
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrocapsules with a permeable hydrogel shell and an aqueous core continuously produced in a 3D microdevice by all-aqueous microfluidics†
Serhii Mytnyk‡
 a,
Iwona Ziemecka‡a,
Alexandre G. L. Olivea,
J. Wim M. van der Meerb,
Kartik A. Totlanib,
Sander Oldenhofac,
Michiel T. Kreutzerb,
Volkert van Steijn*b and
Jan H. van Esch*a
a,
Iwona Ziemecka‡a,
Alexandre G. L. Olivea,
J. Wim M. van der Meerb,
Kartik A. Totlanib,
Sander Oldenhofac,
Michiel T. Kreutzerb,
Volkert van Steijn*b and
Jan H. van Esch*a
aAdvanced Soft Matter Group, Chemical Engineering Department, Delft University of Technology, van der Maasweg 9, 2629 HZ, Delft, The Netherlands. E-mail: j.h.vanesch@tudelft.nl
bProduct and Process Engineering Group, Chemical Engineering Department, Delft University of Technology, van der Maasweg 9, 2629 HZ, Delft, The Netherlands. E-mail: v.vansteijn@tudelft.nl
cThe Netherlands Forensic Institute, Laan van Ypenburg 6, 2497 GB, Den Haag, The Netherlands
First published on 13th February 2017
Abstract
We report the continuous production of microcapsules composed of an aqueous core and permeable hydrogel shell, made stable by the controlled photo-cross-linking of the shell of an all-aqueous double emulsion. While most previous work on water-based emulsions focused on active droplet formation, here double emulsion droplets were spontaneously generated at a three-dimensional flow-focusing junction through the break-up of a double jet formed by immiscible aqueous solutions of polyethylene glycol and cross-linkable dextrans. The capsules obtained with this lipid-free, organic-solvent-free, and surfactant-free approach displayed excellent stability under a variety of harsh conditions (3 < pH < 13, high salinity). Drying and rehydration experiments demonstrate the permeability of the shell, which may enable molecular-weight-dependent release and uptake of polar solutes.
Introduction
Microcapsules with an aqueous core and dispersed in an aqueous environment are omnipresent in nature, cells being perhaps the most well-known example. Aqueous microcapsules find use as vehicles for the delivery of pharmaceuticals and nutrients,1 storage and protection of incompatible components,2 as well as for catalyst control and recovery.3 A common way to make aqueous microcapsules is to mimic nature by separating the aqueous inner and outer parts of the capsule using a lipid bilayer.4–8 Another strategy, which omits the use of lipids, is to employ water-in-oil-in-water double emulsion droplets as templates, and cross-link the shell.9–12 A major limitation of such capsules, however, is the decreased permeability of the shell to most polar solutes, including many biomolecules. Alternatively, a shell can be formed around an aqueous droplet dispersed in an immiscible aqueous environment based on an interfacial reaction, for example, that of alginate with Ca2+, which often relies on the use of surfactants to be efficient in an all-aqueous environment.13,14 Although these strategies enable the control of the release properties by tuning the properties of the shell, the use of an organic phase or interface stabilizers may degrade the material to be encapsulated in the core of the capsule. To overcome the aforementioned limitations, we developed a lipid-free, organic-solvent-free, and surfactant-free approach to produce all-aqueous microcapsules with a cross-linked permeable hydrogel shell.Our approach is based on the use of aqueous two-phase systems (ATPS), which constitute a bio-friendly class of fluids especially relevant for the partitioning and separation of polar constituents, biomolecules, and even living cells.15–17 Particularly popular is the well-known and thoroughly characterized ATPS that forms upon mixing aqueous solutions of dextran (DEX) and polyethylene glycol (PEG). At sufficiently high polymer concentrations, phase separation occurs, resulting in two immiscible phases: a phase rich in one polymer exists in equilibrium with a phase low in that same polymer.15–17 Recently, we and others explored the use of ATPS for the continuous generation of single and double emulsion microdroplets in a microfluidic device.14,18–28 However, the lack of methods to stabilize the shell of all-aqueous double emulsion microdroplets under continuous flow, until now, precluded the ATPS approach from being applied for the production of microcapsules with an aqueous core and permeable hydrogel shell.
Here, we introduce and detail permeable microcapsules produced from all-aqueous double emulsion droplets, which are stabilized through thiol–yne-based photo-cross-linking of the shell. The capsules are stable under a wide range of conditions, presenting a novel alternative to currently existing bioencapsulation techniques, potentially allowing to better preserve the activity of loaded (bio)objects.
Additionally, the permeability of the shell may enable the application of the capsules as micro-reactors with size-dependent uptake/release capabilities.
