 27 cm−1 towards lower frequency compared to the all-carbonyl compound 1 (Fig. 1). The application of ν(CO) bands is regarded to be a great tool for identifying the alteration of electron density on Fe atoms in the model compounds,36,37 the CO stretches of the monomeric complexes 2–3 are lower in wavenumber in comparison to the all-carbonyl compound 1 which is indicative of the strong donating character of PCy3 and PPh3 group.37 The red-shift value order for the ν(CO) bands of 2–3 is in accordance with electron-donation capabilities of diverse phosphine ligands, PCy3 ligand reveals its strong electron-donation character compared to the PPh3 ligand. These showing a distinct ranking of 2 > 3 > 1 in electron density on active sites.
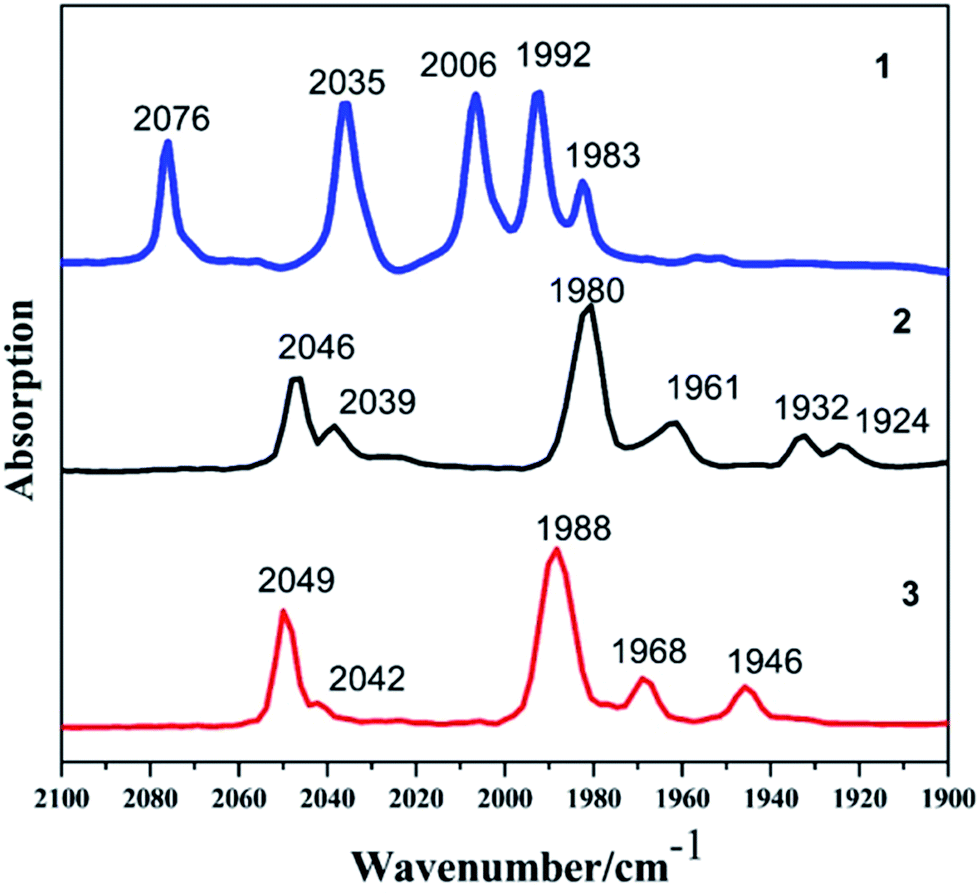 | ||
| Fig. 1 IR spectra of complexes 1–3 in hexane solution (CO region). | ||
To further clarify the electronic effect of the terminal ligands (L) on the activity of the complexes, UV-vis spectra of the compounds 1–3 were recorded in dichloromethane. As shown in the Fig. 2, prominent absorption peaks of complexes 1–3 are observed at 326 nm, 360 nm and 354 nm, respectively. These peaks in the visible region can be attributed to π–π* transition.38 The electronic transitions of compound 2 (360 nm) and 3 (354 nm) were found to be red-shifted compared to compound 1 (326 nm). These indicated more electron-donation ligand will improve the electron densities at the diiron core. The electronic transition signal compound 2 is about 6 nm red-shifted compared to compound 3, which indicated the PCy3 ligand has the strong electron-donation than the PPh3 ligand. These are in accordance with the result derived from the IR spectra analysis and showed a distinct ranking of 2 > 3 > 1.
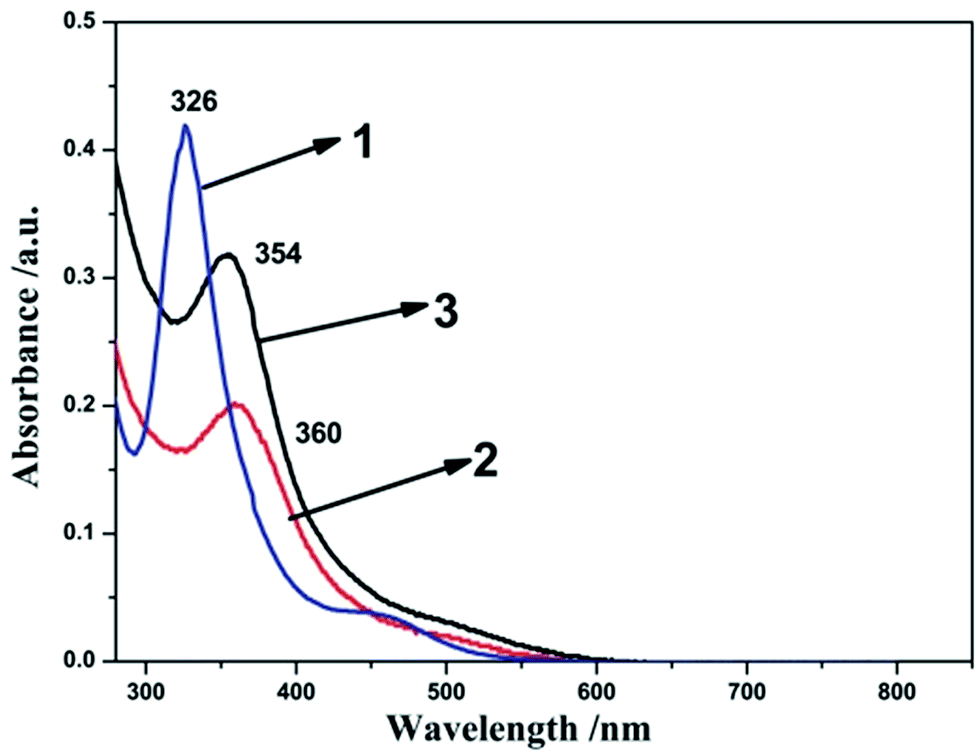 | ||
| Fig. 2 UV-visible spectra of 1 (black), 2 (red) and 3 (blue) recorded in CH2Cl2. | ||
Molecular structures of complexes 1–3
X-ray quality single crystals of complexes 1–3 were obtained by slow evaporation of the respective hexane–dichloromethane solutions at low temperature under normal atmospheric pressure. The structures of the compounds 1–3 are illustrated in Fig. 3. As the coordination locations for phosphine ligands on the diiron compounds are not identifiable only via the spectroscopic data, the X-ray diffraction study was conducted for 1–3. The [Fe2S2] skeleton of compounds 1–3 have the famous butterfly structure where every iron center takes on the twisted square-pyramidal coordination shape.39 Some of the important bond lengths and angles are displayed in Table S1,† while crystallographic data are shown in Table S2.