
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deoxygenation of coal bed methane on LaCoO3 perovskite catalyst: the structure evolution and catalytic performance
Zhenyang Zhao†
,
Li Wang†,
Jian Ma,
Yafen Feng,
Xiaoming Cao ,
Wangcheng Zhan,
Yanglong Guo,
Yun Guo* and
Guanzhong Lu
,
Wangcheng Zhan,
Yanglong Guo,
Yun Guo* and
Guanzhong Lu
Key Laboratory for Advanced Materials and Research Institute of Industrial Catalysis, School of Chemistry & Molecular Engineering, East China University of Science and Technology, Shanghai 200237, People’s Republic of China. E-mail: yunguo@ecust.edu.cn; Fax: +86 21 64253703; Tel: +86 21 64253703
First published on 7th March 2017
Abstract
A series of perovskite-type LaBO3 (B = Fe, Co, Mn, and Ni) materials have been studied as catalysts for coal bed methane (CBM) deoxygenation. Among them, LaCoO3 shows the best catalytic performance and stability, O2 could be completely eliminated by CH4 to produce CO2 and H2O in the range of 400–720 °C, and the complete deoxidization could be maintained at temperatures of 400, 500, 600, and 660 °C for 100 h. Furthermore, the structure of LaCoO3 could transform from perovskite to Co/La2O3 through La2CoO4/LaCoO3 and La2CoO4/Co3O4 during the process of CBM deoxygenation. The results of H2-TPR and O2-TPO showed the perovskite LaCoO3 is like a smart catalyst, whereby the Co species could reversibly move into and out of the perovskite structure depending on the temperature and reaction atmosphere. When Co species exist in an oxidised state (Co3O4, La2CoO4 and/or LaCoO3), the CH4 in CBM is completely oxidized by O2 to produce CO2 and H2O, the results of isotopic tracer experiments and pulse reaction demonstrate that the reaction follows the Mars–van Krevelen mechanism. However, the preferred products of the CBM deoxygenation reaction are CO and H2 on Co/La2O3 through partial oxidation of CH4. With the structure transforming from Co/La2O3 to LaCoO3 after reoxidation by O2, the activity of CBM deoxygenation could be recovered.
1. Introduction
Coal bed methane (CBM), also known as coal mine gas, is a kind of flammable gas whose main component is methane.1 The direct emission of CBM is not only a waste of energy, but also pollutes the environment, because the warming potential of CH4 is over 20 times of CO2 as a greenhouse gas.2 At a typical gassy mine, CBM is mainly emitted in three streams: (1) gas drained from the steam before mining, containing 60–95 vol% CH4 and inert gas, which could be directly used or easily used to produce pure CH4; (2) gas drained from the worked areas of the mine, e.g. goaf, containing 30–95 vol% CH4 and some O2 (2–6 vol%);3,4 (3) CH4 ventilation air (0.1–1 vol% CH4).5For the utilization of CBM with high CH4 concentration and low O2 concentration, it is necessary to remove the O2 from the mixture, because the existence of O2 could be dangerous in the process of storage and transportation. Usually, two main methods are used in CBM deoxygenation: non-catalytic and catalytic deoxidization. The common non-catalytic methods include the adsorption of O2, coke burning and deep freezing methods.6–8 Compared with non-catalytic methods, the catalytic deoxygenation of CBM is a convenient and effective method to eliminate O2 by catalytic combustion of CH4.9–11 However, the catalytic combustion of CH4 is a violent exothermal reaction with a huge ΔH298 of −802.7 kJ mol−1, which could induce a severe temperature runaway of the reactor and sintering of the catalyst. Meanwhile, the high reaction temperature could cause CH4 partial oxidation and a reforming reaction to produce CO and H2 under the conditions of a large excess of CH4.12–14 Therefore, a desirable catalyst used in catalytic deoxygenation of CBM should not only have high activity to remove O2 at low temperatures, but also avoid the production of H2 and CO through side reactions (partial oxidation, cracking and/or reforming reaction) across a wide temperature range.
Furthermore, the composition of the reaction gas in the CBM deoxidization reaction varies from aerobic conditions to reducing conditions with the consumption of O2, which requires the catalyst to maintain high performance under oxidizing and reducing conditions simultaneously. It is another challenge for the deoxygenation catalyst.
Supported noble metal catalysts are widely used in the catalytic combustion of CH4.15–20 However, the excess CH4 in the reaction gas would lead to particle oxidation or cracking reactions, and produce H2 and CO at temperatures as low as 400 °C while providing high CH4 conversion.21–26 Lyubovsky et al.27 prepared Al2O3-supported Pd, Pt and Ru catalysts for CH4 oxidation under both fuel-rich and fuel-lean conditions. The partial oxidation products of H2 and CO appeared under fuel-rich conditions above the light-off temperature, and their concentration increased with increasing temperature. In addition, the chemical state change of the noble metal in the process of CBM deoxygenation could affect the activity of catalyst. Lu et al.28–30 reported CBM deoxygenation on Pd–PdO–NiO/Ni-foam and found the oscillation of O2 conversion to be due to the formation of inert metal Pd under the reducing conditions. The presence of PdNi (alloy) induced by the in situ reaction could eliminate this O2 oscillation, and O2 completely oxidized CH4 to CO2 and H2O in the temperature range of 350–500 °C.
Compared with the supported noble metal catalysts, transition metal oxide catalysts (such as Cu, Co Ni etc.) also have attracted great attention.31–33 For example, Tao et al.34 prepared a nano-NiCo2O4 catalyst via a co-precipitation method, which showed high activity for CH4 combustion under conditions of excess O2 in the temperature range of 350–550 °C due to the integration of nickel cations, cobalt cations and surface lattice oxygen atoms/oxygen vacancies at the atomic scale.
The perovskite-type oxides (ABO3) have high temperature stability in hydrocarbon (CnH2n+2) oxidation35,36 and reforming reactions.37,38 For example, LaCoO3 and partially substituted LaCoO3 have been confirmed to have high activities and stabilities for the partial oxidation of CH4.39–41 Generally, perovskites prepared with La in the A position, and Co, Mn, Fe or Ni in position B, are used in the catalytic combustion of CH4.42–45 Meanwhile, the temperature of partial oxidation or reforming of CH4 over perovskite catalyst usually exceeds 600 °C, which is much higher than that of supported noble metal catalysts and the transition metal oxide catalysts.46,47 For example, Slagtern and Olsbye48 studied the partial oxidation of CH4 to syngas at 800 °C on La–M–O (M = Co, Ni, Rh, and Cr) perovskite catalysts, and found the main product was CO2 on La–Co–O with the main phase of LaCoO3, Co3O4, and La2O3.
Furthermore, the structure of perovskite-type oxides could be reversibly changed depending on the composition of the reaction atmosphere. Nishihata et al.49 reported that LaFe0.57Co0.38Pd0.05O3 exhibited high catalytic activity during long term ageing, and the Pd reversibly moved into and out of the perovskite lattice during the cycle between oxidative and reductive atmospheres. Hence, the perovskite type catalyst may be a good candidate as a catalyst for CBM deoxygenation, and may be able to remove O2 from the CBM via CH4 combustion at a relatively low temperature, and maintain total oxidation across a wide temperature range by prohibiting partial oxidation and other side reactions.
In this work, perovskite-type oxides LaBO3 (B = Co, Mn, Fe and Ni) were prepared, and the activity and stability of LaBO3 for CBM catalytic deoxygenation were investigated. The evolution of LaCoO3 perovskite structure in the reaction and reaction mechanism were also explored.
2. Experimental section
2.1 Catalyst preparation
The perovskite-type oxides (LaBO3, B = Co, Mn, Fe and Ni) were prepared by the co-precipitation method. A stoichiometric amount of metal nitrate mixture solution and sodium hydroxide solution were simultaneously dropped into a NaOH solution with pH of 9–10 under stirring at 60 °C. The pH value of the mixture solution was kept in the range of 9–10 during the whole precipitation process. The obtained precipitate was aged at 60 °C for 2 h. After being washed by deionized water to neutral pH, the precipitate was filtered and dried at 100 °C for 12 h then calcined in air at 750 °C for 3 h to obtain the LaBO3 catalysts. The BET surface areas of the prepared LaBO3 are in the range of 12 to 15 m2 g−1.2.2 Catalyst characterization
The powder X-ray diffraction patterns (XRD) of catalysts were obtained with a RigakuD/max 2550 VB/PC diffractometer with a Cu Kα radiation (λ = 1.54056, scanning step 0.02°). Spectra were collected in a range of 2θ = 10–80° with a scanning rate of 6° min−1. In order to obtain more details about the structure of the sample after reduction, the mapping of the elements was measured on the JOEL 2100 instrument operating at 200 kV.The X-ray photoelectron spectroscopy (XPS) spectra were recorded on an AXIS-Ultra-DLD spectrometer with a Al Kα X-ray source (1486.6 eV). The base pressure inside the analysis chamber was 3 × 10−10 Torr. The XPS spectra of the selected elements were measured with the constant analyzer pass energy of 40 eV. All binding energies (BE) were referenced to the adventitious C 1s peak (BE = 284.8 eV).
The specific surface areas of the catalysts were measured using the N2 adsorption isotherm at −196 °C by using an automatic Micromeritics ASAP 2020 analyzer.
The temperature-programmed reduction of H2 (H2-TPR) experiments were carried out by a conventional flow system equipped with a thermal conductivity detector (TCD). 100 mg catalyst was calcined at 400 °C for 1 h in air before the TPR reaction, and then cooled to room temperature. The pretreated catalyst was heated in a flow of 5 vol% H2/N2 (45 mL min−1) at a heating rate of 10 °C min from room temperature to 800 °C. After H2-TPR, the catalyst was maintained at 800 °C for 1 h in a flow of 5 vol% H2/N2 (45 mL min−1), then purged with pure He for 1 h. After cooling to room temperature in a He flow, the temperature programmed oxidation of O2 (O2-TPO) was performed using the same apparatus; 1 vol% O2/He (50 mL min−1) was used in O2-TPO, and the composition of the outlet gas was monitored by an on-line quadrupole mass spectrometer (IPC 400, INFICON Co. Ltd.).