Results and discussion
The general concept for the continuous production of hydrogel microcapsules is illustrated in Fig. 1a: we focus a stream of an aqueous polyethylene glycol (PEG) solution by a stream of an aqueous dextran (DEX) solution at the junction on the left, and the resulting thread breaks up into droplets when focused by another stream of PEG at the second junction. Introducing both polymers at sufficiently high concentration leads to phase separation while the droplets flow through the channel, thus, forming an all-aqueous double emulsion. The resulting core–shell structure is then stabilized by on-chip photo-cross-linking of the dextran shell using the thiol–yne click-reaction (Fig. 1b). Details on the synthesis and characterization of these polymers are provided in the ESI, Fig. S1–S4.†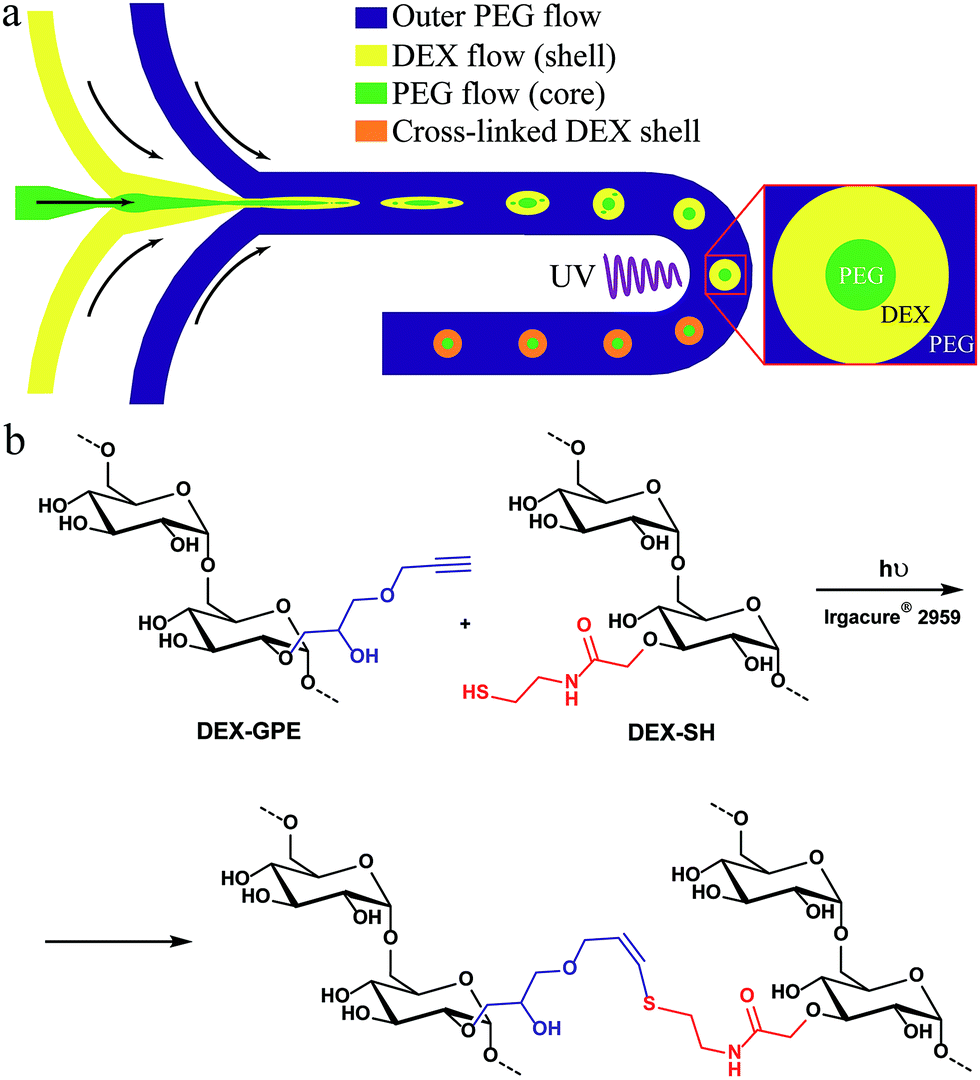 | ||
| Fig. 1 (a) Microfluidic production of particles with a permeable shell and a liquid core from aqueous solutions of PEG and DEX-SH & DEX-GPE; (b) thiol–yne photo-cross-linking of the dextran phase. | ||
The design of the three-dimensional (3D) PDMS device is shown in Fig. 2a and b. The channels have a rectangular cross-section and are 300 μm deep, except for the two 40 μm deep and 80 μm wide nozzles illustrated in the close up. These nozzles are vertically centered with respect to the surrounding channels using the method described by Rotem et al.29 The main channel after the flow focusing section is 550 μm wide. Full details on the design can be found in the AutoCAD file in the ESI.†
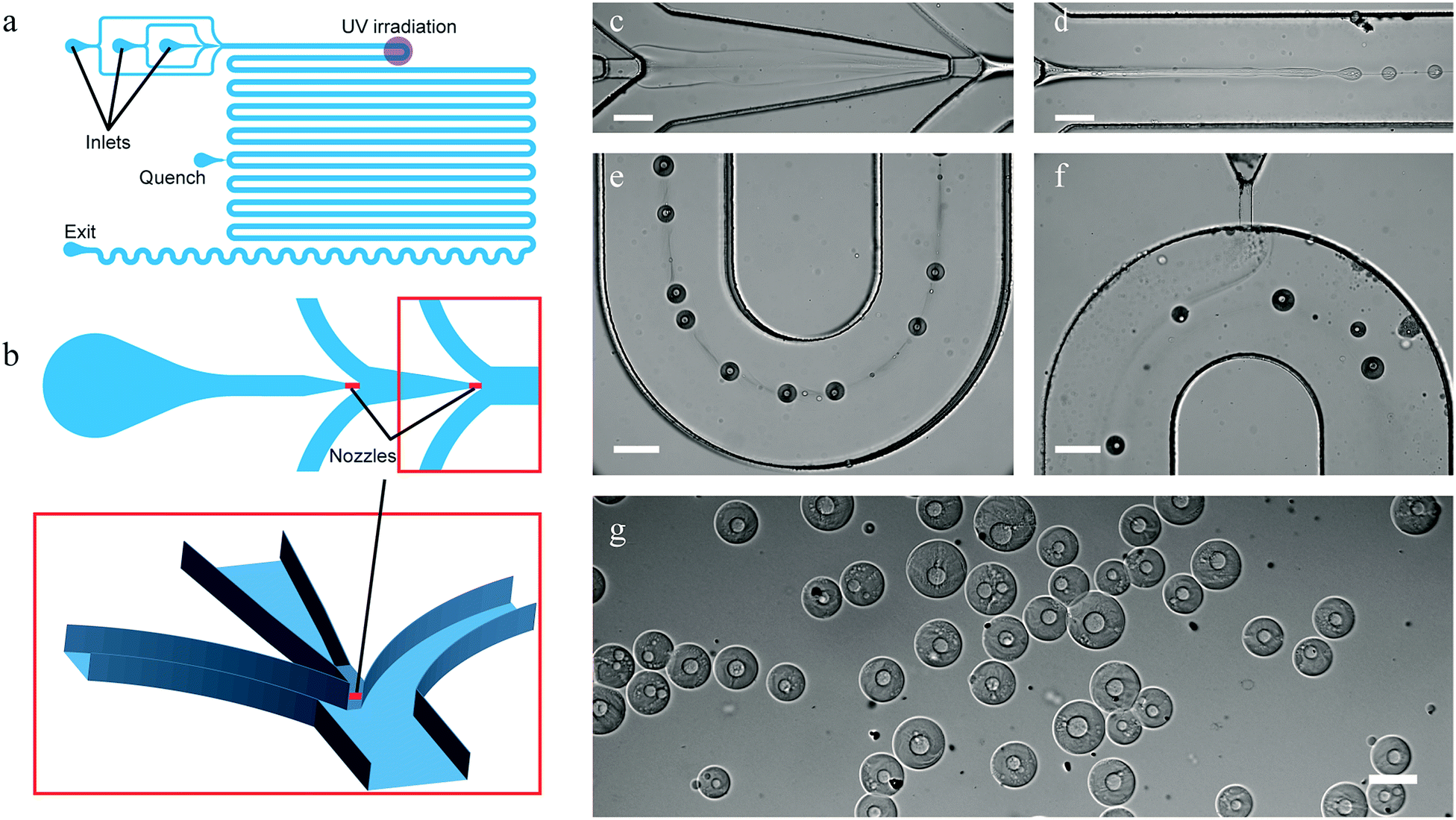 | ||
| Fig. 2 Scheme of the microfluidic device used for producing capsules with a permeable shell and a liquid core (a) (unused inlets not depicted for clarity, detailed scheme can be found in ESI†), and a zoom-in on the 3D structure of its injection nozzles (b). In short, aqueous solutions of PEG and DEX-SH & DEX-GPE were injected at two consecutive flow-focusing junctions (b and c), resulting jet-in-jet spontaneously broke up into core–shell droplets (d), and their