† The Fe–Fe distances (2.5090 (4) Å in 2, and 2.5126(11) Å in 3) were found to be slightly longer compared to the Fe–Fe distances in complex 1 (2.5054 Å in 1). The PCy3 or PPh3 substituent group has a minor impact on the Fe–Fe distances. PCy3 or PPh3 ligand was located in the apical site of Fe atom and was placed trans to the Fe–Fe bond. The Fe–P bond distances of complex 2(2.2734(5) Å) are longer than that of complex 3 (2.2447(15) Å), showing the larger steric effects on the coordination geometry following the order PCy3 > PPh3. X-ray crystallographic analysis for complexes 2 and 3 implied that the replacement of one CO by tertiary phosphine ligands on the μ-(SCH(CH2CH3)CH2S)–Fe2(CO)6 offers solely an apical isomer as illustrated in Fig. 3. The displacement of CO by the tertiary phosphine ligands (PCy3 or PPh3), which are better electron-donating, increases the electron density on the iron centers of a diiron dithiolate model compound, thereby further strengthening the strong back-bonding from the Fe atoms to the CO ligands and simultaneous weakening of CO triple bonds.11,40,41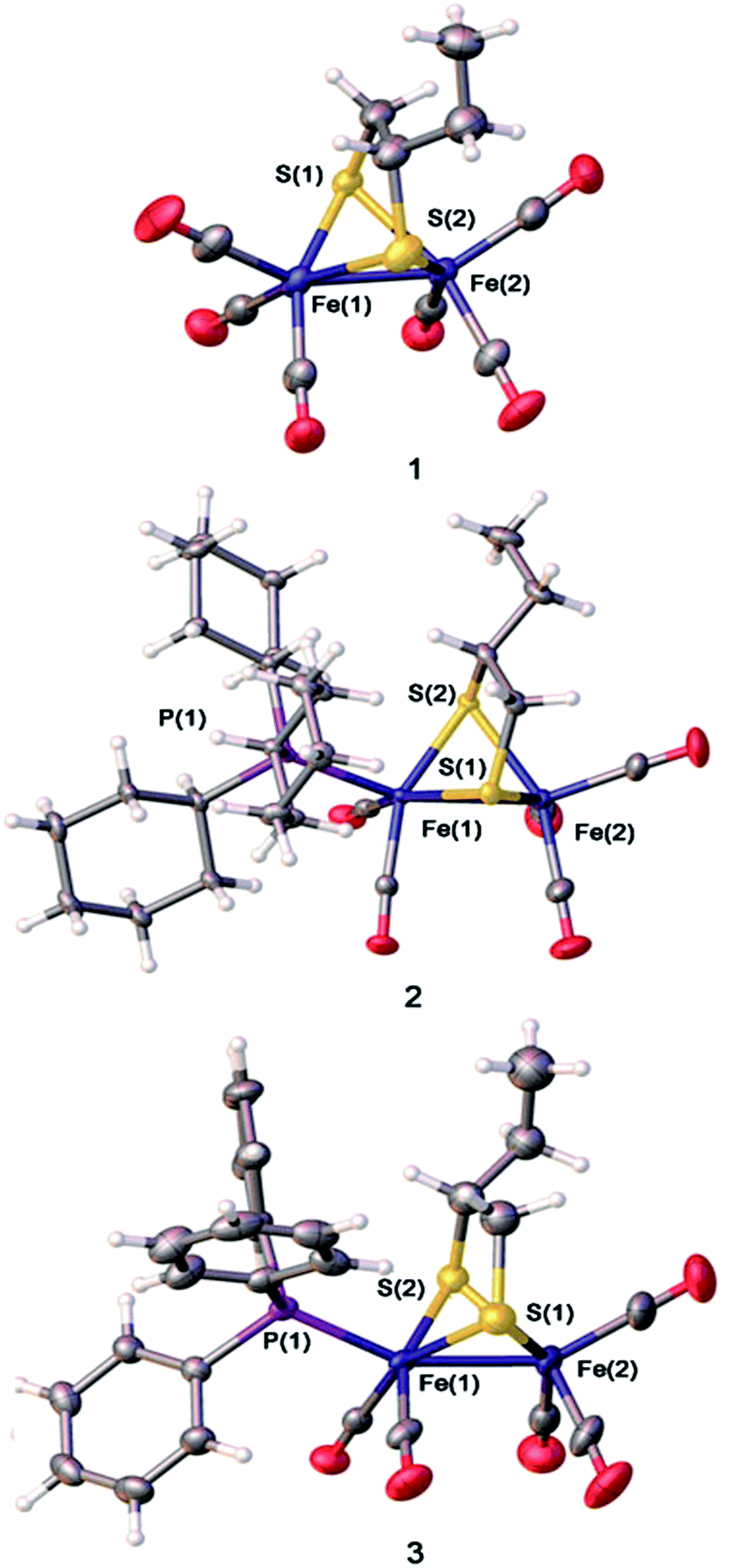 | ||
| Fig. 3 Crystal structures of the complexes (ellipsoids are drawn at the 50% probability level). | ||
Electrochemistry
The electrochemical properties of complexes 1–3 were investigated via cyclic voltammogram (Fig. 4). CV data are shown in Table 1. The cyclic voltammogram of complex 1 displays an irreversible oxidation process at Epa = 0.74 V and a quasi-reversible process at Epc = −1.78 V. The first oxidation process is ascribed to the process from FeIFeI to FeIIFeI, and the initial process is able to be allocated to the FeIFeI/FeIFe0 reduction.11 The CV of the PCy3-substituted complex 2 and PPh3-substituted complex 3 in CH3CN solution display one irreversible reduction process (FeIFeI/FeIFe0) at Epc = −1.94 V and −1.84 V, and a quasi-reversible oxidation process at Epa = 0.6 V and 0.75 V, respectively. Compared with the first reduction and oxidation event of complex 1, the PCy3-substituted complex 2 and PPh3-substituted complex 3, reveal cathodic shifts in the reduction potential, all in accordance with the enlargement of electron density in diiron centers when CO were substituted by better electron donating ligands.11,12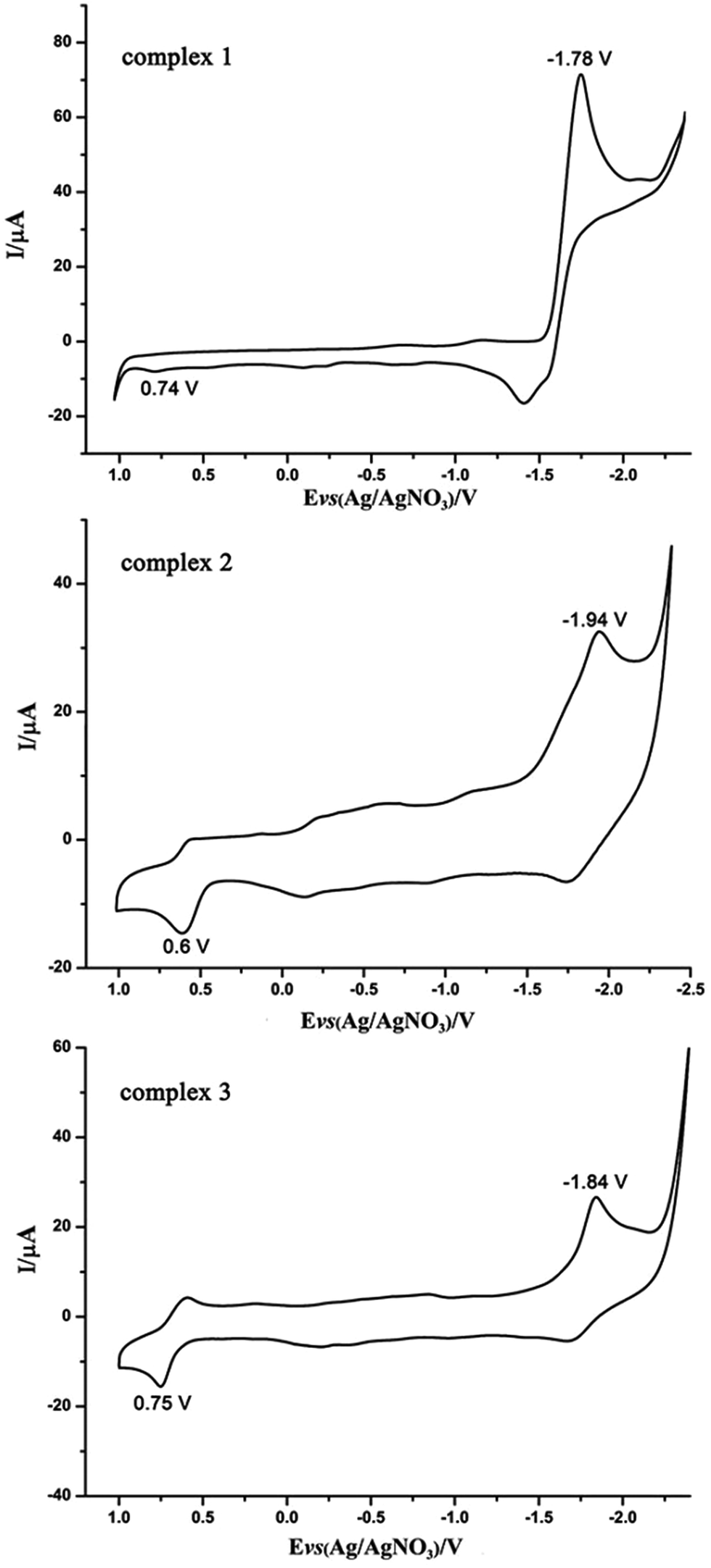 | ||
| Fig. 4 Cyclic voltammograms of the diiron complexes 1–3 (1.0 mM) in CH3CN solution (0.1 M n-Bu4NPF6; scan rate 200 mV s−1). | ||
| Complex | Epc (V) vs. Fc/Fc+ | Epa (V) vs. Fc/Fc+ |
|---|---|---|
| 1 | −1.78 | 0.74 |
| 2 | −1.94 | 0.60 |
| 3 | −1.84 | 0.75 |
Hydroxylation reactions of aromatic compounds
Although many iron-based catalysts have been studied, but the [FeFe]-hydrogenase model complexes have rarely been used for the direct production of phenol from benzene.11 In the present work, the [FeFe]-hydrogenase model compounds 1–3 were used as catalysts in the single-step hydroxylation from benzene into phenol with oxidant. We choose O2, H2O2, PhIO, m-CPBA as oxidants in the single-step hydroxylation. Among the oxidants, H2O2 has a higher phenol yield than m-CPBA and O2, PhIO were not have the ability to catalyze the single-step hydroxylation from benzene into phenol with these complexes under the given experimental conditions, as summarized in Table S3.† Thus, we used H2O2 as the oxidant in the single-step hydroxylation from benzene into phenol with [FeFe]-hydrogenase model compounds. The catalytic performance of complex 1 was assessed by following the procedures well established in literature42–45 in acetonitrile using H2O2 as oxidant. Some parameters which can potentially impact the catalytic properties, like the amount of H2O2, amount of catalyst, the reaction time, as well as the temperature, were also examined.The phenol yield and selectivity is significantly affected by the amount of H2O2 (Fig. 5). The yield increased with increase in amount of H2O2 from 0–5.0 mmol. The maximum yield of phenol was 10.5% when the amount of H2O2 was 5.0 mmol. But with the further raise in the amount of H2O2, the yield and selectivity of phenol has an obvious decreased, because too much H2O2 may be resulted in excessive oxidation of phenol. Therefore, the best amount of H2O2 applicable for this reaction is 5.0 mmol.
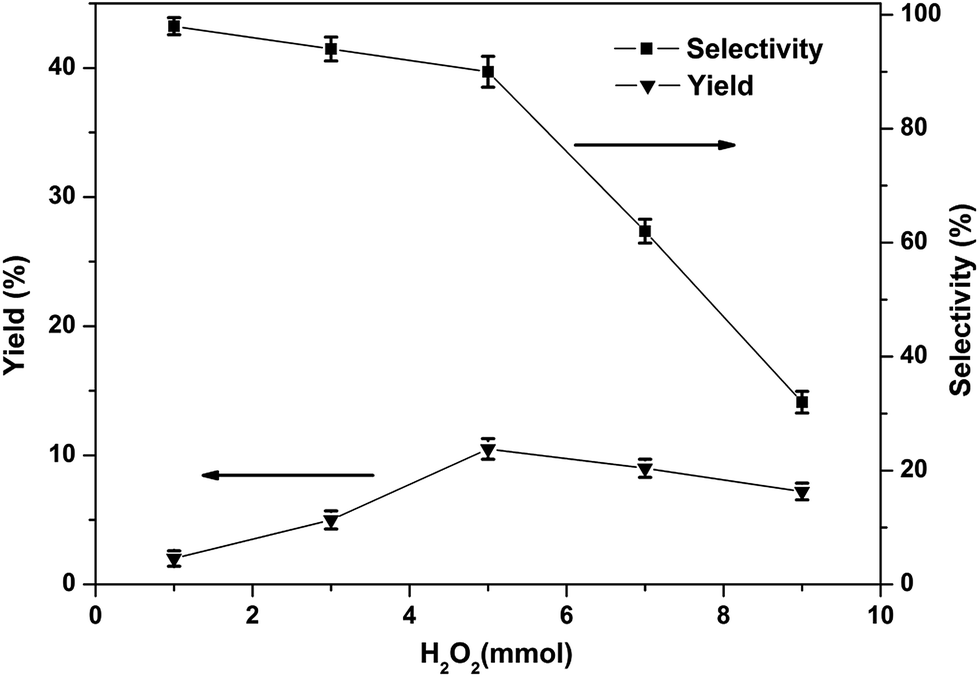 | ||
| Fig. 5 Effects of the amount of H2O2 on the phenol yield in the hydroxylation of benzene (1, 30 μmol; benzene, 1.12 mmol; CH3CN, 2.0 mL; 60 °C; reaction time, 1 h). All the experimental data are the average value of three sets of duplicate experiments. | ||
The phenol yield as well as selectivity is displayed with the variation temperature in Fig. 6. The result showed none of phenol is detected below 40 °C, since induction for a longer period of time at a low-temperature blocks the creation of phenol. The yield of phenol increased with temperatures growing from 40 to 60 °C. But with further rise in temperature, a clear decrease in yield and selectivity of phenol could be noticed, the probable reason being the excessive oxidation of phenol high temperatures, leading to by-products. Summing up from the tests the best appropriate reaction temperature was 60 °C.
