DOI:
10.1039/C7RA00360A
(Paper)
RSC Adv., 2017,
7, 14176-14185
New amphiphilic polycarbonates with side functionalized cholesteryl groups as biomesogenic units: synthesis, structure and liquid crystal behavior†
Received
10th January 2017
, Accepted 22nd February 2017
First published on 2nd March 2017
Abstract
The synthesis of four new amphipathic copolymers with side functionalized-cholesterol based aliphatic polycarbonates is described through the ring-opening polymerization and coupling reaction. The chemical structures, liquid crystal (LC) behavior, and thermal stability of the chiral monomers and copolymers obtained in this study were characterized using Fourier transform infrared (FT-IR) spectroscopy, proton nuclear magnetic resonance (1H NMR) spectroscopy, gel permeation chromatography (GPC), polarizing optical microscopy (POM), differential scanning calorimetry (DSC), X-ray diffraction (XRD), and thermogravimetric analysis (TGA) measurements. The effect of the spacer length on the molecular interaction and mesophase of the chiral monomers and copolymers was investigated. It was found that chiral monomers with longer spacer seemed beneficial for the formation of mesophases, and the additional ordering on polymerization caused mesophases to be more ordered than for the corresponding monomers. The LC copolymers all revealed a smectic A phase with an interdigitated molecular arrangement. The results seemed to show a decreased tendency toward the glass transition temperature, and isotropic temperature for the LC copolymers by increasing the spacer length. In addition, four LC copolymers had a good thermal stability.
Introduction
In recent years, aliphatic polycarbonates have been found to be promising biomaterials because of their good biodegradability and biocompatibility.1–5 Until now, many modification approaches have been used to obtain the desirable properties. Among them, the incorporation of the bioactive or self-assembly functional compounds to the polymer chain has currently become the most important modification strategies.6–10 As known, liquid crystal (LC) materials have been widely used for display devices because their flexible ordered structures can lead to systems displaying switchable optical modulation. Moreover, the LC compounds can potentially be used as new functional materials for electron, ion, molecular transporting, sensory, catalytic, optical and bioactive materials.11.12 As a kind of self-assembled functional soft materials, LC compounds possess both order and mobility at molecular, supramolecular and macroscopic levels.13–16 Moreover, LC compounds also can produce response or sensitively through the self-assembling ability under external stimuli such as temperature, pressure, and electromagnetic field. In fact, LC compounds are also useful in biorelated fields because their self-organizing structures through non-covalent specific interactions are compatible with those in living systems.17–20 Recently, LC compounds in the fields of biomedical application such as controlled drug delivery and protein binding have been demonstrated.21 Because of their dynamic nature, photochemically, thermally or mechanically induced structure changes of LC can be used for the construction of stimuli-responsive multifunctional materials.
As an important structural component in mammalian cells, cholesterol has high thermodynamic affinity toward cell membrane and ability to change the fluidity and permeability of the membrane.22 Therefore, cholesterol can play important roles in the functions of biological system and LC soft matter and become an interesting component with bioactive because of its universal effect, regardless of cell type and receptor map on the membrane. In recent years, cholesterol as a natural biomesogen has also become an attractive candidate to synthesize new smart, responsive and biodegradable LC polymer materials.3,23–29 Stupp and coworkers first reported the synthesis of cholesterol end functionalized oligo(L-lactic acid).3,23 Later, Cheng24,25 and Yang26 et al. also reported cholesteryl end-capped aliphatic polycarbonates and dicholesteryl end functionalized block poly(ε-caprolactone), respectively, as biodegradable polymers. These study results have shown that the cholesterol-functionalized biodegradable polycarbonate block copolymers can play an important role in encapsulation of complex hydrophobic therapeutics for drug application. However, because the functionalized cholesteryl units are located in the terminal groups of macromolecular chain, the copolymers obtained in these study show lower average molecular weight, in other words, they are oligomers, which will affect their application in drug delivery and tissue engineering. Compared with the biodegradable polycarbonates with main chain functionalized cholesteryl mesogenic groups, the polycarbonates based on side functionalized cholesteryl groups have more advantage such as controlled multiple structure and molecular weight. However, to the best of our knowledge, little research on the biodegradable aliphatic polycarbonate based on cholesterol as side chain biomesogenic units is reported.9,10,30 So, it is necessary to synthesize new biodegradable side chain LC polycarbonate derived from cholesterol and study their structure–property relationships and explore the potential applications in drug delivery and tissue engineering templates.
In the present work, we designed and synthesized a series of new side cholesterol-functionalized amphipathic copolymers based aliphatic polycarbonates, and investigated their structure–mesomorphism relationships. A side chain functionalized cholesterol with hydrophobic moiety was added onto the amphiphilic block copolymers with polycarbonate and mPEG backbone for the introduction of physicochemical functionality. The ability of side cholesterol groups to form mesophase will be anticipated to provide a driving force for self-assembly into nanostructure. Herein, we only discussed the synthesis, LC properties, mesophase structure, and thermal stability of the obtained copolymers with Fourier transform infrared (FT-IR) spectrum, proton nuclear magnetic resonance (1H NMR) spectrum, gel permeation chromatographic (GPC), polarizing optical microscopy (POM), differential scanning calorimetry (DSC), X-ray diffraction (XRD), and thermogravimetric analysis (TGA) measurements.
Experimental
Materials
All chemicals were obtained from the indicated sources. Cholesterol was purchased from Xiayi Beier Biological Products Co., Ltd. (Xiayi, China). Adipic acid and sebacic acid were purchased from Shenyang Xinxing Chemical Reagent Plant (Shenyang, China). Glycerol and benzaldehyde were purchased from Tianjin Bodi Chemical Co., Ltd. (Tianjin, China). Succinic anhydride and benzyl chloride were purchased from Shanghai Chemical Reagent Plant (Shanghai, China). Suberic acid, hexadecyl trimethylammonium bromide (HTAB), stannous octoate [Sn(Oct)2], and methoxypolyethylene glycols (mPEG43) were purchased from Sigma-Aldrich. Ethyl chloroformate was purchased from Xinyi Huili Fine Chemical Co., Ltd. (Xinyi, China). Tetrahydrofuran (THF) was dried over potassium hydroxide and then distilled over sodium potassium alloy in argon atmosphere. Toluene was dried by treatment with Na and distilled before use. All other solvents and reagents used were purified by standard methods.
Characterization
FT-IR spectra were measured on a PerkinElmer spectrum One (B) spectrometer (PerkinElmer, Foster City, CA). 1H NMR spectra were obtained using a Bruker ARX 600 (Bruker, Germany) high resolution NMR spectrometer, and chemical shifts were reported in ppm with tetramethylsilane (TMS) as an internal standard. Gel permeation chromatographic (GPC) measurements were carried out at room temperature on a Waters 1515 instrument calibrated with a polystyrene standard, and using THF as an eluent. The phase transition temperature was determined with a Netzsch 204 (Netzsch, Hanau, Germany) differential scanning calorimetry (DSC) equipped with a cooling system at a heating and cooling rate of 10 °C min−1 in a nitrogen atmosphere. The optical texture was observed with a Leica DMRX (Leica, Wetzlar, Germany) polarizing optical microscopy (POM) equipped with a Linkam THMSE-600 (Linkam, London, UK) cool and hot stage. The thermal decomposition temperature was measured under nitrogen atmosphere with a Netzsch 209C thermogravimetric analysis (TGA) at a heating rate of 20 °C min−1. X-ray diffraction (XRD) measurements were performed with a nickel-filtered Cu-Kα radiation with a Bruker D8 Advance (Bruker, Germany) powder diffractometer.
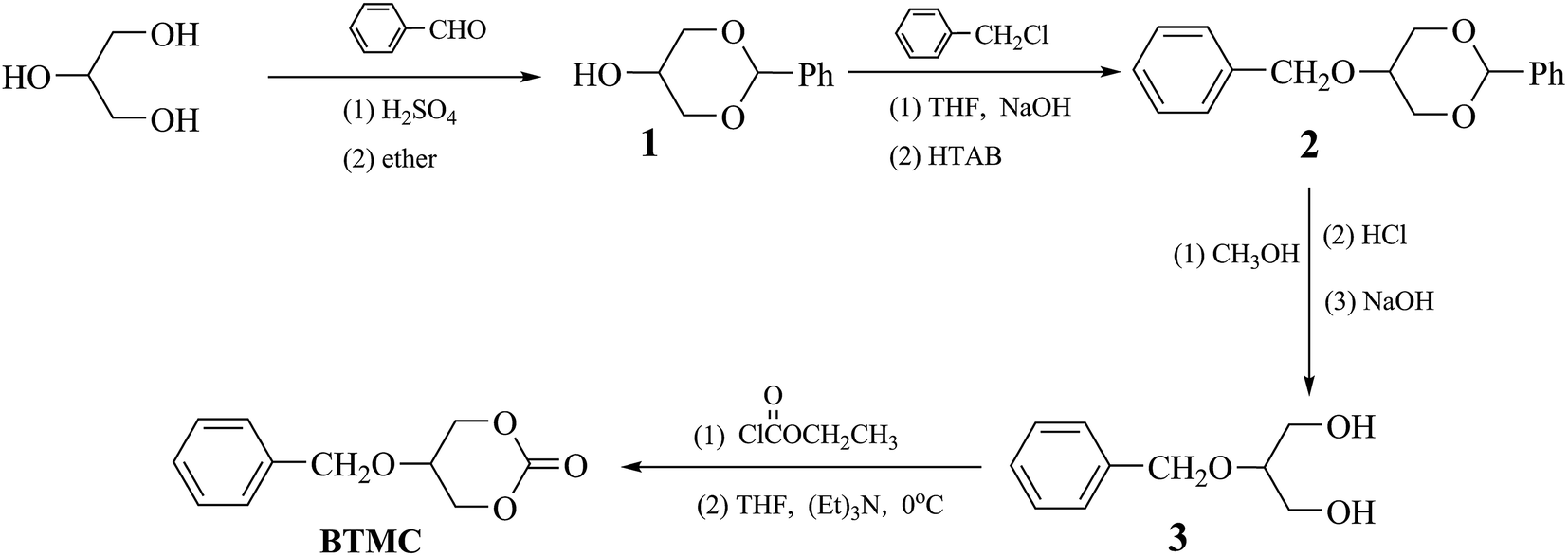
Synthesis of cyclic monomer
The cyclic monomer 5-(benzyloxy)-1,3-dioxan-2-one (BTMC), shown in Scheme 1, was prepared according to the method reported by Hu et al.30 Yield: 43%, mp: 145.8 °C. IR (KBr, cm−1): 3069 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H); 2969, 2872 (–CH2–); 1746 (CO); 1531, 1467 (Ph–), 1192 (C–O). 1H NMR (δ, ppm from TMS in CDCl3): 7.39–7.33 (m, 5H, J = 7.2, Ph–H); 4.66 (s, 2H, Ar–CH2O–); 4.52–4.43 (m, 4H, J = 2.4,
C–H); 2969, 2872 (–CH2–); 1746 (CO); 1531, 1467 (Ph–), 1192 (C–O). 1H NMR (δ, ppm from TMS in CDCl3): 7.39–7.33 (m, 5H, J = 7.2, Ph–H); 4.66 (s, 2H, Ar–CH2O–); 4.52–4.43 (m, 4H, J = 2.4, ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CHCH2O–); 3.91–3.89 (m, 1H, J = 2.4, –CH
CHCH2O–); 3.91–3.89 (m, 1H, J = 2.4, –CH![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) ).
).
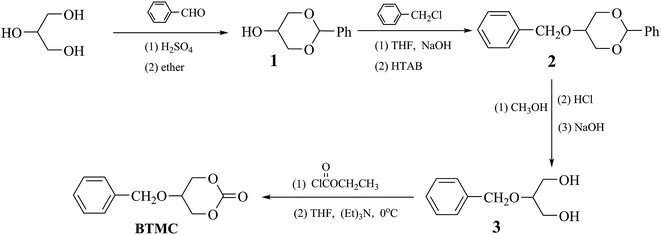 |
| | Scheme 1 Synthetic route of cyclic monomer. | |
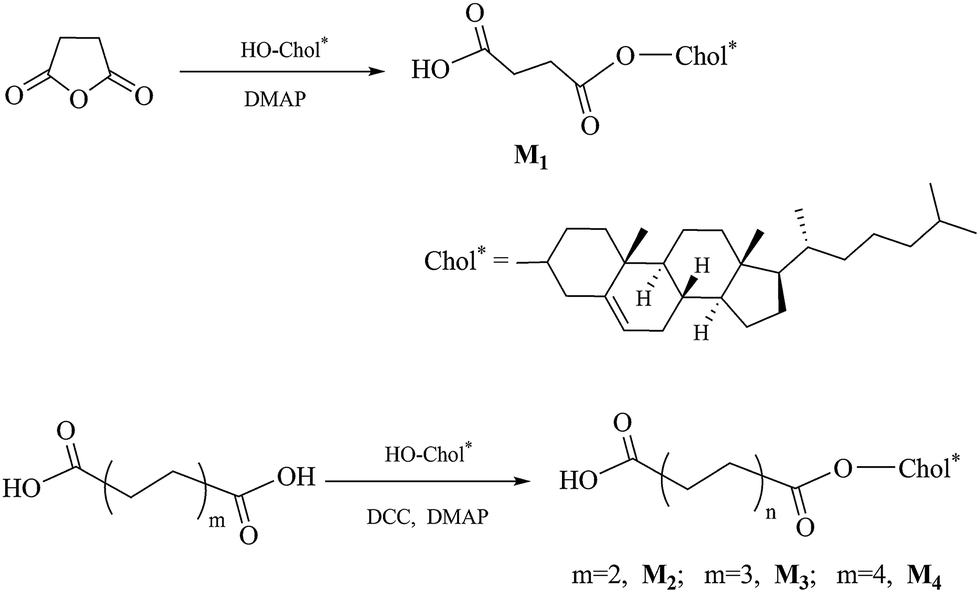
Synthesis of chiral monomers
The synthetic route of the chiral monomers M1–M4 is shown in Scheme 2.
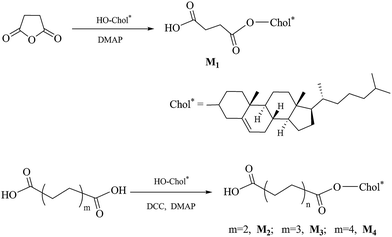 |
| | Scheme 2 Synthetic route of chiral monomers. | |
Synthesis of 4-cholesteroxy-4-oxobutanoic acid (M1). Succinic anhydride (10 g, 0.1 mol), 4-dimethylaminopyridine (DMAP) (1.2 g, 0.01 mol), and anhydrous dichloromethane (150 mL) were placed in a 500 mL three-neck flask with magnetic stir bar. After stirring for 0.5 h, cholesterol (19.3 g, 0.05 mol), dissolved in 70 mL of dichloromethane, was added dropwise to the above mixture. The reaction mixture was stirred for 24 h at 30 °C. The resulting mixture was evaporated to dryness, the crude product was recrystallized with acetic acid, washed with hot water, and then further purified by silica gel column chromatography (petroleum ether/ethyl acetate = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). A white solid was obtained. Yield: 60%, mp: 180.1 °C. IR (KBr, cm−1): 2949, 2862 (–CH2–, –CH3); 1731, 1710 (CO). 1H NMR (δ, ppm from TMS in CDCl3): 5.39–5.37 (d, 1H, J = 4.2 Hz, –CHC in cholesteryl); 4.66–4.62 (m, 1H, –COOCH in cholesteryl); 2.69–2.67 (t, 2H, J = 6.6, HOOCCH2CH2–); 2.61–2.59 (t, 2H, J = 6.6, HOOCCH2CH2–); 2.33–0.68 (m, 43H, rest of the protons from cholesteryl).
:1). A white solid was obtained. Yield: 60%, mp: 180.1 °C. IR (KBr, cm−1): 2949, 2862 (–CH2–, –CH3); 1731, 1710 (CO). 1H NMR (δ, ppm from TMS in CDCl3): 5.39–5.37 (d, 1H, J = 4.2 Hz, –CHC in cholesteryl); 4.66–4.62 (m, 1H, –COOCH in cholesteryl); 2.69–2.67 (t, 2H, J = 6.6, HOOCCH2CH2–); 2.61–2.59 (t, 2H, J = 6.6, HOOCCH2CH2–); 2.33–0.68 (m, 43H, rest of the protons from cholesteryl).
Synthesis of 6-cholesteroxy-6-oxocaproic acid (M2). Adipic acid (14.6 g, 0.1 mol) and dichloromethane (200 mL) were placed in a 500 mL three-neck flask with magnetic stir bar. N,N′-Dicyclohexylcarbodiimide (DCC) (5.2 g, 0.025 mol) and DMAP (1.2 g, 0.01 mol) were dissolved in dichloromethane (20 mL), and then added dropwise to the flask. After stirring for 0.5 h, cholesterol (9.7 g, 0.025 mol), dissolved in 20 mL of dichloromethane, was added dropwise to the above mixture. The reaction mixture was stirred for 24 h at 30 °C. The resulting mixture was washed with 50 mL of water, and a solid, N,N′-dicyclohexyl urea, was precipitated and filtered off. The filtrate was dried with anhydrous magnesium sulfate and evaporated to dryness. The crude product was first recrystallized with ethanol, and then further purified by silica gel column chromatography (petroleum ether/ethyl acetate = 3:1). A white solid was obtained. Yield: 65%, mp: 139.5 °C. IR (KBr, cm−1): 2944, 2863 (–CH2–, –CH3); 1722, 1694 (CO). 1H NMR (δ, ppm from TMS in CDCl3): 5.38–5.36 (d, 1H, J = 6.0 Hz, –CHC in cholesteryl); 4.66–4.57 (m, 1H, –COOCH in cholesteryl); 2.39 (m, 2H, –CH2COO–); 2.35, 2.01 (m, 2H, CHCH2CC in cholesterol); 2.31 (m, 2H, –CH2COOH); 1.99–0.68 (m, 45H, rest of the protons from cholesteryl and –CH2(CH2)2CH2COOH).
Synthesis of 8-cholesteroxy-8-oxooctanoic acid (M3). In a way similar to the synthesis of M2, new cholesteryl derivative M3 was accordingly prepared. Yield: 67%, mp: 129.5 °C. IR (KBr, cm−1): 2956, 2865 (–CH2–, –CH3); 1737, 1705 (CO). 1H NMR (δ, ppm from TMS in CDCl3): 5.39–5.37 (d, 1H, J = 4.8 Hz, –CHC in cholesteryl); 4.64–4.60 (m, 1H, –COOCH in cholesteryl); 2.36–2.34 (m, 2H, J = 7.2 Hz, –CH2COO–); 2.33–0.69 (m, 53H, rest of the protons from cholesteryl and –CH2(CH2)5COOH).
Synthesis of 10-cholesteroxy-10-oxodecanoic acid (M4). In a way similar to the synthesis of M2, new cholesteryl derivative M4 was accordingly prepared. Yield: 40%, mp: 117.3 °C. IR (KBr, cm−1): 2960, 2858 (–CH2–, –CH3); 1737, 1707 (CO). 1H NMR (δ, ppm from TMS in CDCl3): 5.38–5.36 (d, 1H, J = 4.2 Hz, –CHC in cholesteryl); 4.64–4.60 (m, 1H, –COOCH in cholesteryl); 2.36–2.33 (m, 2H, J = 7.8 Hz, –CH2COO–); 2.32–0.68 (m, 57H, rest of the protons from cholesteryl and –CH2(CH2)5COOH).
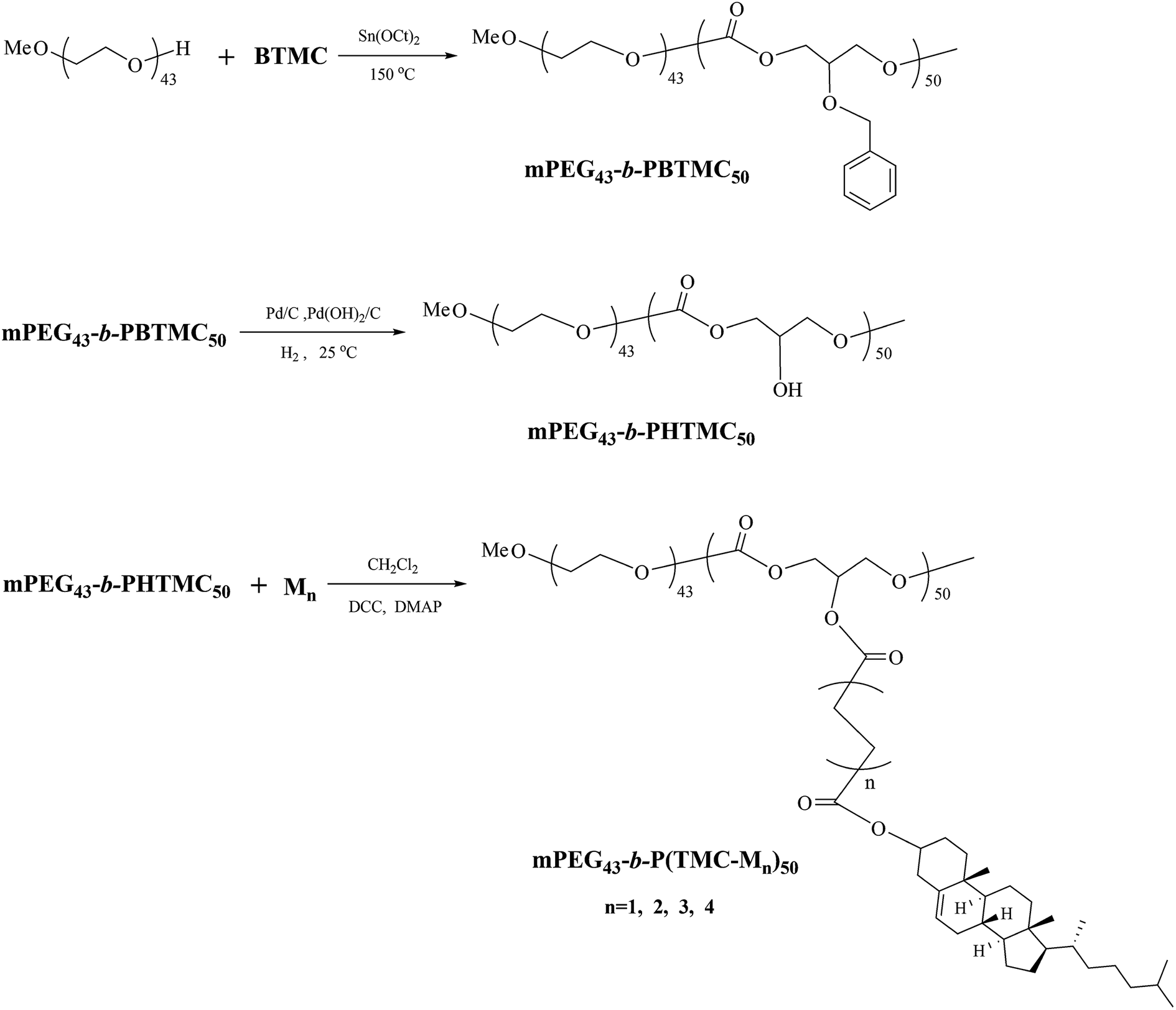
Synthesis of block copolymers
The synthetic route of the block copolymers is shown in Scheme 3.
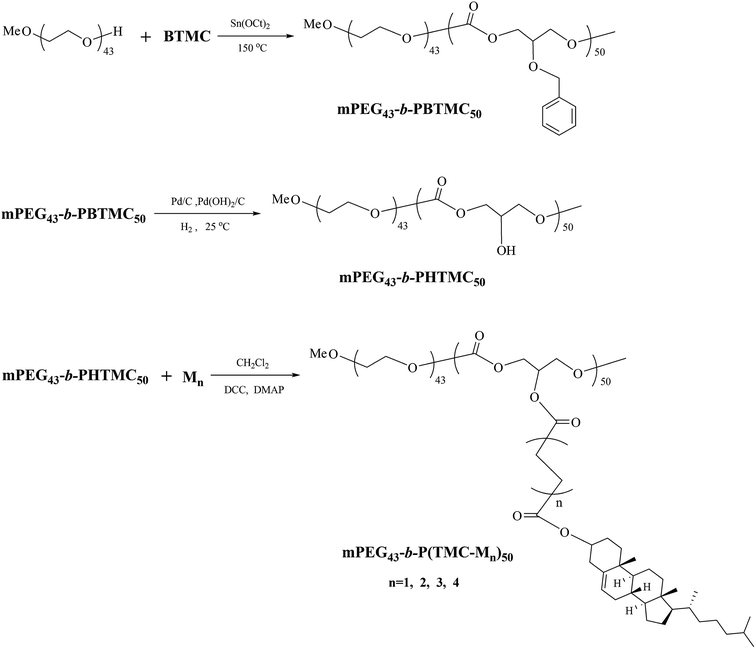 |
| | Scheme 3 Synthetic route of block copolymers. | |
Synthesis of mPEG43-b-PBTMC50. The block copolymer mPEG43-b-PBTMC50 was synthesized by ring-opening polymerization of the cyclic carbonate monomer BTMC and mPEG43 in polymerization flask equipped with a sealing cap using Sn(Oct)2 as a catalyst. The general synthetic procedure for the copolymer was described as follows: BTMC (8.32 g, 40 mmol), mPEG (2.24 g, 1.18 mmol), and Sn(Oct)2 toluene solution (0.8 mL, 0.1 mol L−1) were placed in the glass flask, which was degassed in a vacuum by several vacuum-purge cycles, and then sealed under argon atmosphere. The reaction was held for 24 h at 150 °C in an oil bath. After completion of polymerization, the crude polymer was purified by dissolving it in dichloromethane and precipitated in methanol, and then dried in a vacuum until a constant sample mass was obtained. Yield: 90%. IR (KBr, cm−1): 2961, 2872 (–CH2–, –CH3); 1750 (CO); 1496, 1454 (Ph–), 1239 (C–O). 1H NMR (δ, ppm from TMS in CDCl3): 7.32–7.26 (m, Ph–H); 4.72–4.58 (m, Ph–CH2O–); 4.31–4.21 (m, –OCH2–CH–CH2O–); 3.86–3.83 (m, –OCH2–CH–CH2O–); 3.67–3.62 (m, –OCH2– in mPEG).
Synthesis of mPEG43-b-PHTMC50. mPEG43-b-PBTMC50 (8 g), Pd/C (0.04 g), Pd(OH)2 (0.04 g, 10%), and 150 mL of methanol/tetrahydrofuran (1:1) were placed in a 250 mL three-neck flask with magnetic stir bar under argon atmosphere. The reaction mixture was stirred for 48 h at 25 °C in hydrogen system. After completion of reducing reaction, the Pd/C and Pd(OH)2 were filtered off, the filtrate was evaporated to dryness, and then dried in a vacuum until a constant sample mass was obtained. Yield: 75%. IR (KBr, cm−1): 3376 (–OH); 2961, 2904 (–CH2–, –CH3); 1753 (CO); 1256 (C–O). 1H NMR (δ, ppm from TMS in DMSO-d6): 5.45 (m, –OH); 4.10–4.02 (m, –OCH2–CH–CH2O–); 3.95–3.91 (m, –OCH2–CH–CH2O–); 3.50 (m, –OCH2– in mPEG).
Synthesis of mPEG43-b-P(TMC-M1)50. M1 (4.7 g, 10 mmol) and dichloromethane (50 mL) were placed in a 100 mL three-neck flask with magnetic stir bar. DCC (2.16 g, 10.5 mmol) and DMAP (0.12 g, 1 mmol) were dissolved in 5 mL of dichloromethane, and then added dropwise to the flask. After stirring for 0.5 h, mPEG43-b-PHTMC50 (1.6 g, 10 mmol for –OH groups), dissolved in 5 mL of dichloromethane, was added dropwise to the above mixture. The reaction mixture was stirred for 24 h at 30 °C. The resulting mixture was washed with 20 mL of water, and a solid, N,N′-dicyclohexyl urea, was precipitated and filtered off. The filtrate was dried with anhydrous magnesium sulfate and evaporated to dryness. The crude polymer was purified by dissolving them in dichloromethane and precipitated in methanol, and then dried in a vacuum until a constant sample mass was obtained. Yield: 32%. IR (KBr, cm−1): 2940, 2966 (–CH2–, –CH3); 1737 (CO); 1653 (CC); 1259 (C–O). 1H NMR (δ, ppm from TMS in CDCl3): 5.38–5.36 (t, –CHC in cholesteryl); 5.29–5.26 (m, –OCH2–CH–CH2O–); 4.65–4.60 (m, –COOCH in cholesteryl); 4.36–4.21 (m, –OCH2–CH–CH2O–); 3.65 (m, –OCH2– in mPEG); 2.67–2.60 (m, –COO(CH2)4COO–); 2.32–0.68 (m, rest of the protons from cholesteryl).
Synthesis of mPEG43-b-P(TMC-M2)50. The block copolymer mPEG43-b-P(TMC-M2)50 was prepared by a procedure similar to that for mPEG43-b-P(TMC-M1)50, using M2 instead of M1. Yield: 45%. IR (KBr, cm−1): 2932, 2857 (–CH2–, –CH3); 1735 (CO); 1643 (CC); 1255 (C–O). 1H NMR (δ, ppm from TMS in CDCl3): 5.39–5.37 (t, –CHC in cholesteryl); 5.28–5.25 (m, –OCH2–CH–CH2O–); 4.61–4.55 (m, –COOCH in cholesteryl); 4.42–4.16 (m, –OCH2–CH–CH2O–); 3.65 (m, –OCH2– in mPEG); 2.38–0.69 (m, rest of the protons from PCL, cholesteryl and –COO(CH2)4COO–).
Synthesis of mPEG43-b-P(TMC-M3)50. The block copolymer mPEG43-b-P(TMC-M3)50 was prepared by a procedure similar to that for mPEG43-b-P(TMC-M1)50, using M3 instead of M1. Yield: 38%. IR (KBr, cm−1): 2934, 2859 (–CH2–, –CH3); 1735 (CO); 1647 (CC); 1260 (C–O). 1H NMR (δ, ppm from TMS in CDCl3): 5.39–5.37 (t, –CHC in cholesteryl); 5.28–5.26 (m, –OCH2–CH–CH2O–); 4.63–4.58 (m, –COOCH in cholesteryl); 4.38–4.20 (m, –OCH2–CH–CH2O–); 3.65 (m, –OCH2– in mPEG); 2.39–0.68 (m, rest of the protons from PCL, cholesteryl and –COO(CH2)6COO–).
Synthesis of mPEG43-b-P(TMC-M4)50. The block copolymer mPEG43-b-P(TMC-M4)50 was prepared by a procedure similar to that for mPEG43-b-P(TMC-M1)50, using M4 instead of M1. Yield: 35%. IR (KBr, cm−1): 2936, 2861 (–CH2–, –CH3); 1739 (CO); 1648 (CC); 1263 (C–O). 1H NMR (δ, ppm from TMS in CDCl3): 5.38–5.35 (t, –CHC in cholesteryl); 5.27–5.25 (m, –OCH2–CH–CH2O–); 4.69–4.59 (m, –COOCH in cholesteryl); 4.35–4.09 (m, –OCH2–CH–CH2O–); 3.60 (m, –OCH2– in mPEG); 2.38–0.66 (m, rest of the protons from PCL, cholesteryl and –COO(CH2)8COO–).
Results and discussion
Synthesis and characterization of chiral monomers
To investigate the effect of spacer length on molecular interaction and physical properties of the chiral monomers and block copolymers obtained in this study, as shown in Schemes 2 and 3, a series of chiral LC monomers with two, four, six, and eight methylene segments were synthesized. Their chemical structures were characterized by FT-IR and 1H NMR spectra. To reduce the bulky steric hindrance, improve the reactivities of cholesterol, and obtain mesophase of the cholesteryl derivatives, the commercially available cholesterol was allowed to be structurally modified. The cholesteryl derivatives with different longer spacer and more reactive terminal carboxyl group were successfully synthesized by reacting cholesterol with an excess amount of succinic anhydride, adipic acid, suberic acid and sebacic acid, respectively. The crude products were purified by silica gel column chromatography to result in a white powder as the monomers. FT-IR spectrum of the monomers M1–M4 showed characteristic bands at 2960–2858 and 1737–1694 cm−1 attributable to the C–H and ester CO stretching bands, respectively. 1H NMR spectra of M1–M4 showed peaks at the characteristic resonances values of 5.39–5.36, 4.66–4.57, and 2.69–0.68 ppm corresponding to olefinic, –COOCH, and –CH2– and –CH3 protons, respectively. The spectrum was agreed well with the proposed molecular structure of M1–M4. As an example, the 1H NMR spectra of M1 are shown in Fig. S1.†
Synthesis and characterization of the copolymers
At present, it is known that the ring-opening polymerization is an efficient method for the preparation of biodegradable aliphatic polycarbonates. The ring-opening polymerization of the cyclic carbonate could be carried out in the presence of macroinitiator with hydroxyl groups such as polyethylene glycols.28,31 In this investigation, the block copolymers were synthesized according to the route shown in Scheme 3. First, the copolymer mPEG43-b-PBTMC50 was prepared by the ring-opening polymerization of cyclic monomer BTMC using Sn(Oct)2 as a catalyst in the presence of mPEG43 under argon atmosphere. The optimum reaction conditions were selected to be 24 h at 150 °C, and the molar ratio of BTMC to Sn(Oct)2 was 500:1. Second, mPEG43-b-PHTMC50 was obtained by hydrogenolysis reaction to result in the deprotection of side benzyl groups for mPEG43-b-PBTMC50 using Pd/C and Pd(OH)2 composite catalyst. Third, four LC copolymers mPEG43-b-P(TMC-Mn)50 were synthesized through esterification reaction between side hydroxyl group of mPEG43-b-PHTMC50 and terminal carboxyl group of the cholesteryl derivatives M1–M4, respectively, using DCC as the condensation agent and DMAP as the catalytic system in dry dichloromethane. The copolymers obtained in this study were soluble in common organic solvents such as TFF, dichloromethane, and toluene. Their molecular structures were confirmed by FT-IR and 1H NMR spectra. The number average molecular weight (Mn) and the polydispersity index (PDI) of the block copolymers were detected by GPC. Yields and average molecular weight of the obtained copolymers are summarized in Table 1. The degree of polymerization and molecular composition of the obtained block copolymer were estimated from the 1H NMR spectra by comparing the integrated signals in the –CH2O– regions located at 3.67–3.62 ppm for mPEG43. After the polymerization, the signal peaks of methylene proton at 4.52–4.43 ppm for BTMC shifted to 4.31–4.21 ppm for mPEG43-b-PBTMC50, moreover, the peaks of the chemical shifts corresponding to the polymer were broad and consistent with the expected polymer structure. In addition, compared with mPEG43-b-PBTMC50, the proton signals of mPEG43-b-PHTMC50 at 7.32–7.26 and 4.72–4.58 ppm for benzyl groups completely disappeared, which indicated that the benzyl groups were complete removed by hydrogenolysis reaction. The typical proton signals of mPEG, polycarbonate chains, and side cholesteryl units could be observed. 1H NMR spectra of mPEG43-b-PBTMC50, mPEG43-b-PHTMC50, and mPEG43-b-P(TMC-M1)50 are shown in Fig. S2–S4.†
Table 1 Yields and average molecular weight of the copolymers
| Copolymer |
Yield/% |
Mna (kDa) |
Mnb (kDa) |
PDIc |
| Mn values calculated from 1H NMR. Mn values determined by GPC in THF using PS standards, g mol−1. PDI = Mw/Mn. |
| mPEG43-b-PBTMC50 |
90 |
12.3 |
9.2 |
1.47 |
| mPEG43-b-PHTMC50 |
75 |
7.8 |
5.5 |
1.42 |
| mPEG43-b-P(TMC-M1)50 |
36 |
31.3 |
27.2 |
1.55 |
| mPEG43-b-P(TMC-M2)50 |
45 |
32.7 |
29.1 |
1.61 |
| mPEG43-b-P(TMC-M3)50 |
38 |
34.1 |
29.9 |
1.66 |
| mPEG43-b-P(TMC-M4)50 |
35 |
35.5 |
31.6 |
1.57 |
Liquid crystal properties of chiral monomers
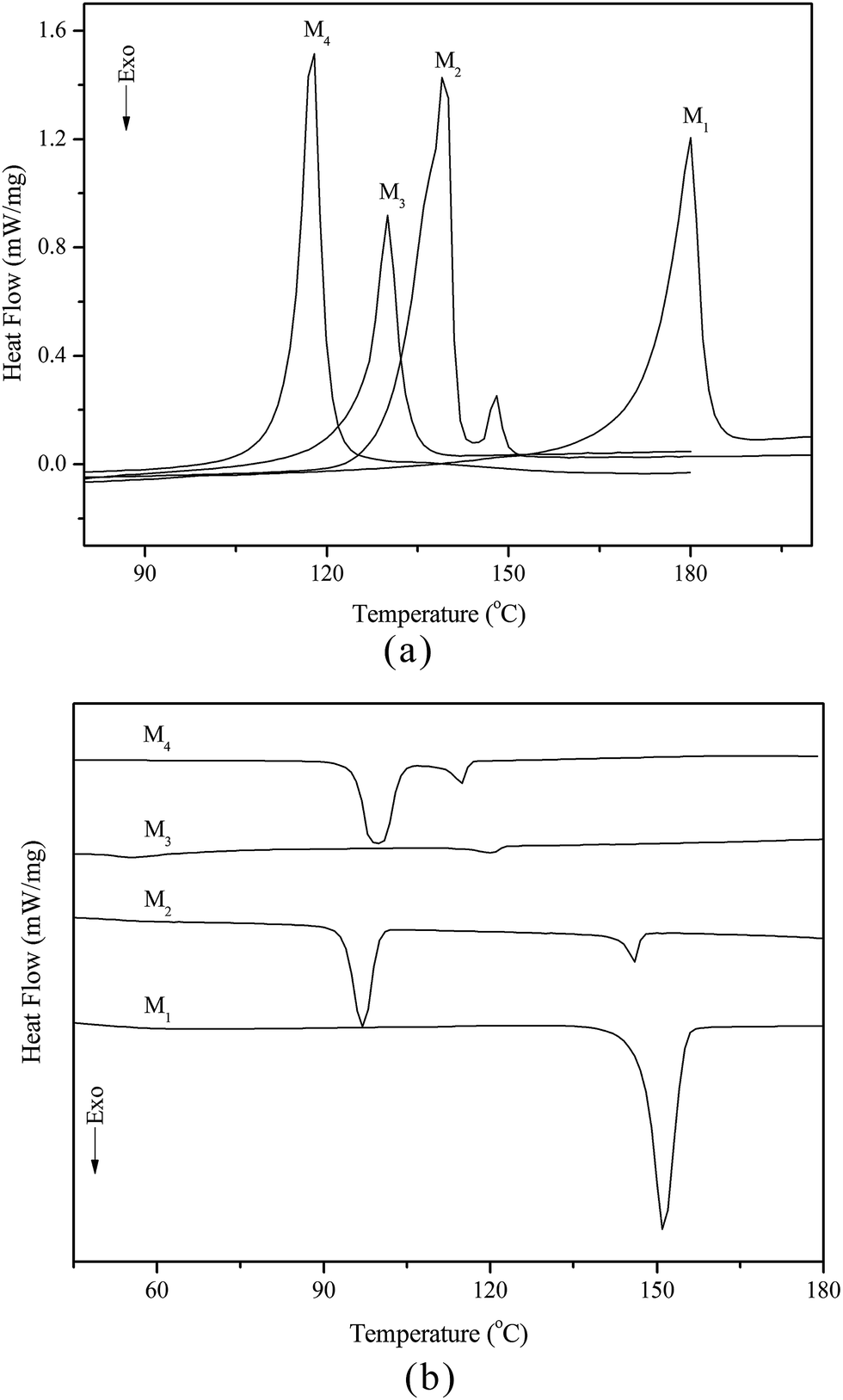
The thermal behaviour and optical texture of the chiral monomers obtained in this study were investigated with DSC and POM. The corresponding phase transition temperatures, enthalpy changes and phase sequence of M1–M4, obtained during first cooling and second heating scans, are summarized in Table 2. Typical DSC curves of M1–M4 are shown in Fig. 1(a and b).
Table 2 The phase transition temperatures of the monomersa
| Monomer |
Phase transition temperature (°C) and enthalpy changes (J g−1) |
| Heating cycle |
Cooling cycle |
| K = crystal; Ch = cholesteric phase; I = isotropic. Isotropic temperature determined by POM. |
| M1 |
K180.1(36.5)I |
I151.3(43.2)K |
| M2 |
K139.5(54.7)Ch147.7(2.7)I |
I146.0(3.6)Ch96.6(21.1)K |
| M3 |
K129.5(20.5)Ch132.6b(—)I |
I120.3(1.2)Ch55.2(10.2)K |
| M4 |
K117.3(37.2)Ch119.8b(—)I |
I115.0(3.7)Ch99.4(33.2)K |
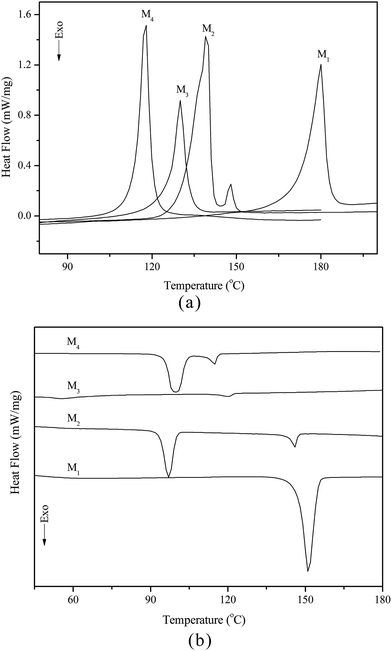 |
| | Fig. 1 DSC curves of M1–M4 on heating (a) and cooling (b) cycles. | |
According to DSC curves of the chiral monomers, M1 only showed an endothermic peak on heating process and an exothermic peaks on cooling process. However, M2–M4 all showed two endothermic peaks and two exothermic peaks, and exhibited enantiotropic cholesteric phase. In general, the formation of mesophases may be affected by aspect ratio, flexible spacer at the center, and polarity of the monomers.32,33 As shown in Table 2, the chiral monomer M1 with shorter flexible spacer length did not reveal any mesophase, which was also confirmed by POM result. However, the chiral monomer M2–M4 with longer flexible spacer length all showed enantiotropic cholesteric phase during heating and cooling cycles. This suggested that the flexible spacer length had a considerable influence on the thermal behaviour, mesomorphism, and molecular arrangements of the chiral monomers. As seen from the data listed in Table 2, with increasing the number of the flexible methylene, the corresponding melting temperature (Tm) decreased from 180.1 °C for M1 to 117.3 °C for M4, while the isotropic temperature (Ti) decreased from 147.7 °C for M2 to 119.8 °C for M4. POM showed that M1 did not exhibit any mesophase texture, while M2–M4 all exhibited enantiotropic oily streak and focal conic textures of cholesteric phase. The typical optical textures of M2 are shown in Fig. 2(a and b).
 |
| | Fig. 2 Optical textures of M2. (a) Oily streak texture of cholesteric phase on cooling to 136 °C; (b) focal conic texture of cholesteric phase on cooling to 134 °C. | |
Liquid crystal properties of the copolymers
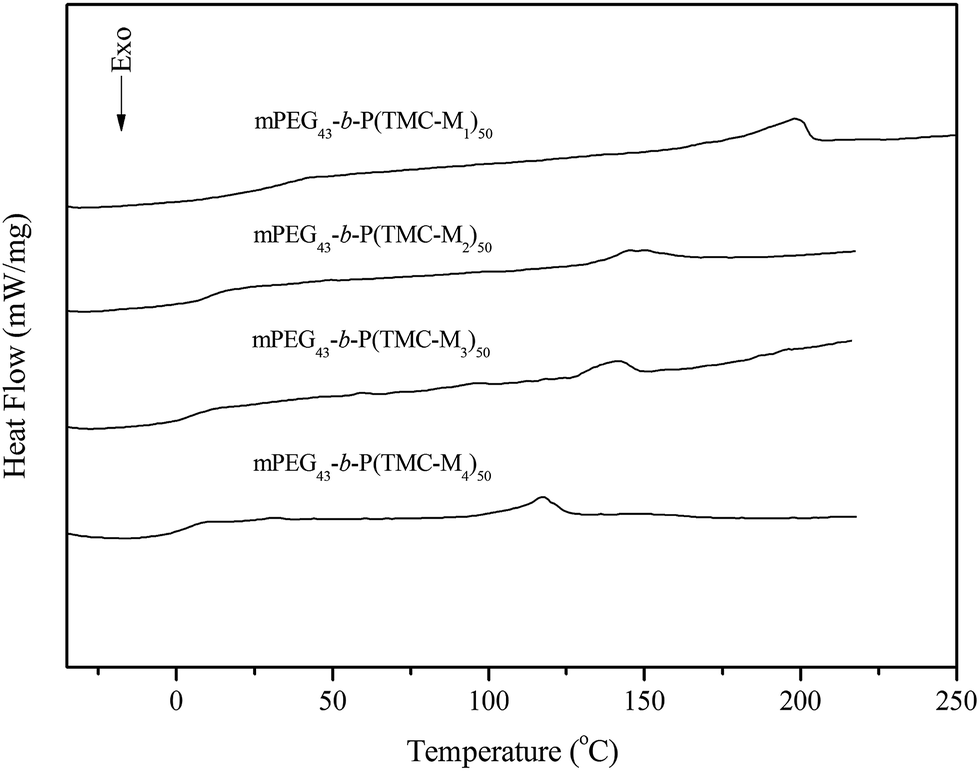
The transition temperatures of the synthesized copolymers are listed in Table 3. Their DSC curves are presented in Fig. 3. As shown in Fig. 3, the copolymers mPEG43-b-PBTMC50 and mPEG43-b-PBTMC50 only showed a glass transition because they did not contain any mesogenic units. However, four copolymers with side functionalized cholesteryl groups all revealed a glass transition at low temperature and a smectic a (SmA) to isotropic phase transition at high temperature. Although the monomer M1 did not exhibit LC property, the corresponding copolymer showed a SmA phase, and the fan-shaped texture was observed by POM. This indicated that the copolymer mPEG43-b-P(TMC-M1)50 had high interaction between side chain units leading to the generation of mesophase. Moreover, this behavior could be attributed to an increased density of the mesogenic units in side chain.34 The data listed in Table 3 revealed that three copolymers incorporating chiral monomer M2–M4 shown cholesteric phase all exhibited SmA phases, which could be further confirmed by fan-shaped texture observed by POM. This indicated in many cases that the mesophase of side chain LC polymers formed by the additional ordering on polymerization was more ordered than that the corresponding monomers. The results also suggested that the polymer chains might hinder the formation of a helical supermolecular mesogenic structure and an ordered organization into the mesophase. As an example, the optical texture of mPEG43-b-P(TMC-M2)50 is shown in Fig. 4.
Table 3 Thermal properties of the copolymersa
| Copolymer |
Phase transition temperature (°C) |
| g = glass state; SmA = smectic A phase; I = isotropic. |
| mPEG43-b-PBTMC50 |
g −20.5 I |
| mPEG43-b-PHTMC50 |
g −28.3 I |
| mPEG43-b-P(TMC-M1)50 |
g 26.6 SmA 198.8 (4.7) I |
| mPEG43-b-P(TMC-M2)50 |
g 10.2 SmA 148.9 (3.3) I |
| mPEG43-b-P(TMC-M3)50 |
g 4.9 SmA 139.5 (3.1) I |
| mPEG43-b-P(TMC-M4)50 |
g 1.5 SmA 123.4 (3.8) I |
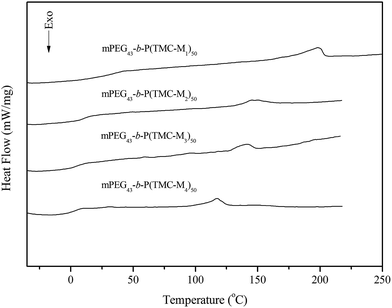 |
| | Fig. 3 DSC curves of block LC copolymers. | |
 |
| | Fig. 4 Optical textures of mPEG43-b-P(TMC-M2)50. | |
In general, the mesomorphic properties of side chain LC polymers depend on the nature of polymer backbone, flexible spacer, and mesogenic units. For four LC copolymers mPEG43-b-P(TMC-Mn)50, their glass transition temperature (Tg) and Ti were mainly affected by the spacer length because of same polycarbonate backbone and cholesteryl mesogenic units. As can be seen in Table 3, the influence of methylene segment as the flexible spacer on the phase transition temperatures of the copolymers revealed same tendency as those mentioned above for the chiral monomers, that is to say, Tg and Ti of four copolymers mPEG43-b-P(TMC-Mn)50 all decreased with increasing methylene segment spacer. Furthermore, the mesophase temperature ranges of the copolymers mPEG43-b-P(TMC-Mn)50 were greater than those of the corresponding monomers, which indicated the polymerization could further stabilize and widen the mesophase ranges.
Mesophase structure
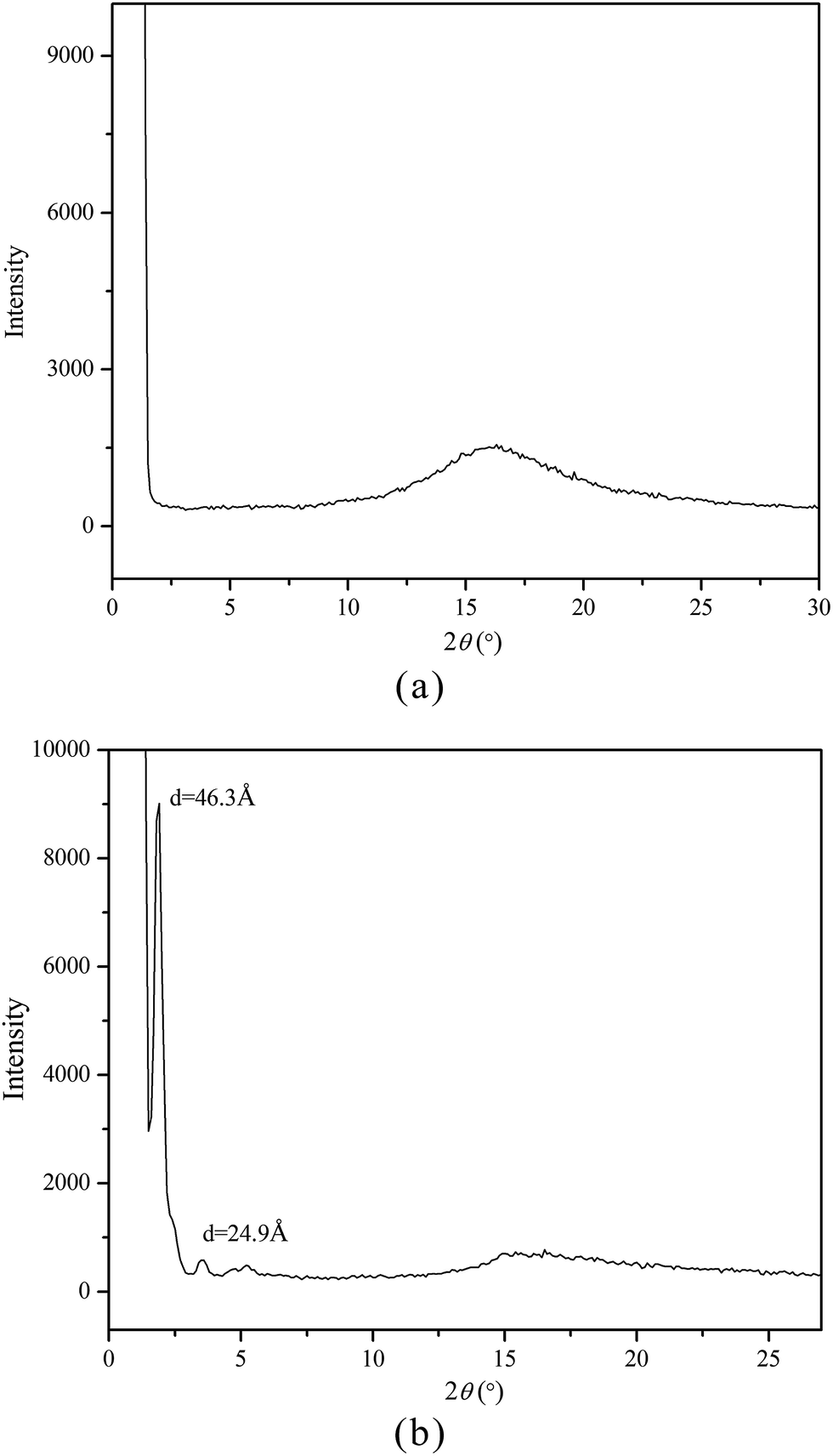
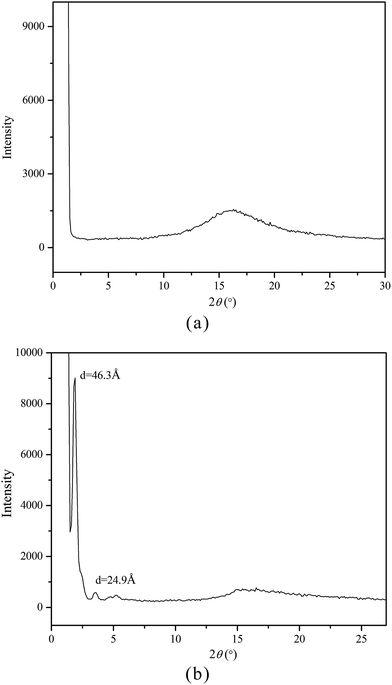
As known, XRD measurement is a powerful tool for recognizing the mesophase organization in LC materials. To further identify the mesophase structure of the synthesized monomers and copolymers, the variable-temperature XRD studies were carried out. Fig. 5(a and b) shows the representative XRD patterns of M2 at 142 °C (a) and mPEG43-b-P(TMC-M2)50 at 100 °C, respectively.
 |
| | Fig. 5 XRD curves of M2 at 142 °C (a) and mPEG43-b-P(TMC-M2)50 at 100 °C (b). | |
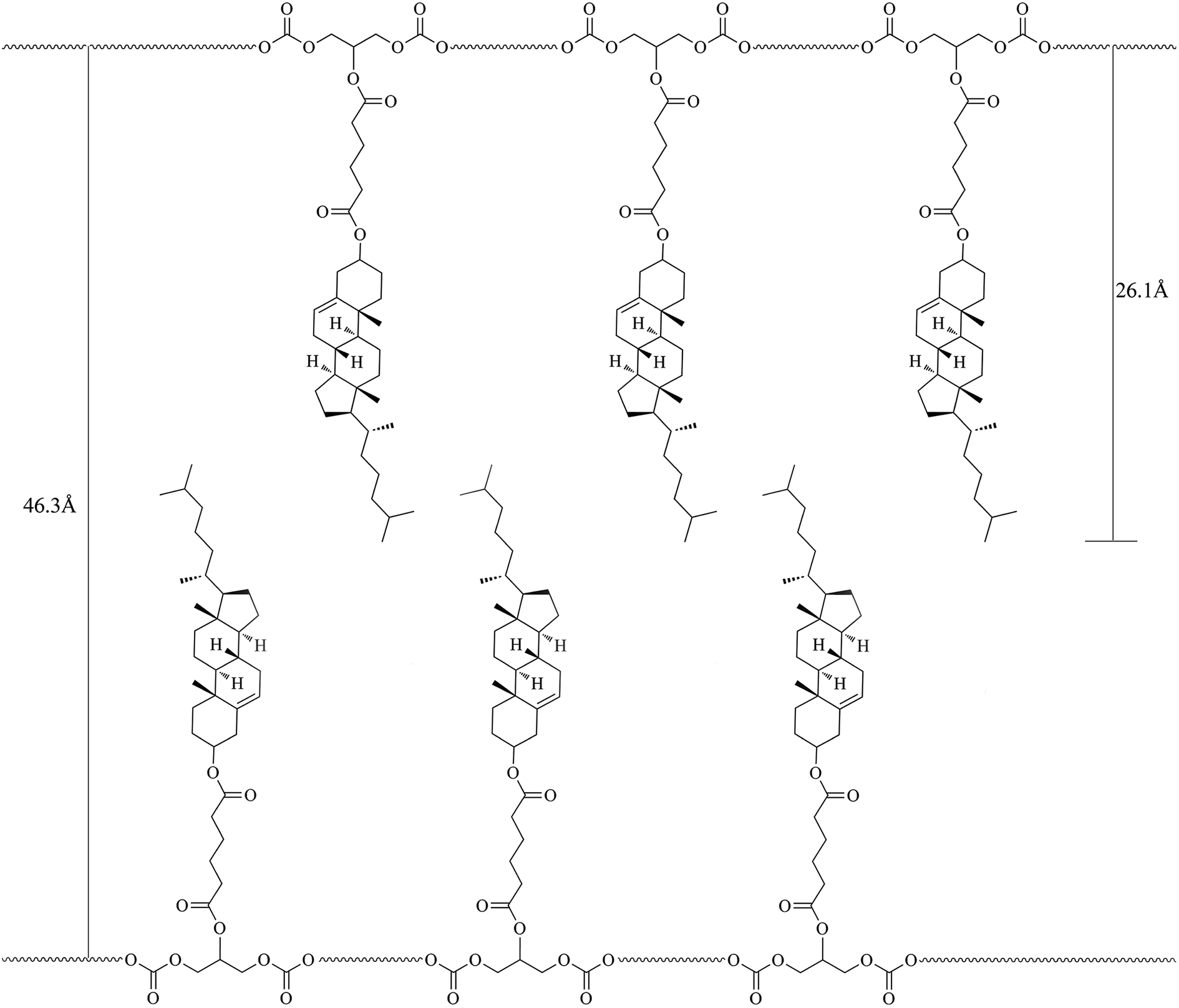
In general, a sharp reflection, corresponding to the periodic distance, is characteristic of a smectic phase in which the molecules are stacked into layer with short-range order within the layers. A broad and weak peak corresponds to the distance of the local arrangement between the neighboring mesogenic side groups. The XRD patterns of M2–M4 showed a broad peak only at around 2θ = 17°, which a typical cholesteric characteristic according to oily streak and focal conic texture observed with POM. However, the XRD patterns of four LC copolymers mPEG43-b-P(TMC-Mn)50 all revealed a sharp and intense peak at a low angle region and a weak and diffuse peak at a wide angle region, respectively. As an example, the XRD pattern of mPEG43-b-P(TMC-M2)50, shown in Fig. 5(b), revealed a sharp and strong peak at 2θ = 2.08°, corresponding to the layer d-spacing value of 46.3 Å at 100 °C, implying a smectic structure of the copolymer. Furthermore, a fan-shaped texture, as shown in Fig. 4, was clearly observed with POM, which is a characteristic texture of a SmA phase. According to the molecular modeling calculation using ChemBio3D-Ultra, employing MM2 energy parameters, the estimated all-trans molecular length L of the most extended conformation of mPEG43-b-P(TMC-M2)50 is about 26.1 Å. A d/L ratio of 1.79 (L < d < 2L) was calculated, indicating an interdigitated molecular arrangement of the mesogenic groups within the smectic layers. This partial bilayer structure was similar to the SmAd phase formed by polar mesogens. Similar results were also reported.35,36 The schematic representation of the molecular arrangement of the pendant LC groups (a) and SmA arrangements model of the interdigitated SmA layer (b) for mPEG43-b-P(TMC-M2)50 are shown in Fig. 6.
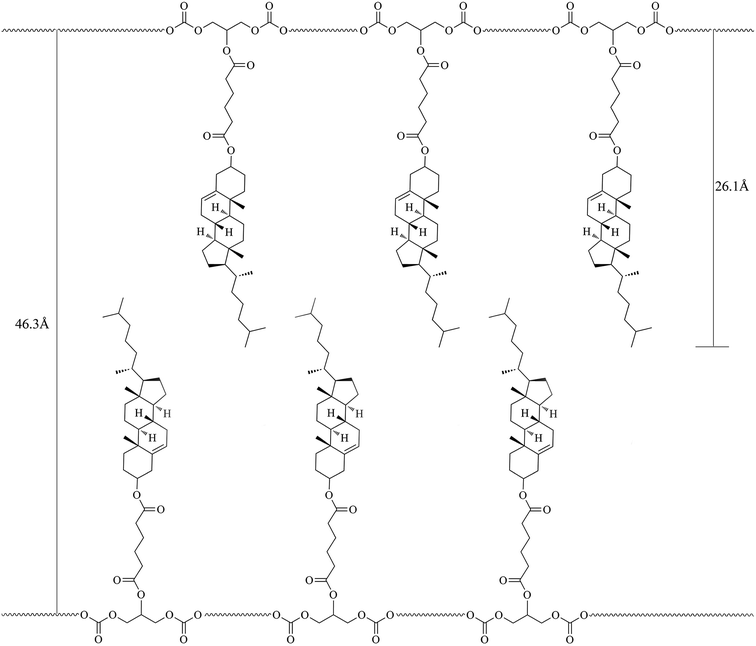 |
| | Fig. 6 Schematic representation of the SmA molecular arrangement of the pendant LC groups for mPEG43-b-P(TMC-M2)50. | |
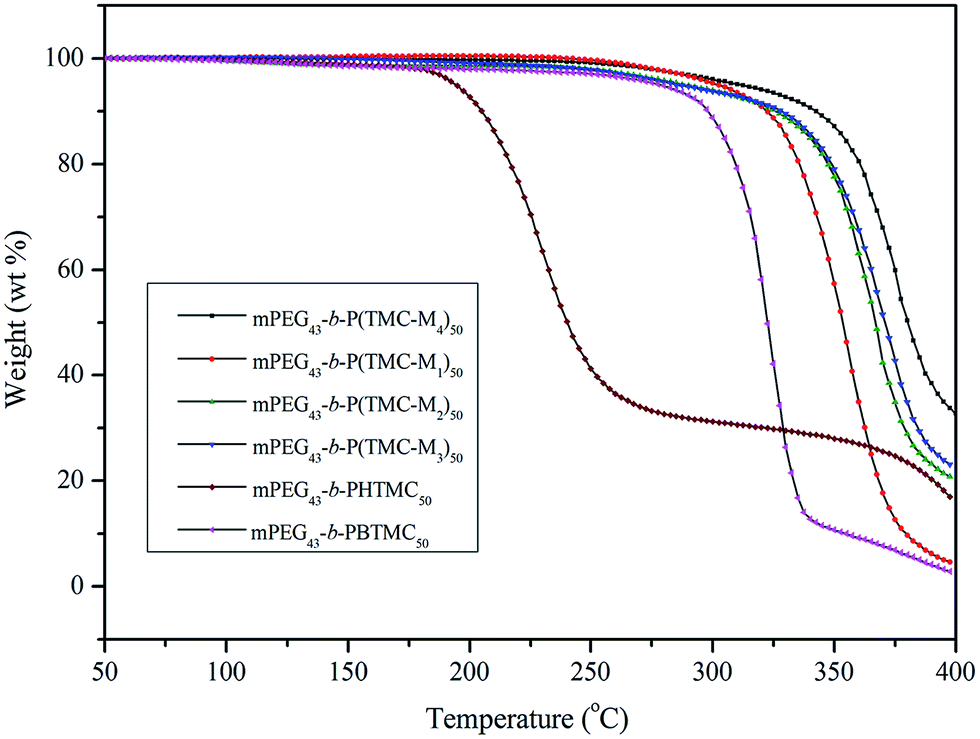
Thermal stability
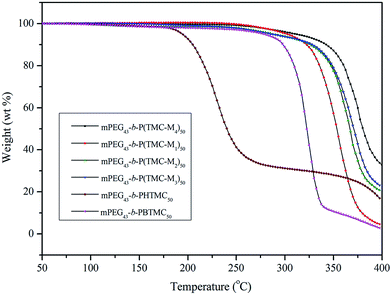
The thermal stability of the copolymers was also studied with TGA under nitrogen atmosphere, and the corresponding thermal decomposition and weight loss data are summarized in Table 4. Fig. 7 shows TGA curves of six copolymers.
Table 4 TGA data of the copolymers
| Copolymer |
Tda/°C |
Weight loss/% |
| 200 °C |
250 °C |
300 °C |
350 °C |
400 °C |
| Temperature at which 5% weight loss occurred. |
| mPEG43-b-PBTMC50 |
279 |
2.0 |
2.9 |
11.8 |
89.3 |
97.5 |
| mPEG43-b-PHTMC50 |
195 |
6.9 |
59.2 |
68.8 |
71.9 |
84.2 |
| mPEG43-b-P(TMC-M1)50 |
303 |
— |
— |
4.6 |
42.2 |
95.6 |
| mPEG43-b-P(TMC-M2)50 |
288 |
1.4 |
2.1 |
6.1 |
32.3 |
79.8 |
| mPEG43-b-P(TMC-M3)50 |
286 |
— |
2.0 |
6.2 |
20.8 |
77.4 |
| mPEG43-b-P(TMC-M4)50 |
312 |
— |
— |
4.0 |
12.8 |
61.2 |
 |
| | Fig. 7 TGA curves of block copolymers. | |
As can be clearly seen in Table 4, the temperatures at which 5% weight loss occurred (Td) of mPEG43-b-PHTMC50 was only 195 °C due to the existence of side active hydroxyl groups, which easily result in the occurrence of thermal decomposition or degradation. However, the Td increased from 195 °C for mPEG43-b-PHTMC50 to 279 °C for mPEG43-b-PBTMC50 with side phenyl groups. This result indicated that the existence of phenyl segments could increase the intermolecular π–π interaction between side repeating units, form resonance structures with the polycarbonate backbones to stabilize or deactivate the radical species, and enhance the resistance of the polymers against thermal degradation. According to Fig. 7, four LC copolymers all had higher thermal stability, moreover, the thermal weight loss or thermal degradation showed a decreased tendency with increasing the flexible spacer length of the side functionalized cholesteryl groups. The fast thermal decomposition of four LC copolymers appeared between 300 and 400 °C. When the temperature increased to 400 °C, the corresponding weight loss was 95.6%, 79.8%, 77.4%, and 61.2%, respectively. The results also suggested that the existence of the longer mesogenic side chain might cause a strong interaction between the repeating units and entanglement of the polymer chains, which lead to an increase of thermal resistance.33,37
Conclusions
Four new LC amphiphilic polycarbonates with side functionalized cholesteryl groups were synthesized and characterized. The LC copolymers obtained in this study all exhibited an enantiotropic fan-shaped texture of a SmA phase, and showed an interdigitated molecular arrangement of the mesogenic groups within the smectic layers. Although these chiral monomers showed the cholesteric phases, the corresponding copolymers showed smectic phases. With increasing the flexible spacer length, the corresponding Tg and Ti all showed a decreased tendency. Furthermore, the mesophase ranges of the copolymers were greater than those of the chiral monomers. In addition, four LC copolymers all had higher thermal stability, and the thermal weight loss showed a decreased tendency with an increase of the flexible spacer length. In summary, the amphiphilic LC copolymers based on polycarbonate backbone revealed mesophase state below body temperature, which was expected to be used clinically as a new self-assemble material for applications in controlled drug delivery to specific tumor site and so on.
Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (51503093), Science and Technology Bureau of Shenyang (F14-231-1-05), Liaoning Provincial Key Laboratory of Functional Textile Materials, and Science and Technology Committee of Liaoning Province (201501116, 2016LD0112 and 210602342).
Notes and references
- G. Rokicki, Prog. Polym. Sci., 2000, 25, 259–342 CrossRef CAS.
- X. L. Wang, R. X. Zhuo, L. J. Liu, F. He and G. Liu, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 70–75 CrossRef CAS.
- H. A. Klok, J. J. Hwang, J. D. Hartgerink and S. I. Stupp, Macromolecules, 2002, 35, 6101–6111 CrossRef CAS.
- J. Yang, Q. Li, Y. Li, L. Jia, Q. Fang and A. Cao, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2045–2058 CrossRef CAS.
- F. Suriano, O. Coulembier, J. L. Hedrick and P. Dubois, Polym. Chem., 2011, 2, 528–533 RSC.
- H. A. Klok, J. J. Hwang and S. I. Stupp, Macromolecules, 2002, 35, 746–759 CrossRef CAS.
- Y. Y. Luk, M. L. Tingey, D. J. Hall, B. A. Israel, C. J. Murphy, P. J. Bertics and N. L. Abbott, Langmuir, 2003, 19, 1671–1680 CrossRef CAS.
- S. Meng, W. Zhong and L. S. L. Chou, J. Appl. Polym. Sci., 2007, 103, 989–997 CrossRef CAS.
- S. Venkataraman, A. L. Lee, H. T. Maune, J. L. Hedrick, V. M. Prabhu and Y. Y. Yang, Macromolecules, 2013, 46, 4839–4846 CrossRef CAS.
- D. S. Yao, P. Li, X. F. Liu, J. S. Hu and L. Q. Yang, Colloid Polym. Sci., 2015, 293, 3049–3059 CAS.
- T. Kato, M. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2006, 45, 38–68 CrossRef CAS PubMed.
- H. K. Bisoyi and S. Humar, Chem. Soc. Rev., 2011, 40, 306–319 RSC.
- M. R. Hammond and R. Mezzenga, Soft Matter, 2008, 4, 952–961 RSC.
- S. H. Yang and C. S. Hsu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2713–2733 CrossRef CAS.
- J. W. Goodby, I. M. Saez, S. J. Cowling, V. Gortz, M. Draper, A. W. Hall, S. Sia, G. Cosquer, S. E. Lee and E. P. Raynes, Angew. Chem., Int. Ed., 2008, 47, 2754–2787 CrossRef CAS PubMed.
- C. Tschierske, Chem. Soc. Rev., 2007, 36, 1930–1970 RSC.
- I. Koltover, T. Salditt, J. O. Rädler and C. R. Safinya, Science, 1998, 281, 78 CrossRef CAS PubMed.
- I. G. Denisov, Y. V. Grinkova, A. A. Lazarides and S. G. Sligar, J. Am. Chem. Soc., 2004, 126, 3477–3487 CrossRef CAS PubMed.
- I. W. Hamley, Soft Matter, 2010, 6, 1863–1871 RSC.
- M. Nakata, G. Zanchetta, B. D. Chapman, C. D. Jones, J. O. Cross, R. Pindak, T. Bellini and N. A. Clark, Science, 2007, 318, 1276–1279 CrossRef CAS PubMed.
- S. J. Woltman, G. D. Jay and G. P. Crawford, Nat. Mater., 2007, 6, 929–938 CrossRef CAS PubMed.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CAS.
- J. J. Hwang, S. N. Iyen, L. S. Li, R. Claussen, D. A. Harrington and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9662–9667 CrossRef CAS PubMed.
- T. Wan, T. Zou, S. X. Cheng and R. X. Zhou, Biomacromolecules, 2005, 6, 524–529 CrossRef CAS PubMed.
- T. Zuo, S. X. Cheng and R. X. Zhuo, Colloid Polym. Sci., 2005, 283, 1091–1099 Search PubMed.
- J. Guo, J. Sun, H. Cao, D. Zhao and H. Yang, J. Appl. Polym. Sci., 2007, 105, 3505–3512 CrossRef CAS.
- K. Nagahama, Y. Ueda, T. Ouchi and Y. Ohya, Biomacromolecules, 2007, 8, 3938–3943 CrossRef CAS PubMed.
- A. L. Z. Lee, S. Venkataraman, S. B. M. Sirat, S. J. Gao, J. L. Hedrick and Y. Y. Yang, Biomaterials, 2012, 33, 1921–1928 CrossRef CAS PubMed.
- F. Z. Hu, S. D. Chen, H. Li, J. J. Sun, R. L. Sheng, T. Luo and A. M. Cao, Acta Chim. Sin., 2013, 71, 351–359 CrossRef CAS.
- Z. H. Guo, Q. Li, X. F. Liu, J. S. Hu and L. Q. Yang, Liq. Cryst., 2016, 43, 93–102 CrossRef.
- L. Yu, H. Zhang, S. X. Cheng, R. X. Zhuo and H. Li, J. Biomed. Mater. Res., Part B, 2006, 77B, 39–46 CrossRef CAS PubMed.
- P. J. Collings and M. Hird, Introduction to Liquid Crystals Chemistry and Physics, Taylor & Francis, London, 1997, ch. 3 Search PubMed.
- J. H. Liu, H. J. Hung, P. C. Yang and K. H. Tien, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6214–6228 CrossRef CAS.
- J. T. Soltysiak, K. Czupryński and W. S. Drzewiński, Polym. Int., 2006, 55, 273–278 CrossRef CAS.
- A. Favier and M. T. Charreyre, Macromol. Rapid Commun., 2006, 27, 653 CrossRef CAS.
- I. Dierking. Textures of liquid crystals. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. 2003 Search PubMed.
- J. H. Liu, Y. K. Wang, C. C. Chen, P. C. Yang, F. M. Hsieh and Y. H. Chiu, Polymer, 2008, 49, 3938–3949 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00360a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Qun Lib,
Jianshe Hu*b,
Qifan Chen*a,
Liqun Yangc and
Yanhua Lud
b,
Qun Lib,
Jianshe Hu*b,
Qifan Chen*a,
Liqun Yangc and
Yanhua Lud