shells were then cross-linked by exposure to UV-light (e). Radicals were quenched further downstream (f), and stable core–shell particles were collected (g) (scale bars 200 μm). The trail observed between the droplets in (e) is caused by redistribution of water and phase-forming polymers leading to local differences in composition and hence refractive index of the carrying phase. These trails slowly disappear due to diffusion (f). | ||
Droplet generation
We introduced a 10% w/w PEG solution as the core-forming phase at the first flow-focusing junction at a flow rate of 0.1 μL min−1 into the shell-forming DEX-phase (solution containing 5% w/w of both cross-linkable dextrans) flowing at 0.3 μL min−1. The resulting PEG-in-DEX thread, shown in Fig. 2c and d, spontaneously broke up into droplets downstream from the second flow-focusing junction, where a second solution of PEG (40% w/w) was introduced at 3.5 μL min−1, see Fig. 2. The mean velocity of all phases combined, downstream of the second junction, was 0.4 mm s−1 and the mean velocity of the droplets was 1.0 mm s−1.In sharp contrast to the highly stable ATPS threads reported previously,19 we observed the spontaneous breakup of a PEG-in-DEX thread into droplets near the nozzle, presenting one of the first examples of non-forced formation of an all-aqueous double emulsion using microfluidics.30,31 The spontaneous breakup is explained by the use of a 3D-device that reduces the stabilizing influence of the top and bottom walls on jet breakup.32 Additionally, the concentration of PEG in the outer phase was chosen to be significantly higher than in previous work19 to prevent the clogging of the channels (see below), with the added benefit of a larger interfacial tension facilitating droplet generation.33 Hence, there was no need to mechanically force the formation of droplets as done previously,14,18–21,26,27 which, besides requiring a simpler device, has the advantage that the encapsulated phases are not rigorously mixed upon encapsulation, resulting in a relatively fast phase separation.