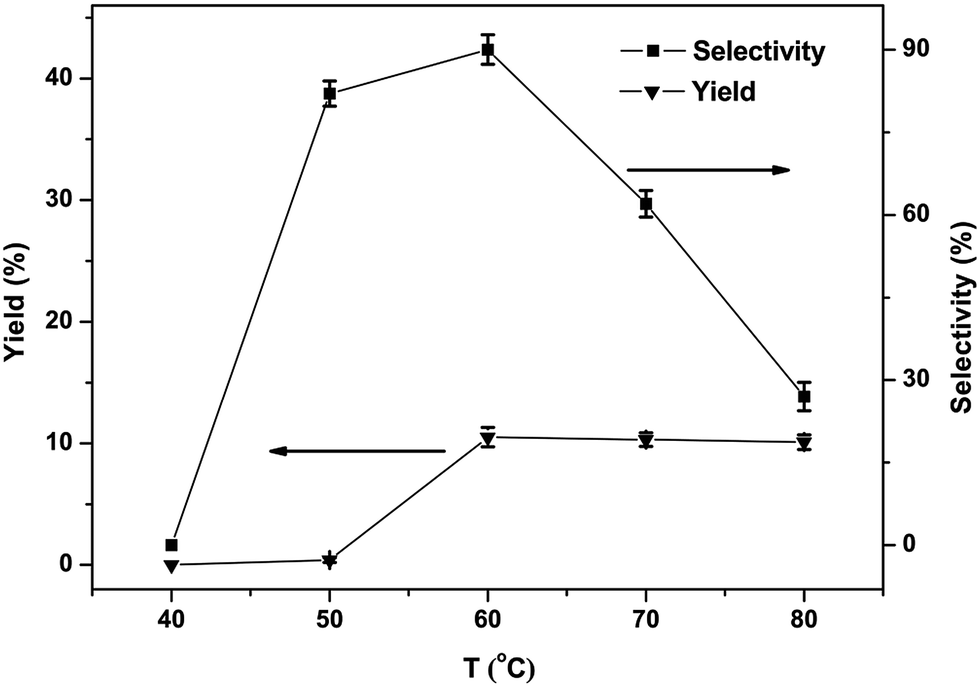 | ||
| Fig. 6 Effects of the reaction temperature on the phenol yield in the hydroxylation of benzene (1, 30 μmol; benzene, 1.12 mmol; CH3CN, 2.0 mL; H2O2, 5.0 mmol; reaction time, 1 h). All the experimental data are the average value of three sets of duplicate experiments. | ||
The effect of amount of catalyst in the one-step hydroxylation of benzene into phenol was tested and the results shown in the Fig. 7. The phenol yield increased from 7.2% to 10.5% when the catalyst amount was changed from 10 μmol to 30 μmol. And the selectivity of phenol was no significant change when the catalyst amount was changed from 10 μmol to 30 μmol. Moreover, with the increased of catalyst amount to 40 μmol caused an obvious decrease in yield and selectivity of phenol which may be attributed to the oxidation on all possible sites of benzene or excessive oxidation of phenol caused by the excessive catalyst. So, the best amount of catalyst is 30 μmol.
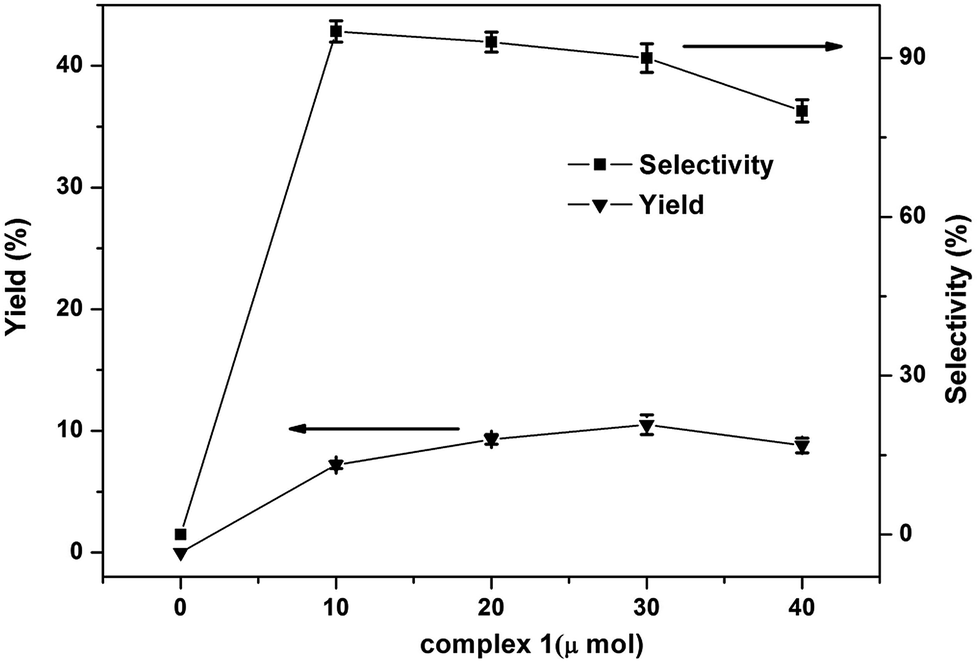 | ||
| Fig. 7 Effects of the amount of complex 1 on the phenol yield in the hydroxylation of benzene (H2O2, 5.0 mmol; benzene, 1.12 mmol; CH3CN, 2.0 mL; 60 °C; reaction time, 1 h). All the experimental data are the average value of three sets of duplicate experiments. | ||
The effect of reaction time on the yield and selectivity of phenol was also studied and the findings are shown in Fig. 8. The yield went to a maximum (12.6%) after 3 h. Declining selectivity due to phenol overoxidation was also discovered with extension in the reaction time.31 Under the optimized circumstances (1, 30 μmol; 60 °C; benzene, 1.12 mmol; CH3CN, 2.0 mL; H2O2, 5.0 mmol; reaction time, 3 h), the selectivity and yield of one-step conversion of benzene to phenol with catalyst complex 1 is 12.6% and 85%, respectively.
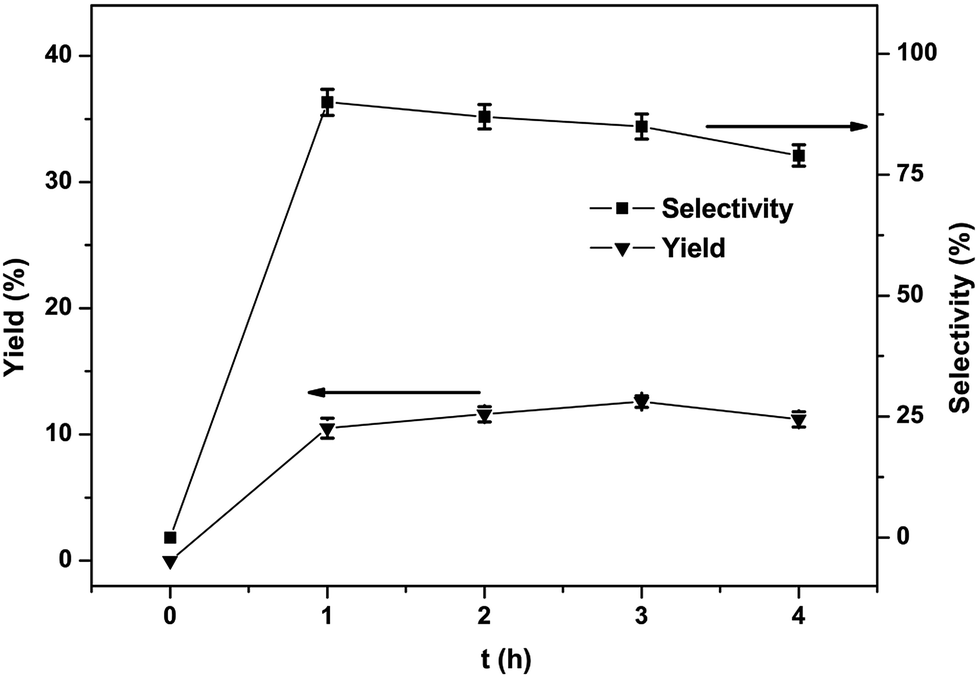 | ||
| Fig. 8 Effects of the reaction time of complex 1 on the phenol yield in the hydroxylation of benzene (H2O2, 5.0 mmol; benzene, 1.12 mmol; CH3CN, 2.0 mL; 60 °C; 1, 30 μmol). All the experimental data are the average value of three sets of duplicate experiments. | ||
According to previous results,12 the catalytic hydroxylation activity of such diiron complexes is based on their special structural and electronic properties, which was noticed firstly in some precious fundamental studies of the oxygen sensitivity of the [FeFe]-hydrogenase active site models, which was carried out by Darensbourg' group.25 They found the iron based oxidized species was thermodynamically favored in the diiron model complexes, which was further proven by the experimental and theoretical studies. Just based on this idea, we designed the catalytic system of these diiron complexes as an oxygen transfer species. Actually, the Fe–(μ-O)–Fe species as the iron–oxygen reactive species in the catalytic oxidation was also studied by Can Li' group.46 They identified the Fe–(μ-O)–Fe sites in Fe/ZSM-35 by in situ resonance Raman spectroscopy. Fe–(μ-O)–Fe species worked as an oxygen transfer species.
Under the optimized reaction conditions, performances of complexes 1–3 as catalysts were investigated and the results are summarized in Table 2, their active properties decline in the ranking of 2 > 3 > 1 under identical conditions. The complexes 1–3 showed better phenol yield and comparable selectivity to that we reported recently,11 namely [(μ-dmedt)-{Fe(CO)3}2] [dmedt = SCH(CH3)CH(CH3)S] (yield 7.5%; selectivity 92.5%), [(μ-dmest)-{Fe(CO)3}2] [dmest = SCH(CH3)CH(CH3)S(O)] (yield 3.7%; selectivity 95.6%). These observations can probably be attributed to the different activities of 1, 2 and 3 correlations with the diverse Fe–Fe electron densities. The Fe–Fe bond of the complex 2 or complex 3 with the electron-donating PCy3 ligand or PPh3 ligand could be oxidized more easily by H2O2 to form the iron–oxygen reactive species, whereas the less electron-donating complexes were found to be less reactive with this oxidant. These proved the electron density of the Fe–Fe bond of the complexes would affect the catalytic performances.43,45
According to the inference of the experimental results and based on previously published theoretical results,25,27,46 a probable mechanism has been proposed for hydroxylation of benzene to phenol. The [FeFe]-hydrogenase model complex has the ability to catalyze the hydroxylation of different substrates to phenol, which revealed that hydroxylation reaction involves an electrophilic addition process and the proposed reaction mechanism for the hydroxylation of benzene to phenol is shown in Fig. 9. In the process of hydroxylation, the nucleophilic diiron center of [Fe+Fe+] complex tended to react with the oxidant of H2O2 due to the high electron density of the diiron center.
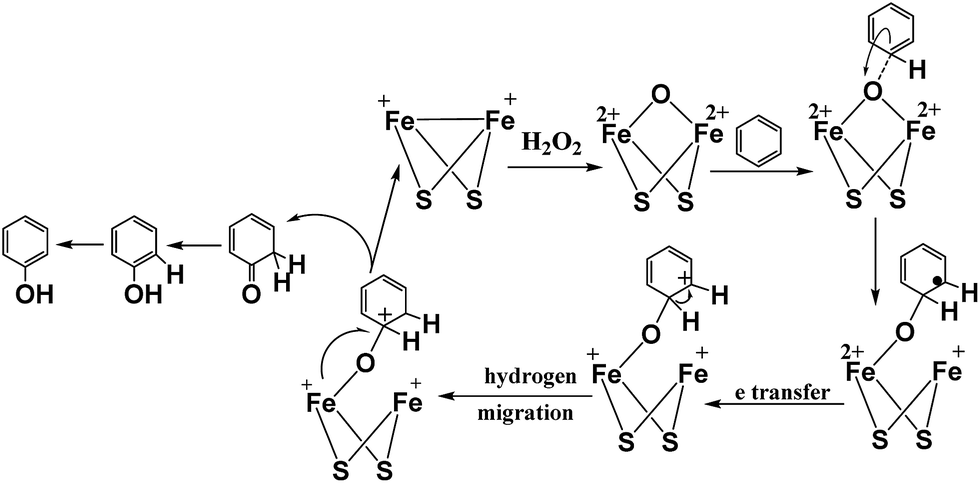 | ||
| Fig. 9 Proposed reaction mechanism for the hydroxylation of benzene to phenol. | ||
In order to investigate the catalysts stability of complexes 1–3, further investigations were carried out through solution IR spectrum before and after the hydroxylation reaction in CH3CN. As shown in Fig. 10, the CO-stretching band positions of complexes 1–3 didn't change, only the intensities of IR spectrum of complexes 1–3 has slightly changes before and after the hydroxylation reaction in CH3CN. This indicates the preservation of molecular structure of complexes 1–3 in the hydroxylation reaction in CH3CN. Further research on the catalysts stability of complexes 1–3 is concluded by the TG analysis.
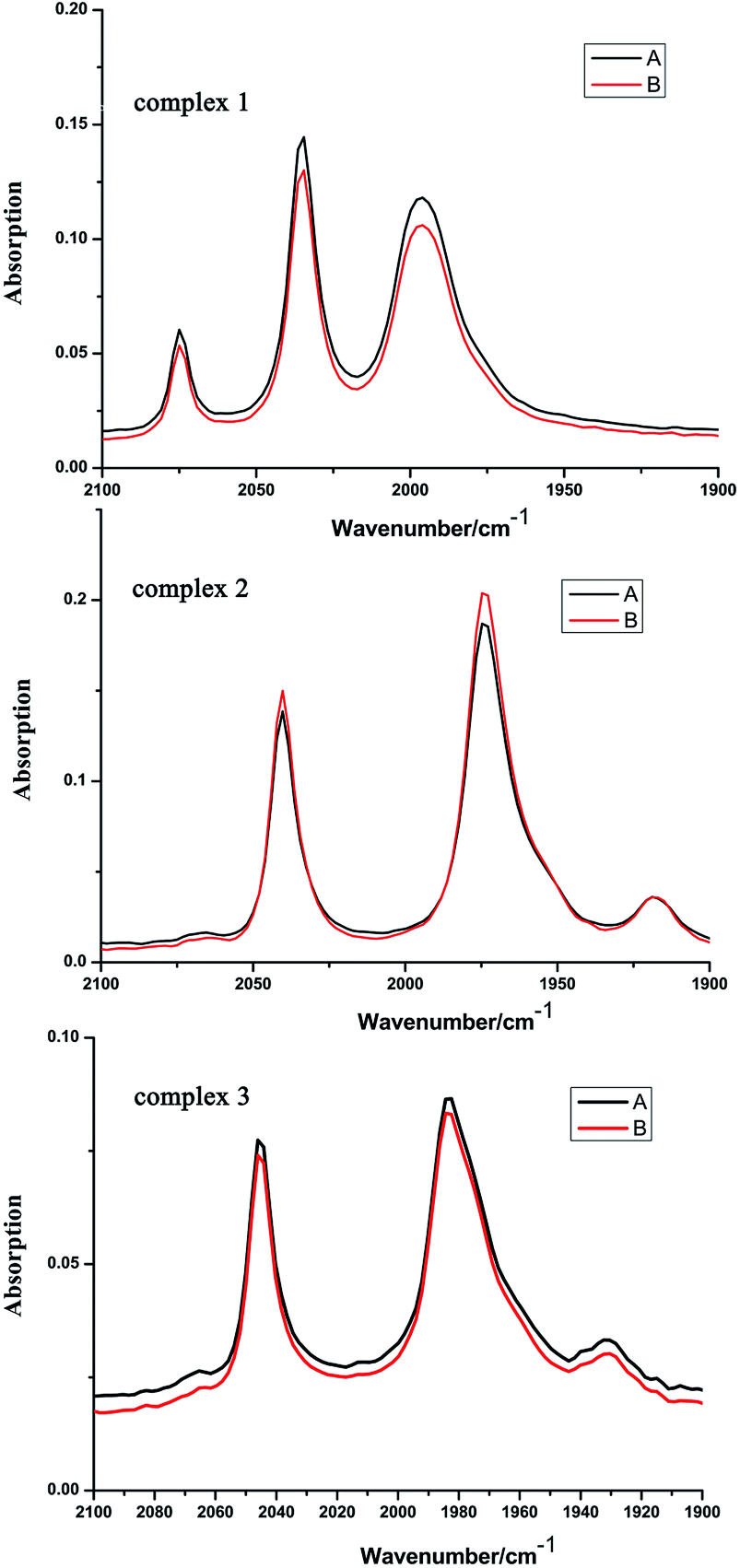 | ||
| Fig. 10 The IR spectra of complexes 1–3 before benzene hydroxylation (A) and after benzene hydroxylation (B) (recorded in CH3CN solution). | ||
The TG analysis patterns of complexes 1–3 are shown in Fig. 11, these showed great heat resistance of complexes 1–3 below 100 °C. Among them complexes 2 and 3 have better heat resistance than complex 1. A rapid weight loss for complexes 2 and 3 originated from the decomposition ranging from 200 to 300 °C. From the results it can be concluded that the structure of the catalyst in hydroxylation reaction has not changed.
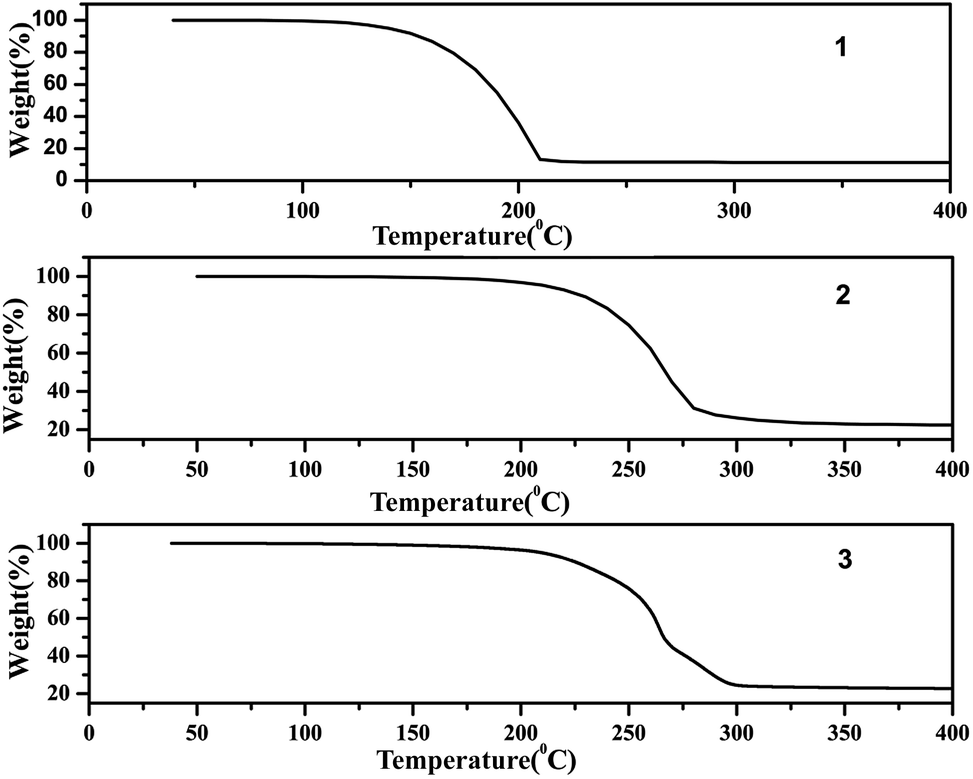 | ||
| Fig. 11 The TG analysis of complexes 1–3. | ||
Conclusions
In this work, we successfully prepared three dinuclear [2Fe–2S] bio-mimetic complexes μ-(SCH(CH2CH3)CH2S)–Fe2(CO)6 (1), μ-(SCH(CH2CH3)CH2S)–Fe2(CO)5PCy3 (2) and μ-(SCH(CH2CH3)CH2S)–Fe2(CO)5PPh3 (3). The molecular structures of 1, 2 and 3 were well characterized by IR, 1H, 13C, 31P NMR, UV-vis spectra and single crystal X-ray analysis, respectively. These complexes were tested as catalysts in the study of the one-step oxidation of benzene by H2O2 to phenol. The study firstly revealed the correlations between the electron densities of the catalytically active sites of the complexes and the catalytic performance. On comparing the catalytic performance of complexes, the complex 2 was found to be leading to highest phenol yield (24.6%) and phenol selectivity (92%), which has the highest electron densities of the Fe–Fe bond. This study demonstrates the catalytic abilities of [FeFe]-hydrogenase model complexes. This is a new research direction for the design and development of new [FeFe]-hydrogenase model complexes applied on the one-step oxidation of benzene by H2O2 to phenol which has the excellent phenol selectivity.Experimental section
Materials
Reactions and measurements related to [FeFe]-hydrogenase model compounds were performed in an inert (N2) atmosphere with Schlenk line technology. THF, hexane, CH2Cl2, toluene, and acetonitrile were purchased from Jiangtian Chemical and dried and distilled by standard methods. THF, hexane, CH2Cl2, toluene, and acetonitrile were deoxygenated before every use. The Fe(CO)5 was acquired from Jiangsu Tianyi Ultra-fine Metal Powder Co., Ltd. H2O2 (30%) and Me3NO (trimethylamine oxide) were directly applied after purchasing from Guangfu Chemical. PPh3, PCy3 CDCl3, CD2Cl2, benzene, and AgNO3, 1,2-butanedithiol, and n-Bu4NPF6 were bought from Sigma-Aldrich.General methods
The 1H NMR, 13C NMR and 31P NMR spectrum are acquired with Bruker AVANCE III 400 MHz NMR spectrometer. Solution IR spectrum was recorded by a Shimadzu FTIR-8400 spectrometer with 0.1 mm KBr sealed battery. Element analysis was conducted in a Heraeus CHN-Rapid, a fully automatic factor analyzer along with TCD inspection, type: TMT CHN, BESTELL-NR 2215001. By using a Rigaku MM-007 diffractometer along with a Saturn 724CCD, the single-crystal X-ray diffraction data were gathered at 293 K or 113 K with a confocal monochromator in Mo-Kα radiation (λ = 0.71073 Å). Data gathering, decrease as well as absorption rectification was conducted in the CRYSTALCLEAR program.47 Structural issues were addressed by direct approaches in SHELXL-97 program47 and also refined by SHELXL-97 on F2. The UV/visible spectra of the compounds determined using the UV-vis diffuse reflectance spectra (UV-3600) at room temperature. TG was performed using a TA SDT-Q600 apparatus and conducted in dynamic temperature at 10 °C min−1 from room temperature to a temperature of 400 °C in nitrogen circumstance. The conductivities of the complexes 1–3 were on a Delsa™ Nano C Particle Analyzer (Beckman Coulter Commercial Enterprise Co., Ltd, USA).Electrochemical examinations were performed with a CHI760B electrochemical workstation. The working electrode (diameter 3 mm) was polished in an α-alumina suspension, washed with CH3CN, and then sonicated with ion-free water for 20 min before usage. The counter electrode referred to a Pt wire. And the reference electrode referred to a non-aqueous Ag/Ag+ (0.01 M AgNO3 and 0.1 M n-Bu4NPF6 in CH3CN) reference electrode. A liquid of 0.1 M n-Bu4NPF6 with CH3CN was applied as auxiliary electrolyte that was degassed with dry N2 for 10 min prior to examination. Ferrocene was applied as an internal criterion in the identical examining situation and each potential was referred to the Cp2Fe+/0 couple at 0 V.
Hydroxylation of benzene with H2O2 was conducted in a 25 mL flask that is equipped by a condenser as well as a magnetic agitator. In a characteristic reaction, the composite 1 (4 mg, 0.01 mmol) was mixed in CH3CN (2.0 mL). After heating to the required temperature, benzene (0.1 mL, 1.12 mmol) was added into the blend. A given mass of H2O2 was added at last for beginning the reaction, and in the meanwhile the blend was agitated for a couple of hours. The research was conducted in atmospheric pressure. Examinations of GC (9890B) equipped by a FID sensor as well as a capillary pillar (OV-1701; 30 m × 0.25 mm × 0.25 μm) were conducted to investigate the product composition. TPD analysis was performed, from 70 °C to 180 °C by a variation of 10 °C, and then from 180 °C to 280 °C by a variation of 30 °C.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (21276187), Tianjin Science and Technology Innovation Platform Program (14TXGCCX00017), and the Tianjin Municipal Natural Science Foundation (16JCYBJC20800) for financial support of this work.Notes and references
- L. Chen, Y. Xiang and T. Feng, Appl. Organomet. Chem., 2012, 26, 108–113 CrossRef.
- N. I. Kuznetsova, L. I. Kuznetsova, V. A. Likholobov and G. P. Pez, Catal. Today, 2005, 99, 193–198 CrossRef.
- Y. K. Zhong, G. Y. Li, L. F. Zhu, Y. Yan, G. Wu and C. W. Hu, J. Mol. Catal. A: Chem., 2007, 272, 169–173 CrossRef.
- J. K. Joseph, S. Singhal, S. L. Jain, R. Sivakumaran, B. Kumar and B. Sain, Catal. Today, 2009, 141, 211–214 CrossRef.
- L. Balducci, D. Bianchi, R. Bortolo, R. DAloisio, M. Ricci, R. Tassinari and R. Ungarelli, Angew. Chem., Int. Ed., 2003, 42, 4937–4940 CrossRef PubMed.
- T. Kusakari, T. Sasaki and Y. Iwasawa, Chem. Commun., 2004, 8, 992–993 RSC.
- J. Yang, G. Sun, Y. Gao, H. Zhao, P. Tang, J. Tan, A. H. Lu and D. Ma, Energy Environ. Sci., 2013, 6, 793–798 Search PubMed.
- H. Xin, A. Koekkoek, Q. Yang, R. van Santen, C. Li and E. J. M. Hensen, Chem. Commun., 2009, 7590–7592 RSC.
- B. Xu, W. Zhong, Z. Wei, H. Wang, J. Liu, L. Wu, Y. Feng and X. Liu, Dalton Trans., 2014, 43, 381–393 Search PubMed.
- D. Bianchi, L. Balducci, R. Bortolo, R. D'Aloisio, M. Ricci, G. Span, R. Tassinari, C. Tonini and R. Ungarellia, Adv. Synth. Catal., 2007, 349, 979–986 CrossRef.
- X. Wang, T. Y. Zhang, Q. S. Yang, S. Jiang and B. Li, Appl. Organomet. Chem., 2014, 28, 666–672 CrossRef.
- Y. H. Wang, T. Y. Zhang, B. Li, S. Jiang and L. Sheng, RSC Adv., 2015, 5, 29022–29031 RSC.
- L. Wu, W. Zhong, B. Xu, Z. Wei and X. Liu, Dalton Trans., 2015, 44, 8013–8020 RSC.
- W. Na, F. Xu, Y. Feng and S. Du, J. Inorg. Biochem., 2011, 105, 1123–1130 CrossRef PubMed.
- C. M. Krest, E. L. Onderko, T. H. Yosca, J. C. Calixto, R. F. Karp, J. Livada, J. Rittle and M. T. Green, J. Biol. Chem., 2013, 288, 17074–17081 CrossRef PubMed.
- S. Friedle, E. Reisner and S. J. Lippard, Chem. Soc. Rev., 2010, 39, 2768–2779 RSC.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr, Chem. Rev., 2004, 104, 939–986 CrossRef PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef PubMed.
- M. E. Carroll, B. E. Barton, T. B. Rauchfuss and P. J. Carroll, J. Am. Chem. Soc., 2012, 134, 18843–18852 CrossRef PubMed.
- R. D. Bethel, M. L. Singleton and M. Y. Darensbourg, Angew. Chem., Int. Ed., 2010, 49, 8567–8569 CrossRef PubMed.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245–2274 CrossRef PubMed.
- J. M. Camara and T. B. Rauchfuss, J. Am. Chem. Soc., 2011, 133, 8098–8101 CrossRef PubMed.
- H. Cui, M. Wang, W. Dong, L. Duan, P. Li and L. Sun, Polyhedron, 2007, 26, 904–910 CrossRef.
- Y. Dong, X. Niu, W. Song, D. Wang, L. Chen, F. Yuan and Y. Zhu, Catalysts, 2016, 6, 74, DOI:10.3390/catal6050074.
- T. Liu, B. Li, M. L. Singleton, M. B. Hall and M. Y. Darensbourg, J. Am. Chem. Soc., 2009, 131, 8296–8307 CrossRef PubMed.
- J. Messelhäuser, K. U. Gutensohn, I.-P. Lorenz and W. Hiller, J. Organomet. Chem., 1987, 321, 377–388 CrossRef.
- B. Li, T. Liu, M. L. Singleton and M. Y. Darensbourg, Inorg. Chem., 2009, 48, 8393–8403 CrossRef PubMed.
- E. Y. Tshuva and S. J. Lippard, Chem. Rev., 2004, 104, 987–1012 CrossRef PubMed.
- D. Lee and S. J. Lippard, Inorg. Chem., 2002, 41, 2704–2719 CrossRef PubMed.
- M.-H. Baik, M. Newcomb, R. A. Friesner and S. J. Lippard, Chem. Rev., 2003, 103, 2385–2419 CrossRef PubMed.
- C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef PubMed.
- X. Wang, T. Zhang, Q. Yang, S. Jiang and B. Li, Eur. J. Inorg. Chem., 2015, 5, 817–825 CrossRef.
- L. Chen, Y. Xiang and T. Feng, Appl. Organomet. Chem., 2012, 26(3), 108–113 CrossRef.
- (a) G. A. N. Felton, C. A. Mebi, B. J. Petro, A. K. Vannucci, D. H. Evans, R. S. Glass and D. L. Lichtenberger, J. Organomet. Chem., 2009, 694, 2681–2699 CrossRef; (b) Y. Si, M. Hu and C. Chen, C. R. Chim., 2008, 11, 932–937 CrossRef.
- L. Schwartz, P. S. Singh, L. Eriksson, R. Lomoth and S. Ott, C. R. Chim., 2008, 11, 875–889 CrossRef.
- N. Wang, M. Wang, K. Jin, J. Liu, L. Chen and L. Sun, Inorg. Chem., 2009, 48, 11551–11558 CrossRef PubMed.
- P. Li, M. Wang, C. He, G. Li, X. Liu, C. Chen, B. Åkermark and L. Sun, Eur. J. Inorg. Chem., 2005, 2506–2513 CrossRef.
- A. L. Haley, L. N. Broadbent, L. S. McDaniel, S. T. Heckman, C. H. Hinkle, N. N. Gerasimchuk, J. C. Hershberger and C. A. Mebi, Polyhedron, 2016, 114, 218–224 CrossRef.
- F. Huo, J. Hou, G. Chen, D. Guo and X. Peng, Eur. J. Inorg. Chem., 2010, 25, 3942–3951 CrossRef.
- P. Zhao, S. Liu, Y. Liu and Y. Liu, J. Cluster Sci., 2014, 25, 1331–1340 CrossRef.
- T. B. Rauchfuss, Acc. Chem. Res., 2015, 48, 2107–2116 CrossRef PubMed.
- D. Bianchi, M. Bertoli, R. Tassinari, M. Ricci and R. Vignola, J. Mol. Catal. A: Chem., 2003, 200, 111–116 CrossRef CAS.
- X. You, Z. Wei, H. Wang, D. Li, J. Liu, B. Xu and X. Liu, RSC Adv., 2014, 4, 61790–61798 RSC.
- A. Conde, M. Mar Diaz-Requejo and P. J. Perez, Chem. Commun., 2011, 47, 8154–8156 RSC.
- H. Ge, Y. Leng, F. Zhang, J. Piao, C. Zhou and J. Wang, Sci. China, Ser. B: Chem., 2009, 52, 1264–1269 CrossRef CAS.
- G. Li, E. A. Pidko, I. A. W. Filot, R. A. van Santen, C. Li and E. J. M. Hensen, J. Catal., 2013, 301, 77–82 CrossRef.
- (a) G. M. Sheldrick, SHELXS-97, Program for crystal structure solution, Göttingen, Germany, University of Göttingen, 1997 Search PubMed; (b) O. Dolomanov and H. Puschmann, Acta Crystallogr., 2007, 63, 74–75 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1470792, 1502264 and 1502265 contains the supplementary crystallographic data of complexes 1–3 for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra27831k |
| This journal is © The Royal Society of Chemistry 2017 |