
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Branched PdAu nanowires with superior electrocatalytic formic acid oxidation activities†
Yingxia Wang *ab,
Zhong Xionga and
Yanzhi Xiaa
*ab,
Zhong Xionga and
Yanzhi Xiaa
aCollaborative Innovation Center for Marine Biomass Fibers, Materials and Textiles of Shandong Province, Qingdao University, Qingdao 266071, PR China. E-mail: yxwang@qdu.edu.cn
bMaterial Science and Engineering College, Qingdao 266071, PR China
First published on 21st August 2017
Abstract
In this work, novel branched palladium–gold (PdAu) bimetallic nanowires supported on graphene (PdAu/graphene) were fabricated at relatively low temperatures for the first time by a one-pot co-reduction method with the aid of poly(diallyldimethyl-ammonium chloride). The structural characteristics of PdAu/graphene were confirmed by X-ray diffraction, X-ray photoelectron spectroscopy, transmission electron microscopy, energy-dispersive X-ray spectroscopy, high-angle annular dark-field scanning transmission electron microscopy and element mapping observations. The electrochemical properties of the PdAu/graphene were characterised by cyclic voltammetry and chronoamperometry (CA) in the formic acid oxidation reaction. Compared with a commercial Pd/C catalyst, the PdAu/graphene catalyst prepared in this work exhibited enhanced electrocatalytic activity including a higher oxidation current density, enhanced CO tolerance, and a more negative onset potential. Moreover, CA analyses demonstrated that PdAu/graphene shows higher stability than the Pd/C catalyst. The PdAu/graphene catalyst exhibited higher activity and strong poisoning resistance, which could be attributed to its special branched structure from PdAu nanowires.
1. Introduction
Given increasing concerns over the environment and accelerated depletion of fossil fuels, over the past few decades the development of new energy sources as alternatives to fossil fuel has resurged. Fuel cells have attracted intense research activity due to ever-increasing environmental issues and the impending depletion of fossil fuels.1–7 Low temperature fuel cell types, including direct methanol fuel cells (DMFCs) and direct formic acid fuel cells (DFAFCs), present unique properties, such as low operating temperatures and relatively high energy densities. As such, these fuel cell types hold great promise as potential alternative power sources for portable electronic devices.8–10 DFAFCs operating near room temperature are potential power sources for stationary or portable electronic devices because of their relatively high electromotive force, relatively low fuel crossover flux through a Nafion membrane, competitive energy density and facile power-system integration.11–14 In general, the performance and efficiency of DFAFCs greatly depend on the intrinsic properties of the applied electrode catalysts, as well as the chemical and electrochemical stability of these materials in an electrolytic medium.Most fuel cells at low temperatures use Pt as a catalyst.15–17 Pt-based alloys with other transition metals, especially nanostructured materials, exhibit higher performance because of the so-called bi-functional mechanism.18–20 However, the surface of Pt as an anode catalyst is usually heavily poisoned by the strong adsorption of CO intermediates, which are produced during the oxidation of organic fuels, thereby resulting in lower catalytic performance.21–23 Among the metal-based catalysts that have been studied to date, palladium (Pd) is considered a promising alternative because of its similar properties but much lower cost compared with platinum.24 Recent studies revealed that nanoscale Pd can catalyse formic acid oxidation at the anode of a polymer electrolyte membrane fuel cell.25 Oxidation of HCOOH on Pd mainly proceeds via the desired direct dehydrogenation pathway without forming poisonous CO species as a reaction intermediate. However, the catalytic performance of pure Pd is unstable over time, particularly with high concentrations of formic acid, which may be attributed to the progressive dissolution of Pd in the oxidized state. The addition of other metals to Pd/C, such as Au has beneficial effect on the oxidation of HCOOH.26–28 Suo and Hsing found that PdAu alloys exhibited higher Pd-specific activities towards the oxidation of HCOOH compared with individual Pd catalyst and easier removal of adsorbed oxygen species (e.g. Oads or OHads) was observed from the surface of the PdAu alloy with higher Au content.29 Yuan and Liu found the enhanced catalytic activity of PdAu nanoparticles resulted from the Pd site containing Au and the atomic distribution of the alloy.30
For nanostructured electrocatalysts, catalytic performance is largely dependent on their surface structures, such as the surface facets, particle size, particle shape, etc. Xia and coworkers have shown that one-dimensional bimetallic nanowires exhibit enhanced electrocatalytic activity for anodic oxidation fuels because of the large specific areas.31 However, in the published studies, the seed-based epitaxial synthetic route is the most used method to prepare bimetallic nanowires. First the metal seeds were synthesized, and then the nanowires grew subsequently onto the metal core nanoparticles.32–34 Therefore, synthesis of PdAu bimetallic nanowires using a facile method is promising for designing novel nanostructured fuel cell electrocatalysts.
The catalytic performance of catalysts is also tightly associated with their support. Reduced graphene oxide (GO) with an interesting 2-dimensional plane structure has attracted considerable attention as a carbon support because of its convenient large-scale preparation. GO has been extensively studied in recent years because of its high conductivity (103 to 104 S m−1), large theoretical surface area (calculated value, 2600 m2 g−1), unique graphitized basal plane structure, and potentially low manufacturing cost.35–37 Functionalized GO has also attracted special attention because it enables customization of the properties of the hybrids of nanoparticles and graphene while preserving nearly all of the intrinsic properties of graphene.38,39 Functionalized GO has been demonstrated to be a promising candidate material for supporting bimetallic nanoparticles (e.g. PtRu,40 PtPd,41 and PtCu42) in fuel cells.
In this study, we report a facile, efficient, and controllable route to disperse branched palladium–gold (PdAu) nanowires on reduced GO with the aid of poly(diallyldimethyl-ammonium chloride) (PAC). Adsorption of PAC on GO can lead to a highly dense and homogeneous distribution of positive charges on the GO surface and preserve the intrinsic properties of GO without any chemical oxidation treatment. The structure of PdAu/graphene was characterised in detail by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HR-TEM), energy-dispersive X-ray spectroscopy (EDS), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), and element mapping. The electrocatalytic activity of PdAu/graphene during formic acid oxidation was examined by electrochemical cyclic voltammetry (CV) and chronoamperometry (CA), and results showed that the synthesised PdAu/graphene exhibits better activity including a higher current density and durability than commercial Pd/C.
2. Experiments
2.1. Reagents
Graphite flake (99.8%, 325 mesh) was provided by Alfa. Poly(diallyldimethyl-ammonium chloride) (PAC, 20 wt% solution in water, average molecular weight 200–350 K) was purchased from Aladdin Chemical Company, China. Analytical grade HAuCl4 and Na2PdCl4 were obtained from Tianjin Chemical Reagents, China. NaNO3, H2SO4, KMnO4 and other chemicals were of analytical grade and used without further purification. A 0.02 mol L−1 HAuCl4 solution was prepared by dissolving HAuCl4·4H2O power in twice-distilled water. High-purity nitrogen and twice-distilled water were used in all experiments. Sodium borohydride (NaBH4) was purchased from commercial sources in China and used as supplied. All solutions were prepared with deionized water.2.2. Preparation of PAC-functioned graphene
The GO was firstly synthesized according to a modified Hummers method.43,44 Then GO (40 mg) was sonicated in 50 mL distilled water for 1 h, and then 0.1 g of PAC was added. The solution was sonicated for another 1.5 h. Fastly, the suspension was centrifuged to isolate the solid product which was then washed with distilled water. This washing process was repeated three times to remove excess PAC. The PAC-functioned GO powders were dried in a vacuum oven at 60 °C for 24 h and the final product is denoted as PAC-functioned GO.2.3. Preparation of PdAu/graphene
20 mg PAC-functioned GO was dissolved in 50 mL water by ultrasonic treatment 2 h. Then 3.8 mg Na2PdCl4 and 650 μL 0.02 M HAuCl4 were added into the mixture, and the solution was sonicated for 0.5 h. Then an aqueous solution of NaBH4 (40 mL, 0.05 M) in a water-ice bath was then slowly added dropwise into the above solution under magnetic stirring, and the color of mixture changed from yellow to black. After continuous reaction for 2 h under stirring, the black product was collected by centrifugation through washing with ethanol and deionized water (V![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V = 1:1) to remove the ions possibly remaining in the final products, and then vacuum-dried 24 h at 60 °C. For Comparison, Pd/graphene was prepared with a similar method for PdAu/graphene.
:V = 1:1) to remove the ions possibly remaining in the final products, and then vacuum-dried 24 h at 60 °C. For Comparison, Pd/graphene was prepared with a similar method for PdAu/graphene.
2.4. Characterization
X-ray diffraction (XRD) analysis was carried out on Bruker D8-ADVANCE diffractometer with Cu Kα radiation (λ = 1.5406 Å, 40 kV and 40 mA). The composition of the product was measured by the inductively coupled plasma-atomic emission spectrometer (ICP-AES; USA Thermo Jarrell-Ash Corp. ICP-9000 (N + M)). Field-emission scanning electron microscopy (FESEM; JEOL, JSM-7500F, 8 kV) was used to analyze the morphology of the samples. Transmission electron microscopy (TEM), energy-dispersive X–ray (EDS) and EELS elemental mapping observations were performed on a Philips Tecnai F20 microscope, working at 200 kV. All samples subjected to TEM measurements were dispersed in ethanol ultrasonically and were dropped on copper grids. X-ray photoelectron spectroscopy (XPS) measurement was conducted with Kratos Axis Ultra DLD spectrometer with an Al Kα radiation as the X-ray source (hν = 1486.7 eV). The peak positions were corrected for sample charging by setting the C 1s binding energy at 284.8 eV.2.5. Electrochemical measurements
All electrochemical measurements were carried out with a CHI 660D electrochemical workstation system (CH Instruments, Chenhua Co., Shanghai, China) and a conventional three-electrode system. The working electrode was a Glassy Carbon Electrode (GCE) with a diameter of 3 mm. Ag/AgCl (in saturated KCl) electrode was employed as reference via a salt bridge and a Pt wire was used as counter electrode. Prior to use, the glassy carbon electrode was first polished subsequently with a 1.0, 0.3, and 0.05 μm alumina slurry and was ultrasonically cleaned with 1:1 nitric acid, ethanol and distilled water, and then dried in a stream of nitrogen gas. For electrochemical measurements, 4 mg of catalyst were dispersed in 1 mL ethanol under ultrasonication for 1 h and 10 μL of the catalyst suspension was dropped onto the GCE. After drying in air for 2 hours, a 5 μL Nafion (Sigma Aldrich) solution (0.2%) was added, followed by drying at ambient temperature. The dried GCE were cleaned again by sequentially washing with water. Prior to electrochemical measurements, the electrolytes were deoxygenated by bubbling with N2 for 30 min. The formic acid oxidation reaction (FAOR) experiments were carried out by sweeping the potential from −0.20 V to +0.90 V at a scan rate of 50 mV s−1 in a solution containing 0.5 M H2SO4 and 0.5 M HCOOH. CA curves were recorded at +0.2 V in a solution containing 0.5 M H2SO4 and 0.5 M HCOOH for 7 h to investigate the durability of the catalysts. Prior to CO adsorption, the electrolytes were deoxygenated by bubbling with N2 for 30.0 min. In the CO stripping experiments, CO was adsorbed on the pre-cleaned electrode by holding the potential at 0.10 V for 5.0 min while bubbling CO gas through the electrolyte solution. CO dissolved in the electrolyte solution and was removed by purging nitrogen through the solution for 15.0 min. All electrochemical experiments were carried out at room temperature.
For the electrochemical surface area (ECSA) study, results were calculated by integrating the charge passing the electrode during palladium oxide reduction and the CO stripping. For example, ECSA values were calculated by integrating the area under curves for the oxygen adsorption charge (QO) region between −0.2 V and +0.4 V of the reverse sweep in the CV and the QO can be determined using QO = 0.5 × Q, where Q is the charge in the oxygen adsorption/desorption area obtained after double-layer correction. The specific ECSA was calculated based on the following relation:
| Specific ECSA = QO/m × qO | (1) |
3. Results and discussion
3.1. Physicochemical properties of the samples
The morphology and structure of the as-synthesized materials were characterised by TEM (Fig. 1). Fig. 1A and B show TEM micrographs of the final products obtained from one-pot preparation methods at different magnifications. In Fig. 1A, several branched PdAu nanowires formed, and PdAu intermetallics were well-dispersed on graphene. Upon closer examination at higher magnification (Fig. 1B), a high yield of distinct nanobranches were observed from the proposed one-pot preparation method. The selected-area electron-diffraction (SAED) pattern of the product (inset of Fig. 1B) illustrates that the PdAu nanowires were crystalline in nature. Fig. 1C depicts an HRTEM micrograph of the branched PdAu nanowires. Here, the worm-like nanowires were well dispersed and a well-resolved lattice fringe can be clearly observed. Careful measurements revealed that primarily one type of lattice fringe with an interplanar spacing of 2.35 Å, which can be ascribed to the Au (111) plane, was present in the sample. To investigate the crystal structure and the element distribution of the branched PdAu nanowires, HAADF-STEM analysis was performed. Fig. 1D–F show the HAADF-STEM micrographs and selected-area element analysis maps of Pd and Au. The element maps revealed that Pd and Au were homogeneously dispersed in the PdAu nanobranches, thereby indicating the formation of an alloy structure. The HRTEM and elements analysis results strongly suggested that the PdAu alloy nanoparticles were first formed, after which the branched structure was developed. The STEM-EDS line-scan profiles (Fig. S1A–D†) of two regions of the sample showed that the distribution of Pd and Au in the branched nanowires coincided well with their alloy structures, and agreed with the results of HAADF-STEM. Fig. S2A–C† show the Pd/graphene TEM, HRTEM, and EDS results, where Pd was observed to form branched nanowires. | ||
| Fig. 1 (A, B) TEM micrographs of PdAu/graphene at different magnifications. The scale bars are (A) 100 nm and (B) 20 nm, inset of (B) is SAED of PdAu/graphene. (C) High-resolution TEM micrographs (D) the high angle annular dark field (HAADF) STEM image of PdAu and the corresponding elemental mapping of Pd (E) and Au (F) in the PdAu alloy. | ||
The EDS of PdAu/graphene shown in Fig. 2A, revealed that both Pd and Au, in addition to the Cu substrate, are present in the branched nanowires. EDS revealed that the average atomic ratio of Pd:Au was 0.45:0.55, which agrees well with the value (Pd:Au = 0.48:0.52) obtained by inductively coupled plasma-atomic emission spectrometry (ICP-AES). The weight percentage of metal species in the PdAu/graphene, Pd/graphene and Pd/C were measured to be approximately 26, 22.3 and 17.3 wt%, respectively with thermogravimetric analysis (TGA, Fig. S3†). The crystalline phase of the PdAu nanowires was studied by powder XRD measurements. The XRD patterns of GO and PdAu/graphene are shown in curves b and d of Fig. 2B. The X-ray (002) peak of graphite is observed at 2θ value of 26.5° (curve a, Fig. 2B). This peak shifted to 10.0° (curve b, Fig. 2B) after oxidation treatment. Once PdAu/graphene was formed, the 2θ value of (002) shifted to approximately 23.6° (curve d, Fig. 2B). Therefore, GO was reduced to graphene after the reduction process via sodium borohydride. XRD analysis revealed the better crystallinity of PdAu/graphene (Fig. 2B, curve d), the four representative diffraction peaks corresponding to the (111), (200), (220), and (311) planes of the PdAu alloy. Pd/graphene showed clear diffraction peaks in curve c. All of the diffraction peaks from PdAu/graphene were located between the expected positions of pure Pd (no. 65-2867) and Au (no. 65-2870). This XRD phenomenon has also been observed in other bimetallic alloy nanostructures.45–47
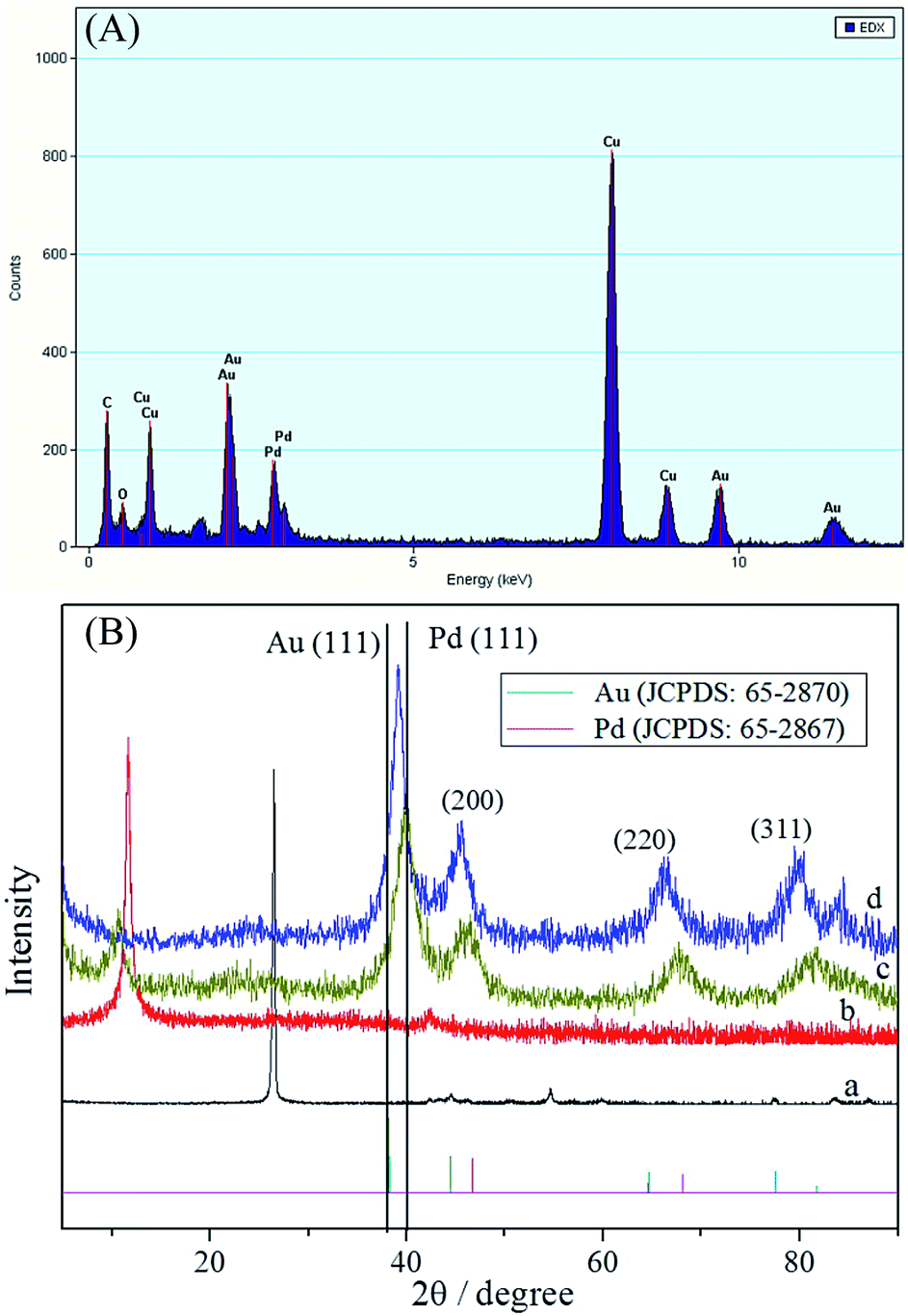 | ||
| Fig. 2 (A) EDS of PdAu/graphene. (B) XRD patterns of (a) graphite, (b) GO, (c) Pd/graphene, and (d) PdAu/graphene. | ||
XPS measurements were performed to investigate C 1s, Pd 3d, and Au 4f. The high-resolution spectra of the C 1s peak are shown in Fig. 3A: four components namely, C–C (284.5 eV), C–O (286.3 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.7 eV), and OC–O (289.7 eV) could be observed. The intensities of carbon atoms became predominant compared to the C 1s for GO (inset pattern of Fig. 3A), whereas the intensities of carbon atoms bonded to oxygen were obviously reduced. The XPS spectra of the Au 4f and Pd 3d regions of PdAu/graphene were shown in Fig. 3B and C, respectively. The results revealed the binding states of AuIII and PdII could not be observed, thereby demonstrating the AuIII and PdII precursors were successfully reduced in our synthesis. The two 4f zero valence Au0 were identified to be 4f7/2 and 4f5/2 and were located at 83.7 eV and 87.4 eV, respectively. By comparison, the Pd0 3d5/2 and 3d3/2 states were located at 335.6 and 340.8 eV, respectively. This trend indicates a small positive shift relative to pure polycrystalline Au (Au 4f7/2 at 82.2 eV and Au 4f5/2 at 86.1 eV) and Pd (Pd 3d5/2 at 334.9 eV and Pd 3d3/2 at 340.4 eV), which confirms the expected electronic interaction between Au and Pd upon the formation of an intermetallic compound.
O (288.7 eV), and OC–O (289.7 eV) could be observed. The intensities of carbon atoms became predominant compared to the C 1s for GO (inset pattern of Fig. 3A), whereas the intensities of carbon atoms bonded to oxygen were obviously reduced. The XPS spectra of the Au 4f and Pd 3d regions of PdAu/graphene were shown in Fig. 3B and C, respectively. The results revealed the binding states of AuIII and PdII could not be observed, thereby demonstrating the AuIII and PdII precursors were successfully reduced in our synthesis. The two 4f zero valence Au0 were identified to be 4f7/2 and 4f5/2 and were located at 83.7 eV and 87.4 eV, respectively. By comparison, the Pd0 3d5/2 and 3d3/2 states were located at 335.6 and 340.8 eV, respectively. This trend indicates a small positive shift relative to pure polycrystalline Au (Au 4f7/2 at 82.2 eV and Au 4f5/2 at 86.1 eV) and Pd (Pd 3d5/2 at 334.9 eV and Pd 3d3/2 at 340.4 eV), which confirms the expected electronic interaction between Au and Pd upon the formation of an intermetallic compound.
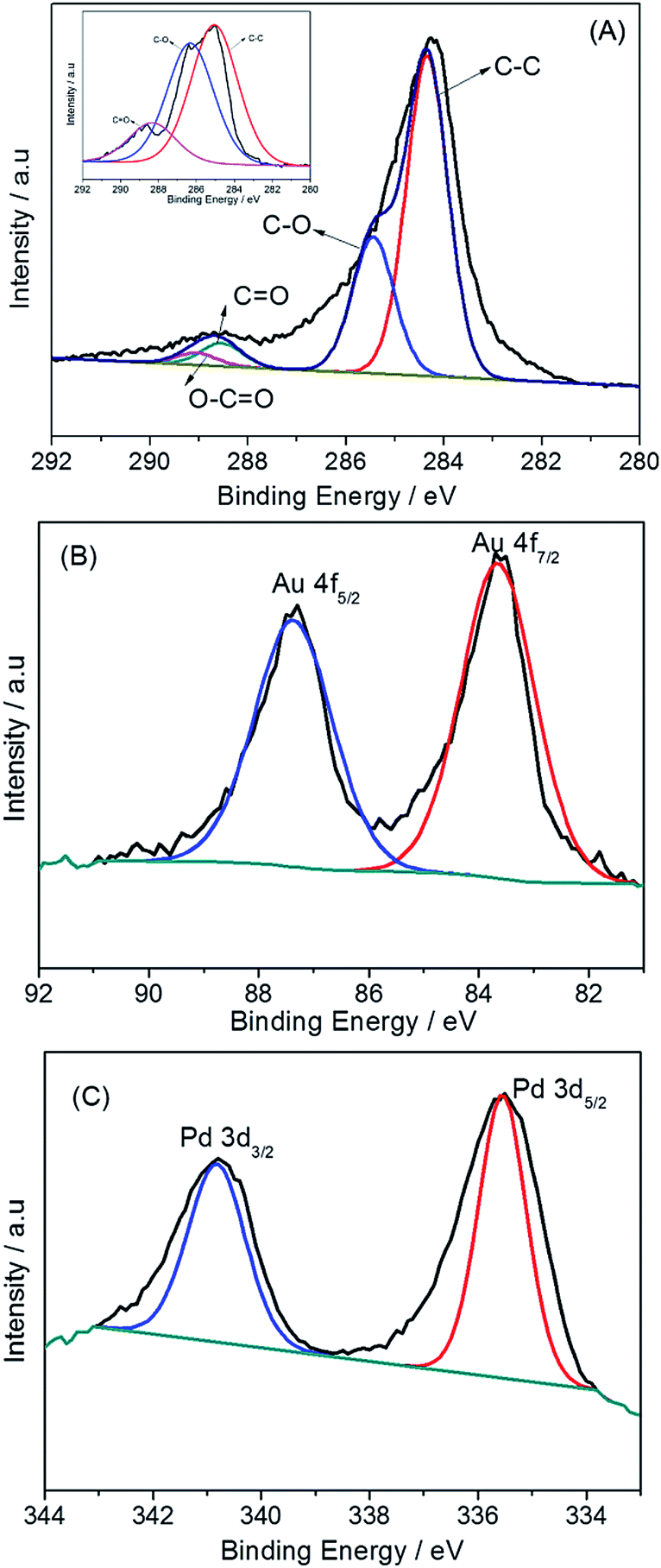 | ||
| Fig. 3 XPS spectrum of PdAu/graphene in (A) C 1s regions (B) Au 4f regions and (C) Pd 3d regions. Inset of (A) shows the C 1s XPS pattern of GO. | ||
Based on these experiments, we suggest a formation mechanism of branched PdAu nanowires on GO (Scheme 1). The steps of the synthetic route of the PdAu/graphene are as follows: first, the GO nanosheets are functionalised with amine moieties carrying PAC, thereby producing a large amount of positively charged sites on the GO sheets. The amine groups function as preferential reactive sites in electrostatic conjunction with PdCl42− and AuCl4−. Finally, the reduction of the Pd and Au precursors by NaBH4 causes the formation of nanoparticles, and GO is simultaneously reduced to graphene. The PAC layer is a polyamine with fine biocompatibility, and abundant amine that help stabilize the metal nanoparticles. Moreover, the sites of the longer PAC chains cause PdAu to form branched nanowires.
 | ||
| Scheme 1 Synthetic scheme of nanobranched palladium–gold nanowires supported on graphene. | ||
3.2. Electrocatalytic behavior of the PdAu/graphene catalyst
The ECSA of an electrocatalyst can provide important information on the availability of its active sites. The CV curves of the PdAu/graphene and Pd/C electrocatalysts in a 0.5 M H2SO4 solution were in the range of −0.2 V to 1.2 V (vs. Ag/AgCl), with a sweep rate of 0.1 V s−1. The CV curves show potential regions in the ranges −0.2–0.15 V, 0.15–0.4 V and 0.4–1.0 V which are characteristics of hydrogen adsorption–desorption, a double layer charge, and the formation and reduction of PdO, respectively. The ECSA was calculated by integrating the charge passing the electrode during palladium oxide reduction and CO stripping (by assuming a qO value of 424 μC cm−2).48 Fig. 4A also demonstrates that the area of the reduced peak of the palladium oxide for PdAu/graphene, Pd/graphene, and Pd/C, respectively. The ECSA per unit weight of Pd was estimated from the palladium oxide reduction charge to be 99.6 m2 g−1 Pd for PdAu/graphene and 93.6 m2 g−1 Pd for Pd/graphene, both of which are higher than that of the Pd/C catalyst (79.3 m2 g−1 Pd, Fig. 4B). By calculating the stripping charges of pre-adsorbed CO, the ECSA of PdAu/graphene, Pd/graphene and Pd/C were 40.6 m2 g−1, 43.5 m2 g−1 and 51.8 m2 g−1, respectively.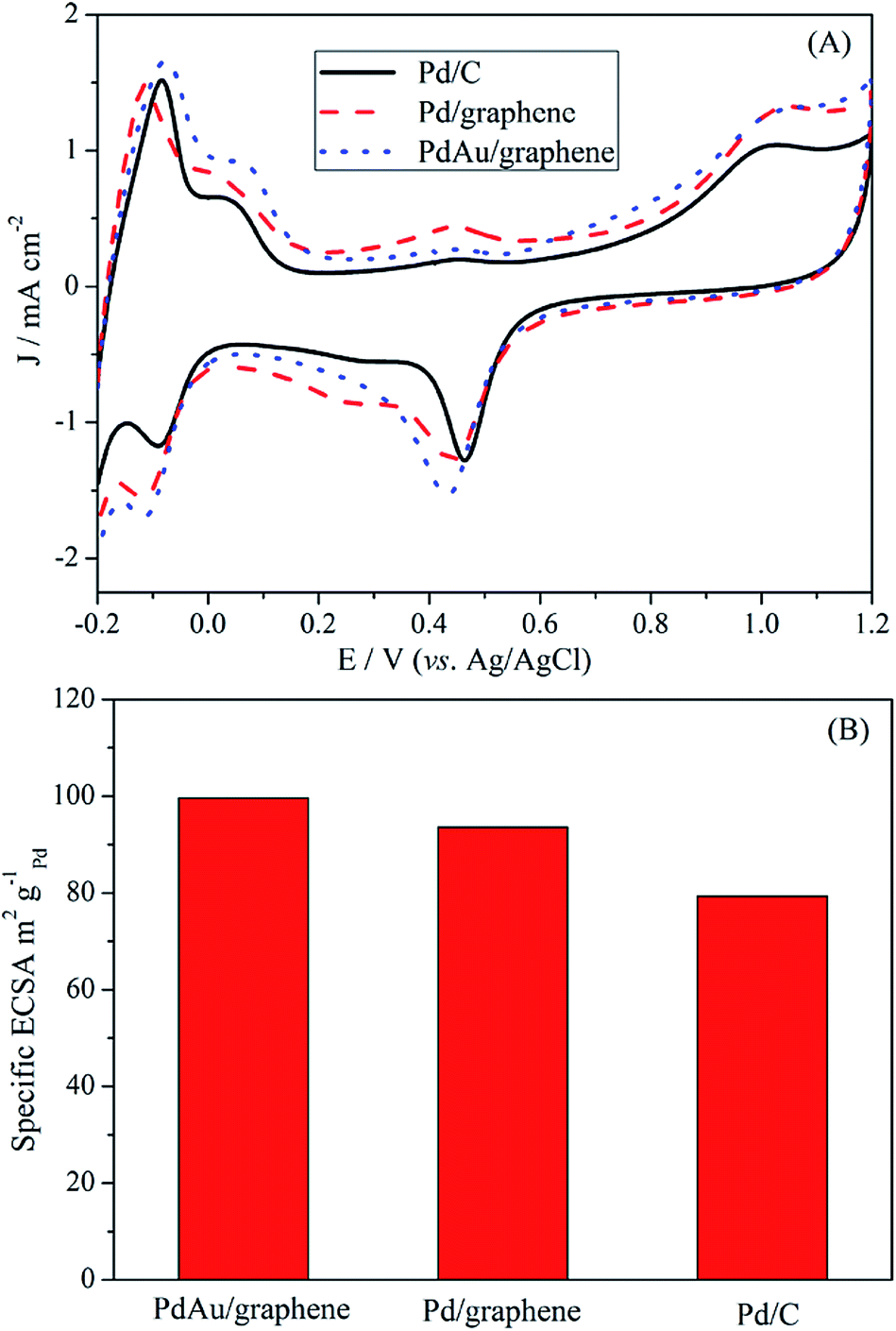 | ||
| Fig. 4 Fig. 4 (A) CV profiles of commercial Pd/C, Pd/graphene and PdAu/graphene catalysts in a 0.5 M H2SO4 solution. (B) Specific ECSA for the PdAu/graphene, Pd/graphene and commercial Pd/C catalysts. | ||
The large ECSA of the PdAu/graphene catalyst may be attributed to its branched nanostructure. The loss of ECSA with cycling was plotted in Fig. S5.† After 1000 cycles, the PdAu/graphene catalyst loss only 25% of its initial ECSA, while the commercial Pd/C catalyst lost 50% of its initial ECSA.
The catalytic activity of PdAu/graphene, Pd/graphene, and Pd/C in formic acid oxidation was investigated. To compare the mass activity of different catalysts, the kinetic current was calculated from the formic acid oxidation curve by mass-transport correction. Fig. 5A shows the steady CV curves of formic acid oxidation with PdAu/graphene and commercial Pd/C in 0.5 M H2SO4/0.5 M HCOOH at a potential scan of 50 mV s−1. The voltammetric currents were normalized to the actual active surface area (i.e. the ECSA by calculating the stripping charges of pre-adsorbed CO) of the respective electrodes. The current density of the anode peak (Ja) was larger than that of the cathodic peak (Jc) during HCOOH electro-oxidation by PdAu/graphene, thereby qualitatively indicating the improved CO tolerance of the Pd nanomaterials compared with that of Pd-based catalysts.49,50 At least two aspects of the CV formic acid oxidation curve at the PdAu/graphene electrode warrant special attentions. First, the onset potential of formic acid oxidation on PdAu/graphene was more negative than that on the Pd/C catalyst. Second, the current density of formic acid oxidation in the forward and reverse potential sweeps obtained from the Pd catalysts was much higher than that obtained from the commercial Pd/C catalysts. The current density and onset potential results indicated that PdAu/graphene has much better catalytic activity than the commercial Pd/C catalyst. The specific activity of the kinetic current was calculated from the ECSA and mass activity. The peak current densities of formic acid oxidation were: PdAu/graphene > Pd/graphene > Pd/C.
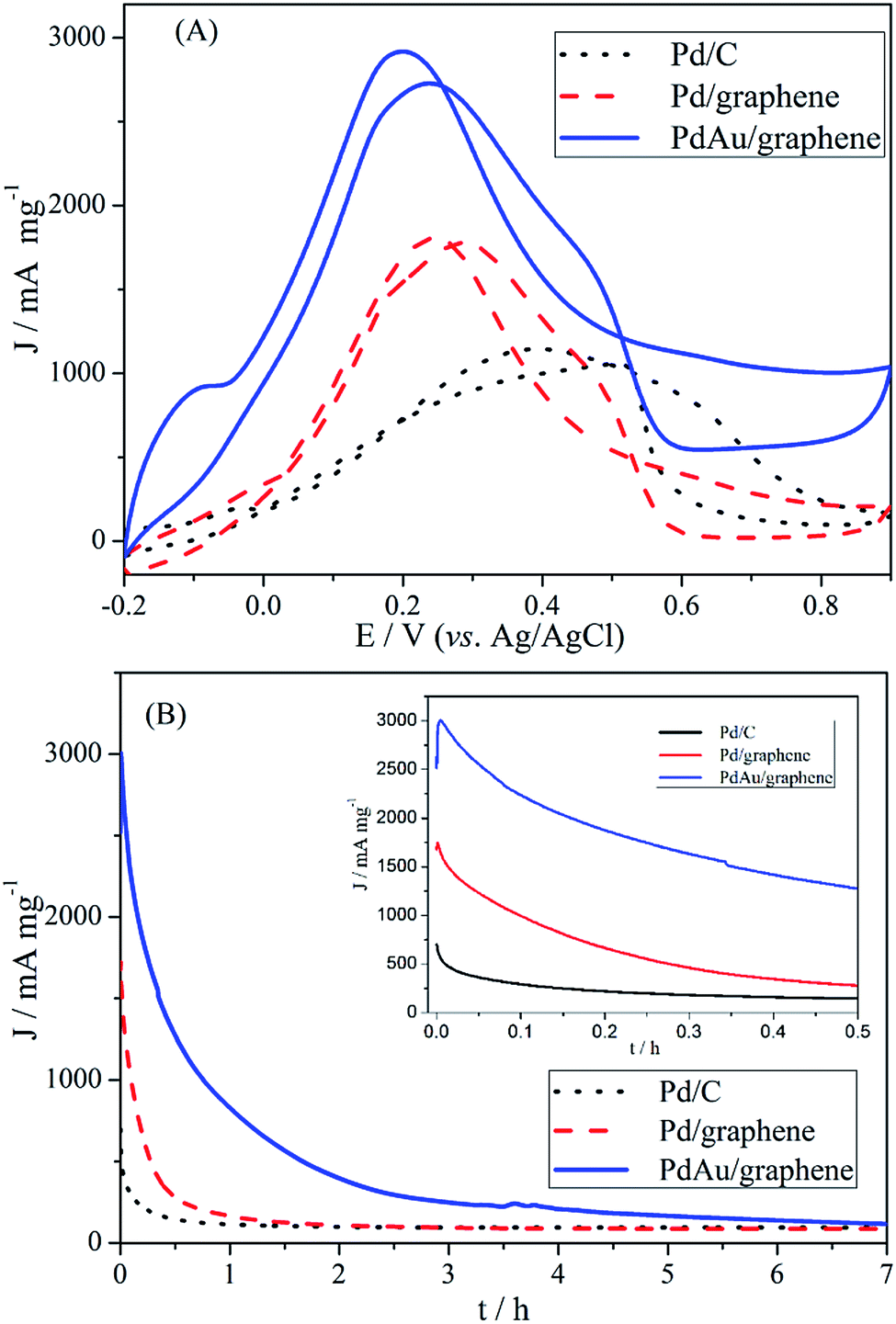 | ||
| Fig. 5 (A, B) CV and CA results of HCOOH on PdAu/graphene, Pd/graphene and Pd/C catalysts (50 mV s−1) for formic acid electro-oxidation in 0.5 M H2SO4/0.5 M HCOOH solutions. CA curves were recorded at 0.2 V. | ||
To further explore the observed enhancements in the electrocatalytic activity and CO-tolerance of the proposed catalyst, the activity and stability of PdAu/graphene, Pd/graphene and Pd/C catalysts in formic acid oxidation were evaluated by CA in 0.5 M H2SO4/0.5 M HCOOH. The CA curves at a potential of +0.20 V for the PdAu/graphene, Pd/graphene, and Pd/C electrodes are shown in Fig. 5B. The maximum initial and steady-state current densities obtained with PdAu/graphene were much larger than that on Pd/C, which agreed well with the observed CV curves (Fig. 5A). According to the literature, Pd/C catalysts suffer from poor durability in formic acid oxidation, although they show higher catalytic activity. Pd electrocatalysts could easily aggregate, dissolve from the carbon support into the electrolyte, or become poisoned by the intermediates, thereby resulting in poor durability.51 We also performed TEM on PdAu/graphene after 300 electrochemical cycles. The structure of the PdAu nanowires did not change and aggregated partly (Fig. S4†). The comparison of the catalytic activities to the previously reported results are shown in Table S1.† Compared to some earlier catalysts, which were Pd-based catalysts, the PdAu/graphene catalysts investigated in this work demonstrated enhanced activity.
Accordingly, the long-term durability of the electrocatalyst was evaluated by continuously scanning the CV curves to follow the changes in the anode peak current density with cyclic number, as shown in Fig. 6. Similar to the literature,52,53 the peak current density of commercial Pd/C sharply decreased and only 20% of the initial current remained after 60 cycles, thereby confirming the poor durability of Pd/C (Fig. 6). By contrast, the peak current density of PdAu/graphene initially increased (via electrochemically activation) and decreased to 40% after 300 cycles, thereby showing that the PdAu/graphene is relatively more stable.
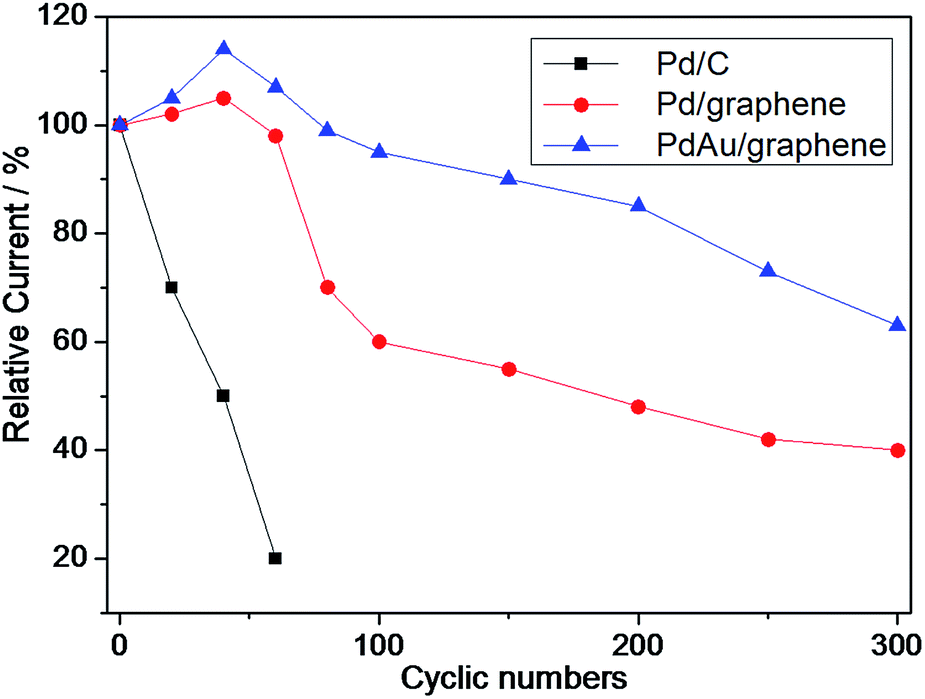 | ||
| Fig. 6 Durability of PdAu/graphene and Pd/C electrocatalysts for formic acid electro-oxidation in 0.5 M H2SO4 + 0.5 M HCOOH solutions. | ||
CO stripping experiments were also performed and the results are shown in Fig. 7. For the Pd/C catalyst, a sharp peak was observed at approximately 0.69 V. Compared with that of the Pd/C catalyst, the CO stripping voltammograms of the PdAu/graphene and Pd/graphene catalysts presented sharp peaks with potentials of 0.60 and 0.67 V, respectively, thereby suggesting higher CO oxidation activity on the PdAu/graphene and Pd/graphene catalysts because of their slightly more negative potential. Furthermore, the onset potentials of PdAu/graphene and Pd/graphene catalysts were lower than that of Pd/C, thus indicating the higher CO tolerance of the former compared with that of the latter.
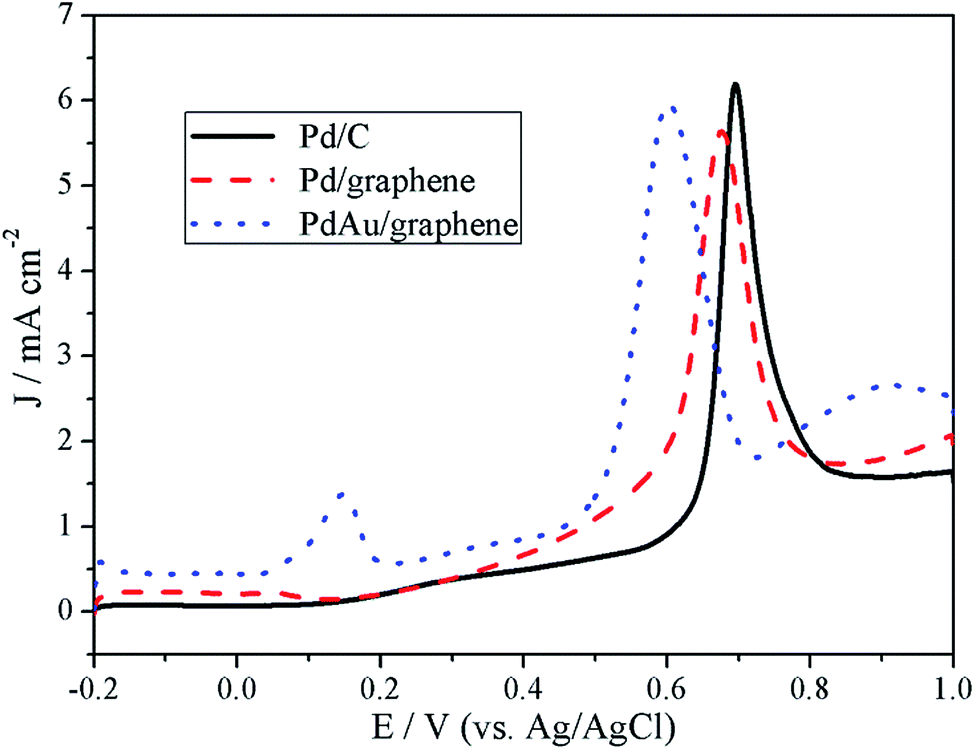 | ||
| Fig. 7 CO stripping curves of the Pd/C, PdAu/graphene and Pd/graphene catalysts in 0.5 M H2SO4. | ||
The enhanced electrocatalytic performance of branched PdAu nanowires for formic acid oxidation should be attributed to the following reasons. (1) Special nanostructures: the branched nanowires networks can improve electron transport characteristics during the catalysis. What's more, graphene can offer high electron conductivity and a large specific surface. (2) The Au co-metals on Pd can provide an opportunity to improve the adsorption of OHads onto the surface of the catalyst. Additionally, the Au can result in higher CO tolerance on the surface of the catalyst and thus activate the catalyst's surface and help to enhance the formic acid process.
4. Conclusions
Branched PdAu bimetallic nanowires enclosed by a stepped surface supported on reduced GO (PdAu/graphene) catalysts with the aid of PAC were prepared using a one-pot method. The nanostructures and electronic properties of the catalysts were investigated with XRD, XPS, TEM, HR-TEM, and HAADF-TEM. Based on its electrocatalytic performance for formic acid electro-oxidation, PdAu/graphene is a highly durable and more efficient electrocatalyst compared with commercial Pd/C in formic acid electro-oxidation. The improved performance of PdAu/graphene may be attributed to its PdAu alloy structure and the increased electrical conductivity of its graphene support. The superior overall electrocatalytic performance, especially the higher CO tolerance, suggests that a PdAu/graphene catalyst could be a promising anode catalyst for fuel cell applications. The enhanced performance of the proposed catalyst for the formic acid oxidation could be attributed to its special structure, which includes branched nanowires enclosed by a stepped surface and possible synergetic effects among its Pd and Au components. Further product analyses by in situ FTIR and HPLC are required to better understand the enhancement effects of the branched PdAu/graphene nanowires.Acknowledgements
This work is supported by the Natural Science Foundation of China (No. 51203083, 21373116), Special Fund for Self-directed Innovation of Shandong Province of China (No. 2013CXB80201), Natural Science Foundation of Shandong Province, China (No. ZR2016EMB21) and the Qingdao Postdoctoral Application Research Project.Notes and references
- R. Elosato, G. Cordaro, D. Stucchi, C. Cristiani and G. Dotelli, J. Power Sources, 2015, 298, 46–67 CrossRef.
- J. K. Wang, J. Zhou, T. Wang, G. Chen, K. Wu and Y. H. Cheng, J. Alloys Compd., 2016, 689, 581–586 CrossRef CAS.
- Z. J. He, H. Li and E. Birgersson, Renewable Energy, 2016, 85, 1301–1305 CrossRef CAS.
- G. R. Molaeimanesh, H. S. Googarchin and A. Q. Moqaddam, Int. J. Hydrogen Energy, 2016, 41, 22221–22245 CrossRef CAS.
- A. Baldinelli, L. Barelli and G. Bidni, Energy, 2015, 90, 2070–2084 CrossRef CAS.
- L. Duclos, L. Svecova, V. Laforest, G. Mandil and P. X. Thivel, Hydrometallurgy, 2016, 160, 79–89 CrossRef CAS.
- S. Dresp, F. Luo, R. Schmack, S. Kuhl, M. Gliech and P. Strasser, Energy Environ. Sci., 2016, 9, 2020–2024 CAS.
- M. M. Liu, S. J. He and W. Chen, Electrochim. Acta, 2016, 199, 218–226 CrossRef CAS.
- S. Zhang, Y. Shao, G. Yin and Y. Lin, Angew. Chem., Int. Ed., 2010, 49, 2211–2214 CrossRef CAS PubMed.
- Z. Q. Niu, D. S. Wang, R. Yu, Q. Peng and Y. D. Li, Chem. Sci., 2012, 3, 1925–1929 RSC.
- Y. Z. Lu, Y. Y. Jiang, H. B. Wu and W. Chen, J. Phys. Chem. C, 2013, 117, 2926–2938 CAS.
- Z. L. Xin, S. H. Wang, J. Wang, X. Huang, X. B. Ji, Y. Y. Yao and L. D. Shao, Electrochem. Commun., 2016, 67, 26–30 CrossRef CAS.
- Z. L. Xin, J. Wang, X. Huang, Y. Y. Yao and L. D. Shao, J. Electrochem. Soc., 2015, 162, H898–H902 CrossRef CAS.
- M. D. Obradovic and S. L. Gojkovic, Electrochim. Acta, 2013, 88, 384–389 CrossRef CAS.
- L. Zhang, L. Wang, C. M. B. Holt, B. Zahiri, Z. Li, K. Malek, T. Navessin, M. H. Eikerling and D. Mitlin, Energy Environ. Sci., 2012, 5, 6156–6172 CAS.
- C. Alegre, D. Sebastian, M. E. Galvez, R. Moliner and M. J. Lazaro, Appl. Catal., B, 2016, 192, 260–267 CrossRef CAS.
- N. Doan, M. C. Figueiredo, C. Johans and T. Kallio, Int. J. Electrochem. Sci., 2016, 11, 7631–7643 CrossRef CAS.
- Y. X. Wang, H. J. Zhou, P. C. Sun and T. H. Chen, J. Power Sources, 2014, 245, 663–670 CrossRef CAS.
- X. L. Sui, D. M. Gu, Z. B. Wang, J. Liu, L. Zhao and L. M. Zhang, RSC Adv., 2016, 6, 79254–79262 RSC.
- C. H. Wan, M. T. Lin and B. F. Su, Int. J. Hydrogen Energy, 2016, 39, 7447 CrossRef.
- S. J. Yoo, T. Y. Jeon, K. S. Kim, T. H. Lim and Y. E. Sung, Phys. Chem. Chem. Phys., 2010, 12, 15240–15246 RSC.
- A. X. Yin, X. Q. Min, W. Zhu, W. C. Liu, Y. W. Zhang and C. H. Yan, Chem.–Eur. J., 2012, 18, 777–782 CrossRef CAS PubMed.
- J. Kugai, T. Moriya, S. Seino, T. Nakagawa, Y. Ohkubo, H. Nitani, H. Daimon and T. A. Yamamoto, Int. J. Hydrogen Energy, 2012, 37, 4787–4797 CrossRef CAS.
- C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183–4206 CrossRef CAS PubMed.
- D. D. Sun, L. Si, G. T. Fu, C. Liu, D. M. Sun, Y. Chen, Y. W. Tang and T. H. Lu, J. Power Sources, 2015, 280, 141–146 CrossRef CAS.
- S. G. da Silver, J. C. M. Silva, G. S. Buzzo, E. V. Spinace, A. O. Neto and M. H. M. T. Assumpcao, Electrocatalysis, 2015, 6, 442–446 CrossRef.
- J. Nandenha, R. F. B. de Souza, M. H. M. T. Assumpcao, E. V. Spinace and A. O. Neto, Ionics, 2013, 19, 1207–1213 CrossRef CAS.
- S. X. Zhang, M. Qing, H. Zhang and Y. N. Tian, Electrochem. Commun., 2009, 11, 2249–2252 CrossRef CAS.
- Y. Suo and I.-M. Hsing, Electrochim. Acta, 2011, 56, 2174–2183 CrossRef CAS.
- D. W. Yuan and Z. R. Liu, J. Power Sources, 2013, 224, 241–249 CrossRef CAS.
- Y. Wang, S. I. Choi, X. Zhao, S. F. Xie, H. C. Peng, M. F. Chi, C. Z. Huang and Y. N. Xia, Adv. Funct. Mater., 2014, 24, 131–139 CrossRef CAS.
- Q. Yuan, Z. Y. Zhou, J. Zhuang and X. Wang, Chem. Mater., 2010, 22, 2395–2402 CrossRef CAS.
- S. J. Guo, S. J. Dong and E. K. Wang, Energy Environ. Sci., 2010, 3, 1307–1310 CAS.
- X. W. Teng, M. Feygenson, Q. Wang, J. Q. He, W. X. Du, A. I. Frenkel, W. Q. Han and M. Aronson, Nano Lett., 2009, 9, 3177–3184 CrossRef CAS PubMed.
- Y. P. Yang, P. Cheng and S. P. Huang, J. Alloys Compd., 2016, 688, 1172–1180 CrossRef CAS.
- W. L. Wang, Q. H. Zhang, J. L. Li, X. Liu, L. J. Wang, J. J. Zhu, W. Luo and W. Jiang, RSC Adv., 2015, 5, 8988–8995 RSC.
- L. Li, Y. Y. Dou, L. F. Wang, M. Luo and J. Liang, RSC Adv., 2014, 4, 25658–25665 RSC.
- J. D. Qiu, G. C. Wang, R. P. Liang, X. H. Xia and H. W. Yu, J. Phys. Chem. C, 2011, 115, 15639–15645 CAS.
- S. D. Yang, C. M. Shen, X. J. Lu, H. Tong, J. J. Zhu, X. G. Zhang and H. J. Gao, Electrochim. Acta, 2012, 62, 242–249 CrossRef CAS.
- H. Wang, J. Du, Z. Yao, R. Yue, C. Zhai, F. Jiang, Y. Du, C. Wang and P. Yang, Colloids Surf., A, 2013, 436, 57–61 CrossRef CAS.
- S. S. Li, J. J. Lv, Y. Y. Hu, J. N. Zheng, J. R. Chen, A. J. Wang and J. J. Feng, J. Power Sources, 2014, 247, 213–218 CrossRef CAS.
- F. Li, Y. Guo, Y. Liu, H. Qiu, X. Sun, W. Wang, Y. Liu and J. Gao, Carbon, 2013, 64, 11–19 CrossRef CAS.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771–778 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- B. N. Wanjala, J. Luo, R. Loukrakpam, B. Fang, D. Mott, P. N. Njoki, M. Engelhard, H. R. Naslund, J. K. Wu, L. C. Wang, O. Malis and C. J. Zhong, Chem. Mater., 2010, 22, 4282–4294 CrossRef CAS.
- J. C. Huang, Z. L. Liu, C. B. He and L. M. Gan, J. Phys. Chem. B, 2005, 109, 16644–16649 CrossRef CAS PubMed.
- Y. Z. Lu and W. Chen, Chem. Commun., 2011, 47, 2541–2543 RSC.
- M. Hara, U. Linke and T. Wandlowski, Electrochim. Acta, 2007, 52, 5733–5748 CrossRef CAS.
- W. Chen, J. Kim, S. H. Sun and S. W. Chen, Phys. Chem. Chem. Phys., 2006, 8, 2779–2786 RSC.
- W. Chen, J. M. Kim, S. H. Sun and S. W. Chen, Langmuir, 2007, 23, 11303–11310 CrossRef CAS PubMed.
- S. Y. Wang and A. M. Manthiram, Electrochim. Acta, 2013, 88, 565–570 CrossRef CAS.
- X. Yu and P. G. Pickup, Electrochem. Commun., 2009, 11, 2012–2014 CrossRef CAS.
- X. Yu and P. G. Pickup, J. Power Sources, 2009, 187, 493–499 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02115a |
| This journal is © The Royal Society of Chemistry 2017 |