Analysis of the core–shell droplets at the exposure location (Fig. 2e) showed that they had a core diameter of 22 ± 6 μm and a total diameter of 82 ± 22 μm (Fig. S5†). Operation of the microfluidic device in a jetting regime due to the low interfacial tension results in simultaneous growth of multiple jet instabilities, explaining the relatively broad droplet size distribution. As previously shown for double emulsion droplets,26 the core diameter and shell thickness can be controlled by the relative flow rates of the core and the shell phases. Varying this ratio in the range between 0.33 and 3.0, the ratio of shell to core diameter varied from 3.6 to 2.2, while the total droplet diameter decreased from 82 μm to 60 μm (Fig. S6–S8†). Since a constant total droplet diameter was expected for a fixed outer PEG flow rate and a fixed combined flow rate of the inner PEG and DEX phases, the observed decrease in diameter suggests a partial escape of the core phase during droplet formation and its subsequent phase separation into a core–shell structure.
Finally, by increasing the flow rate of the outer phase up to 10.5 μL min−1, the total droplet diameter could be reduced to 44 ± 16 μm, but the residence time in the UV beam was then too short to cross-link the droplets and form stable particles. We anticipate that the polydispersity reported here can be reduced by (mild) mechanical actuation of the forming droplets.14,18–21,26,27
Photo-cross-linking of the shells of the droplets
The core–shell droplets, obtained after phase separation, were stabilized by polymerizing the DEX shell using UV-light focused through the microscope objective lens. In order to maximize the exposure duration, droplets were irradiated in one of the channel bends (18 mm downstream from the nozzle, or approx. 15 s after the break-up), where they remained the longest in the field of irradiation (Fig. 2e). On average droplets remained in the irradiated area for about 5 s. To prevent agglomeration of still reactive particles through cross-linking of their surfaces with one another downstream from the irradiation point, we introduced a third solution of PEG (40% w/w) containing a radical quenching agent (5% w/w sodium ascorbate) at 2.0 μL min−1. The flow rate and PEG concentration (viscosity) were chosen to be sufficiently high to ensure proper contact between the injected stream and the particles as shown in Fig. 2f. Quenched particles consisting of a liquid PEG core and a hydrogel DEX shell were then collected off-chip in a magnetically-stirred vial filled with demineralized water (Fig. 2g).Before characterizing the particles, we had to address two challenges involved in stabilizing the microdroplets by photo-cross-linking in an all-aqueous environment. Initial cross-linking experiments were unsuccessful either due to incomplete polymerization of the particles or rapid clogging of the devices upon irradiation.
The issue on incomplete polymerization was caused by a generic feature of aqueous multi-phase systems, i.e. the largely nonspecific partitioning of especially low molecular weight components over all phases, compared to the much more specific partitioning in oil/water emulsions. Consequently, the photoinitiator can partition out of the cross-linkable phase. The resulting concentration of the photoinitiator in the cross-linkable dextran phase then can become insufficient for cross-linking of the dextran shell upon UV exposure. Initial polymerization tests, with the photoinitiator added solely to the DEX phase, indeed failed to produce stable particles, as noted from their immediate dissolution after exposure to pure water. Further tests, with the photoinitiator added in equal concentrations to all solutions to maintain a sufficient amount of the photoinitiator in the DEX phase, confirmed that the problem of insufficient cross-linking was resolved as demonstrated by the production of stable capsules.
The issue of clogging turned out to be more complicated. Although a full study on clogging mechanisms is beyond the scope of the present work, one mechanism observed in initial experiments is that still reactive particles sediment and stick to the bottom wall either immediately in the irradiation area, or several millimeters downstream, resulting in the rapid accumulation of partially cured particles and blocking of the main channel. This challenge was overcome by increasing the viscosity of the outer phase by increasing the outer phase PEG concentration from 15% to 40% w/w. This slowed down the sedimentation of the droplets, precluding them from contacting the bottom of the channel before their exposure to UV-light. Initial experiments in which we varied the concentration of PEG in the outer phase indeed showed that the distance needed for droplets to sediment to the bottom of the channel, increased with increasing PEG concentration. For a PEG concentration of 40%, we observed no droplets near the bottom wall over the length of the device. This observation agrees with the prediction that the sedimentation distance (estimated as uH/2u∞, with u the droplet velocity, H the height of the channel, and u∞ the terminal fall velocity) exceeds the length of the microchannel. We note that the increase in PEG concentration had an additional effect: it also reduced the amount of cross-linkable dextrans that partitioned in the outer phase, which further reduced the chance of clogging through the polymerization of the outer PEG flow.
The issues of incomplete cross-linking and clogging make stabilization of particles in an all-aqueous environment significantly more difficult compared to the stabilization of oil/water (double) emulsions. Above-mentioned considerations have allowed us to select polymer and photoinitiator concentrations that do allow complete cross-linking of the shell without clogging. Finally, we note that the time necessary for droplet formation, core–shell phase separation, UV exposure, and cross-linking must all be considered in the stabilization of all-aqueous double (and higher order) emulsions.
Characterization of the capsules
Once collected in water, the particles swelled as is common to polymeric hydrogels.34,35 Swollen particles were about twice the size they had at the irradiation point, while the core–shell structure remained intact. They had a core diameter of 44 ± 28 μm and a total diameter of 180 ± 50 μm (Fig. 2g).To study the structure of the particles in greater detail, we added dextran–FITC to the DEX-phase and imaged the collected particles using confocal microscopy. As can be seen in Fig. 3a–c, dextran indeed forms the shell and the particles have clearly defined cores. A small percentage of the particles have multiple cores, which we primarily attribute to merging of core–shell droplets prior to cross-linking. All particles displayed micro pores in their shell, as shown in Fig. 3c. Similar behavior has been observed previously for ATPS droplets22,26 and has been explained as incomplete phase separation. We envision that arresting the intermediate stages of this process, for instance by changing the irradiation location, provides a way to tune the porosity of the shell.
 | ||
| Fig. 3 Confocal microscopy images of the collected core–shell particles. Scale bars: (a) 400 μm (b) 200 μm (c) 100 μm. | ||
Permeability and stability of the capsules
Core–shell particles displayed a great resistance to various external factors once the protective and stabilizing shell was formed. No changes in size and shape were observed during their storage in water for at least one month. The structure stayed intact in drying and rehydration experiments. After 12 hours in a dry state, they recovered their initial size and shape upon rehydration, without any visible disintegration (Fig. 4a–c). These drying and rehydration experiments together with the observation of swelling, when the capsules were collected in water, clearly demonstrate the permeability of the shell, enabling their use in future release/uptake applications. Additionally, entrapment of non-cross-linkable dextran–FITC in the shell, even after dilution, suggests a potential for molecular-weight/size-dependent control of permeability.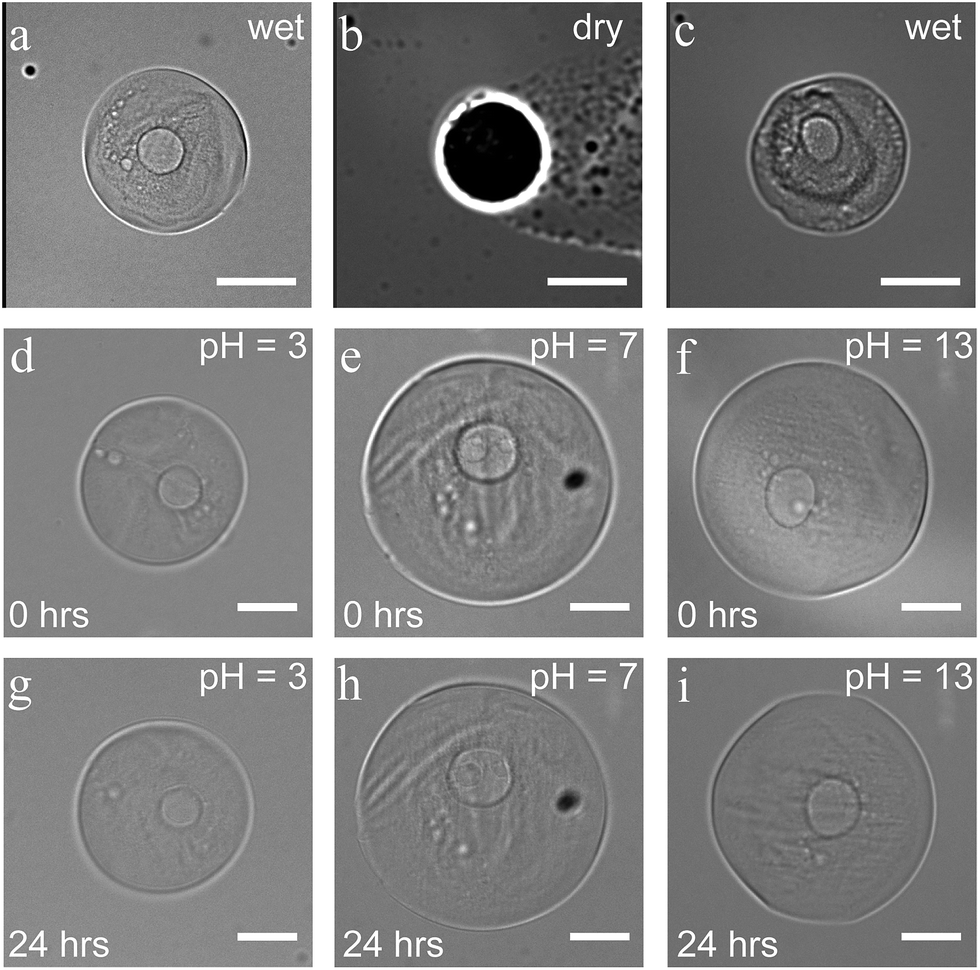 | ||
| Fig. 4 Core–shell capsule before drying (a), dried (b), and rehydrated (c). Particles immersed in solutions with pH = 3 (d and g), pH = 7 (e and h) and pH = 13 (f and i), imaged upon immersion (d–f) and 24 hours later (g–i) via bright field microscopy (scale bars 50 μm). | ||
Capsules also showed good stability upon exposure to various harsh conditions. No changes were observed when the particles were submerged for 24 hours in solutions with a pH between 3 and 13 (see Fig. 4d–i). In a more acidic environment (pH < 3), the 1,6-glycosidic bonds of dextran are expected to slowly hydrolyze, leading to chain fragmentation.36 At pH = 1, capsules indeed remained intact for 30 minutes, while the shell visibly changed within 5 hours followed by complete dissolution within 24 hours. Besides the stability in basic and acidic environments (3 < pH < 13), the particles also showed excellent stability in high-salinity medium (1 M KCl). Comparing these conditions to physiological ones, such as the salinity of blood (0.150 M of NaCl), gastric acid (pH ∼ 1.5 to 3.5), and fluids in the small intestine (pH 7.0 to 9.0), demonstrates the potential use of the all-aqueous microcapsules for biomedical applications.
Conclusions
We have reported stable and permeable hydrogel microcapsules, obtained using a novel continuous approach comprising of the selectively cross-linking of the shell of a core–shell ATPS double emulsion produced in a microfluidic device. We do acknowledge the challenging nature of selecting the appropriate combination of ATPS and cross-linking chemistry which enables (1) a favorable partitioning of the material to be encapsulated, (2) the desired release properties of the shell, and, at the same time, (3) spontaneous formation of core–shell droplets. This process may require a great deal of optimization for a specific application. Fortunately, powerful tools to efficiently screen the huge parameter space (types of polymers and salts, their concentration, and partitioning coefficients) are already available. For example, several recently published reviews summarize known partitioning trends and outline the selection criteria of ATPS formulations for specific applications.37,38 Additionally, Mace et al. have studied a large number of polymer combinations and successfully predicted the formation of more than 200 aqueous multiphase systems,39 and Lee et al. used droplet microfluidics to dynamically control the morphology of the phases.22 Given the large number of known aqueous two-phase systems, we believe that the method demonstrated here for polyethylene glycol and dextran provides access to the continuous water-based production of a wide variety of compartmentalized water-permeable microcapsules. Our future work aims at controlling molecular-weight-dependent release from the PEG core to the outer environment and application of the developed method for creating aqueous enzymatic micro reactors.Materials and methods
Materials
All reagents were used as received. Polyethylene glycol (PEG, MW = 10 kDa), sodium hydroxide, sodium ascorbate, glycidyl propargyl ether, sodium chloroacetate, N-hydroxysuccinimide (NHS), cysteamine hydrochloride, 2-hydroxy-4′-(2-hydroxyethoxy)-2-methyl-propiophenone (Irgacure® 2959) and sodium azide were purchased from Sigma-Aldrich (Steinheim, Germany). Dextrans (MW = 500 and 20 kDa) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) were purchased from Alfa-Aesar (Karlsruhe, Germany).Preparation of modified dextrans
The alkyne-functionalized dextran (DEX-GPE, MW = 500 kDa) was synthesized using the procedure of Nielsen et al.40 The degree of substitution (DS) of DEX-GPE was determined by 1H NMR as 7%.The thiol-functionalized dextran (DEX-SH, MW = 20 kDa) was prepared in two steps. First, commercial dextran (20 kDa) was carboxymethylated by sodium chloroacetate following the procedure of Brunsen et al.41 Next, an aqueous solution of the resulting carboxymethyl dextran (CM-DEX, DS = 21%) was reacted with cysteamine hydrochloride using an EDC-NHS coupling to produce DEX-SH with DS = 10%. Detailed synthetic procedures and characterization data of the prepared polymers can be found in the ESI.†
Preparation of the fluids
Polymers were freshly dissolved in demineralized water to obtain solutions with the desired concentrations. Before use, photoinitiator (Irgacure 2959) was dissolved in all phases to obtain a concentration of 5 mg mL−1 for the outer PEG phase, and 3 mg mL−1 for both the core and shell phases. Solutions were then filtered through syringe filters (Sartorius, MicroSart, 0.45 μm) and injected in the microfluidic device using individual syringe pumps (Harvard Apparatus, 11 PicoPlus).Microfabrication
The device was fabricated out of PDMS (Dow Corning, Sylgard 184 elastomer kit) using soft lithography. In short, a 4 inch silicon wafer was coated with a 40 μm thick layer of SU-8 photoresist. This layer was exposed to UV-light through a photomask, transferring the full two-dimensional design to the photoresist layer. After baking, the resulting wafer was coated with a second, 130 μm thick, layer of photoresist. It was exposed through a second photomask that only differs from the first one in having two non-transparent nozzle channels. The resulting photoresist structures on the wafer were 170 μm high except for the 40 μm high nozzles. Replica moulding in PDMS hence resulted in 170 μm deep channels and 40 μm deep nozzles. Bonding this piece of PDMS to another piece of PDMS in which all channels have a depth of 130 μm yields the desired three-dimensional devices with vertically centered nozzles. For further details, we refer to Rotem et al.29Setup
All microfluidic experiments were performed on an Axio Observer A1 inverted microscope (Zeiss) and imaged using a Zyla 5.5 sCMOS camera (Andor) at 50 fps. The microscope was equipped with an EC-Plan Neofluar objective (10×, 0.3 NA) and mercury-arc light source (HXP 120 V, 120 W), which was used with a band pass filter 300–400 nm (peak intensity at 365 nm, Zeiss, filter set 02) to initiate the thiol–yne cross-linking of the shells of the particles. In this way, the core–shell droplets were simultaneously irradiated and imaged. Total irradiance at the cross-linking spot was calculated to be 315 mW cm−2.Microfluidic experiments
Before connecting the tubes with the polymer solutions to the chip, the chip was flushed with filtered demineralized water to minimize the formation of air bubbles. Initially, all flows were set to a relatively high rate (5–20 μL min−1) to ensure that the supply is stable and the formed jets are not in contact with the walls. After this was achieved, flow rates were lowered to the desired values.Once the stable formation of core–shell droplets was observed, we cross linked the DEX shell via a thiol–yne “click”-reaction. Shortly before the start of irradiation, we put the tube (PEEK®, 1.6 mm outer and 0.5 mm inner diameter) connected to the exit of the device in a collection vial filled with 40 mL of demineralized water and stirred with a magnetic stirring bar (100–200 rpm). The reason to do this prior to the start of irradiation is that the mechanical disturbance induced by handling the tube affects the stability of jet break-up.
At the end of the experiment, the contents of the collection vial were well-stirred magnetically (700–800 rpm), transferred to a centrifugation tube, and repeatedly diluted and centrifuged (3000 rpm, ∼800g, 10 min × 3) to ensure the removal of all dissolved polymers and additives. For prolonged storage of the capsules, sodium azide (0.01%) was added to prevent bacterial and algae growth.
Size distribution analysis
Bright-field microscopy was used to collect images of droplets and particles. For each set, the diameters of 250–400 droplets or particles were measured. The resulting data was plotted as histograms and fitted to a normal distribution, see ESI† for further details. Values are reported as the mean plus/minus two times the standard deviation, μ ± 2σ, which captures over 95% of the droplets or particles.Characterization of the capsules and their stability
Aqueous dispersions of collected particles were transferred into microwells (μ-Slide 8 Well Glass Bottom, Ibidi®), and analysed using bright-field and confocal laser-scanning microscopy (Zeiss LSM 710).For the stability tests, excess of solutions with various pH and ionic strength was added, and particles were imaged using bright-field light microscopy.
For the drying and rehydration tests, the collected particles were drop-casted on a microscopy slide and allowed to dry at 20 °C while being imaged at 5 minute intervals for 12 hours. After complete drying, an excess of demineralized water was added and the rehydration process was imaged.
Acknowledgements
The authors gratefully acknowledge the financial support of the EC 7th Framework Programme Marie Curie Actions via the European ITN SMARTNET No. 316656 (S. M.), Netherlands Organization for Scientific Research (I. Z. and J. H. v. E. through NWO-VICI grant; V. v. S. through NWO-STW VENI grant; A. G. L. O. through the NWO-ECHO grant) and the NCTV (S. O.).Notes and references
- S. Mitragotri and J. Lahann, Adv. Mater., 2012, 24, 3717–3723 CrossRef CAS PubMed.
- M. A. Augustin and L. Sanguansri, in Food Materials Science: Principles and Practice, ed. J. M. Aguilera and P. J. Lillford, Springer, New York, NY, 2008, DOI:10.1007/978-0-387-71947-4_24, pp. 577–601.
- Y. Wei, S. Soh, M. M. Apodaca, J. Kim and B. A. Grzybowski, Small, 2010, 6, 857–863 CrossRef CAS PubMed.
- C. E. Stanley, K. S. Elvira, X. Z. Niu, A. D. Gee, O. Ces, J. B. Edel and A. J. deMello, Chem. Commun., 2010, 46, 1620–1622 RSC.
- M. S. Long, C. D. Jones, M. R. Helfrich, L. K. Mangeney-Slavin and C. D. Keating, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5920–5925 CrossRef CAS PubMed.
- D. C. Dewey, C. A. Strulson, D. N. Cacace, P. C. Bevilacqua and C. D. Keating, Nat. Commun., 2014, 5, 4670 CrossRef CAS PubMed.
- M. Yanagisawa, S. Nigorikawa, T. Sakaue, K. Fujiwara and M. Tokita, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15894–15899 CrossRef CAS PubMed.
- S. Deshpande, Y. Caspi, A. E. Meijering and C. Dekker, Nat. Commun., 2016, 7, 10447 CrossRef CAS PubMed.
- B. Kim, T. Y. Jeon, Y. K. Oh and S. H. Kim, Langmuir, 2015, 31, 6027–6034 CrossRef CAS PubMed.
- C.-H. Choi, H. Wang, H. Lee, J. H. Kim, L. Zhang, A. Mao, D. J. Mooney and D. A. Weitz, Lab Chip, 2016, 16, 1549–1555 RSC.
- S. H. Kim, J. W. Kim, J. C. Cho and D. A. Weitz, Lab Chip, 2011, 11, 3162–3166 RSC.
- S. S. Lee, A. Abbaspourrad and S. H. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 1294–1300 CAS.
- N. Bremond, E. Santanach-Carreras, L. Y. Chu and J. Bibette, Soft Matter, 2010, 6, 6207 RSC.
- H. C. Shum, J. Varnell and D. A. Weitz, Biomicrofluidics, 2012, 6, 012808 CrossRef PubMed.
- M. W. Beijerinck, Z. Chem. Ind. Kolloide, 1910, 7, 16–20 CrossRef.
- H. Walter and G. Johansson, Anal. Biochem., 1986, 155, 215–242 CrossRef CAS PubMed.
- P. A. Albertsson, Nature, 1958, 182, 709–711 CrossRef CAS PubMed.
- S. D. Geschiere, I. Ziemecka, V. van Steijn, G. J. Koper, J. H. Esch and M. T. Kreutzer, Biomicrofluidics, 2012, 6, 22007–2200711 CrossRef PubMed.
- I. Ziemecka, V. van Steijn, G. J. M. Koper, M. Rosso, A. M. Brizard, J. H. van Esch and M. T. Kreutzer, Lab Chip, 2011, 11, 620–624 RSC.
- S. Hardt and T. Hahn, Lab Chip, 2012, 12, 434–442 RSC.
- D. Lai, J. P. Frampton, S. A. Hari and S. Takayama, Lab Chip, 2011, 11, 3551–3554 RSC.
- S. Hui Sophia Lee, P. Wang, S. Kun Yap, T. Alan Hatton and S. A. Khan, Biomicrofluidics, 2012, 6, 22005–220058 Search PubMed.
- S. H. Ma, J. Thiele, X. Liu, Y. P. Bai, C. Abell and W. T. S. Huck, Small, 2012, 8, 2356–2360 CrossRef CAS PubMed.
- K. Vijayakumar, S. Gulati, A. J. deMello and J. B. Edel, Chem. Sci., 2010, 1, 447–452 RSC.
- M. Yasukawa, E. Kamio and T. Ono, ChemPhysChem, 2011, 12, 263–266 CrossRef CAS PubMed.
- I. Ziemecka, V. van Steijn, G. J. M. Koper, M. T. Kreutzer and J. H. van Esch, Soft Matter, 2011, 7, 9878–9880 RSC.
- Y. Song and H. C. Shum, Langmuir, 2012, 28, 12054–12059 CrossRef CAS PubMed.
- Y. Song, Y. K. Chan, Q. M. Ma, Z. Liu and H. C. Shum, ACS Appl. Mater. Interfaces, 2015, 7, 13925–13933 CAS.
- A. Rotem, A. R. Abate, A. S. Utada, V. Van Steijn and D. A. Weitz, Lab Chip, 2012, 12, 4263–4268 RSC.
- B. U. Moon, N. Abbasi, S. G. Jones, D. K. Hwang and S. S. H. Tsai, Anal. Chem., 2016, 88, 3982–3989 CrossRef CAS PubMed.
- L. Y. Zhang, L. H. Cai, P. S. Lienemann, T. Rossow, I. Polenz, Q. Vallmajo-Martin, M. Ehrbar, H. Na, D. J. Mooney and D. A. Weitz, Angew. Chem., Int. Ed., 2016, 55, 13470–13474 CrossRef CAS PubMed.
- P. Guillot, A. Colin and A. Ajdari, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 016307 CrossRef PubMed.
- D. Forciniti, C. K. Hall and M. R. Kula, J. Biotechnol., 1990, 16, 279–296 CrossRef CAS.
- F. Ganji, S. Vasheghani-Farahani and E. Vasheghani-Farahani, Iran. Polym. J., 2010, 19, 375–398 CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- M. A. Swanson and C. F. Cori, J. Biol. Chem., 1948, 172, 797–804 CAS.
- R. R. G. Soares, A. M. Azevedo, J. M. Van Alstine and M. R. Aires-Barros, Biotechnol. J., 2015, 10, 1158–1169 CrossRef CAS PubMed.
- M. Iqbal, Y. Tao, S. Xie, Y. Zhu, D. Chen, X. Wang, L. Huang, D. Peng, A. Sattar, M. A. Shabbir, H. I. Hussain, S. Ahmed and Z. Yuan, Biol. Proced. Online, 2016, 18, 18 CrossRef PubMed.
- C. R. Mace, O. Akbulut, A. A. Kumar, N. D. Shapiro, R. Derda, M. R. Patton and G. M. Whitesides, J. Am. Chem. Soc., 2012, 134, 9094–9097 CrossRef CAS PubMed.
- T. T. Nielsen, V. Wintgens, C. Amiel, R. Wimmer and K. L. Larsen, Biomacromolecules, 2010, 11, 1710–1715 CrossRef CAS PubMed.
- A. Brunsen, U. Ritz, A. Mateescu, I. Hofer, P. Frank, B. Menges, A. Hofmann, P. M. Rommens, W. Knoll and U. Jonas, J. Mater. Chem., 2012, 22, 19590–19604 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details on the synthesis and characterization of the polymers, fabrication and operation of the devices. See DOI: 10.1039/c7ra00452d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |