
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The enhanced uranyl–amidoxime binding by the electron-donating substituents†
Zhen Qin a,
Yiming Rena,
Siwei Shia,
Chuting Yangb,
Jie Yuc,
Shaofei Wanga,
Jianping Jiaa,
Haizhu Yu*a and
Xiaolin Wang*ab
a,
Yiming Rena,
Siwei Shia,
Chuting Yangb,
Jie Yuc,
Shaofei Wanga,
Jianping Jiaa,
Haizhu Yu*a and
Xiaolin Wang*ab
aInstitute of Materials, China Academy of Engineering Physics, Mianyang, 621900, China. E-mail: xlwang@caep.cn
bInstitute of Nuclear Physics and Chemistry, China Academy of Engineering Physics, Mianyang, 621900, China
cDepartment of Chemistry, Center for Atomic Engineering of Advanced Materials, Anhui University, Hefei, 230601, China. E-mail: yuhaizhu@ahu.edu.cn
First published on 27th March 2017
Abstract
The enhanced binding of aryl amidoximes with uranyl due to the electron-donating substituents was identified by mass spectrometry, fluorescence quenching experiments, and theoretical calculations.
The sequestering of uranium from the ocean is an attractive choice for clean energy due to its vast inventory (about 4.5 billion tons).1,2 In this regard, the extractant with high binding affinity to uranyl (UO22+) is desired.3 In the past few decades, amidoxime (AO) has been screened as the most promising candidate, and various absorption materials bearing this ligand or its derivatives have been developed and applied in both mimic seawater and field experiments.4–6 To date, sequestering uranium from ocean at the industrial level is still limited.7 Enhancing the binding capability of the extractant towards uranyl has always been an issue of top-priority.8
The binding modes with uranyl were recognized for AO, as presented in Scheme 1 (η1 and η2, and chelated).9–11 Among these, the η2 mode was identified as the most favorable, and modification of the ligands at their side chains is a promising strategy to enhance their binding with uranyl. Regarding the electronic effect, actinides are strong electron acceptors,12 and the binding of the ligand with uranyl can be enhanced by incorporating electron-donating groups into the ligand molecules. This concept has been suggested based on theoretical calculations.13 Experimental studies dealing with this issue, however, are scarce due to the lack of model AO derivatives.
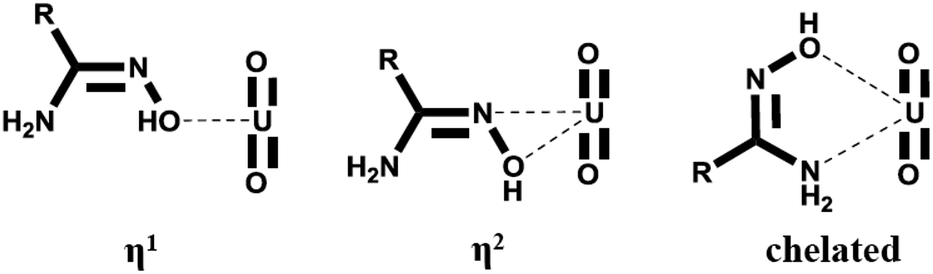 | ||
| Scheme 1 The binding modes between AO and uranyl. | ||
Recently, we reported the one-pot synthesis of aryl amidoxime (AO1–AO5 in Fig. 1) using Pd catalyst14 and investigated their complexion (AO2–AO4) with uranyl by density functional theory (DFT) calculations15 and mass spectrometric experiments.16 The synthetic strategy allows us to investigate the aforementioned substitutive effect by providing a series of AO derivatives. In this study, selected AO ligands bearing different substituent groups were studied. The results from electrospray ionization mass spectrometry (ESI-MS), fluorescence quenching experiments, and theoretical calculations provide consistent conclusions on the electronic effect of the substituent on AO–UO2 bonding.
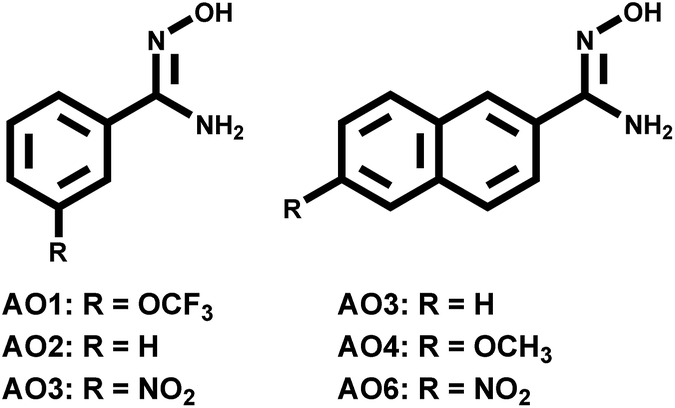 | ||
| Fig. 1 The structures and the substitutive groups of the investigated aryl-amidoxime ligands. | ||
The structures of the AO ligands are shown in Fig. 1. In the following experiments, AO2 was utilized as the reference compound with no substituent groups. The AO1 ligand has a poor electron-withdrawing effect because the strong electronegativity of fluorine atoms overcomes the slight electron donation effect of the side group itself. In comparison, the electron donation effect increases in AO3 due to an extension of the aryl ring and further increases in AO4 due to the substitutive methoxy group. Although AO5 and AO6 containing electron-withdrawing nitro-groups were expected to poorly bind with uranyl and were hard to be experimentally studied, they were investigated together with the other AO ligands using DFT calculations. As a result, the increasing trend of the electron donating abilities of the substitutive groups along with the series of AO ligands was achieved, facilitating a study of the electronic effect within uranyl–AO binding.
As we have previously reported, the main ESI-MS generated uranyl–AO species were [UO2(AO)n(NO3)]+ (n = 1–3), with the intrinsic nitrate stemmed from solid uranyl nitrate crystal. Considering the structural similarities among the AO homologues (Fig. 1) as well as the constant instrumental and solution parameters (see the ESI material†), the response factors of them in ESI-MS are expected to be similar.17 With this assumption, the signals of the UO2–AO complexes will reflect the yield of the complexes during ESI-MS process. Indeed, increasing MS intensities of all the investigated complexes (Fig. 2) were observed along the AO ligand series. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complexes, i.e., [UO2(AO)2(NO3)]+ demonstrates stronger MS signals than the 1:1 or 1:3 complexes across the AO ligand series because the equatorial plane of uranyl in 1:2 complexes was saturated with the most stable η2 binding mode of AO;18 however, it was unsaturated in 1:1 complexes or partially ligated by AO with the less stable η1 binding mode.16 Note that uranyl–AO5 complexes show a much poorer MS intensity; hence, they were not further experimentally investigated.
:2 complexes, i.e., [UO2(AO)2(NO3)]+ demonstrates stronger MS signals than the 1:1 or 1:3 complexes across the AO ligand series because the equatorial plane of uranyl in 1:2 complexes was saturated with the most stable η2 binding mode of AO;18 however, it was unsaturated in 1:1 complexes or partially ligated by AO with the less stable η1 binding mode.16 Note that uranyl–AO5 complexes show a much poorer MS intensity; hence, they were not further experimentally investigated.
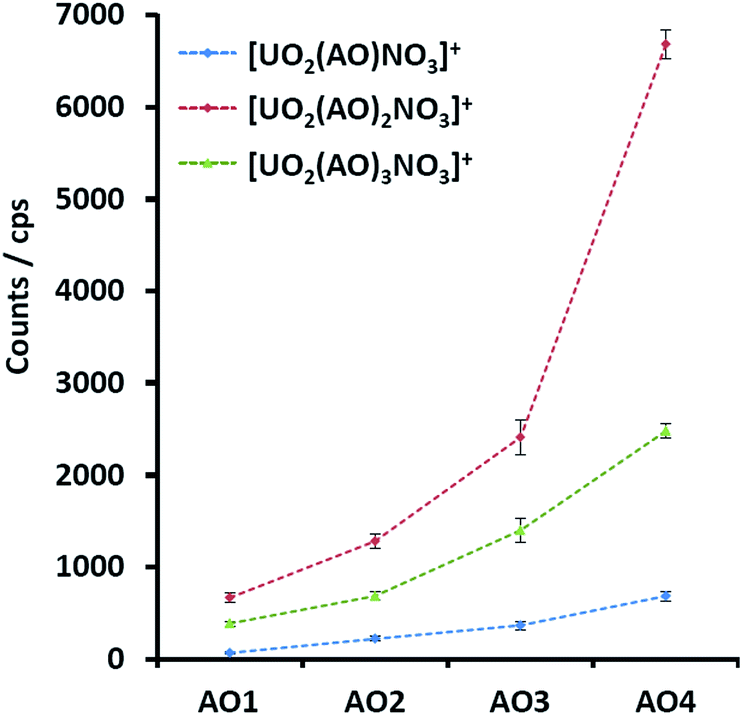 | ||
| Fig. 2 MS signals of the uranyl complexes with AO1, AO2, AO3, and AO4 with the same experimental parameters. | ||
The further exploration of the dissociation of uranyl–AO complexes by tandem MS also revealed the same trend. In these experiments, the precursor ions were mass-selected and fragmented upon collision with the buffer gas (He) in the ion trap. The dissociation profiles were plotted by obtaining the abundance of the precursor ion relative to the summed ions including both the precursor and product ions (Fig. 3). The dissociation energy (D.E.) associated with 50% relative abundance19 was utilized to evaluate the relative binding affinity of the corresponding complexes. The selection of the precursor ion, however, must be taken into account. Although there are two competing fragmentation pathways for the ESI-MS generated [UO2(AO)n(NO3)]+ (n = 1–3) complexes, viz. the loss of nitric acid or the loss of AO ligand, the first pathway was predominant.16 Consequently, the D.E. of these complexes mainly reflected the binding between uranyl and nitrate. To preclude this interference, [UO2(AO)n–H]+ (n = 2–3), which was the fragment ion of [UO2(AO)n(NO3)]+ in CID, was alternatively chosen as the target complexes (Fig. 3). Because its main fragmentation pathway was the loss of AO ligand, the corresponding D.E. reflected the uranyl–AO binding affinity. The results show that the D.E. increased from about 16% (AO1) to 22% (AO4) for [UO2(AO)2–H]+ (Fig. 3(a)), with the arbitrary unit (normalized collision energy, NCE) defined by the instrument manufacturer. The same trend was also obtained in [UO2(AO)3–H]+ complexes (Fig. 3(b)). These results indicated that breaking the UO2–AO interaction required higher energy for the AO ligands with electron-donating substituents.
| [UO2(AO2)n(NO3)]+ + nAOm (m = 1, 3, 4–6) → [UO2(AOm)n(NO3)]+ nAO2 (n = 1–3) | (1) |
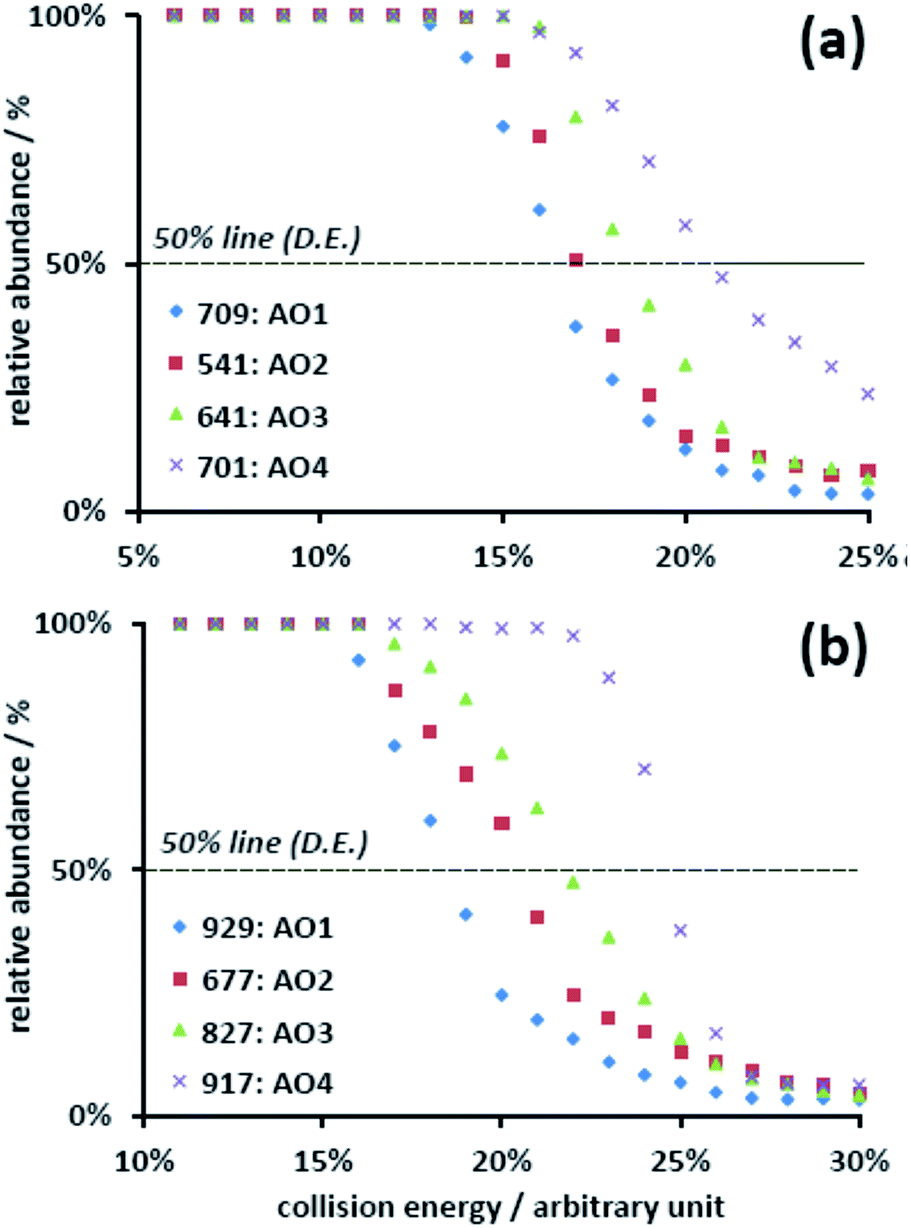 | ||
| Fig. 3 Dissociation profiles of (a) the [UO2(AO)2–H]+ and (b) [UO2(AO)3–H]+ complexes for AO1, AO2, AO3, and AO4. | ||
The selection of [UO2(AO)n–H]+ was necessary for the abovementioned experiments, but their MS signals decreased by at least one magnitude during the mass isolation. Consequently, an investigation of the D.E. of the 1:1 complexes, viz. [UO2(AO–H)]+ failed because of the low MS signals of their parent ions (Fig. 2), viz. [UO2(AO)(NO3)]+. The electronic effect of the substituents, however, could still be concluded from the abovementioned tandem MS results.
Although the solvation condition was different in the gas phase (MS) from that in the aqueous phase, the binding modes and the trend due to the binding affinity in both phases were similar.20 With this fact, the electronic effect of the substituents was also confirmed by the fluorescence quenching experiments in an aqueous solution.21,22 Under the neutral condition (pH = 7.0), the uranyl solution was titrated by the AO ligands. As presented in Fig. 4, the fluorescence of uranyl decreased upon the addition of the ligands (see details in the ESI†), which was attributed to the binding between them. Using the Stern–Volmer equation,23 the conditional stability constants (logKSV) were obtained from the slopes of the plots. The increasing conditional stability constants across the AO ligand series (3.4 for AO1, 3.6 for AO2, 3.7 for AO3, and 3.9 for AO4) clearly suggests the same trend for the binding affinity and thus for the electronic effect.
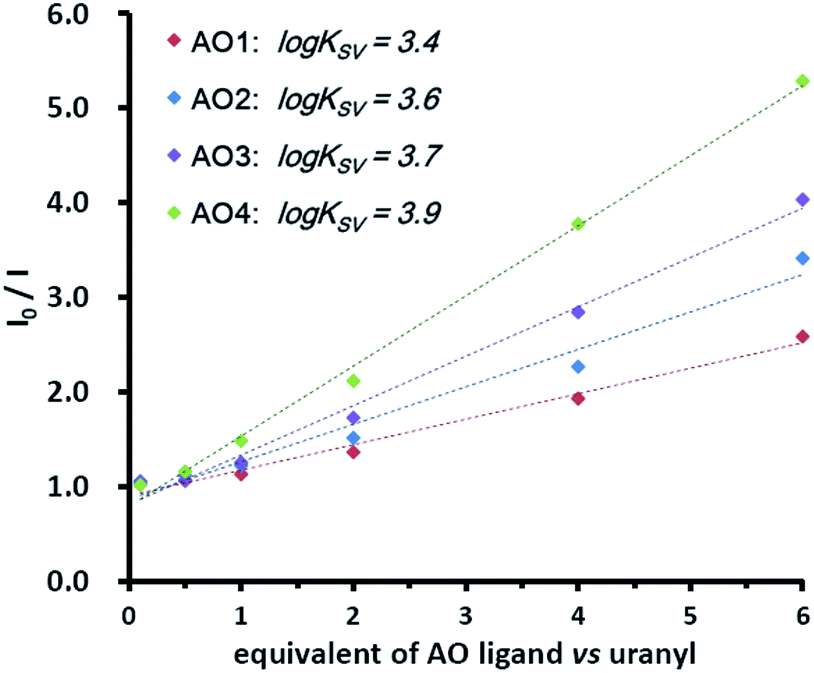 | ||
| Fig. 4 Titration of uranyl (0.1 mM, pH = 7.0 by MES buffer) by 0.1, 0.5, 1.0, 2.0, 4.0, and 6.0 equivalents of AO ligands. The excitation and emission wavelength was 310 nm and 330–580 nm, respectively. The conditional stability constants (logKSV) were calculated from the slopes of the plots. | ||
We also made an attempt to explore the electronic effect of the substituents by vibrational spectroscopy, in which the symmetric (Raman) and asymmetric (IR) stretching bands may shift upon binding with different ligands.24,25 Unfortunately, inconspicuous changes (ESI†) were obtained for the ligands, possibly due to the low resolution of the instruments.
In addition to the abovementioned experimental results, the energetics of the MS observed species were calculated using the reported density functional theory (DFT) methods.15 After geometric optimization and frequency calculations, [UO2(AO2)nNO3]+ was chosen as the reference complex, and the exchange reactions by the other AO ligands were calculated (eqn (1)). As shown in Table 1, the enthalpy changes of the corresponding exchange reactions were positive for AO5, AO6, and AO1, illustrating its poorer binding affinity with uranyl relative to that of AO2. In contrast, the exchange reactions of AO3 and AO4 were exothermic with decreasing enthalpies. In addition, according to natural bond orbital (NBO) analysis on different UO2(AOm)nNO3 (m = 1–6) complexes, the energy of the highest occupied molecular orbital (HOMO) of AOm (m = 1–6) gradually increased. As shown in Table 1, the lower HOMO energies of the electron-donating ligands result in the weaker orbital interaction between the HOMO of ligand part and the LUMO of the uranyl part, and thus weaken the binding affinities. Similar to this observation, the charge characteristic of AOn parts in the [UO2(AOm)nNO3]+ complexes also follow similar trends (Table 1). The more positive charge characters of AO2 and AO4 indicates that they donate more electrons to the uranyl center and result in stronger uranyl–AO binding strengths. Therefore, all the aforementioned discussions suggest that the electron-donating substituent benefits the stronger U–AO binding (compared to the electron-withdrawing ones). These results proved the electronic effect of the substituents.
| n = 1 | n = 2 | n = 3 | HOMO | Charge | |
|---|---|---|---|---|---|
| AO5 | 8.15 | 11.64 | 13.70 | −5.59 | 0.497 |
| AO6 | 4.83 | 8.66 | 11.88 | −5.57 | 0.518 |
| AO1 | 4.08 | 5.91 | 7.27 | −5.40 | 0.509 |
| AO2 | 0.00 | 0.00 | 0.00 | −5.12 | 0.518 |
| AO3 | −3.07 | −2.15 | −1.74 | −5.11 | 0.536 |
| AO4 | −7.50 | −6.50 | −6.40 | −4.96 | 0.559 |
In summary, the substitution effect on the binding ability of the amidoxime ligand with uranyl was qualitatively explored using the formation yields in the gas phase and the dissociation energy (D.E.) in tandem mass spectrometry. Furthermore, the fluorescence quenching experiments and theoretical calculations of the energetics presented quantitative results of this effect. This study suggested that the incorporation of the electron-donating substituents into the ligand is a promising strategy to improve the future practices in sequestering uranium from the seawater. This study also provides a protocol for evaluation of the binding between the ligands and metal ions with simplicity.
Acknowledgements
The authors appreciate the support received from the Natural Science Foundation of China (No. 21401172, 21402179, 21672001, 21507118) and the Radiochemistry 909 Project (XK 09201401) in China Academy of Engineering Physics.Notes and references
- I. Tabushi, Y. Kobuke and T. Nishiya, Nature, 1979, 280, 665–666 CrossRef CAS.
- J. Kim, C. Tsouris, R. T. Mayes, Y. Oyola, T. Saito, C. J. Janke, S. Dai, E. Schneider and D. Sachde, Sep. Sci. Technol., 2013, 48, 367–387 CrossRef CAS.
- C. J. Leggett, F. Endrizzi and L. Rao, Ind. Eng. Chem. Res., 2016, 55, 4257–4263 CrossRef CAS.
- Y. Wei, J. Qian, L. Huang and D. Hua, RSC Adv., 2015, 5, 64286–64292 RSC.
- S. Das, Y. Oyola, R. T. Mayes, C. J. Janke, L. J. Kuo, G. Gill, J. R. Wood and S. Dai, Ind. Eng. Chem. Res., 2016, 55, 4110–4117 CrossRef CAS.
- X. Lu, S. N. He, D. X. Zhang, A. T. Reda, C. Liu, J. Feng and Z. Yang, RSC Adv., 2016, 6, 101087–101097 RSC.
- J. Kim, C. Tsouris, Y. Oyola, C. J. Janke, R. T. Mayes, S. Dai, G. Gill, L. J. Kuo, J. Wood, K. Y. Choe, E. Schneider and H. Lindner, Ind. Eng. Chem. Res., 2014, 53, 6076–6083 CrossRef CAS.
- Y. Zhao, X. Wang, J. Li and X. Wang, Polym. Chem., 2015, 6, 5376–5384 RSC.
- G. Tian, S. J. Teat, Z. Zhang and L. Rao, Dalton Trans., 2012, 41, 11579–11586 RSC.
- G. Tian, S. J. Teat and L. Rao, Dalton Trans., 2013, 42, 5690–5696 RSC.
- C. Z. Wang, J. H. Lan, Q. Y. Wu, Q. Luo, Y. L. Zhao, X. K. Wang, Z. F. Chai and W. Q. Shi, Inorg. Chem., 2014, 53, 9466–9476 CrossRef CAS PubMed.
- J.-P. Dognon, Coord. Chem. Rev., 2014, 266, 110–122 CrossRef.
- C. W. Abney, S. Liu and W. Lin, J. Phys. Chem. A, 2013, 117, 11558–11565 CrossRef CAS PubMed.
- C. T. Yang, J. Han, J. Liu, M. Gu, Y. Li, J. Wen, H. Z. Yu, S. Hu and X. Wang, Org. Biomol. Chem., 2015, 13, 2541–2545 CAS.
- C. Yang, S. Pei, B. Chen, L. Ye, H. Yu and S. Hu, Dalton Trans., 2016, 45, 3120–3129 RSC.
- Z. Qin, S. Shi, C. Yang, J. Wen, J. Jia, X. Zhang, H. Yu and X. Wang, Dalton Trans., 2016, 45, 16413–16421 RSC.
- N. B. Cech and C. G. Enke, Mass Spectrom. Rev., 2001, 20, 362–387 CrossRef CAS PubMed.
- S. Vukovic, L. A. Watson, S. O. Kang, R. Custelcean and B. P. Hay, Inorg. Chem., 2012, 51, 3855–3859 CrossRef CAS PubMed.
- J. M. Daniel, S. D. Friess, S. Rajagopalan, S. Wendt and R. Zenobi, Int. J. Mass Spectrom., 2002, 216, 1–27 CrossRef CAS.
- Y. Gong, G. Tian, L. Rao and J. K. Gibson, Inorg. Chem., 2014, 53, 12135–12140 CrossRef CAS PubMed.
- C. Lebrun, M. Starck, V. Gathu, Y. Chenavier and P. Delangle, Chem.–Eur. J., 2014, 20, 16566–16573 CrossRef CAS PubMed.
- S. G. Thangavelu, S. J. A. Pope and C. L. Cahill, CrystEngComm, 2015, 17, 6236–6247 RSC.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, US, New York, 3rd edn., 2006 Search PubMed.
- D. D. Schnaars and R. E. Wilson, Inorg. Chem., 2013, 52, 14138–14147 CrossRef CAS PubMed.
- P. Di Pietro and A. Kerridge, Inorg. Chem., 2016, 55, 573–583 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, MS data, and DFT calculations. See DOI: 10.1039/c7ra00404d |
| This journal is © The Royal Society of Chemistry 2017 |