Isotope tracer experiments were conducted in the quartz tube reactor and the effluent gas was monitored by an on-line quadrupole mass spectrometer (MS, IPC 400, INFICON Co. Ltd.).
The catalyst was pretreated at 700 °C for 2.5 h in pure He at 50 mL min−1. 500 mg catalyst was used in the pulse experiments, 517.3 μL of 12 vol% 18O2/6 vol% CH4 was pulsed into the reactor 20 times. 200 mg catalyst was used in the continuous isotope tracer experiments at different designated temperatures, 12 vol% 18O2/6 vol% CH4 was used as the reaction gas.
The reaction orders of O2 and CH4 were measured in the temperature range of 340 to 410 °C with feed steams of 4.0–12.0 kPa O2, CH4 and N2. 1.0 kPa CH4 was used to investigate the catalytic combustion of CH4, and 50.0 kPa CH4 was used in the deoxidization of CBM. The O2/CH4 conversion was adjusted to below 15% by varying the space velocity in the range of 6000–72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1 so as to eliminate the thermal effect and diffusion effect.
000 mL g−1 h−1 so as to eliminate the thermal effect and diffusion effect.
CH4 and O2 pulse experiments were conducted on the same apparatus as that for the isotope tracer experiments. The procedures were as follows: (1) 10 vol% CH4/He was pulsed into 500 mg catalyst 20 times (CH4-1st); (2) 20 pulses of pure O2 was passed through the catalyst bed; (3) step 1 was repeated again (CH4-2nd). The pulse volume was 517.3 μL.
2.3 Evaluation of the catalytic performance
The catalytic activities of LaBO3 (B = Co, Mn, Fe and Ni) catalysts for the simulated deoxygenation of CBM were tested in a fixed bed quartz tubular reactor at atmospheric pressure, 300 mg catalyst (40–60 mesh) diluted with 2 g silica sand (20–40 mesh) was used. The feed gas, containing 50 vol% CH4, 6 vol% O2 and N2 to balance, was passed through the catalytic bed at a flow rate of 30 mL min−1. The temperature of the catalyst bed was measured by a thermocouple inserted in the top of the catalyst bed, and the heating rate was 4 °C min−1. An on-line gas chromatograph (Agilent 7890) was used to monitor the composition of the outlet gas. The catalyst activity was expressed by T10 and T90 of O2, which corresponded to the reaction temperatures required for 10% and 90% O2 conversion, respectively.Because the excess CH4 in the feed gas could lead to partial oxidation or the reforming reaction to produce CO and H2 at high temperature, the temperature range between the lowest temperature of complete conversion (LTCC) of O2 and the initial temperature of H2 formation is defined as the operation window of the catalyst for the deoxygenation reaction.
3. Results
3.1 Catalytic activities of LaBO3
The catalytic activities of LaBO3 (B = Co, Fe, Mn and Ni) for CBM deoxygenation are shown in Fig. 1. The type of transition metal in the B-site shows a significant effect on the catalytic activity of the perovskites. LaFeO3 shows the lowest activity for O2 elimination, T10 and T90 are 350 and 450 °C, respectively. Meanwhile, LaCoO3 exhibits the highest catalytic activity, the T10 and T90 are 300 and 390 °C, respectively. Combined with the results of the BET surface area in Table 1, the activity of the catalyst is not directly related to its surface area.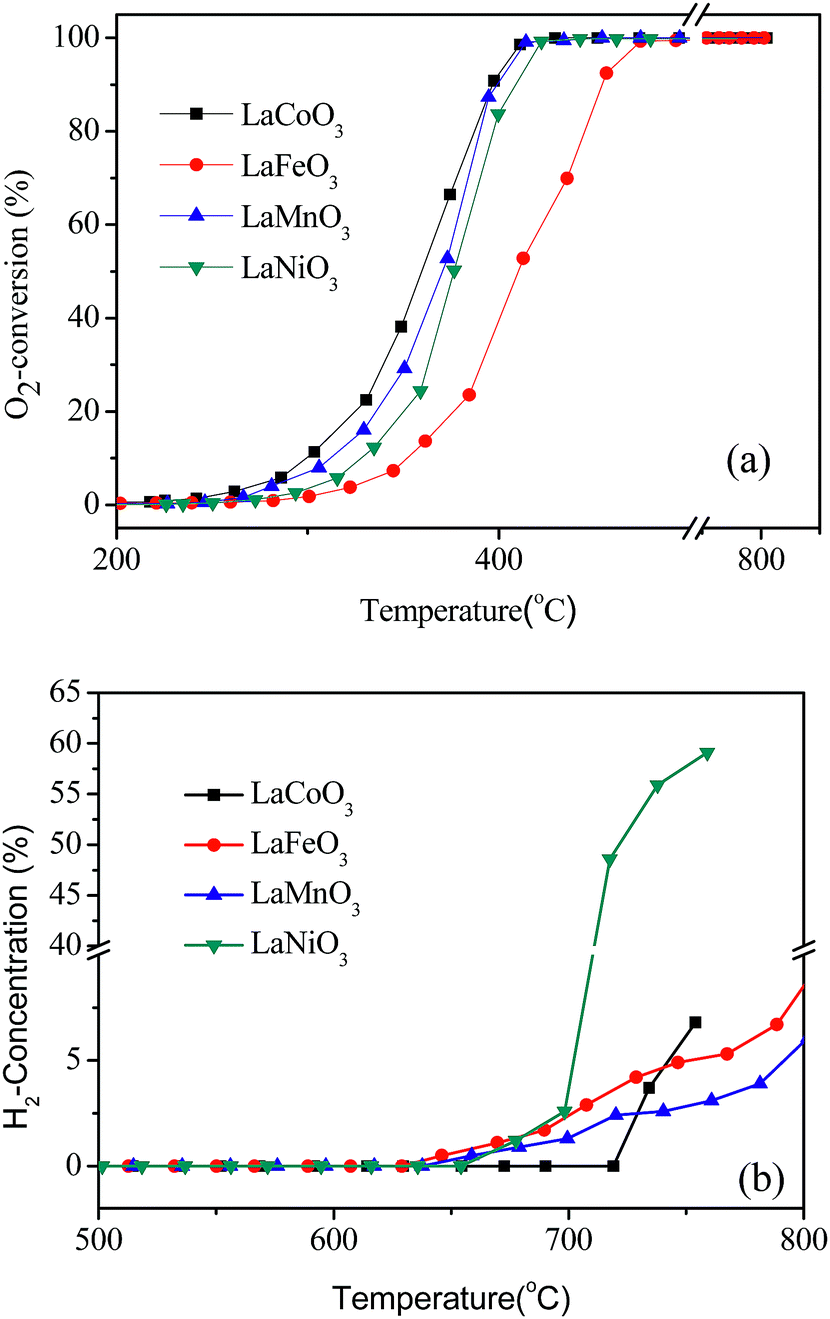 | ||
| Fig. 1 The conversion of O2 (a) and production of H2 (b) in the deoxygenation reaction by LaBO3 catalysts as a function of temperature. | ||
| LaCoO3 | LaFeO3 | LaMnO3 | LaNiO3 | |
|---|---|---|---|---|
| a The cell parameters were obtained by Rietveld refinement calculations from the diffractogram of the structures.b The crystallite size was calculated by the Debye–Scherrer formula. | ||||
| Spatial group | Hexagonal | Orthorhombic | Hexagonal | Hexagonal |
| a (Å) | 5.4358 | 5.4672 | 6.0731 | 5.5953 |
| b (Å) | 5.4358 | 6.7968 | 6.0731 | 5.5953 |
| c (Å) | 13.0643 | 28.8799 | 13.4010 | 5.6679 |
| Crystallite size (nm) | 39.1 | 53.72 | 10.7 | 26.4 |
| BET area (m2 g−1) | 11 | 16 | 22 | 14 |
The excess CH4 in the feed gas could produce H2 and CO by the partial oxidation or reforming reaction at high reaction temperature (>700 °C). The production of H2 following the reaction temperature in CBM deoxygenation on different catalysts is shown in Fig. 1b. CO was observed simultaneously but is not shown. The results in Fig. 1b show the initial sequence of H2 formation is LaFeO3 < LaMnO3 < LaNiO3 < LaCoO3, and the H2 formation on LaNiO3 increases more rapidly than the others when the temperature exceeds 700 °C. Combined with the results in Fig. 1a, LaCoO3 shows the widest operation window, where O2 could be completely eliminated by CH4 in the temperature range of 400 to 720 °C. Continuously increasing the reaction temperature, the amount of H2 and CO increased rapidly. Therefore, catalyst LaCoO3 was selected for the further investigation in the following sections.
The stability of LaCoO3 for deoxygenation reaction was investigated at temperatures of 360, 400, 500, 600, and 660 °C for 100 h. The results in Fig. 2 show O2 conversion was maintained at 360 °C at about 75% for 100 h, and O2 could be completely eliminated at 400 and 660 °C for 100 h, and H2 and CO were not detected during the whole experiments. The same results were obtained at 500 and 600 °C, which are not shown. After reaction at 660 °C for 100 h, the light-off activity of the aged catalyst is nearly consistent with the fresh one (Fig. 2b), which indicates that LaCoO3 has high stability for the deoxygenation reaction in the temperature range of 400–660 °C.
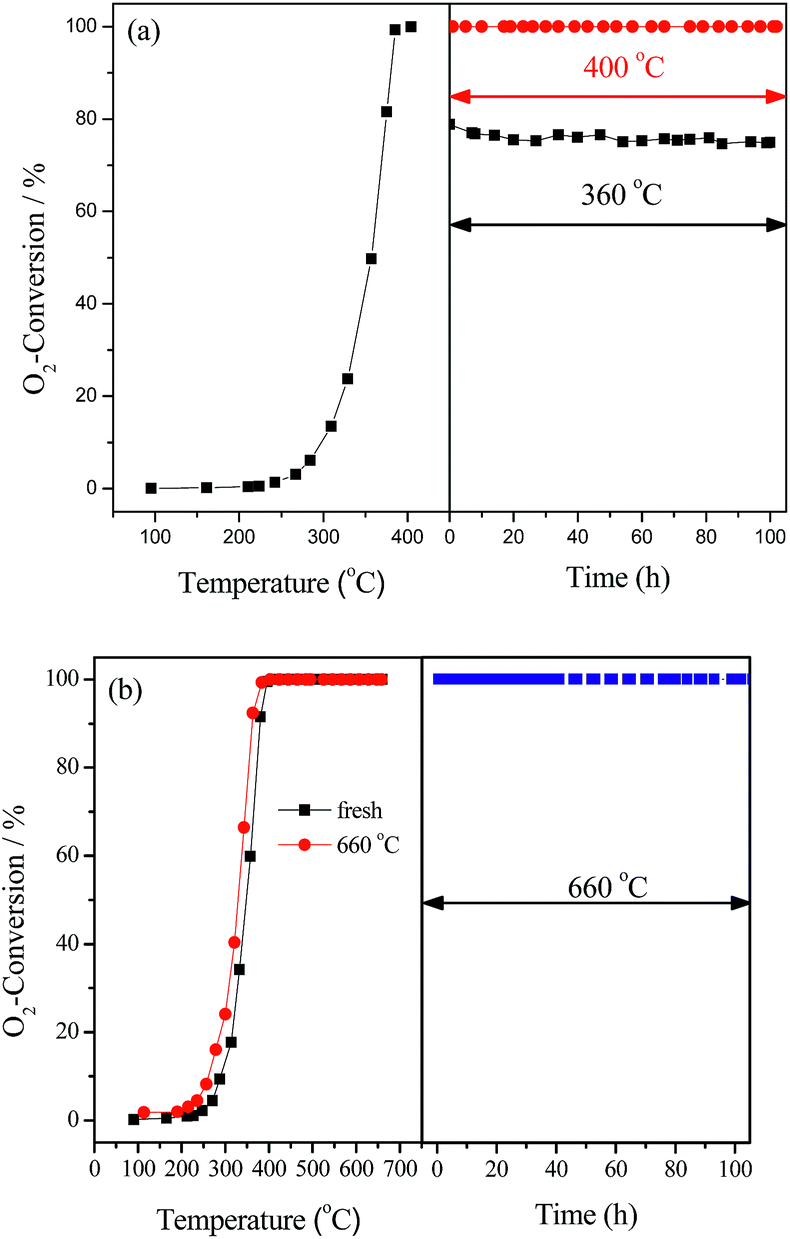 | ||
| Fig. 2 The stability of LaCoO3 measured at 360, 400 °C (a) and 660 °C (b). In part (b) the catalytic activity of fresh LaCoO3 (■) and after reaction at 660 °C for 100 h (●) are compared. | ||
3.2 XRD
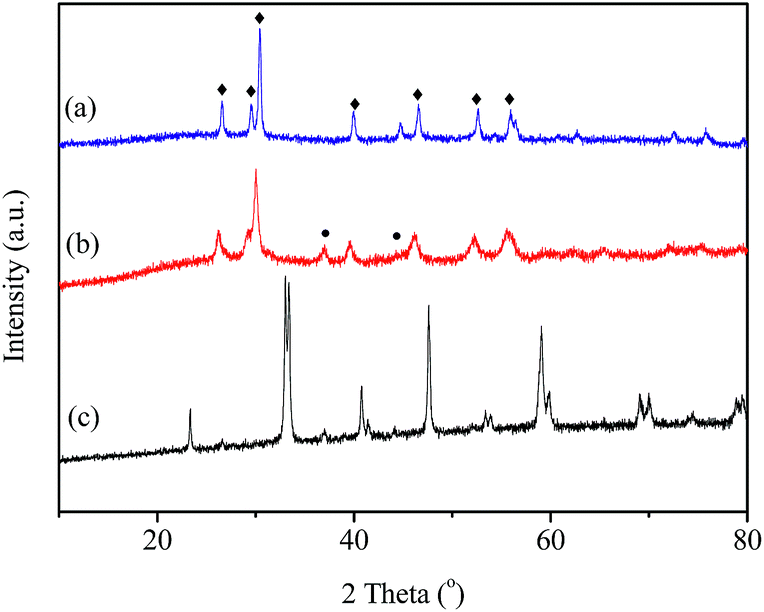
The XRD patterns of the fresh LaBO3 catalysts are exhibited in Fig. 3. The prepared LaBO3 (Ni, Mn and Co) show a typical hexagonal perovskite structure. For catalyst LaFeO3, the major phase is orthorhombic perovskite structure, and some weak diffraction peaks corresponding to Fe2O3 and La2O3 are also detected at 2θ = 33, 35, 49 and 54° and 2θ = 26, 30, 39 and 52°, respectively. The cell parameters, crystallite size and BET surface area of the perovskites are list in Table 1.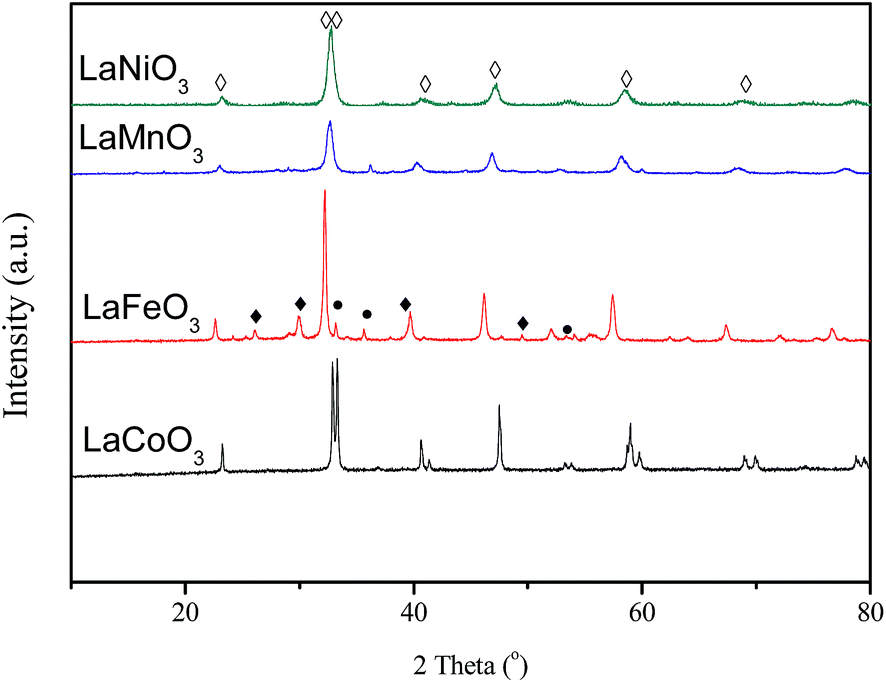 | ||
| Fig. 3 XRD patterns of LaBO3 (◊: perovskite; ●: Fe2O3; ♦: La2O3). | ||
Fig. 4 shows the XRD patterns of LaCoO3 after stability tests at different temperatures. Compared with the results in Fig. 3, there is no observable difference in the LaCoO3 structure after reaction at 400 and 500 °C for 100 h (Fig. 4a and b), which indicates the stability of the perovskite structure. However, after reaction at 600 °C for 100 h (Fig. 4c), the structure of LaCoO3 transforms from perovskite into a mixture of perovskite (LaCoO3, 2θ = 23, 33, 40, 53 and 59°) and perovskite-like (La2CoO4, 2θ = 24, 32, 43, 47 and 65°). Continuously increasing the reaction temperature to 660 °C, only perovskite-like La2CoO4 and Co3O4 crystal phases are detected after 100 h reaction (Fig. 4d). Combined with the results in Fig. 2, it could be concluded that the structure change of LaCoO3 is dependent on the reaction temperature, but this structure evolution does not bring an apparent difference in the catalytic performance of LaCoO3 for CBM deoxygenation.
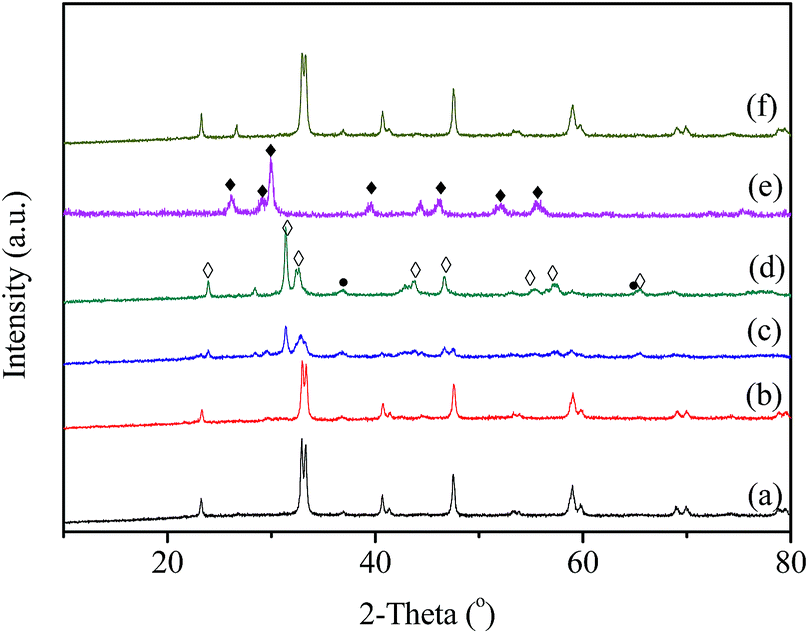 | ||
| Fig. 4 The XRD patterns of the LaCoO3 after reaction at 400 (a), 500 (b), 600 (c), and 660 °C (d) for 100 h and 800 °C (e), the re-oxidation of sample (e) at 750 °C in air (f) (◊: La2CoO4; ●: Co3O4; ♦: La2O3). | ||
When the deoxygenation reaction is finished at 800 °C, the perovskite structure of LaCoO3 is completely destroyed, and only La2O3 is detected at 26, 30, 39 and 52° (Fig. 4e). The results in Fig. 1 show the production of H2 and CO when the reaction temperature was higher than 720 °C, which could lead to the reduction of LaCoO3.46 The diffraction peaks of Co species cannot be observed, which means Co species are highly dispersed or below the detection limit of XRD. However, it is worth noting that the completely destroyed LaCoO3 could be reverted back to the perovskite structure after reoxidation at 750 °C in air (Fig. 4f).
3.3 XPS characterization
XPS characterization is performed to investigate the surface chemical state of the catalysts. Fig. 5 shows the Co 2p spectra of LaCoO3 after reaction at different temperatures (400, 500, 600, and 660 °C) for 100 h. The resolution of the asymmetrical spectra of Co 2p shows the co-existence of two species at BE of 779.8 and 782.1 eV, which could be ascribable to Co3+ and Co2+, respectively.50,51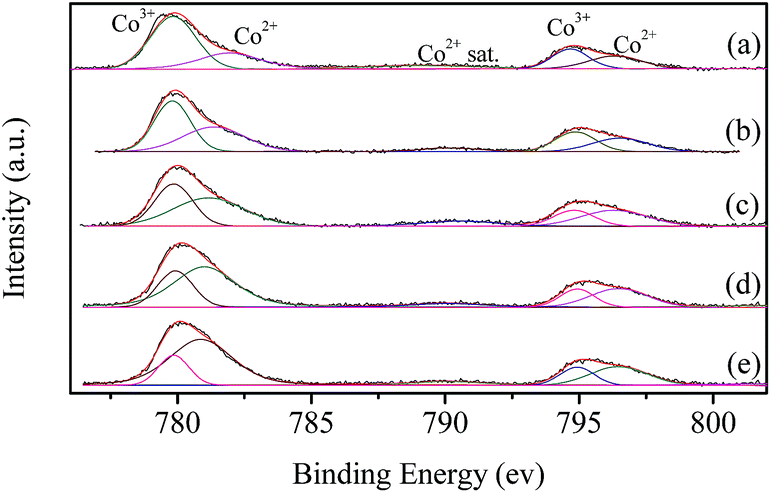 | ||
| Fig. 5 XPS spectra of fresh LaCoO3 (a) and aged LaCoO3 after stability tests at temperatures of 400 (b), 500 (c), 600 (d) and 660 °C (e) for 100 h. | ||
The surface Co2+/Co3+ ratio of LaCoO3 after reaction is much higher than that of fresh catalyst, and the Co2+/Co3+ ratio increases with the increase in the reaction temperature as shown in Table 2, which indicates the partial reduction of LaCoO3 during the reaction, and coincides with the results of XRD shown in Fig. 4. The predominant crystal phase changes from perovskite to a mixture of La2CoO4 and Co3O4 through the mixed phase of LaCoO3 and La2CoO4, the average chemical state of the surface Co species is gradually reduced during this process.
3.4 Temperature programmed reaction
In order to further investigate the effects of reaction gas and temperature on structure evolution of LaCoO3 during the reaction, experiments of H2-TPR and O2-TPO are carried out.The H2-TPR profile of fresh LaCoO3 shows three reduction peaks in Fig. 6a. The peaks in the temperature range of 200–500 °C correspond to the reduction of the oxygen adsorbed on the catalyst surface and reduction of Co3+ to Co2+, the high temperature peak at 500–800 °C could be assigned to the reduction of Co2+ to Co.46,52,53 The XRD pattern of Fig. 7a demonstrates that the perovskite structure of LaCoO3 has been completely destroyed and converted to a mixture of metallic Co and La2O3 after H2-TPR, which coincides with that of LaCoO3 after reaction at 800 °C (Fig. 4).
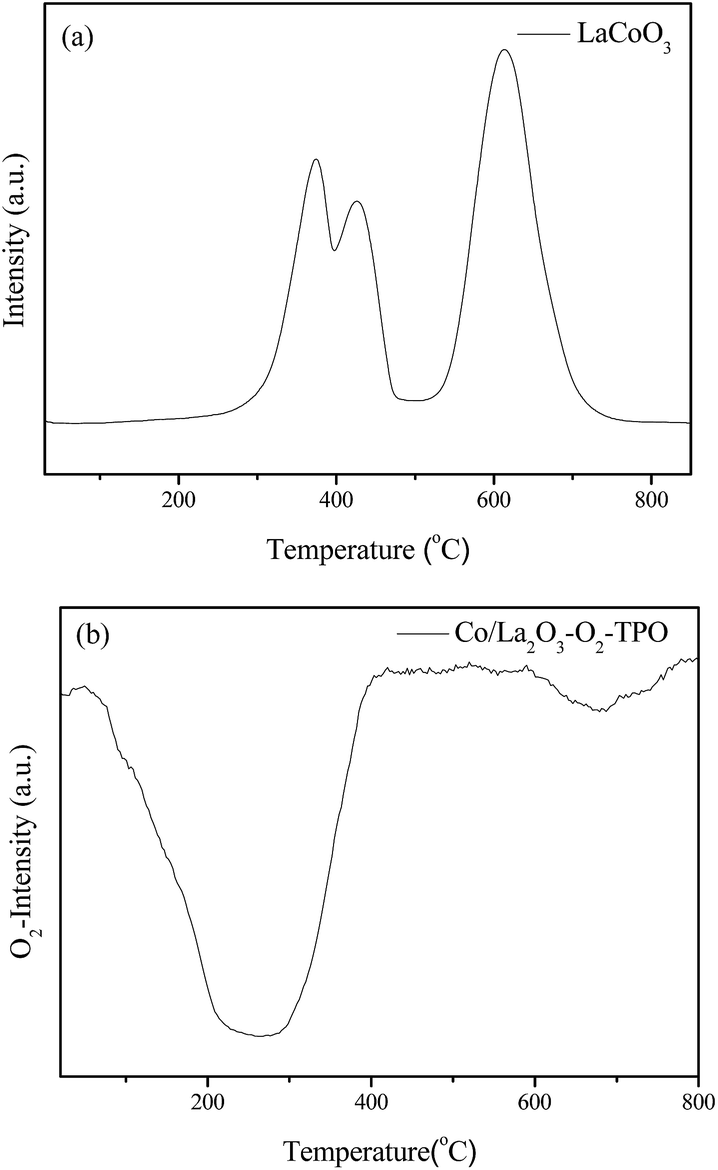 | ||
| Fig. 6 H2-TPR (a) and O2-TPO (b) profiles of LaCoO3. | ||
 | ||
| Fig. 7 (a) XRD patterns of LaCoO3 reduced by H2 at 800 °C (a); reoxidation by O2 at 300 °C (b) and 750 °C (c). ●, Co3O4; ♦, La2O3. | ||
After H2-TPR, O2-TPO of the reduced sample is performed. The result in Fig. 6b shows there are two O2 consumption peaks: a significant peak is located at the range of 200–300 °C and a weak peak is observed at near 700 °C.
For the sample after H2-TPR, XRD results show that the main phases are Co3O4 and La2O3 after reoxidation at 300 °C for 0.5 h (Fig. 7b), which indicates the O2 consumption peak at range of 200–300 °C in Fig. 6b should correspond to the oxidation of metal Co to Co3O4 (Co/La2O3 + O2 → Co3O4 + La2O3). Because the oxidation of metal Co is a strong exothermic reaction, the accumulated heat could result in the direct oxidation of some metallic Co species to Co3+. When the oxidation temperature is increased to 750 °C, the perovskite structure of LaCoO3 was recovered (Co3O4 + La2O3 + O2 → LaCoO3), the main phase of the sample is perovskite with a minor Co3O4 phase observed at 2θ = 37° (Fig. 7c), which corresponds to the O2 consumption at the high temperature range in Fig. 6b.
3.5 Kinetic analysis
Fig. 8 shows the pressure-dependent reaction rates on the partial pressure of O2 (PO2) from 4.0 to 12.0 kPa while keeping the partial pressure of CH4 (PCH4) at 1.0 o 50.0 kPa, which corresponds to the catalytic combustion of CH4 and deoxidization of CBM, respectively.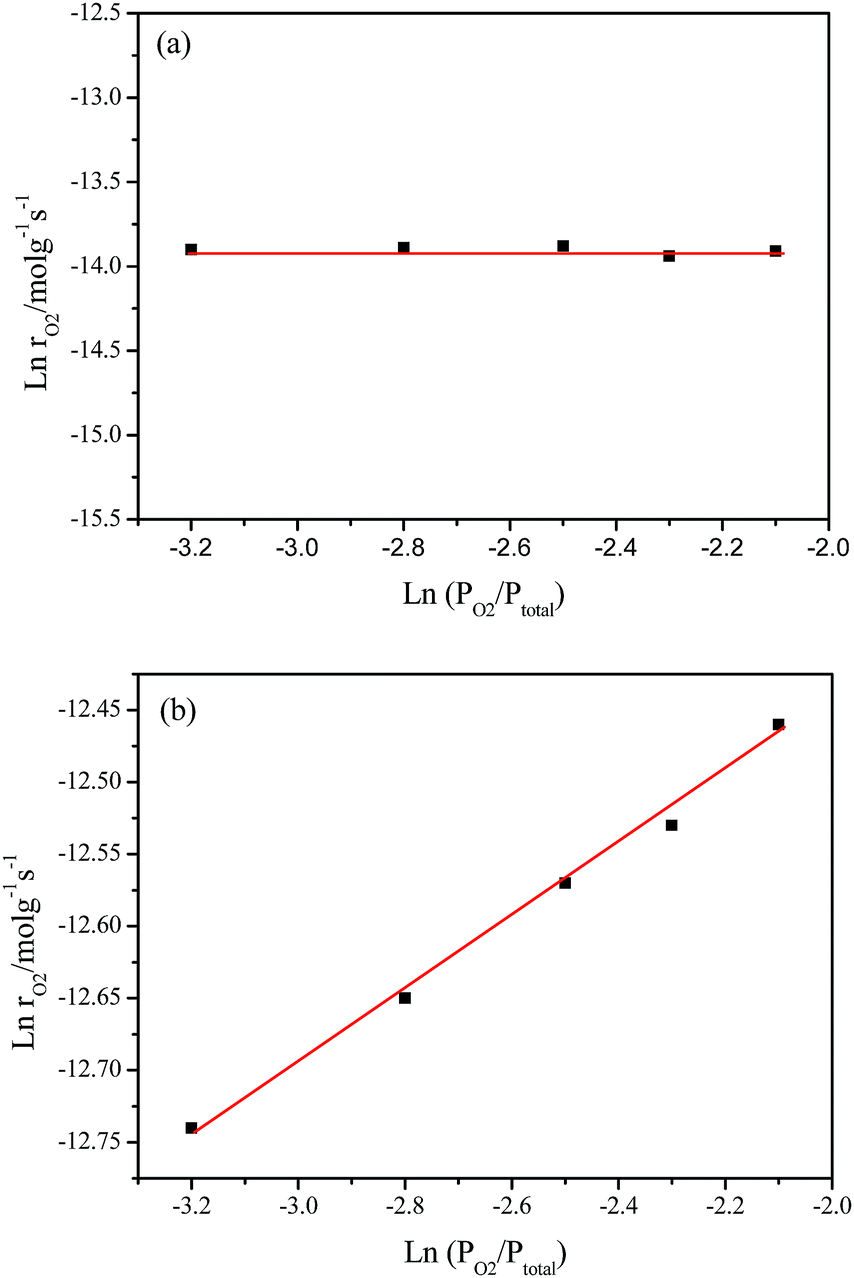 | ||
| Fig. 8 lnrO2 as a function of lnPO2 over LaCoO3: (a) at 420 °C, the feed gas consisted of 4–12% O2 and 1% CH4 at a space velocity of 30000 mL g−1 h−1; (b) at 370 °C, the feed gas consisted of 4–12% O2 and 50% CH4 at a space velocity of 48000 mL g−1 h−1. | ||
Under O2 rich conditions, the O2 reaction rate doesn’t change with an increase in PO2, yielding an order of 0 with respect to O2 at 420 °C. The fact that reaction order of O2 is about zero shows the O2 concentration hardly affect the rate of O2 consumption, which implies the activation of CH4 is the rate determination step for CH4 combustion under O2 excess.54,55
However, under oxygen lean conditions, the O2 reaction rates increases with an increase in PO2, yielding an order of 0.25 with respect to O2 at 370 °C, which demonstrates that the activation of O2 is a key factor for CBM deoxygenation reaction. The apparent activation energy (Ea) of deoxygenation reaction on LaCoO3 is 121 kJ mol−1, as shown in Fig. 9.
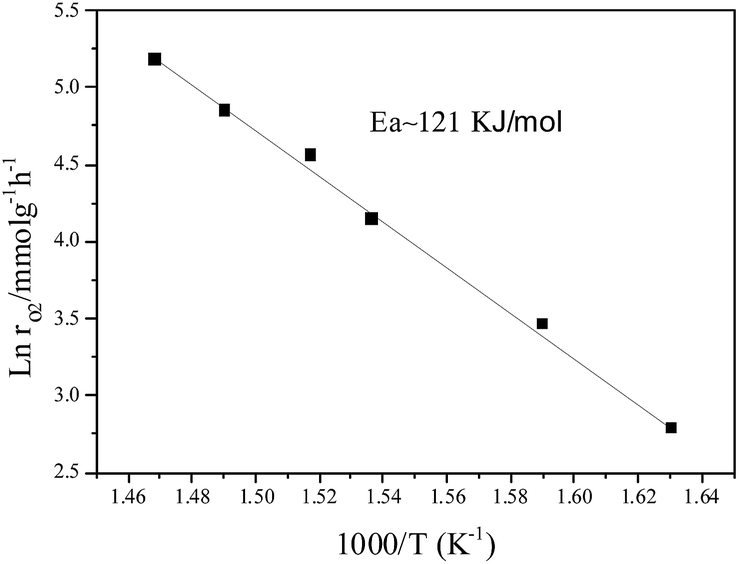 | ||
| Fig. 9 Arrhenius plot of the reaction rate (lnr) vs. 1/T for O2 deoxygenation over LaCoO3. | ||
3.6 Isotopic tracer experiments
The isotopic tracer pulse reaction results of 18O2 + CH4 are shown in Fig. 10. When the temperature is 600 °C, 18O2 (O2-36) is completely consumed in the 20 pulses on the LaCoO3 (Fig. 10a), and the production of C16O2 (m/z = 44) could be observed at the same time. Furthermore, any CO2 containing 18O (m/z = 46 and 48) are not detected, which indicates CH4 reacts with the lattice oxygen rather than gas 18O2. The same results are obtained at the temperature of 700 °C (Fig. 10b).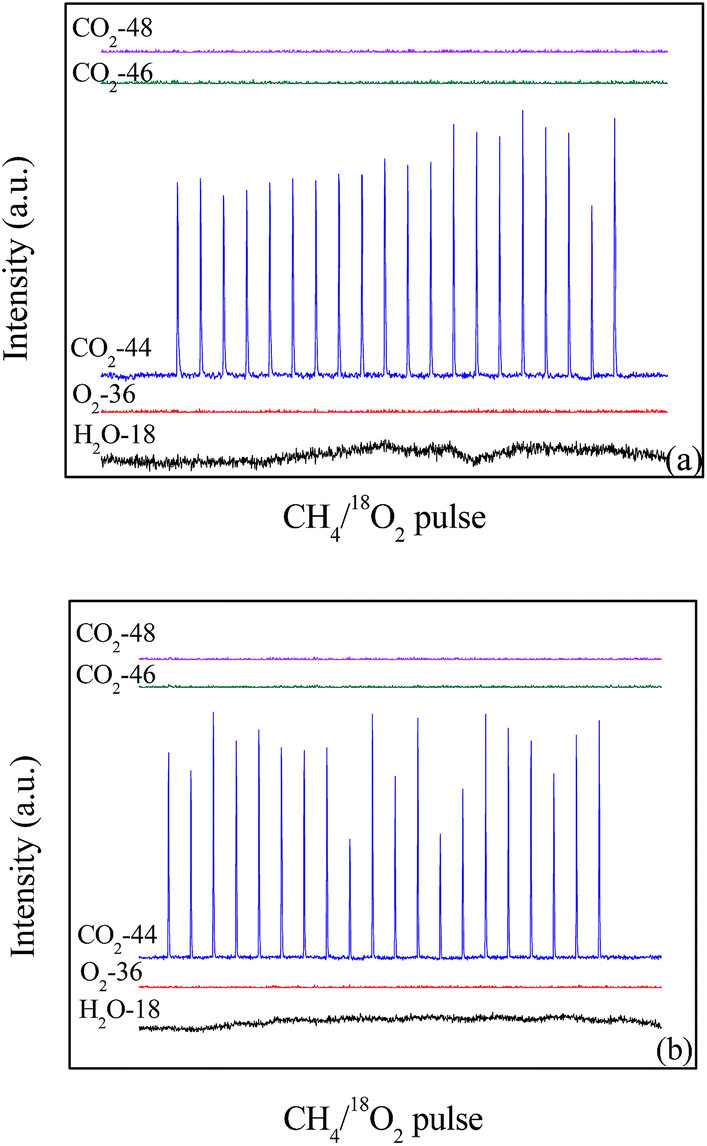 | ||
| Fig. 10 The pulses test of catalyst LaCoO3 under the conditions of 12 vol% 18O2/6 vol% CH4 at 600 (a) and 700 °C (b). | ||
When the feed gas of 12 vol% 18O2/6 vol% CH4 continuously passes through the catalyst bed at 600 °C, the result in Fig. 11a shows that C16O2 is produced immediately and exists as the dominant product in the first 300 s, then the content of C16O2 obviously decreases with the increase of reaction time. Meanwhile, the content of C16O18O (46) and C18O2 (48) increases gradually. After 40 min, C18O2 becomes the main product, next is C16O18O. Similar results are obtained at 700 °C, as shown in Fig. 11b. Combined with the results in Fig. 10, it may be suggested that the deoxidization reaction of CBM may follow the Mars–van Krevelen mechanism: the CH4 in the feed gas firstly reacts with lattice oxygen and creates oxygen vacancies, which could be replenished by the diffusion of lattice oxygen from bulk to surface and the adsorption and activation of gas O2.
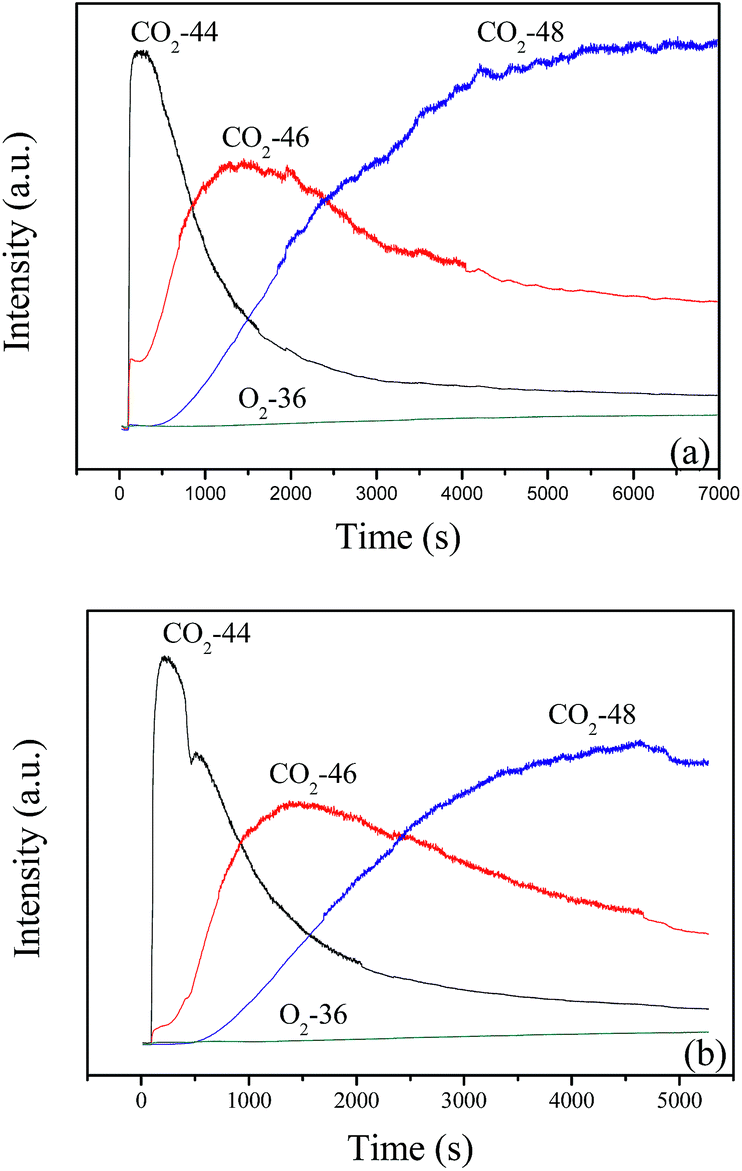 | ||
| Fig. 11 The continuous reaction of 12 vol% 18O2/6 vol% CH4 balanced with N2 on LaCoO3 at 600 (a) and 700 °C (b). | ||
3.7 Pulse reaction
The results in Fig. 4 showed structure transformation of LaCoO3 during the CBM deoxygenation reaction. Separated CH4 and O2 pulse reactions on LaCoO3 and pre-reduced LaCoO3 at different temperatures are performed to explore the effects of LaCoO3 structure on the CBM deoxygenation reaction and the formation mechanism of byproducts (H2 and CO); the results are shown in Fig. 12 and 13.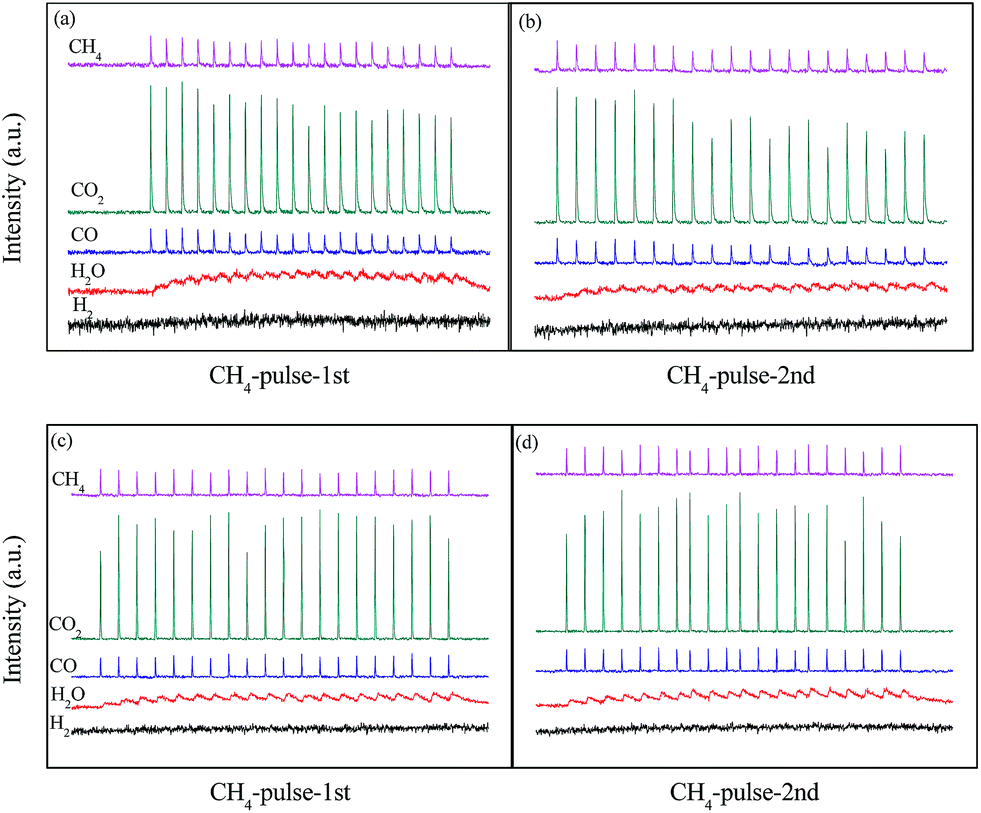 | ||
| Fig. 12 CH4 pulse reaction at 700 (a, b) and 800 °C (c, d) on LaCoO3 (a and c are the first run of CH4 pulse reaction, b and d are the second run). | ||
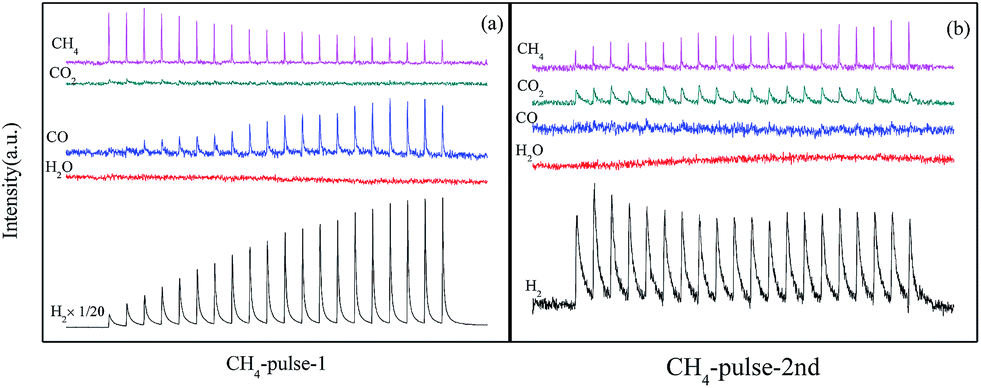 | ||
| Fig. 13 CH4 pulse reaction on Co/La2O3 at 700 °C (a: first run, b: second run). | ||
For the CH4 pulse reaction on LaCoO3 at 700 °C (CH4-1st, Fig. 12a), most of the CH4 is consumed in 20 pulses, accompanied by the production of CO2 and H2O simultaneously. At the same time, weak signals of CO (m/z = 28) are also detected. Based on the standard spectra of CO2, the CO signals may be induced by dissociative ionisation of CO2 in the chamber of mass spectrometer. After the CH4 pulses, 20 pulses of O2 were passed through catalyst bed, CO2/CO and H2O were not detected during this process (not shown).
For the second run of CH4 pulse reactions (CH4-2nd, Fig. 12b), the similar results to those in CH4-1st are obtained, which indicates CH4 is oxidized by the lattice and/or adsorbed oxygen on LaCoO3 to produce CO2 and H2O, but insufficient lattice oxygen or limited diffusion rate of lattice oxygen from bulk to surface leads to the residual CH4.
The signals of CO (m/z = 28) in the pulse reaction are induced by the dissociative ionisation of CO2 in the chamber of mass spectrometer, while not from the reaction production. We have already explained this phenomenon in the CH4 pulse reaction on LaCoO3 at 700 °C. However, the results in Fig. 1 show the production of CO and H2 in the deoxidization reaction when the temperature is higher than 720 °C. It should be noted that the structure of the perovskite LaCoO3 transforms to Co/La2O3 in the deoxidization reaction at a temperature higher than 720 °C (Fig. 4).
In order to further investigate CBM deoxygenation reaction on Co/La2O3, the LaCoO3 with perovskite structure is pretreated with 10% H2/N2 at 800 °C to obtain Co/La2O3 (Fig. 7), then the pulse reactions are performed after purging with He for 0.5 h. In the first run of CH4 pulse reaction on Co/La2O3 at 700 °C (Fig. 13a), most of the CH4 is consumed while H2 and CO is generated, and the amount of H2 and CO significantly increases with an increase in the number of pulses. Meanwhile, a trace amount of CO2 is detected at the beginning of the pulse reaction, and the amount of CO2 decreases gradually. After 3 pulses, the CO2 could hardly be detected.
During the following O2 pulse reaction, O2 is completely consumed in the 20 pulses due to the oxidation of Co/La2O3. CO/CO2, H2 and H2O are not observed during this process, the results are not shown.
After the O2 pulse reaction, the second run of the CH4 pulse reaction (CH4-2nd) is performed. Fig. 13b shows the production of CO2, CO and H2, and their amount remains nearly constant during the 20 pulses, which is significantly different from the results in Fig. 13a. It may be induced by the partial Co/La2O3 oxidation to CoOx/La2O3 by O2 during the process of the O2 pulse reaction. The formation of CoOx/La2O3 decreases the amount of Co/La2O3, which leads to the significant decrease of CO and H2.
4. Discussion
The results in Fig. 1 and 2 show LaCoO3 behaves with high activity and stability for CBM deoxygenation across a wide temperature range, O2 could be completely eliminated by CH4 to produce CO2 and H2O in the range of 400–720 °C, and the activity of LaCoO3 could be maintained after reaction at 400, 500, 600 or 660 °C for 100 h.Roseno et al.39 investigated the structure change of LaCoO3 in partial oxidation of CH4, and found that high temperature reduction could decompose the perovskite structure of LaCoO3 to Co/La2O3, and metallic Co was oxidized to CoO in O2, and further reacted with La2O3 to form La2CoO4 with spinel structure. During CBM deoxidization reaction, the structure of LaCoO3 gradually transfers from perovskite to Co/La2O3 depending on the reaction temperature (Fig. 4). The H2-TPR also showed the structure evolution of LaCoO3 induced by the reduction of H2 in the feed gas. Meanwhile, the destructed perovskite structure could be recovered from Co/La2O3 by calcination or reoxidation (Fig. 4 and 7). The structure evolution of LaCoO3 depending on the temperature and reaction gas is shown schematically in Fig. 14, which demonstrates that Co species could reversibly move into and out of the perovskite structure depending on the temperature and reaction atmosphere.
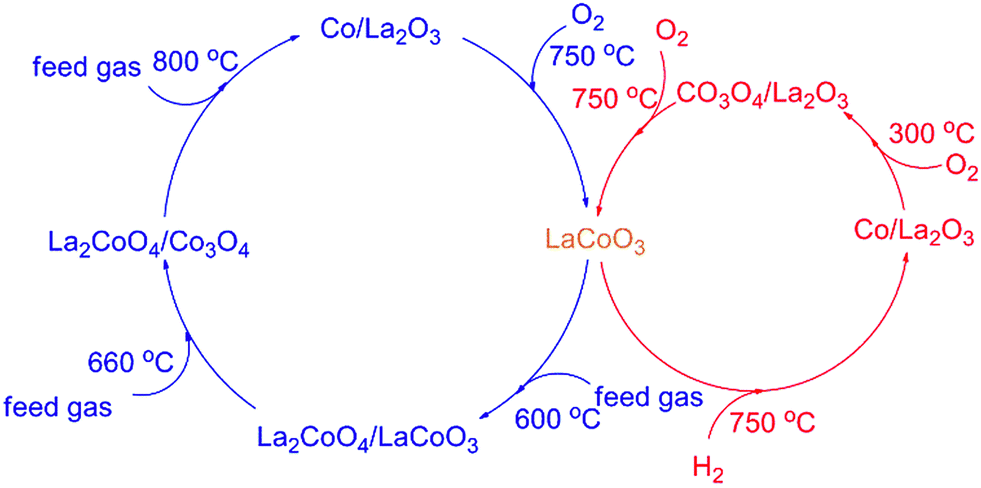 | ||
| Fig. 14 The structure evolution of LaCoO3 depending on the reaction gas and temperature. | ||
Based on the results in Fig. 7, the LaCoO3 has been reduced by 5 vol% H2/N2 (45 mL min−1) at 750 °C for 30 min to obtain Co/La2O3 (LaCoO3-R), then Co/La2O3 is reoxidized to perovskite LaCoO3 (LaCoO3-R-O). The activities of CBM deoxygenation in Fig. 15 show that LaCoO3-R behaves with much higher activity than LaCoO3-R-O, and there are no by-products of CO and H2 before 720 °C, as with LaCoO3-R-O. Compared with the result in Fig. 1, LaCoO3-R-O shows nearly the same activity as fresh LaCoO3. Combined with the results in Fig. 6 and 7, the apparently enhanced activity of LaCoO3-R in the low temperature range may be derived from the oxidation of metallic Co by O2. The above results indicate that even if the structure of LaCoO3 with perovskite is completely destroyed when the CBM deoxygenation temperature exceeds 720 °C, the structure and activity could be recovered after calcination at 750 °C in O2. Therefore, LaCoO3 like a smart catalyst, its structure could be reversibly transformed between Co/La2O3, La2CoO4 and LaCoO3 depending on the temperature and reaction atmosphere. This reversible structure evolution of LaCoO3 could meet the challenge of the shift between oxidative and reductive atmosphere typically encountered in CBM deoxygenation.49,56,57
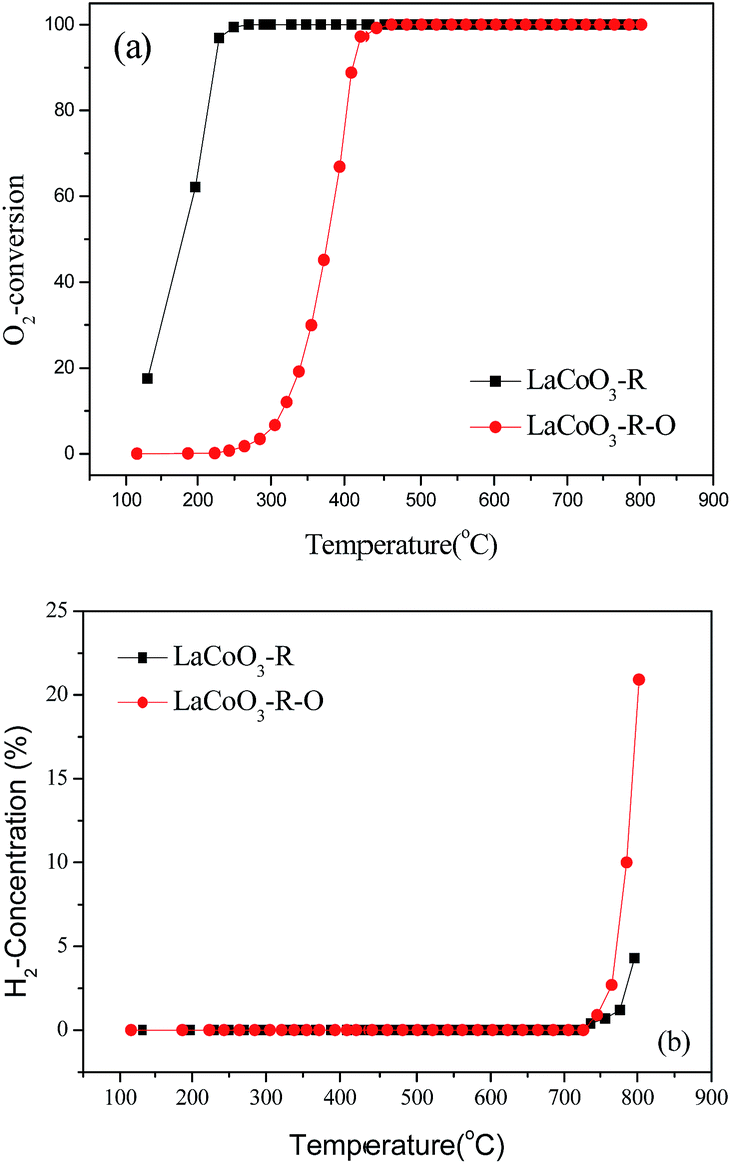 | ||
| Fig. 15 The conversion of O2 (a) and production of H2 (b) over pretreated LaCoO3 under different conditions. | ||
CH4 combustion over metal oxides catalysts is known to follow a redox mechanism, and a variety of kinetic models for the catalytic combustion of methane, such as the Eley–Rideal, Langmuir–Hinshelwood or Mas–van Krevelen mechanism.58,59 The results of isotopic tracer experiments in Fig. 10 and 11 confirms the deoxidization reaction of CBM on LaCoO3 following the Mas–van Krevelen mechanism: the lattice oxygen reacts with CH4 to produce CO2, H2O and oxygen vacancies, and the surface vacancies could be replenished by bulk lattice oxygen and gas O2, which indicates the activation of O2 should be a key factor for CBM deoxygenation reaction. The kinetic data in Fig. 8 also confirmed this. As shown in Fig. 14, LaCoO3 could continuously provide lattice oxygen, accompanying the reduction of perovskite structure to Co/La2O3; meanwhile, O2 gas could be adsorbed and dissociated on the surface, and incorporated into the lattice of the crystal as O2− species.60,61 And therefore, the perovskite LaCoO3 acted as an oxygen pump toward CBM deoxidization reaction.62
The results in Fig. 1 show the O2 could be completely eliminated by CH4 in the temperature range of 400 to 720 °C. As shown in Fig. 14, LaCoO3 could exist as perovskite, La2CoO4/Co3O4 and La2CoO4/LaCoO3 in this temperature range, which indicates that the total oxidation of CH4 by O2 will take place on the catalyst despite the structure transformation of LaCoO3 from perovskite to La2CoO4/Co3O4.
When the reaction temperature exceeds 720 °C, the CO and H2 begin to form and their amounts increase significantly with continuously increasing the temperature (Fig. 1). However, the CO and H2 could not be observed during the CH4 pulse reaction on LaCoO3 even when reaction temperature is 800 °C; the CH4 pulse reaction on reduced LaCoO3 (Co/La2O3) only produces CO and H2 at 700 °C (Fig. 13a). Meanwhile, when Co/La2O3 is partially oxidized to CoOx/La2O3, the co-existence of CoOx/La2O3 and Co/La2O3 results in the formation of CO2 and a significant decrease of CO/H2.
These results show the products of CBM deoxygenation reaction mainly depend on the structure of LaCoO3. When the Co species exists in an oxidised state, such as perovskite, La2CoO4 or CoOx/La2O3, the CBM deoxygenation only produces CO2 and H2O by the total oxidation of CH4. If Co species exists as metal, such as Co/La2O3, the preferred reaction is partial oxidation of CH4, which would lead to the formation of CO and H2.
Therefore, the CBM deoxygenation reaction on LaCoO3 at different temperatures is shown schematically in Fig. 16. When the reaction temperature is below 720 °C, CH4 reacts with the lattice oxygen to generate CO2 and H2O despite the structure transformation from perovskite to the mixture of Co3O4 and La2CoO4. With further increasing the reaction temperature, the lattice oxygen will be depleted due to the limited amount of O2 in the feed gas and the perovskite structure of LaCoO3 will be completely destroyed. Then, the partial oxidation of CH4 could take place on the surface of metallic Co to produce by-products of CO and H2.
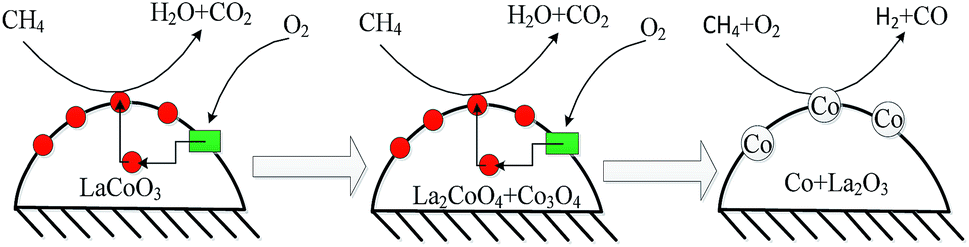 | ||
Fig. 16 CBM deoxygenation reaction on the LaCoO3 catalyst ( : lattice oxygen; : lattice oxygen;  : oxygen vacancy). : oxygen vacancy). | ||
5. Conclusions
The catalyst LaCoO3 prepared by the co-precipitation method exhibits high activity and catalytic stability for the CBM deoxidization reaction across a wide temperature range. The O2 could be completely eliminated by CH4 to produce CO2 and H2O in the range of 400–720 °C, and complete deoxidization could be maintained in the temperature range of 400–660 °C for 100 h.The perovskite LaCoO3 acts as a smart catalyst during the process of CMB deoxidization; the structure of LaCoO3 gradually transforms from perovskite to Co/La2O3 through La2CoO4/LaCoO3 and La2CoO4/Co3O4 with the increasing reaction temperature, and these different structures could be transformed into each other depending on the reaction temperature and reaction gas.
When Co species exists as Co3O4, La2CoO4 and/or LaCoO3, CH4 is completely oxidized by O2 to produce CO2 and H2O. The deoxidization of CBM on catalysts follows the Mars–van Krevelen mechanism, and the activation of O2 was a key factor in the deoxidization of CBM. When Co species exist as metal Co (Co/La2O3), the preferred reaction in CBM deoxygenation would be partial oxidation, which generates CO and H2. However, the complete oxidation of CH4 could be recovered with the structure transformation of Co/La2O3 to LaCoO3 after reoxidation by O2.
Acknowledgements
This project was supported financially by the National Key Research and Development Program of China (2016YFC0204300), the National High Technology Research and Development Program of China (2015AA034603), NSFC of China (21171055, 21333003, 21571061), the “Shu Guang” Project of the Shanghai Municipal Education Commission (12SG29), and the Commission of Science and Technology of Shanghai Municipality (15DZ1205305).Notes and references
- L. I. Guo-jun, Procedia Earth Planet. Sci., 2009, 1, 94–99 CrossRef.
- M. N. Debbagh, C. S. M. d. Lecea and J. Pérez-Ramírez, Appl. Catal., B, 2007, 70, 335–341 CrossRef CAS.
- A. Olajossy, A. Gawdzik, Z. Budner and J. Dula, Chem. Eng. Res. Des., 2003, 81, 474–482 CrossRef CAS.
- D. Zhong and P. Englezos, Energy Fuels, 2012, 26, 2098–2106 CrossRef CAS.
- S. Su and J. Agnew, Fuel, 2006, 85, 1201–1210 CrossRef CAS.
- X. Guo, J. Ren, C. Xie, J. Lin and Z. Li, Energy Convers. Manage., 2015, 100, 45–55 CrossRef CAS.
- J. Ren, C. Xie, J.-Y. Lin and Z. Li, Process Saf. Environ. Prot., 2014, 92, 896–902 CrossRef CAS.
- D. W. X. C. W. Peng, Y. Z. X. Z. Keda and D. Yiying, Coal Conversion, 2009, 4, 021 Search PubMed.
- C. Ö. Karacan, F. A. Ruiz, M. Cotè and S. Phipps, Int. J. Coal Geol., 2011, 86, 121–156 CrossRef CAS.
- C. J. Bibler, J. S. Marshall and R. C. Pilcher, Int. J. Coal Geol., 1998, 35, 283–310 CrossRef CAS.
- T. Thielemann, B. Cramer and A. Schippers, Org. Geochem., 2004, 35, 1537–1549 CrossRef CAS.
- C. H. Bartholomew, Appl. Catal., A, 1993, 107, 1–57 CrossRef CAS.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- F. Yin, S. Ji, P. Wu, F. Zhao and C. Li, J. Catal., 2008, 257, 108–116 CrossRef CAS.
- D. Kim, S. Woo, J. Lee and O. B. Yang, Catal. Lett., 2000, 70, 35–41 CrossRef CAS.
- R. D. Waters, J. J. Weimer and J. E. Smith, Catal. Lett., 1994, 30, 181–188 CrossRef CAS.
- L. F. Liotta, G. Di Carlo, A. Longo, G. Pantaleo and A. M. Venezia, Catal. Today, 2008, 139, 174–179 CrossRef CAS.
- A. Maione, F. André and P. Ruiz, Appl. Catal., A, 2007, 333, 1–10 CrossRef CAS.
- T. V. Choudhary, S. Banerjee and V. R. Choudhary, Appl. Catal., A, 2002, 234, 1–23 CrossRef CAS.
- M. Cargnello, J. J. Delgado Jaen, J. C. Hernandez Garrido, K. Bakhmutsky, T. Montini, J. J. Calvino Gamez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef CAS PubMed.
- A. E. York, T. Xiao and M. H. Green, Top. Catal., 2003, 22, 345–358 CrossRef CAS.
- Y. H. Hu and E. Ruckenstein, Adv. Catal., 2004, 48, 297–345 CAS.
- S. Yang, J. N. Kondo, K. Hayashi, M. Hirano, K. Domen and H. Hosono, Appl. Catal., A, 2004, 277, 239–246 CrossRef CAS.
- H. Y. Wang and E. Ruckenstein, J. Catal., 2001, 199, 309–317 CrossRef CAS.
- V. R. Choudhary, B. Prabhakar, A. M. Rajput and A. S. Mamman, Fuel, 1998, 77, 1477–1481 CrossRef CAS.
- V. R. Choudhary, A. M. Rajput and V. H. Rane, Catal. Lett., 1992, 16, 269–272 CrossRef CAS.
- M. Lyubovsky, L. L. Smith, M. Castaldi, H. Karim, B. Nentwick, S. Etemad, R. LaPierre and W. C. Pfefferle, Catal. Today, 2003, 83, 71–84 CrossRef CAS.
- Q. Zhang, X.-P. Wu, Y. Li, R. Chai, G. Zhao, C. Wang, X.-Q. Gong, Y. Liu and Y. Lu, ACS Catal., 2016, 6, 6236–6245 CrossRef CAS.
- Q. Zhang, Y. Li, R. Chai, G. Zhao, Y. Liu and Y. Lu, Appl. Catal., B, 2016, 187, 38–248 CrossRef.
- Q. Zhang, X.-P. Wu, G. Zhao, Y. Li, C. Wang, Y. Liu, X.-Q. Gong and Y. Lu, Chem. Commun., 2015, 51, 12613–12616 RSC.
- D. Dissanayake, M. P. Rosynek, K. C. C. Kharas and J. H. Lunsford, J. Catal., 1991, 132, 117–127 CrossRef CAS.
- A. C. Ferreira, A. P. Gonçalves, T. A. Gasche, A. M. Ferraria, A. M. B. d. Rego, M. R. Correia, A. M. Bola and J. B. Branco, J. Alloys Compd., 2010, 497, 249–258 CrossRef CAS.
- B. Christian Enger, R. Lødeng and A. Holmen, Appl. Catal., A, 2008, 346, 1–27 CrossRef.
- F. F. Tao, J.-j. Shan, L. Nguyen, Z. Wang, S. Zhang, L. Zhang, Z. Wu, W. Huang, S. Zeng and P. Hu, Nat. Commun., 2015, 6, 7798 CrossRef CAS PubMed.
- A. J. Zarur and J. Y. Ying, Nature, 2000, 403, 65–67 CrossRef CAS PubMed.
- L. Fabbrini, A. Kryukov, S. Cappelli, G. L. Chiarello, I. Rossetti, C. Oliva and L. Forni, J. Catal., 2005, 232, 247–256 CrossRef CAS.
- R. Pereñíguez, V. M. González-DelaCruz, J. P. Holgado and A. Caballero, Appl. Catal., B, 2010, 93, 346–353 CrossRef.
- A. G. Bhavani, W. Y. Kim and J. S. Lee, ACS Catal., 2013, 3, 1537–1544 CrossRef CAS.
- K. T. C. Roseno, R. Brackmann, M. A. da Silva and M. Schmal, Int. J. Hydrogen Energy, 2016, 41(40), 18178–18192 CrossRef CAS.
- M. R. Goldwasser, M. E. Rivas, M. L. Lugo, E. Pietri, J. Pérez-Zurita, M. L. Cubeiro, A. Griboval-Constant and G. Leclercq, Catal. Today, 2005, 107–108, 106–113 CrossRef CAS.
- R. M. Navarro, M. C. Alvarez-Galvan, J. A. Villoria, I. D. González-Jiménez, F. Rosa and J. L. G. Fierro, Appl. Catal., B, 2007, 73, 247–258 CrossRef CAS.
- J. Li, L. Zhao and G. Z. Lu, Ind. Eng. Chem. Res., 2008, 48, 641–646 Search PubMed.
- L. Fabbrini, I. Rossetti and L. Forni, Appl. Catal., B, 2010, 93, 346–353 CrossRef.
- L. Marchetti and L. Forni, Appl. Catal., B, 1998, 15, 179–187 CrossRef CAS.
- J. G. McCarty and H. Wise, Catal. Today, 1990, 8, 231–248 CrossRef CAS.
- R. Lago, G. Bini, M. A. Peña and J. L. G. Fierro, J. Catal., 1997, 167, 198–209 CrossRef CAS.
- V. R. Choudhary, B. S. Uphade and A. A. Belhekar, J. Catal., 1996, 163, 312–318 CrossRef CAS.
- Å. Slagtern and U. Olsbye, Appl. Catal., A, 1994, 110, 99–108 CrossRef.
- Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto and N. Hamada, Nature, 2002, 418, 164–167 CrossRef CAS PubMed.
- C. V. Schenck, J. G. Dillard and J. W. Murray, J. Colloid Interface Sci., 1983, 95, 398–409 CrossRef CAS.
- Z. Gao and R. Wang, Appl. Catal., B, 2010, 98, 147–153 CrossRef CAS.
- B. Białobok, J. Trawczyński, W. Miśta and M. Zawadzki, Appl. Catal., B, 2007, 72, 395–403 CrossRef.
- J. A. Villoria, M. C. Alvarez-Galvan, S. M. Al-Zahrani, P. Palmisano, S. Specchia, V. Specchia, J. L. G. Fierro and R. M. Navarro, Appl. Catal., B, 2011, 105, 276–288 CrossRef CAS.
- Y. Han, L. Chen, K. Ramesh, E. Widjaja, S. Chilukoti, I. Kesumawinatasurjami and J. Chen, J. Catal., 2008, 253, 261–268 CrossRef CAS.
- J. Xu, Y. Q. Deng, Y. Luo, W. Mao, X. J. Yang and Y. F. Han, J. Catal., 2013, 300, 225–234 CrossRef CAS.
- N. Guilhaume, S. D. Peter and M. Primet, Appl. Catal., B, 1996, 10, 325–344 CrossRef CAS.
- H. Tanaka, I. Tan, M. Uenishi, M. Taniguchi, M. Kimura, Y. Nishihata and J. i. Mizuki, J. Alloys Compd., 2006, 408–412, 1071–1077 CrossRef CAS.
- N. Bahlawane, Appl. Catal., B, 2006, 67, 168–176 CrossRef CAS.
- V. Belessi, A. Ladavos, G. Armatas and P. Pomonis, Phys. Chem. Chem. Phys., 2001, 3, 3856–3862 RSC.
- S. Royer, H. Alamdari, D. Duprez and S. Kaliaguine, Appl. Catal., B, 2005, 58, 273–288 CrossRef CAS.
- G. Saracco, G. Scibilia, A. Iannibello and G. Baldi, Appl. Catal., B, 1996, 8, 229–244 CrossRef CAS.
- R. Hammami, S. B. Aïssa and H. Batis, Appl. Catal., A, 2009, 353, 145–153 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |