
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrophilic surface molecularly imprinted naringin prepared via reverse atom transfer radical polymerization with excellent recognition ability in a pure aqueous phase†
Xuexue Feng ,
Tielei Wu,
Bingqing Yu,
Yan Wang* and
Shian Zhong*
,
Tielei Wu,
Bingqing Yu,
Yan Wang* and
Shian Zhong*
School of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China. E-mail: 1316365506@qq.com; Tel: +86 15111244989
First published on 26th May 2017
Abstract
An easy and highly efficient method using hydrophilic surface molecularly imprinted polymers (SMIPs) for the selective recognition of naringin from pure aqueous solution was developed. A macroporous support using metal organic gels (MOGs) as the porogen was initially synthesized. Then, surface-initiated reverse atom-transfer radical polymerization (R-ATRP) was performed on the support surface. Finally, hydrophilic SMIPs with abundant molecular imprinting recognition sites were obtained. The SMIPs were characterized by infrared spectroscopy, high performance liquid chromatography, thermogravimetry, scanning electronic microscopy, static contact angle analysis, and nitrogen adsorption–desorption measurements. The aqueous extraction solution can be directly absorbed by SMIPs without adjusting pH and adsorption can occur under neutral conditions. Equilibrium adsorption was reached rapidly within 3 min, and the maximum equilibrium binding capacities of SMIPs were found to be 63.54 μmol g−1 for naringin. Selective analysis indicated a high selectivity of SMIPs towards template naringin over hesperidin and rutin. Extraction experiments suggested excellent separation and purification of naringin in an aqueous grapefruit extract, with a recovery rate of 93% and at least thrice usage of SMIPs.
1. Introduction
Naringin, a flavonoid, has many pharmacological activities such as anticancer, antidepression, antioxidation, diabetes treatment, and osteoporosis therapy.1,2 Naringin has wide ranging applications in medicine and food. The main approach to obtain naringin is to extract it from the fruit of a citrus plant of the rutaceae family. A variety of methods have been used in the separation and purification of naringin, such as silica gel column chromatography, liquid–liquid extraction, and recrystallization.3,4 The methods are always used in combination to achieve better purification due to the lack of naringin specificity, resulting in a long process and high costs. In addition, use of volatile toxic organic solvents results in production safety and environmental and solvent residue problems. Thus, a method with high efficiency of special recognition, mild operational conditions, and environmental friendliness should be developed. This work develops surface molecularly imprinted polymers (SMIPs) with strong specificity to solve these problems.Molecular imprinted polymers (MIPs) as facile adsorption materials are developed for the preparation of specific template molecules.5–8 MIPs have shown desired selectivity and physical stability and have been widely applied in solid-phase extraction, catalysis, drug development and so on.9–11 Functional monomers, initiator, and cross-linkers are copolymerized in the presence of template molecules.12,13 After the removal of template molecules from the polymer, the binding sites within a rigid cavity are achieved.
Surface molecular imprinting technology is based on the molecular imprinting technology, obtaining the molecular recognition sites on the surface of the polymers. Most of the reported SMIPs are synthesized by free radical initiation polymerization and sol–gel polymerization,14,15 which have some drawbacks. The polymer film may be too thick, and the template molecules are buried in the polymer due to the uncontrollable rate of polymerization. In addition, the imprinting sites are heterogeneous, which results in the unsuitable kinetics of the adsorption process.16,17 Therefore, reverse atom-transfer radical polymerization (R-ATRP) is introduced to solve these problems. Atom-transfer radical polymerization (ATRP) is a kind of living/controlled polymerization method, showing some merits, such as controlled molecular weight and narrow distribution. However, the initiator used in ATRP is usually an organic halide (R–X) with high toxicity and the catalysts used are easily oxidized by air.18,19 R-ATRP can solve these problems. R-ATRP uses conventional radical polymerization initiators—higher oxidation state transition metal compounds which is easy to preserve, making it possible to simplify the controllable reaction.20,21 The thickness of the SMIPs prepared by R-ATRP can be adjusted conveniently by controlling the polymerization time to make the template molecules not to be buried in the polymer and the imprinting sites available to the template molecule. In addition, the molecular imprinting process of our materials is exothermic due to the form of the cyclic boronic ester. Therefore, the conventional heating polymerization or thermal initiator was not conducive to the following reaction. The SMIPs can be obtained at room temperature via R-ATRP, which is beneficial to the molecular imprinting process. In this article, azobis-(4-cyanopentanoic acid) (ACPA) immobilized on the surface of the macroporous supports is used as the photo-initiator to initiate the graft polymerization of the polymers.
Moreover, SMIPs prepared by traditional free radical polymerization technique are liable for aggregation due to the hydrophobicity, and they recognize the target molecule in the organic or organic-rich phase.14,15 Therefore, the specific adsorption performance is weakened in the aqueous phase, which limits their application in food, environmental and biological fields.22–24 In addition, a poor hydrophilicity is generated due to the use of the monomer and cross-linker. Though the SMIPs with molecular recognition ability in the water phase have been prepared.25–27 The molecularly imprinting polymers obtained indicated a low molecularly imprinted efficiency. Therefore, the development of SMIPs with high efficiency of molecular recognition in pure aqueous phase remains a challenge. To solve the problem of water solubility, the hydrophilic SMIPs were formed by using the boronic acid group. The boronic acid group was found to be of great advantage in a variety of biomedical applications for derivatives of mannose, galactose, and sialic acids.3 A cis-diol existed in the molecular structure of naringin, which was available for forming boronate ester. Therefore, the hydrophilic 4-vinylphenylboronic acid (4-VPBA) was chosen to react with naringin in the pre-assembly process. In addition, the appropriate proportions of cross-linker ethyleneglycol dimethacrylate (EDMA) were used in the preparation process of SMIPs (EDMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4-VPBA = 20:1), which can make the template naringin and SMIPs have similar polarity. As a result, the template molecule naringin can be adsorbed by SMIPs in the pure aqueous phase.
:4-VPBA = 20:1), which can make the template naringin and SMIPs have similar polarity. As a result, the template molecule naringin can be adsorbed by SMIPs in the pure aqueous phase.
However, the number of the functional monomers can be reduced due to the use of a high proportions of cross-linker, resulting in a reduced number of imprinting sites. To solve the problem, the macroporous supports with a diameter of several tens of nanometers were used, providing more attachment surface for the formation of SMIPs and ensuring the number of imprinted sites. The metal–organic gels (MOGs) were introduced for use as the porogen to form a macroporous support in this article. MOG is a gel form of metal organic framework (MOF). They are special due to easy tunability of the pore size and shape,28–30 and have been used in many fields such as gas storage, catalysis, separation, purification and drug delivery.31–34 The SMIPs with abundant molecular imprinting recognition sites can be obtained and the resulting SMIPs possess a relatively fast adsorption dynamic and special recognition ability of the naringin. Additionally, the use of MOGs can further solve the problem of the embedding of template, which may also contribute to the improvement of the hydrophilicity of SMIPs.
First, a macroporous support using MOGs as porogen was synthesized. Then, the ACPA was grafted onto the surface of the macroporous support to obtain pGMA–EDMA–ACPA microspheres. At last, SMIPs were fabricated with surface-initiated R-ATRP by using naringin as a template, 4-vinylphenylboronic acid as the functional monomer, EDMA as the cross-linker and the ACPA that was grafted onto pGMA–EDMA–ACPA microspheres as photo-initiator, respectively, by UV irradiation at room temperature.
To the best of our knowledge, SMIPs with excellent recognition performance are formed for the first time by surface-initiated R-ATRP on the surface of macroporous supports.
2. Experimental
2.1 Materials
Naringin, nesperidin, rutin, glycidyl methacrylate (GMA), ethyleneglycol dimethacrylate (EDMA), 3-aminophenylboric acid (APBA), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O), trimesinic acid (H3BTC), azobis-(4-cyanopentanoic acid) (ACPA), 2,2-azobisisobutyronitrile (AIBN), 4-vinylphenylboronic acid (4-VPBA), cupricchloride (CuCl2), 2,2′-dipyridyl (2,2′-bpy), triethylamine, dimethyl sulfoxide (DMSO), were purchased from Aladdin Company (Shanghai, China). Phosphate buffer solution (PBS, 0.01 M). Triethylamine (TEA) was dried by potassium hydroxide. All reagents were of analytical grade.2.2 Instrumentation and characterizations
An ultraviolet (UV) spectrophotometer (SHIMADZU, UV-2450) was employed to obtain UV adsorption spectra. FT-IR spectrometer (Nicolet, 6700, USA) was applied to detect the spectral line of the products in each procedure between 4000 cm−1 and 400 cm−1 with a resolution of 4 cm−1, by accumulating 32 scans. The scanning electron microscopy (FEI Sirion 200) was employed to characterize the morphology of polymers. NMR spectra were recorded using Bruker AV-400 spectrometer at room temperature. HPLC analysis was performed on an LC 1060 HPLC system. TGA was performed using a Perkin-Elmer TGA-7 with a heating rate of 10 °C min−1 under nitrogen. The contact angle machine was used to test the hydrophilicity of SMIPs. Static contact angle measurements were carried out on a JC2001C contact angle measurement system with DI water drops of 3 mL on the surface of the materials at room temperature. Specific areas were determined by the nitrogen adsorption–desorption method using Quantachrome Instruments, NOVA 1000 e Surface Area & Pore size analyser.2.3 Preparation of SMIPs
In the pre-assembly process, the naringin (0.1 mmol and 58.1 mg) and 4-VPBA (0.1 mmol and 14.8 mg) were added into water and ethanol (10 mL and 1:1, v/v) in a quartz flask. The mixture was stirred at 15 °C for 75 min. The 1H NMR spectroscopy of the pre-assembly solution was shown in Fig. S1.† To optimize the pre-assembly conditions, the pGMA–EDMA–APBA polymers were composed. The synthesis of pGMA–EDMA–APBA polymers was shown in Fig. S2.† The effect of adsorption pH, adsorption time, adsorption temperature on the binding capacity in self-assembly process were shown in Tables S2, S3, and S4,† respectively. CuCl2 (0.03 mmol and 4.03 mg) and 2,2′-bpy (0.06 mmol and 9.37 mg) were added to the pre-assembly solution and dissolved fully. Then, macroporous pGMA–EDMA supports (100 mg) and EDMA (0.19 mL and 2 mmol) were added, and polymerization was carried out by UV-irradiation with continuous stirring for 6 h (r.t., N2). The obtained products were washed with EtOH and AcOH to remove the template until no naringin was detected in the eluent. Then, the product was dried under a vacuum at 50 °C, and the SMIPs were obtained.
The SNIPs were prepared as a blank without naringin template for comparison.
2.4 Binding experiments
The adsorption kinetics was explored by changing the adsorption time from 0 min to 45 min while keeping the same initial concentration of naringin at 20 mg L−1. The effect of pH on the adsorption capacity was investigated in the pH ranging from 4.0 to 9.0, and pH was adjusted with PBS. The isothermal adsorption was studied by changing the concentrations of naringin from 15 mg L−1 to 100 mg L−1. The adsorption thermodynamics was explored by keeping the shaking temperature at preset temperature (288, 298, and 308 K) for 5 min. Adsorption selectivity experiments were verified using a naringin solution (hesperidin and rutin) at a concentration of 30 mg L−1. The extraction applied experiments ware studied by controlling the temperature at 298 K for 5 min. In all of the above experiments, the mass of SMIPs or SNIPs was 30 mg, the volume of the adsorption solution was 10 mL, and polymers were separated by centrifugation at 9000 rpm for 5 min. The concentration of naringin or its analogs in the supernatant was determined using a UV spectrometer at 282 nm. The centrifugation sediment was washed with water until no naringin was detected. Then, the sediment was washed and shook three times with 10 mL acetic acid solution (5%) and ethanol (1:1, v/v) for 5 min at 298 K. After centrifugation separation, the amount of naringin released to the supernatant was also quantified with UV spectrophotometry at 282 nm. The recovery rate of naringin was obtained by comparing the released amount with the absorbed amount of naringin.
2.5 Extraction-applied experiments
Grapefruit peel (2.0 g) and ethanol (70%, 40 mL) were stirred at 60 °C for 1 h37 and filtered. The solution was removed of ethanol by distillation under reduced pressure and then kept at 50 mL with water. Of the solution, 1 mL was accurately taken out and diluted to 50 mL with water to obtain the grapefruit extract.
3. Results and discussion
3.1 Preparation of the SMIPs
The synthesis strategy of SMIPs is exhibited in Fig. 1. The porogen used to prepare macroporous pGMA–EDMA supports in general is a mixed solvent38 or ionic liquids.39 However, the pore size of the obtained polymers is very small, leading to the embedding of the imprinted sites. In addition, the preparation process and the selection and optimization of the porogenic system are complex. This article used MOGs as porogen for preparing macroporous pGMA–EDMA supports, and the imprinting sites of the obtained SMIPs were not easily embedded, which is beneficial to the recognition and resolution of template molecules.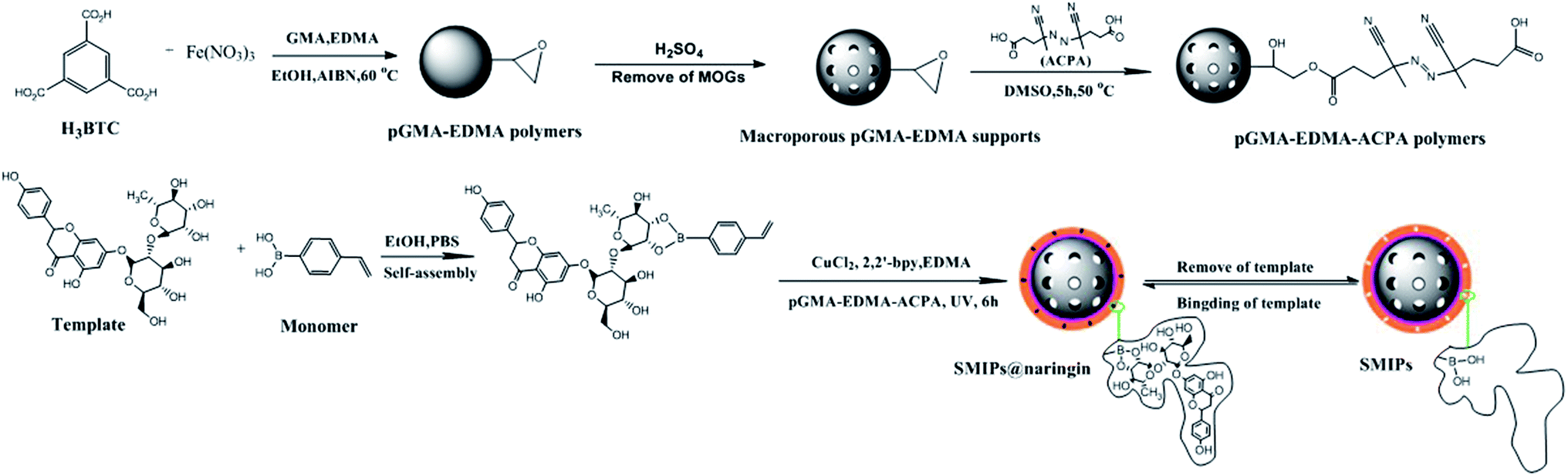 | ||
| Fig. 1 Scheme of the preparation of SMIPs. | ||
4-VPBA was selected as the functional monomer and self-assembled with the template naringin. The boronic acid group of the 4-VPBA is able to strongly interact with the dihydroxy of naringin to form a stable five-ring borate ester. The affinity is pH-dependent, and naringin is bound to a boronic acid group in the weak alkaline condition and released into the eluate in acidic conditions,40 which is beneficial to the assembly and removal of template molecules. The formation of the borate ester was exothermic; therefore, the conventional heating polymerization or thermal initiator was not conducive to the following reaction. In the end, ACPA as photo-initiation was chosen, and the reaction was carried at room temperature. ACPA was grafted onto the surface of the macroporous pGMA–EDMA supports through opening the epoxy ring, initiating the polymerization on the surface of the supports, achieving surface molecular imprinting. In addition, SMIPs@naringin were obtained by the surface R-ATRP. The thickness of the molecularly imprinted membrane can be controlled by adjusting the polymerization time (Fig. S3†), enabling the controlled polymerization of SMIPs.
The IR spectra of macroporous pGMA–EDMA supports (a), pGMA–EDMA–ACPA (b), naringin (c), naringin@4-VPBA (d), 4-VPBA (e), SMIPs (f), and SMIPs@naringin (g), respectively, is shown in Fig. 2. In the FT-IR spectrum of macroporous pGMA–EDMA supports (a), the peaks at 860 cm−1 and 753 cm−1 contributed to epoxy stretching vibrations. In the FT-IR spectrum of pGMA–EDMA–ACPA (b), the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N deformation vibration appeared at about 2170 cm−1. Furthermore, the peaks at around 860 cm−1 and 753 cm−1 corresponding to the epoxy disappeared, proving that ACPA was successfully grafted on the macroporous pGMA–EDMA supports by reaction with epoxy groups.
N deformation vibration appeared at about 2170 cm−1. Furthermore, the peaks at around 860 cm−1 and 753 cm−1 corresponding to the epoxy disappeared, proving that ACPA was successfully grafted on the macroporous pGMA–EDMA supports by reaction with epoxy groups.
 | ||
| Fig. 2 FT-IR spectra of macroporous pGMA–EDMA supports (a); pGMA–EDMA–ACPA (b); naringin (c), naringin@4-VPBA (d); 4-VPBA (e); SMIPs (f); SMIPs@naringin (g). | ||
In the naringin spectra (c), the band at 1080 cm−1 and 1641 cm−1 was assigned to the peak corresponding to C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. In the 4-VPBA spectra (e), the band at 1600 cm−1, 1561 cm−1, and 1500 cm−1 belongs to the benzene ring stretching vibrations of 4-VPBA. In the naringin@4-VPBA spectra (d), the appearance of the band at 1360 cm−1 of was assigned the characteristic absorption peak of B–O stretching vibration, and the peak at 1450 cm−1 and 1080 cm−1 belongs to the benzene ring stretching vibrations. It indicates the successfully introduced naringin and the presence of the double bond of the 4-VPBA in the polymer.
O, respectively. In the 4-VPBA spectra (e), the band at 1600 cm−1, 1561 cm−1, and 1500 cm−1 belongs to the benzene ring stretching vibrations of 4-VPBA. In the naringin@4-VPBA spectra (d), the appearance of the band at 1360 cm−1 of was assigned the characteristic absorption peak of B–O stretching vibration, and the peak at 1450 cm−1 and 1080 cm−1 belongs to the benzene ring stretching vibrations. It indicates the successfully introduced naringin and the presence of the double bond of the 4-VPBA in the polymer.
Compared with the pGMA–EDMA–ACPA spectra (b), the CN deformation vibration disappeared at 2170 cm−1 in the spectra of SMIPs (f) and SMIPs@naringin (g). This further proves that the initiator ACPA has been consumed in the reaction and naringin has been successfully assembled on the 4-VPBA. Furthermore, the band at 1641 cm−1 in the SMIPs@naringin spectra (g) disappeared in the SMIPs spectra (f), which further indicates the successful coating and desorption of naringin. The SMIPs spectra (f) and SMIPs@naringin spectra (g) did not exhibit a significant difference, demonstrating that the PBS would not negatively affect the polymer structure.
3.2 Characterization
The thermo gravimetric analysis (TGA) curves of naringin (a), SMIPs@naringin (b), and SMIPs (c) is shown in Fig. 3. Line naringin (a) shows naringin weight loss (90%) at >200 °C. SMIPs@naringin (line b) shows enhanced thermal stability. The weight loss was observed between 200 °C and 300 °C (from 5% to 40%), attributing to the thermal decomposition of naringin. When the sample was heated up to 400 °C, the mass declined dramatically to 20%, which is due to the decomposition of the polymer framework. Line SMIPs (c) shows a slight drop in the mass of SMIPs (5%) below 200 °C and shows no weight loss between 200 °C and 300 °C, indicating that naringin was totally removed and that the imprinting process belongs to the surface molecular imprinting. The curve was flat, indicating that the SMIPs are extremely stable in this range. When the temperature rises to 300 °C, the SMIPs lost much weight (55%), indicating that the polymer were decomposed. The data measured by TGA were in keeping with those of FTIR, suggesting the successful coating of the naringin on the supports.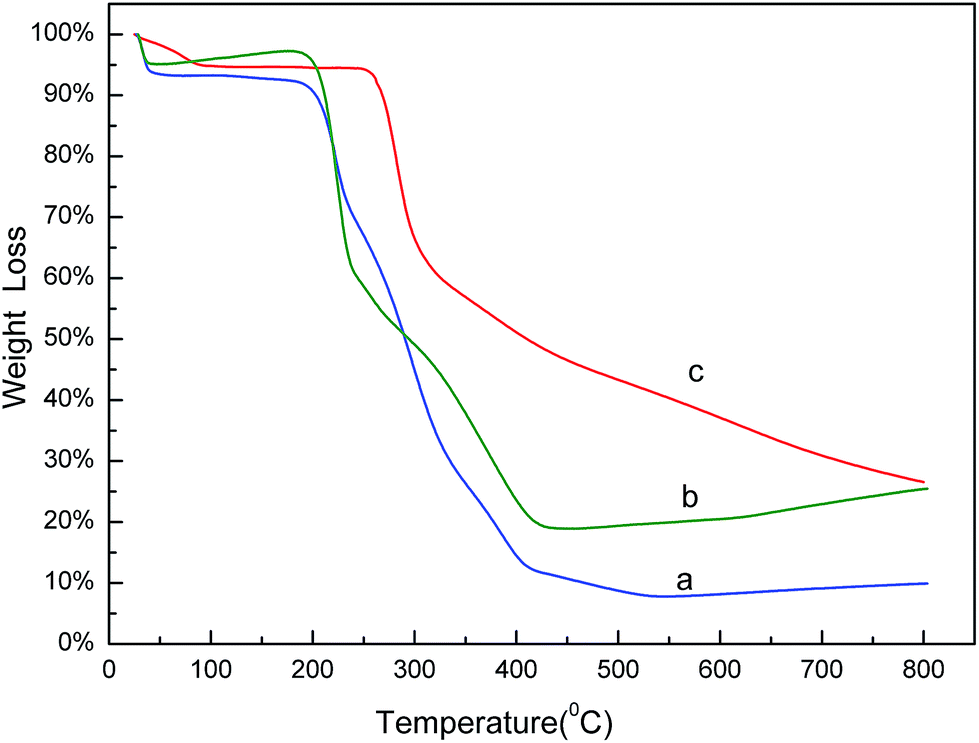 | ||
| Fig. 3 TGA curves of naringin (a), SMIPs@naringin (b), SMIPs (c). | ||
The SEM images of macroporous pGMA–EDMA supports (a), SMIPs (b), and SNIPs (c) are shown in Fig. 4. Compared with the SEM image of macroporous pGMA–EDMA supports (a), SMIPs (b) and SNIPs (c) did not show much difference. The reason is that the imprinting film on the surface of the support is very thin. It further shows that the SMIPs surfaces were rougher and fluffier than that of SNIPs, indicating the successful grafting of naringin on the supports.
 | ||
| Fig. 4 SEM images of macroporous pGMA–EDMA supports (a); SMIPs (b); SNIPs (c) (magnification: 100000×). | ||
Nitrogen adsorption–desorption measurements (Fig. 5) demonstrated that the specific surface area, pore volume, and average pore size of the pGMA–EDMA supports were 41.87 m2 g−1, 0.11 cm3 g−1, and 56.5 nm, respectively. As for the SMIPs, the number fell down to 4.04 m2 g−1, 0.027 cm3 g−1, and 25.7 nm, respectively, indicating a macroporous structure of the pGMA–EDMA support and also suggesting not only the surface of the pGMA–EDMA support but also the inner cavities were grafted with a thin layer of organics. The film thickness was calculated to be 5 nm. As for the SNIPs, the number fell down to 8.38 m2 g−1, 0.073 cm3 g−1, and 35.1 nm, respectively. The film thickness was calculated to be 5 nm. In addition, the specific surface area of SMIPs and SNIPs with the polymerization time of 5, 6, 7, and 8 h were 5.49, 4.04, 2.82 and 1.95 m2 g−1, respectively and 10.79, 8.38, 4.52 and 3.93 m2 g−1, respectively, demonstrating a controllable surface imprinting.
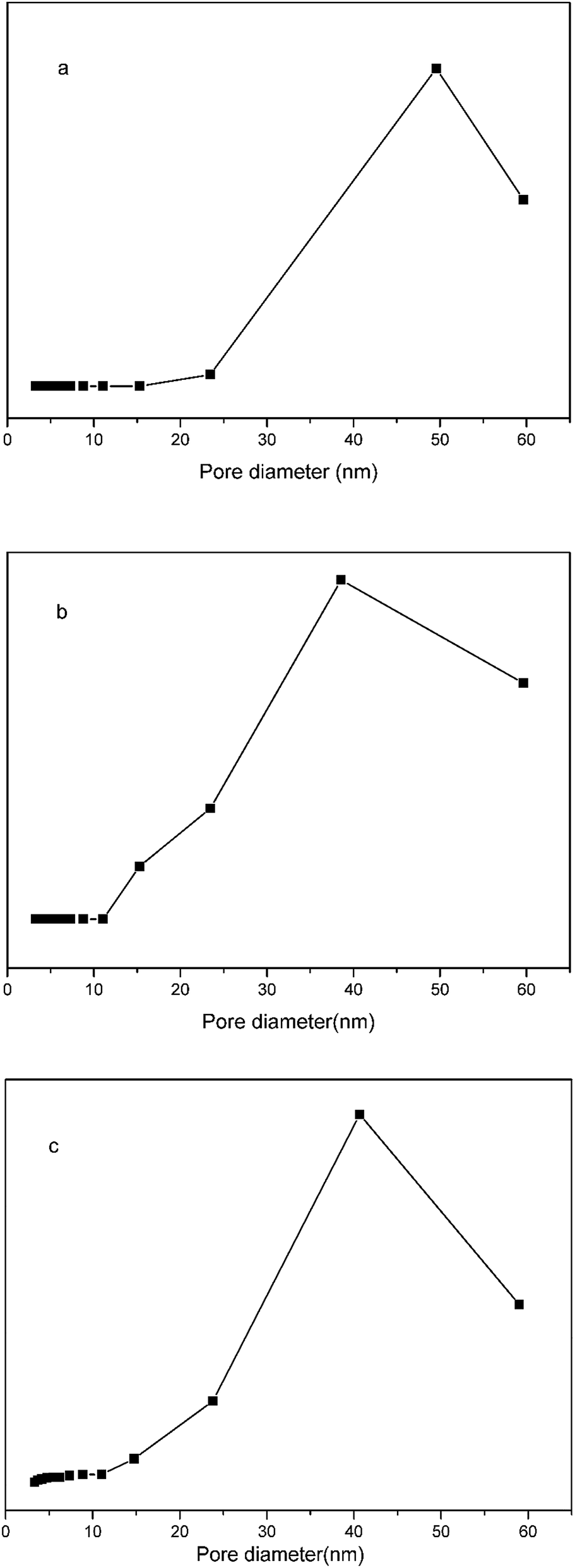 | ||
| Fig. 5 Nitrogen adsorption isotherms for the macroporous pGMA–EDMA supports (a), the SMIPs (b) and the SNIPs (c). | ||
The static contact angle measurements of the SMIPs are shown in Fig. S4.† The hydrophilicity of the SMIPs can be accurately evaluated by performing water contact angle experiments. Therefore, the static water contact angles of the SMIPs were determined to obtain more insight into their surface properties. The use of MOGs can solve the problem of the embedding of the template, which may contribute to the improvement of the hydrophilicity of SMIPs. The static water contact angle was 49.5°, indicating that the SMIPs can be used in aqueous phase.
3.3 Effect of pH on the binding capacity
The adsorption curves of the naringin onto SMIPs and SNIPs are shown in Fig. 6. Adsorption capacity was strongly influenced by the pH of the solution. An increase was observed with increased pH of the PBS in the two curves, as shown in Fig. 6. Adsorption capacity sharply increased at the beginning of the process but was unchanged with increased pH when the pH was 5.0–7.0. However, adsorption ability decreased with further increased pH to 9.0. As a result, pH 7.0 was chosen as the optimum adsorption medium. In addition, water can be used in the adsorption process, and the extraction solution can be directly absorbed by SMIPs without adjusting pH.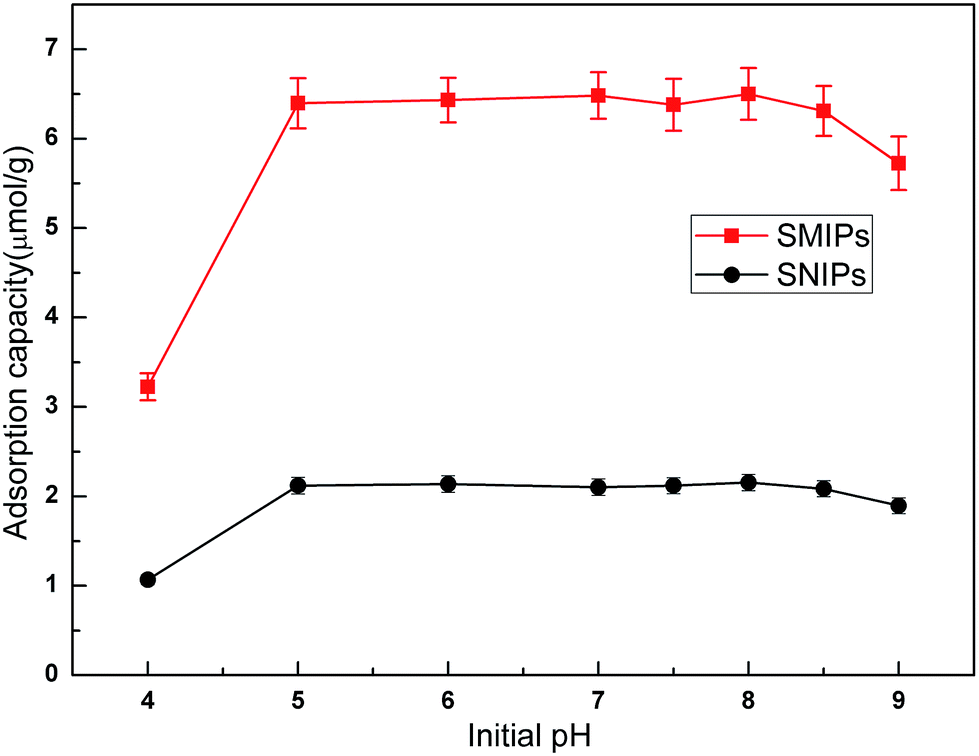 | ||
| Fig. 6 Effect of initial pH on the adsorption of naringin onto SMIPs and SNIPs. Initial concentration of naringin = 20 mg L−1, T = 298 K, contact time = 5 min. | ||
3.4 Adsorption kinetics, isotherm, and thermodynamics
The adsorption kinetics curves of naringin onto SMIPs and SNIPs are shown in Fig. 7. The curves are uncommon for adsorption processes. A sharp increase was observed in the two curves within the first 3 min because of the presence of vast accessibility and empty binding sites. In addition, a higher adsorption capacity was reached for SMIPs, compared with SNIPs, demonstrating that the specific recognition sites were successfully formed on the surface of the polymers. After 3 min, the adsorption equilibrium of SMIPs and SNIPs were both reached due to the occupancy of the majority of the binding sites. The SMIPs as-prepared clearly exhibited a shorter equilibrium time, compared to the traditional SMIPs (about 3 h).3,41 This was attributed to the surface molecule imprinting of the macroporous polymers and the recognition sites that were not embedded.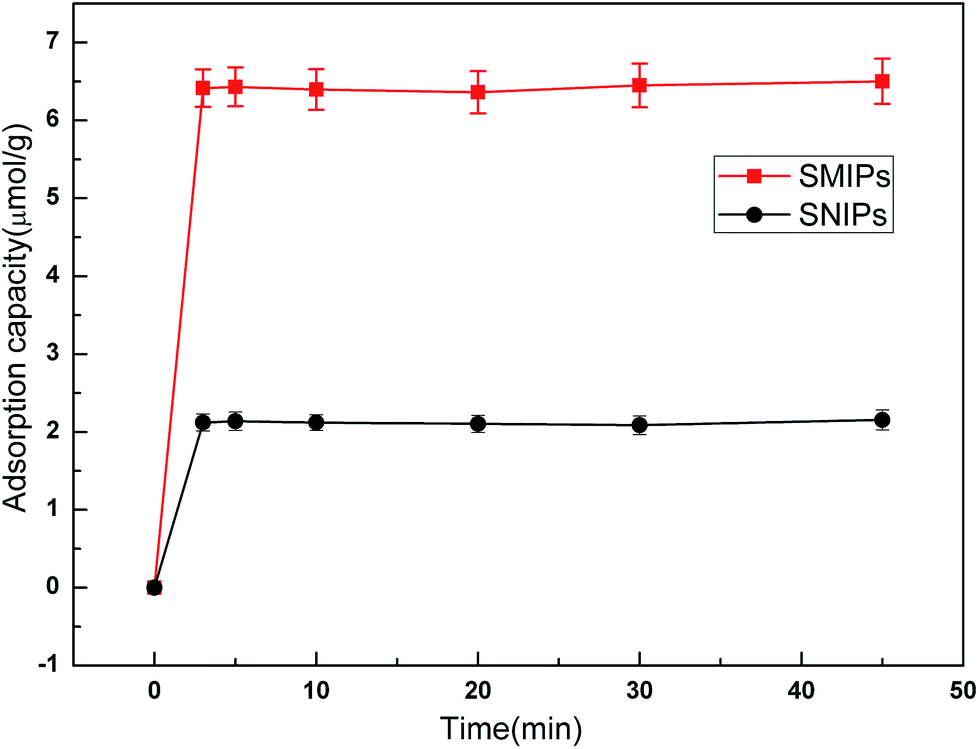 | ||
| Fig. 7 The adsorption dynamic of naringin onto SMIPs and SNIPs. Initial concentration of naringin = 20 mg L−1, T = 298 K, pH = 7.0. | ||
Adsorption isotherm curves provided valuable information to investigate the adsorption capacity. The equilibrium adsorption of naringin onto SMIPs and SNIPs is shown in Fig. 8. With a certain amount of SMIPs (SNIPs), adsorption capacity increased with increased naringin concentration. Compared with SNIPs, the SMIPs exhibited favorable adsorption performance, indicating the molecular imprinting effect.
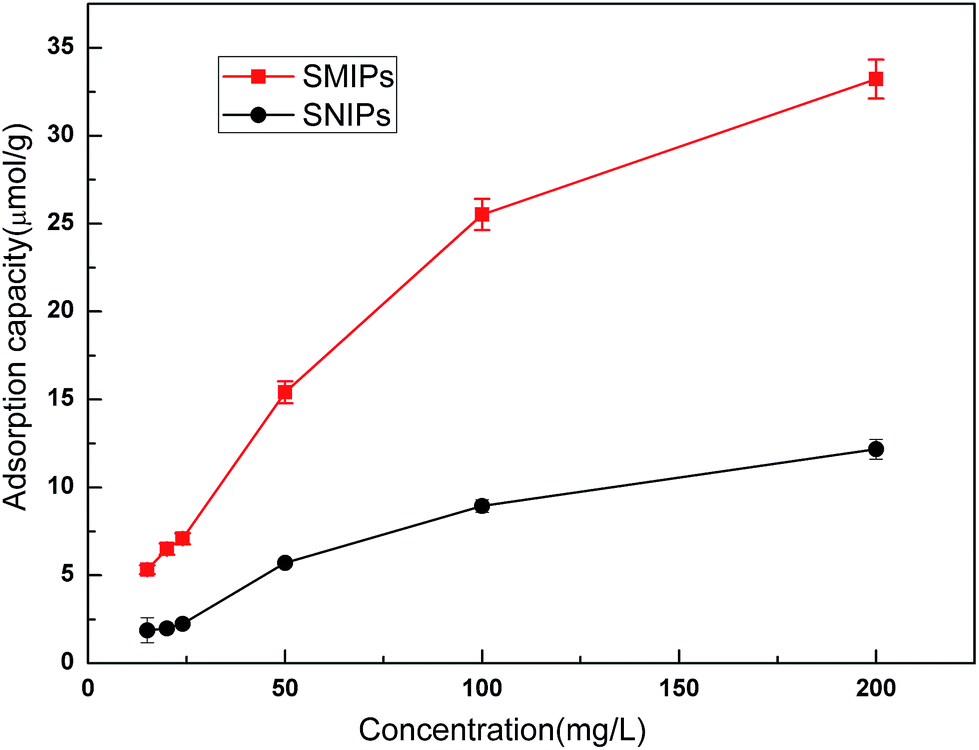 | ||
| Fig. 8 The adsorption isotherm curves of SMIPs and SNIPs. Contact time = 5 min, T = 298 K, pH = 7.0. | ||
The Scatchard curve of SMIPs and SNIPs was obtained according to the equation:
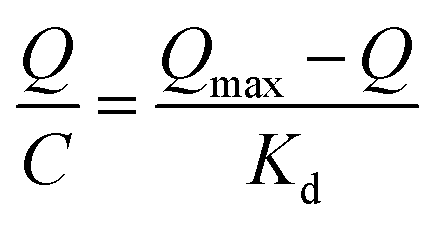
As shown in Fig. S5a,† the Scatchard plot showed a single straight line. The equation was Q/C = 226.06 − 3.558Q (R2 = 0.902). The Kd and Qmax of SMIPs were calculated to be 0.281 mmol L−1 and 63.54 μmol g−1, respectively. However, Fig. S5b† is not consistent with the Scatchard model. The equation was Q/C = 68.83 − 2.34Q (R2 = 0.487). These suggested that there is a significant difference in the spatial structure of the SMIPs and SNIPs. The adsorption of naringin on SNIPs is mainly non-specific adsorption, indicating relatively weak adsorption capacity. The SMIPs contain a functional group of fixed holes and the size and functional groups of the holes were complementary with naringin, resulting in high selectivity and strong adsorption capacity.
The adsorption thermodynamics curves of the naringin onto SMIPs and SNIPs are shown in Fig. 9. The adsorption thermodynamics behavior was determined by operating the adsorption experiment at temperatures of 288, 298, and 313 K. A higher adsorption capacity was reached on SMIPs, compared with SNIPs. In addition, the adsorption ability decreased with increased temperature, indicating that the adsorption process is exothermic. The performance was due to the interaction of covalent bond between naringin and 4-VPBA. However, as for SNIPs, the adsorption capacity slowly decreased with increased temperature. The reason was that the physical adsorption was dominated in the adsorption process.
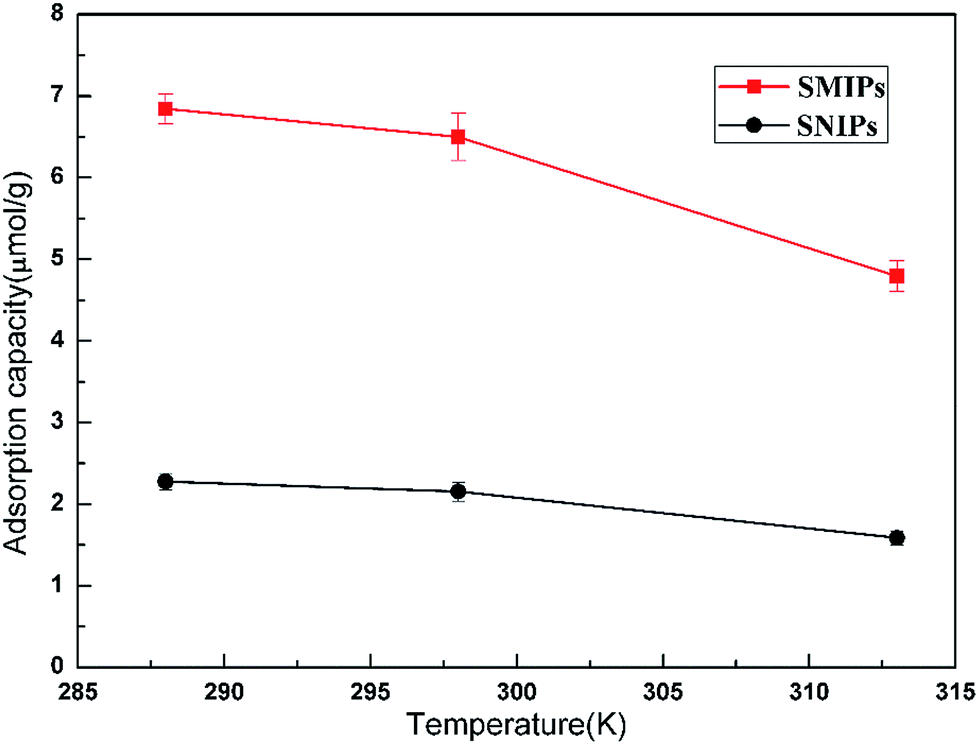 | ||
| Fig. 9 The effect of temperature on the adsorption of naringin onto SMIPs and SNIPs. Initial concentration of naringin = 20 mg L−1, contact time = 5 min, pH = 7.0. | ||
3.5 Selective adsorption
To evaluate the selectivity of the SMIPs for naringin, two structural homologues, hesperidin and rutin, were selected for adsorption experiments and the data are shown in Fig. 10. The adsorption capacity for naringin was 10.09 μmol g−1, as shown in Fig. 10, whereas those of hesperidin and rutin were 0.24 and 0.41 μmol g−1, respectively. Furthermore, the naringin adsorbed onto SMIPs was six times more than the SNIPs. Obviously, the adsorption of SMIPs towards naringin was higher than hesperidin and rutin, which indicated a significant selective property. This phenomenon can be explained by the fact that naringin had specific combination with SMIPs. The SMIPs with highly selective adsorption and specific binding sites were successfully obtained. | ||
| Fig. 10 Selectivity adsorption of SMIPs and SNIPs. | ||
3.6 Application of extraction
To gain more insight into the purification abilities of the imprinted polymers, SMIPs were used as the adsorbent material in the extraction application.The chromatograms of the grapefruit extracts (detection wave length, 254 nm) (a); the grapefruit extract (detection wave length, 282 nm) (b); the desorption solution of SMIPs (detection wave length, 254 nm) (c); the desorption solution of SMIPs (detection wave length, 282 nm) (d); the desorption solution of SNIPs (detection wave length, 254 nm) (e); the desorption solution of SNIPs (detection wave length, 282 nm) (f); the pure naringin (detection wave length, 282 nm) (g) are shown in Fig. 11. The naringin peak was identified by the relative retention time in these chromatograms. The relative retention time of naringin is about 2.8 min, as shown in Fig. 11e. The maximum absorption wave length of naringin is different from the impurities in the extract, as shown in Fig. 11a and b. Fig. 11a shows that the components existing in the solution of grapefruit extract were complex. When the SMIPs were subsequently eluted with ethanol–acetic acid (5%) (1/1, v/v), a clear single peak of naringin appeared in Fig. 11d. A few impurities existed in the desorption solution, as shown in Fig. 11c. When the SNIPs were eluted subsequently with ethanol–acetic acid (5%) (1/1, v/v), it could be seen from Fig. 11e and f that there are few impurities existed in the desorption solution. These findings all suggested the outstanding purification effect of naringin.
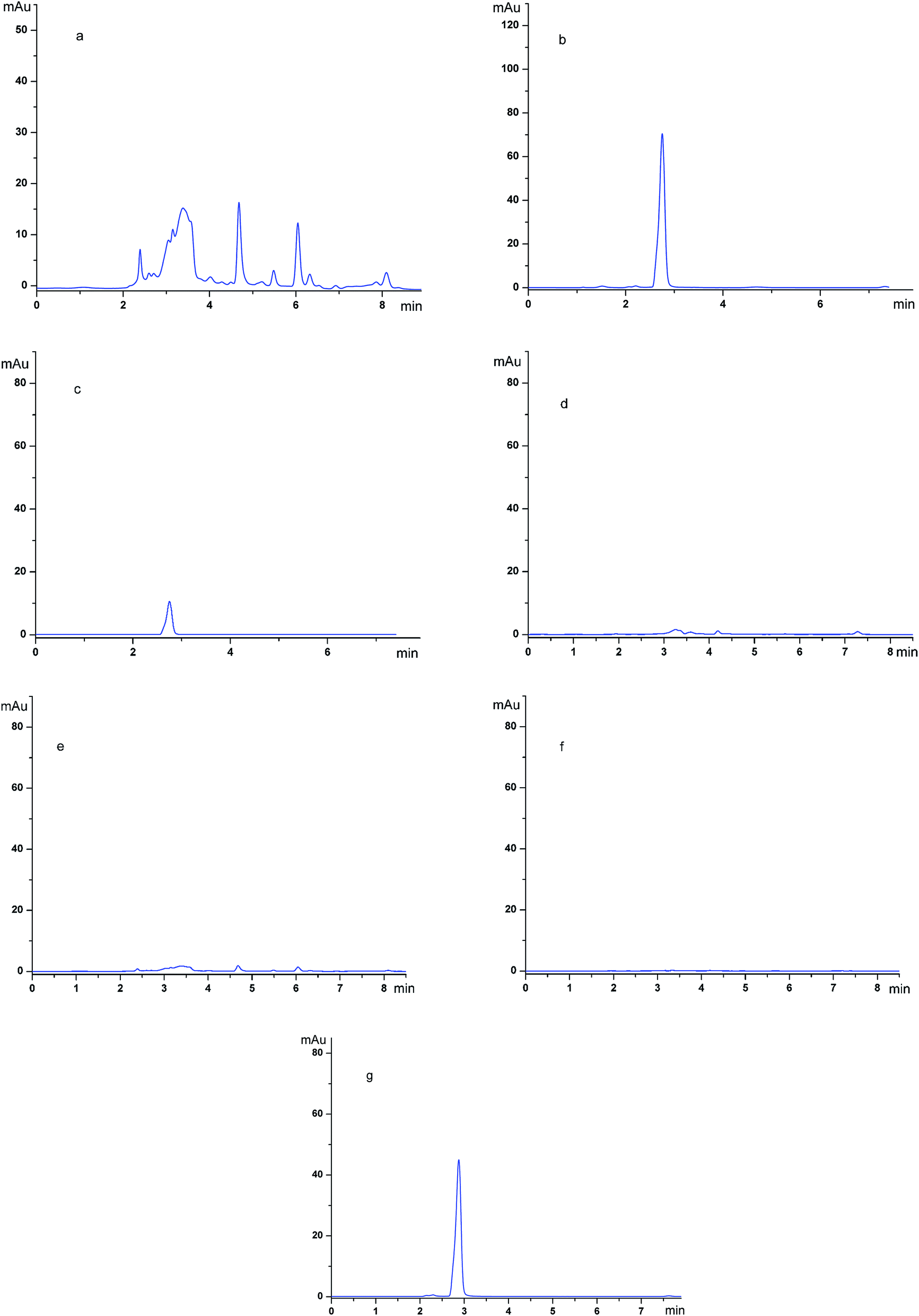 | ||
| Fig. 11 Chromatograms of grapefruit extract and the desorption solution from the SMIPs: before the adsorption (detection wave length, 254 nm) (a); before the adsorption (detection wave length, 282 nm) (b); the desorption solution of SMIPs (detection wave length, 254 nm) (c); the desorption solution of SMIPs (detection wave length, 282 nm) (d); the desorption solution of SNIPs (detection wave length, 254 nm) (e); the desorption solution of SNIPs (detection wave length, 282 nm) (f); the pure naringin (detection wave length, 282 nm) (g). HPLC column: Agilent C18 column. Mobile phase: water–acetonitrile (40/60). Flow rate: 1.0 mL min−1. | ||
The UV absorption spectra of the grapefruit extract (a), the solution after the first adsorption of SNIPs (b), the pure naringin (c), the solution after the third adsorption of SMIPs (d), the solution after the second adsorption of SMIPs (e), and the solution after the first adsorption of SMIPs (f) are shown in Fig. S7.† The grapefruit extract had almost the same waveform with the standard naringin, as shown in Fig. S7a and c,† and their maximum absorption peak position is roughly the same, indicating that the component existing in the solution of grapefruit extract was mainly naringin. After the first adsorption of SMIPs, an obvious decrease in the peak of naringin was observed in Fig. S7f.† When the materials were eluted subsequently with acetic acid (5%)–ethanol solvent (1/1, v/v) and extract was absorbed the second and third times, a slightly increasing peak of naringin was observed in Fig. S7e and d.† According to the standard curve of naringin (Fig. S6†), the naringin that existed in the grapefruit extract was calculated to be 1.33 mg. The first adsorption capacity of naringin bound to SMIPs (30 mg) was calculated to be 0.37 mg, which was 28% of the naringin in the 10 mL grapefruit extract. The first adsorption capacity of naringin bound to SNIPs (30 mg) was calculated to be 0.082 mg, which was 6% of the naringin in the 10 mL grapefruit extract. The recoveries of the three cycles of adsorption and desorption experiment were 95.9%, 82.8%, and 101.3%, respectively, with the average results of 93.3% (RSD 0.93%). It turns out that SMIP is a kind of ideal adsorption material which has good reusability and can be used to separate and purify naringin in pure aqueous solution.
4. Conclusions
Hydrophilic SMIPs with excellent recognition performance toward naringin were prepared for the first time by surface-initiated R-ATRP on the surface of macroporous pGMA–EDMA supports. These supports were prepared using MOGs as porogen, ACPA grafted onto the surface of macroporous pGMA–EDMA supports as photoinitiator and 4-vinylphenylboronic acid as a functional monomer. The thickness of SMIPs can be adjusted by changing the polymerization time. The pure aqueous extraction solution can be directly absorbed by SMIPs without adjusting pH. Experimental data furthermore showed that absorption equilibrium was reached rapidly within 3 min under the optimum polymerization time ∼6 h. The selective absorption was much higher for naringin than for hesperidin and rutin. Additionally, as a novel adsorbent, SMIPs showed excellent separation and purification ability of naringin in complex aqueous extraction. This approach can enable the development of advanced SMIP materials for the selective separation of substances with two adjacent hydroxide groups, (e.g., flavanone compounds like naringin) in pure aqueous systems.Acknowledgements
The authors were grateful to the financial support from the National Natural Science Foundation of China (No. 21276283) and the Fundamental Research funds for the Central South University and the NMR measurements by The Modern Analysis and Testing Center of the Central South University.References
- G. Jagetia and T. Reddy, Life Sci., 2005, 77, 780–794 CrossRef CAS PubMed
.
- Y. Zhai, Z. Li, K. Chen, N. Zhang and G. Cheng, China J. Chin. Mater. Med., 2013, 38, 105–111 CrossRef CAS PubMed
- W. Yang, S. Huang, Q. Wu and J. He, J. Polym. Res., 2014, 21, 383 CrossRef
- Y. Li, J. Liu, R. Cao, S. Deng and X. Lu, J. Chem. Eng. Data, 2013, 58, 2527–2537 CrossRef CAS
- J. Yin, Y. Cui, G. Yang and H. Wang, Chem. Commun., 2010, 46, 7688–7690 RSC
- Z. Zhang, Z. Wang, F. Wang, J. Ren and X. Qu, Small, 2015, 11, 6172–6178 CrossRef CAS PubMed
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495–2504 CrossRef CAS PubMed
- X. Shen, C. Xu and L. Ye, Soft Matter, 2012, 8, 7169–7176 RSC
- S. Yaqub, U. Latif and F. L. Dickert, Sens. Actuators, B, 2011, 160, 227–233 CrossRef CAS
- Y. Liu, R. Liu, C. Liu, S. Luo, L. Yang, F. Sui, Y. Teng, R. Yang and Q. Cai, J. Hazard. Mater., 2010, 182, 912–918 CrossRef CAS PubMed
- M. Resmini, Anal. Bioanal. Chem., 2012, 402, 3021–3026 CrossRef CAS PubMed
- J. Chen, S. Lei, Y. Xie, M. Wang, J. Yang and X. Ge, ACS Appl. Mater. Interfaces, 2015, 7, 28606–28615 CAS
- D. Tai, C. Lin, T. Wu, J. Huang and P. Shu, Clin. Chem., 2006, 52, 1486–1491 CAS
- M. Zhang, J. Huang and P. Yu, Talanta, 2010, 81, 162–166 CrossRef CAS PubMed
- R. Makote and S. Dai, Anal. Chim. Acta, 2001, 435(1), 169–175 CrossRef CAS
- F. Tamayo, J. Casillas and A. Martin-Esteban, Anal. Bioanal. Chem., 2005, 381, 1234 CrossRef CAS PubMed
- B. Dirion, Z. Cobb, E. Schillinger, L. Andersson and B. Sellergren, J. Am. Chem. Soc., 2003, 125, 15101–15109 CrossRef CAS PubMed
- A. Vaughan, S. Sizemore and M. Byrne, Polymer, 2007, 48, 81 CrossRef
- G. Yue, C. Liu, D. Wang, Y. Wang, Q. Yuan and R. Xu, Mater. Res. Bull., 2010, 45, 1319–1323 CrossRef CAS
- J. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117(20), 5614–5615 CrossRef CAS
- J. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS
- Z. Guo, T. Guo and M. Guo, Anal. Chim. Acta, 2008, 612, 136–143 CrossRef CAS PubMed
- L. Hu, L. Xie, J. Guo, X. Li, Y. Jiang, S. Zhang and S. Shi, Food Chem., 2015, 179, 206–212 CrossRef PubMed
- H. Niu, Y. Yang and H. Zhang, Biosens. Bioelectron., 2015, 74, 440–446 CrossRef CAS PubMed
- X. Shen and L. Ye, Chem. Commun., 2011, 47, 10359–10361 RSC
- S. A. Piletsky, H. Matuschewski, U. Schedler, A. Wilpert and M. Ulbricht, Macromolecules, 2000, 33, 3092–3098 CrossRef CAS
- X. Luo, Y. Zhan, Y. Huang, L. Yang and X. Tu, J. Hazard. Mater., 2011, 187, 274–282 CrossRef CAS PubMed
- d. V. B. Van, B. Bueken, J. Denayer and V. D. De, Chem. Soc. Rev., 2014, 43, 5766 RSC
- Q. Wei and S. James, Chem. Commun., 2005, 1555–1556 RSC
- I. Luz, F. X. L. I. Xamena and A. Corma, J. Catal., 2012, 285, 285–291 CrossRef CAS
- L. Murray, M. Dinca and J. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC
- M. Faustini, J. Kim, G. Y. Jeong, J. Y. Kim and H. R. Moon, J. Am. Chem. Soc., 2013, 135, 14619–14626 CrossRef CAS PubMed
- K. Sugikawa, Y. Furukawa and K. Sada, Chem. Mater., 2011, 23, 3132–3134 CrossRef CAS
- B. Preinerstorfer, M. Lammerhofer and W. Lindner, J. Sep. Sci., 2009, 32, 1673–1685 CrossRef CAS PubMed
- Q. Wei and S. L. James, Chem. Commun., 2005, 1555–1556 RSC
- L. Ma, L. Tang, R. Li and Y.-P. Huang, RSC Adv., 2015, 5, 84601 RSC
- Y. Xu, R. Zhang and H. Fu, Journal of Nature and Science, 2005, 3, 43–46 Search PubMed
- E. C. Peters, M. Petro, F. Svec and J. M. Fréchet, Anal. Chem., 1998, 70, 2288–2295 CrossRef CAS PubMed
- X. Sun, J. He, G. Cai, A. Lin, W. Zheng, X. Liu, L. Chen, X. He and Y. Zhang, J. Sep. Sci., 2010, 33, 3786–3793 CrossRef CAS PubMed
- Y. Wang, C. Zhou, L. Sun, B. Yu, M. Cao and S. Zhong, Appl. Surf. Sci., 2015, 353–357 CrossRef
- X. Wang, J. Pan, W. Guan, J. Dai, X. Zou, Y. Yan, C. Li and W. Hu, J. Chem. Eng. Data, 2011, 56, 2793–2801 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00202e |
| This journal is © The Royal Society of Chemistry 2017 |