
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSyntheses, characterisation, and catalytic role of (η5-C5Me5)Rh(III) guanidinato complexes in transfer hydrogenation (TH) and TH–etherification†
Robin Kumar and
Natesan Thirupathi*
and
Natesan Thirupathi*
Department of Chemistry, University of Delhi, Delhi, 110 007, India. E-mail: tnat@chemistry.du.ac.in; thirupathi_n@yahoo.com
First published on 5th July 2017
Abstract
A family of air stable half sandwich meal guanidinato complexes ([(η5-Cp*)MCl{κ2(N,N′)((ArN)2C–N(H)Ar)}]) (M = Rh and Ir; Cp* = C5Me5; Ar = aryl) were synthesized in good yield and characterised by elemental analyses, IR, and NMR (1H, 13C, and 19F) spectroscopy. The geometry of the metal and the conformations of the guanidinate ligands in the complexes were studied by single crystal X-ray diffraction. The solution behaviour of representative complexes was investigated by detailed NMR studies including variable temperature and variable concentration 1H NMR measurements. The new complexes were screened as catalysts for transfer hydrogenation (TH) of acetophenone under basic and base free conditions and from these experiments, ([(η5-Cp*)RhCl{κ2(N,N′)((ArN)2C–N(H)Ar)}]) (Ar = 3,5-(CF3)2C6H3; 3) was chosen as the preferred catalyst due to its slightly better catalytic activity than other complexes. The utility of 3 in TH of a variety of carbonyl compounds was explored under basic and base free conditions. Tandem catalysis involving TH of a carbonyl group and etherification of the resulting –CH2OH group in reduction products of salicylaldehyde, 2-hydroxy-1-naphthaldehyde and 5-(hydroxymethyl)furfural was achieved in the presence of 3 under base free conditions. The role of the guanidinate ligands in the complexes for basic and base free TH of carbonyl compounds and TH–etherification tandem catalysis is discussed. Plausible mechanisms for TH and TH–etherification are outlined.
Introduction
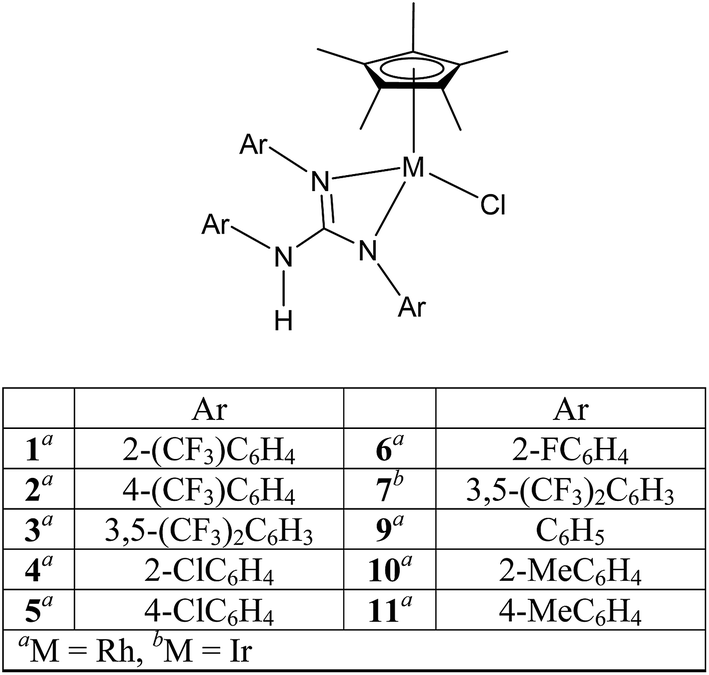
Half sandwich complexes of the type [(η5-Cp*)MCl(NN)] (Cp* = C5Me5; M = Rh and Ir; NN = κ2 monoanionic N-donor ligands) are well known organometallic compounds due to their interesting structural and bonding features, and reactivity patterns, and their utility as homogeneous catalysts in numerous organic transformations.1–12 The utility of the aforementioned complexes when one of the coordinating N atoms of the ligand contains a proton in organometallic catalysis such as hydrogenation and transfer hydrogenation (TH) is well known.4,13–15 Closely related 16-electron complexes of the type [(η5-Cp*)M(NN)] (M = Rh and Ir; NN = κ2 dianionic N-donor ligands) are known for their catalytic value in TH of carbonyl compounds.5a,13Sym N,N′,N′′-trisubstituted guanidines, (RNH)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR (Sym = symmetrical; R = alkyl and aryl) are one of the interesting classes of N-donor ligands due to not only their ability to act as monoanion, [R(H)N–C(NR)2]− (guanidinate(1−) or guanidinato) and dianion, [C(NR)3]− (guanidinate(2−)) but also the ready tunability of the R group so that distinct donor characteristics and coordination modes can be realised for these anions in their metal complexes.16,17 In our previous publications,18,19 we studied structural aspects and the utility of half sandwich (η6-p-cymene)Ru(II) complexes that contain guanidinate ligand or guanidinium cation in TH of carbonyl compounds. Herein, we report syntheses and characterisation of half sandwich (η5-Cp*)M(III) guanidinato complexes, 1–8 by analytical, spectroscopic (IR and NMR) techniques and single crystal X-ray diffraction (SCXRD; see Charts 1 and 2). Structurally characterised half sandwich (η5-Cp*)Rh(III) guanidinato complexes are sparse in the literature.20,21 We wanted to investigate whether electron poor guanidinate ligands such as those present in 1–7 are beneficial in their performance as catalysts in TH of carbonyl compounds. The results obtained from our efforts are outlined in the following sections.
NR (Sym = symmetrical; R = alkyl and aryl) are one of the interesting classes of N-donor ligands due to not only their ability to act as monoanion, [R(H)N–C(NR)2]− (guanidinate(1−) or guanidinato) and dianion, [C(NR)3]− (guanidinate(2−)) but also the ready tunability of the R group so that distinct donor characteristics and coordination modes can be realised for these anions in their metal complexes.16,17 In our previous publications,18,19 we studied structural aspects and the utility of half sandwich (η6-p-cymene)Ru(II) complexes that contain guanidinate ligand or guanidinium cation in TH of carbonyl compounds. Herein, we report syntheses and characterisation of half sandwich (η5-Cp*)M(III) guanidinato complexes, 1–8 by analytical, spectroscopic (IR and NMR) techniques and single crystal X-ray diffraction (SCXRD; see Charts 1 and 2). Structurally characterised half sandwich (η5-Cp*)Rh(III) guanidinato complexes are sparse in the literature.20,21 We wanted to investigate whether electron poor guanidinate ligands such as those present in 1–7 are beneficial in their performance as catalysts in TH of carbonyl compounds. The results obtained from our efforts are outlined in the following sections.
 | ||
| Chart 1 | ||
 | ||
| Chart 2 | ||
Results and discussion
Syntheses
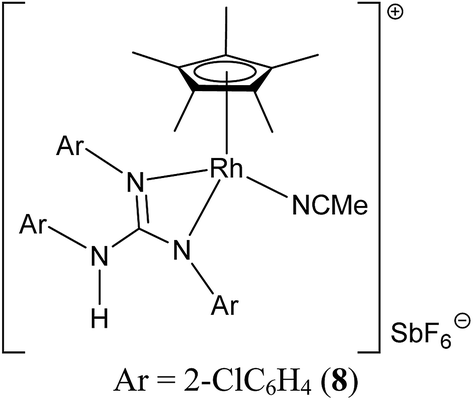
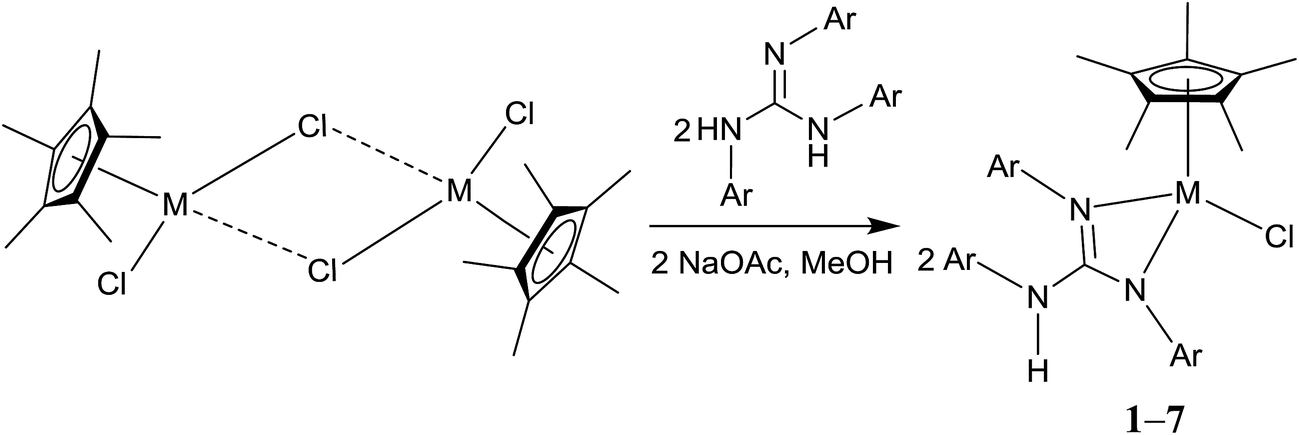
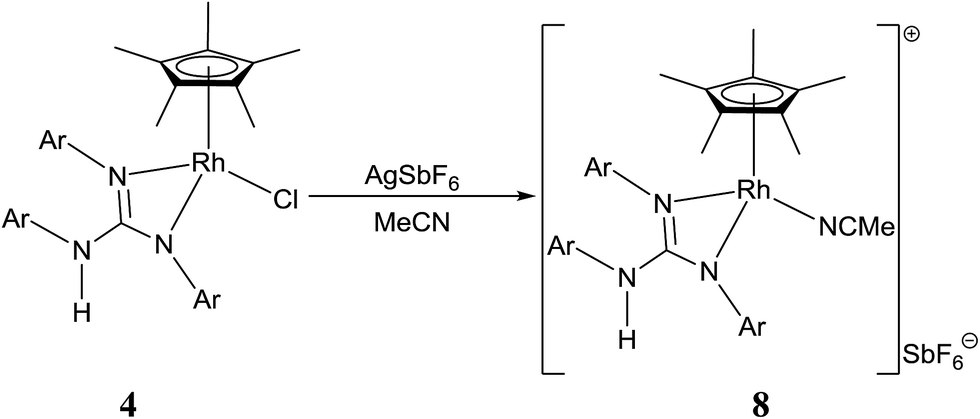
Treatment of [(η5-Cp*)M(μ-Cl)Cl]2 (M = Rh and Ir) with two equiv. of the respective sym N,N′,N′′-triarylguanidines, (ArNH)2CNAr (sym = symmetrical) in the presence of two equiv. of NaOAc in MeOH at RT for 24 h afforded 1–6 as orange solid and 7 as green solid in high yields (see Scheme 1). The metathesis reaction of 4 with one equiv. of AgSbF6 in MeCN at RT for 6 h in the absence of light afforded 8 in high yield (see Scheme 2). Complexes 1–8 are stable to air indefinitely.
 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
Molecular structures
Complexes 1, 2, 3·MeOH and 4–8 revealed a pseudo octahedral three legged piano stool structure, a feature previously noted in 9 (ref. 20) (see Fig. 1, Chart 1 and Fig. S1 and S2 in the ESI†). When the guanidinate ligand contains ortho substituted aryl rings as those present in 1, 4, and 6, then one could, in principle, observe four conformers namely, syn–syn, syn–anti, anti–syn and anti–anti and these conformers arise due to four distinct orientations of ortho substituent of the aryl ring in the –NcoordAr moieties with respect to the orientation of ortho substituent of the aryl ring in the –Nnoncoord(H)Ar moiety (see Fig. S3 in the ESI†).18,19 Thus, guanidinato ligands in 1, 4 and 6 revealed syn–syn conformation. The position of the chloro ligand in 4 is occupied by MeCN in 8 with SbF6− as the counter anion in the latter. The guanidinato ligand in 8, unlike in 4, revealed anti–syn conformation.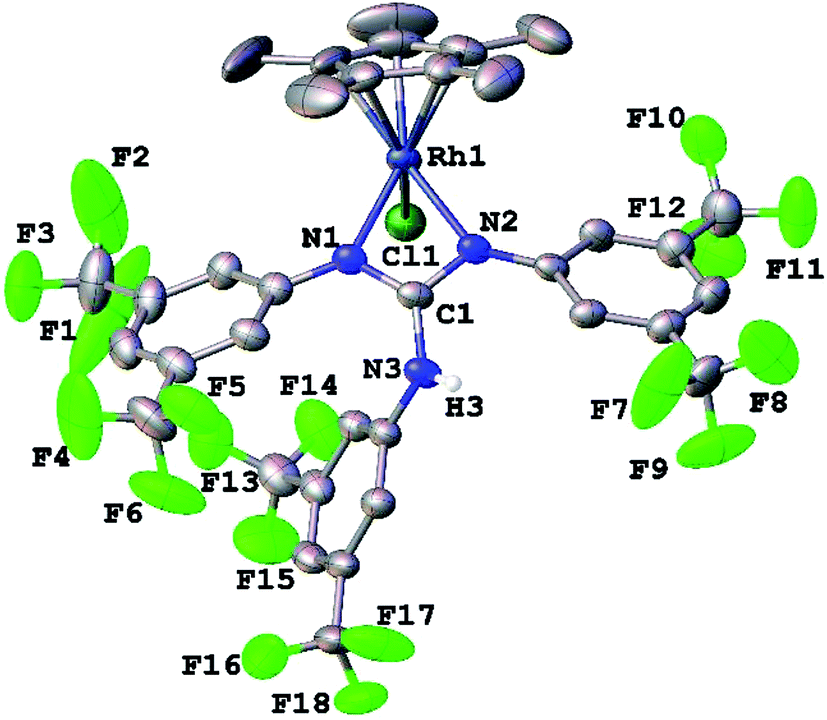 | ||
| Fig. 1 Molecular structure of 3·MeOH at the 30% probability level. | ||
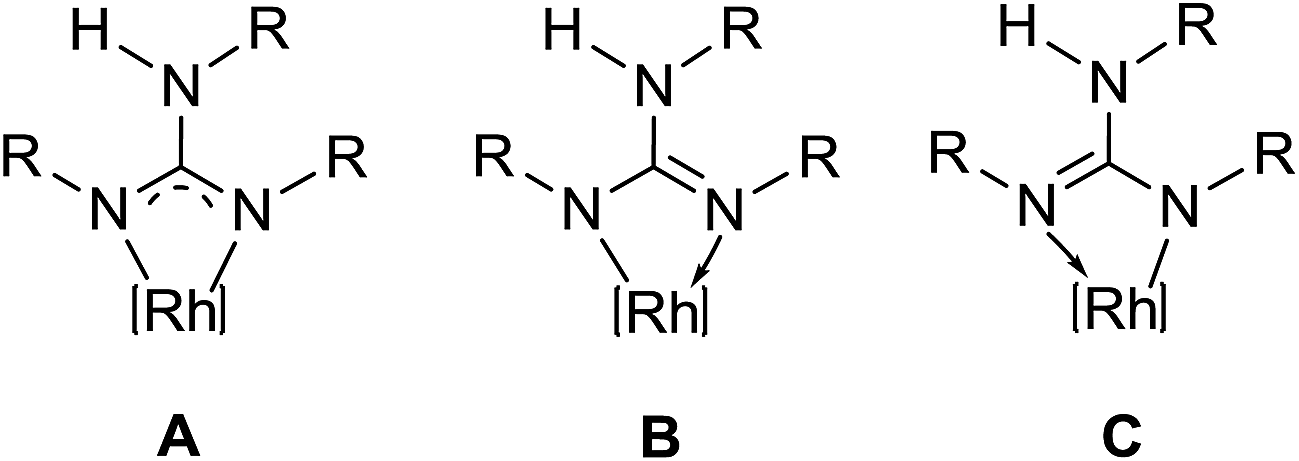
The key structural parameters of 1, 2, 3·MeOH and 4–8 are listed in Table 1. The values of ΔCN = d(C–N)endo − d(C–N)endo and ΔCN′ = d(C–N)endo − d(C–N)exo and angle sums around the N atoms (∑N) can be used to understand the degree of interaction of the N lone pair with the antibonding p-orbital of the C in the CN3 unit of the guanidinato ligand.19 The values of ΔCN and ΔCN′ are comparable within 3 σ cut off in 1 and 4–8 while in 2 and 3·MeOH, the value of ΔCN is smaller than the value of ΔCN′. Three resonance forms are possible for guanidinate ligand in metal guanidinate complexes (see Chart 3). Thus, the contribution of resonance form A is greater in 1 and 4–8 while that of form B is greater in 2 and 3·MeOH. Further, one of the coordinated N atoms in new complexes reported herein is more planar than the other while the non-coordinated N atom is planar. The degree of interaction of the lone pair on the N atoms with the antibonding p-orbital of the C in the CN3 unit of the guanidinato ligand in the aforementioned complexes varies as Nnoncoord > Ncoord, planar > Ncoord, less planar as indicated from the values of ∑N.
| Complex | ΔCN (Å) | ΔCN′ (Å) | ∑N(coord.) (deg) | ∑N(noncoord.) (deg) | φa (deg) |
|---|---|---|---|---|---|
| a φ = Dihedral angle between the HNC(Ar) plane and the chelate NCN plane. | |||||
| 1 | 0.038(12) | 0.056(12) | 354.0, 357.7 | 360.0 | 75.3(4) |
| 2 | 0.020(6) | 0.081(7) | 354.2, 359.0 | 360.0 | 33.0(4) |
| 3·MeOH | 0.003(7) | 0.074(6) | 357.7, 359.5 | 359.9 | 48.7(4) |
| 4 | 0.019(11) | 0.055(11) | 356.2, 359.8 | 360.0 | 23.3(3) |
| 5 | 0.018(7) | 0.042(7) | 354.9, 359.9 | 360.0 | 37.9(3) |
| 6 | 0.015(5) | 0.040(5) | 345.4, 359.9 | 360.0 | 39.2(2) |
| 7 | 0.002(8) | 0.033(8) | 354.1, 360.0 | 360.0 | 29.9(3) |
| 8 | 0.046(10) | 0.065(13) | 352.7, 359.9 | 359.5 | 27.5(5) |
 | ||
| Chart 3 | ||
The coordinated N atoms in resonance forms A–C can be either σ-donor (planar N) or σ/π-donor (less planar) as has been shown for metal amidinato complexes.22 In 1, both the coordinated N atoms revealed predominantly σ-donor character with a slight π-donor character. In 2, 3·MeOH and 4–8, one of the coordinated N atoms is σ-donor while the other is predominantly σ-donor with either a slight amount of π-donor character (4, 5, 7 and 8) or a significant amount of π-donor character (6). The difference in the level of interaction of the N lone pair with the antibonding p orbital of the C in the CN3 unit of guanidinate ligand in complexes could partly arise due to the difference in substitution pattern of the aryl rings of the guanidinate ligands, its conformational properties and packing forces.
In 9, not only does one of the coordinated N atoms deviate from planarity but also the non-coordinated N atom as can be seen from the values of angle sum around the N atoms (∑Ncoord = 354.4° and 359.9°; ∑Nnoncoord = 351.2°). The angle between HNC(Ar) plane and the NCN plane involving coordinated N atoms in 1, 75.3(4)° is significantly greater than the respective value, 42.2(2)° reported for 9 (ref. 20) and the larger value in the former complex is to minimise the repulsive interaction between o-CF3 group in the NnoncoordAr unit with o-CF3 group in one of the NcoordAr units.
Solution behaviour
1H NMR spectra of 3, 5, 6, and 7 and 19F NMR spectra of the latter two complexes revealed the presence of a single isomer in solution. 1H and 19F NMR spectra of 1 revealed the presence of three isomers in about 0.34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.00:0.12 and 0.33:1.00:0.13 ratios respectively in solution which are assigned to syn–syn, syn–anti, and anti–syn conformers. The latter two conformers could possibly arise from the former via guanidine centered rearrangements and the driving force for the isomerisation is to relieve steric strain in syn–syn conformer (see Fig. S4 in ESI†).18,19 In solution, anti–anti isomer is less likely for 1 as o-CF3 group in both NcoordAr units would point towards the chloro ligand causing a destabilisation.
:1.00:0.12 and 0.33:1.00:0.13 ratios respectively in solution which are assigned to syn–syn, syn–anti, and anti–syn conformers. The latter two conformers could possibly arise from the former via guanidine centered rearrangements and the driving force for the isomerisation is to relieve steric strain in syn–syn conformer (see Fig. S4 in ESI†).18,19 In solution, anti–anti isomer is less likely for 1 as o-CF3 group in both NcoordAr units would point towards the chloro ligand causing a destabilisation.
1H and 19F NMR spectra of 2 showed the presence of a single species at 1.309 × 10−2 M concentration in CDCl3 as anticipated due to the presence of a symmetrically substituted aryl substituent in the guanidinato ligand while 1H and 19F NMR spectra measured at 13.09 × 10−2 M concentration revealed the presence of two species in about 1.0:0.7 ratio (see Experimental section). The presence of two solution species at higher concentration was also confirmed by 13C{1H} NMR spectroscopy. In dilute solution, the N2C–N(H)Ar single bond rotation of the guanidinato ligand would be faster than the NMR time scale and thus only one species was detected. In concentrated solution, the rate of N2C–N(H)Ar single bond rotation could be comparable with the NMR time scale as the N(H)Ar proton could be involved in intermolecular hydrogen bonding. This permits the NMR spectrometer to identify two species in concentrated solution and the line drawing structures of these two species are illustrated in Fig. 2. Concentration dependent aggregation of [(η6-p-cymene)RuX{κ2(N,N)(aminoamidate)}] (X = Cl and H) has been observed in solution as evidenced by PGSE diffusion NMR experiments, calculations and also by ESI mass spectrometry.23
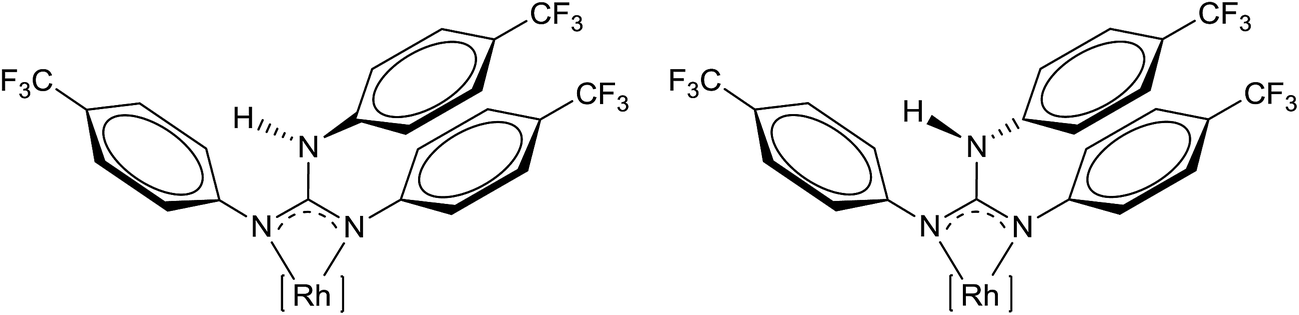 | ||
| Fig. 2 Two rotamers of 2. [Rh] = [(η5-Cp*)RhCl]. The two rotamers shown are distinct from each other when one considers the Cl as the reference atom. | ||
The 19F NMR spectrum of 3 revealed the presence of two species in about 1.0:0.05 ratio at 1.240 × 10−2 M concentration and this observation is unanticipated since aryl substituents in the guanidinate ligand are symmetrically substituted and further even at 12.40 × 10−2 M concentration, both 1H and 13C{1H} NMR revealed the presence of only one species. The two solution species of 3 as detected by 19F NMR spectroscopy, could have originated from the restricted N2C–N(H)Ar single bond rotation and thus leads to the formation of two rotamers. The driving force for the formation of two rotamers is believed to be a repulsive interaction between CF3 group of one of the NcoordAr units and CF3 group of the NuncoordAr unit wherein aryl substituent of these two units lie on the same side (see Fig. S5 in the ESI†). We measured variable temperature (VT) 19F NMR spectra of 3 to identify coalescence temperature between the signals of two rotamers but the signals did not coalesce below the boiling point of CDCl3 (see Fig. S6 in the ESI†).
Complex 4 was subjected to VT 1H NMR measurements to better understand its solution behaviour. The stack plot for CH3 protons of the Cp* ring is illustrated in Fig. 3. At 303 K, a broad and a sharp peaks appeared at δ 1.53 and 1.56 ppm respectively, in about 1.0:0.07 ratio. Upon lowering the temperature down to 273 K, the broad peak merged with the base line and started resolving at ≤253 K as two separate broad peaks at δ 1.53 and 1.41 ppm respectively while the sharp peak at δ 1.56 ppm shifted to δ 1.52 ppm. Subsequently, the two new broad peaks mentioned above gradually became sharper upon lowering the temperature. At 213 K, three sharp singlets were observed at δ 1.50, 1.48, and 1.37 ppm in about 1.00:0.28:1.12 ratio, which we assign to syn–syn, syn-anti, and anti-syn conformers as analogously discussed for 1 (see above). The broad signal observed at δ 1.53 ppm at 303 K is ascribed to the presence of an equilibrating mixture of two conformers formed via guanidine centered rearrangements as the rate of this process is greater than the NMR timescale. The presence of three conformers of 4 was also apparent from three distinct signals of NH protons of the guanidinato ligand (see Fig. S7 in the ESI†). At temperatures ≤253 K, the rate of guanidine centered rearrangements is comparable with the NMR timescale and as a result, the 1H NMR signals of two isomers are resolved. From the 1H NMR spectral behaviour of 1 and 4, it appears that rate of guanidine centered rearrangements can be reduced significantly either by introducing sterically more hindered aryl substituents in the guanidinato ligand as in the former complex or freezing the solution to lower temperatures when the aryl substituent of the guanidinato ligand is sterically less hindered as found in the latter complex.
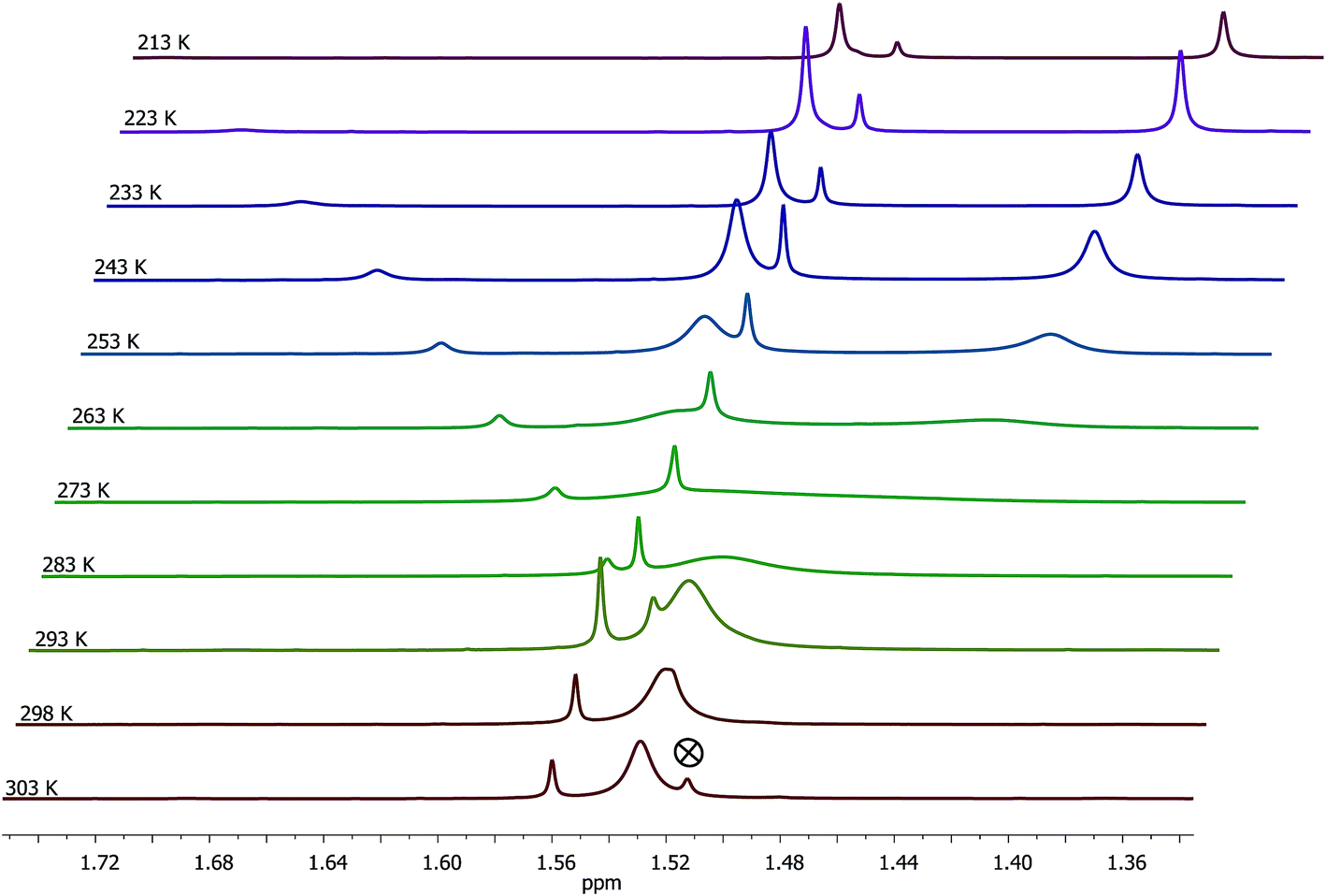 | ||
| Fig. 3 VT 1H NMR (400 MHz, CD2Cl2) spectra of 4 illustrated for CH3 protons of the Cp* ring. The symbol ⊗ refers to signal of water protons. | ||
1H NMR spectra of 6 at 1.629 × 10−2 M and 8.470 × 10−2 M concentrations in CDCl3 revealed the presence of a single isomer. Upon addition of a drop of D2O to the more concentrated solution resulted in the emergence of a new species with the ratio of two species being about 0.1:0.8 as estimated from the integrals of NH proton located at δ 6.02 and 4.75 ppm, respectively. These two species are assigned to 6 (minor) and 6·D2O (major) with the latter possessing an intermolecular N–H⋯O hydrogen bond with the added D2O. The 1H NMR spectrum of 8 revealed the presence of two species in about 1.00:0.13 ratio as estimated from the integrals of Cp* protons and the presence of two solution species was also supported by 13C{1H} NMR spectroscopy. These two species are assigned to any two isomers out of syn–syn, syn–anti and anti–syn.
Transfer hydrogenation
TH of carbonyl compounds is a safer reduction procedure than hydrogenation using H2 gas.4,14,24–28 This methodology became popular since the discovery of bifunctional catalysts of the type [(η6-arene)RuCl(NN)] in 1995 by Noyori and co-workers.29 Since then, the aforementioned half sandwich complexes in conjunction with [(η5-Cp*)MCl(NN)] (M = Rh and Ir) complexes have been used as catalysts in TH of carbonyl compounds with iPrOH/base, HC(O)OH/Et3N or HC(O)ONa/water mixtures, for example, as hydrogen sources. The activity and chemoselectivity of two types of the aforementioned complexes in TH were modulated by changing the metal, arene cap and aminoamidate bidentate N donor ligands. Only a few phosphine free Ru(II)/Os(II) and Ir(III) complexes are known to work as catalysts in TH under base free condition.3,12,13,30–32 In phosphine free Ru(II)/Ir(III) amido complexes, the more basic amido N atom in the ligand skeleton was presumed to act as an internal base in TH of carbonyl compounds and hence it was not necessary to use an external base. This aspect was not elaborated in detail in base free TH catalysed by phosphine free metal amido complexes in the literature. Hence, 1–8, 9–11,20,33 12,19 13 (ref. 34) and [(η5-Cp*)Rh(μ-Cl)Cl]2 were screened in TH of acetophenone both under basic and base free conditions and the results of this study are listed in Table 2 (see Charts 1, 4 and 5).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Conversiona (%) | |||
| In presence of KOHb | TON | In absence of KOHc | TONd | ||
| a Conversion estimated by 1H NMR spectroscopy and reported as an average of two trials.b Substrate/catalyst/KOH = 100/1/100.c Substrate/catalyst = 100.d TON = Number of mmoles of product/number of mmoles of catalyst used.e Ref. 19. | |||||
| 1 | 1 | 97 ± 2 | 97 | 98 ± 1 | 98 |
| 2 | 2 | 98 ± 1 | 98 | 98 ± 1 | 98 |
| 3 | 3 | 100 ± 0 | 100 | 99 ± 0 | 99 |
| 4 | 4 | 99 ± 0 | 99 | 88 ± 2 | 88 |
| 5 | 5 | 98 ± 1 | 98 | 96 ± 3 | 96 |
| 6 | 6 | 99 ± 0 | 99 | 98 ± 0 | 98 |
| 7 | 7 | 98 ± 1 | 98 | 93 ± 1 | 93 |
| 8 | 8 | 99 ± 0 | 99 | 50 ± 9 | 50 |
| 9 | 9 | 97 ± 2 | 97 | 93 ± 2 | 93 |
| 10 | 10 | 94 ± 5 | 94 | 90 ± 0 | 90 |
| 11 | 11 | 97 ± 1 | 97 | 78 ± 2 | 78 |
| 12 | 12 | 71 ± 1e | 71 | 71 ± 6 | 71 |
| 13 | 13 | 84 ± 3 | 84 | 16 ± 0 | 16 |
| 14 | [(η5-Cp*)Rh(μ-Cl)Cl]2 | 98 ± 1 | 98 | <1 | 0 |
 | ||
| Chart 4 | ||
 | ||
| Chart 5 | ||
Under basic condition, all complexes except 12 exhibited high activity while the latter exhibited only 71% conversion. Pleasingly, TH of acetophenone carried out under base free condition afforded the reduction product in good conversion (1–7, 9 and 10), moderate conversion (11, and 12), poor conversion (13) and no conversion ([(η5-Cp*)Rh(μ-Cl)Cl]2). The much better performance of 3 than 13 under base free condition suggests greater robustness of ‘[(η5-Cp*)Rh(III)]’ moiety in the former complex than ‘[(η6-p-cymene)Ru(II)]’ moiety in the latter (entries 3 and 13). The fact that [(η5-Cp*)Rh(μ-Cl)Cl]2 did not catalyse TH under base free condition while 1–11 catalysed with different efficiencies indicates two reasons for the success of the latter complexes as catalysts in TH. The presence of less planar amido N in guanidinate ligands as internal base in conjunction with the ability of the ligands to ligate the metal in neutral and monoanionic forms under base free TH are anticipated to position the NH proton of the guanidinate/guanidine ligands parallel to the M–H functionality in the reactive M–H intermediate for substrate activation. The slightly better performance of 3 than 7 in TH of acetophenone under base free condition illustrates the more suitability of Rh than Ir (entries 3 and 7). To investigate the reusability of 3, a second equiv. of acetophenone was introduced to the reaction mixture and catalysis experiment was repeated under base free condition. From this experiment, only 58% of acetophenone was reduced thereby indicating partial catalyst deactivation.
The reduction product was obtained in 99% yield under basic condition when TH was carried out in the presence of 8 and only in 50% yield under base free condition. This difference is ascribed to arise from two different catalytic pathways operating under basic and base free conditions (see later). We speculate the presence of coordinated MeCN in 8 slows down the formation of Rh–H intermediate under base free condition. Complex 3 was chosen as the catalyst of choice for TH of several other carbonyl compounds as shall be discussed in the following paragraphs although 1, 2, 5 and 6 are comparable or only slightly inferior in their performance.
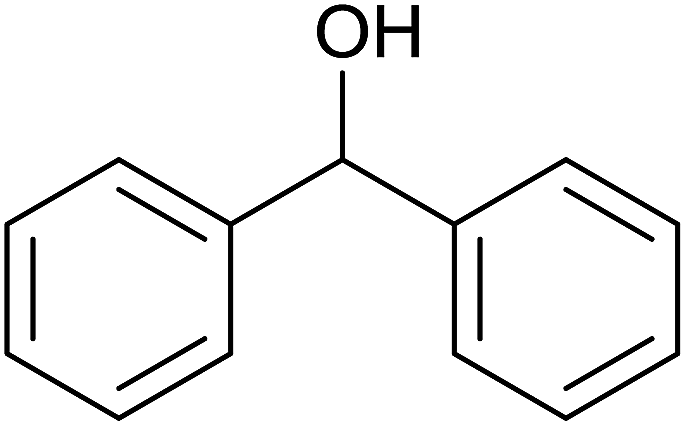
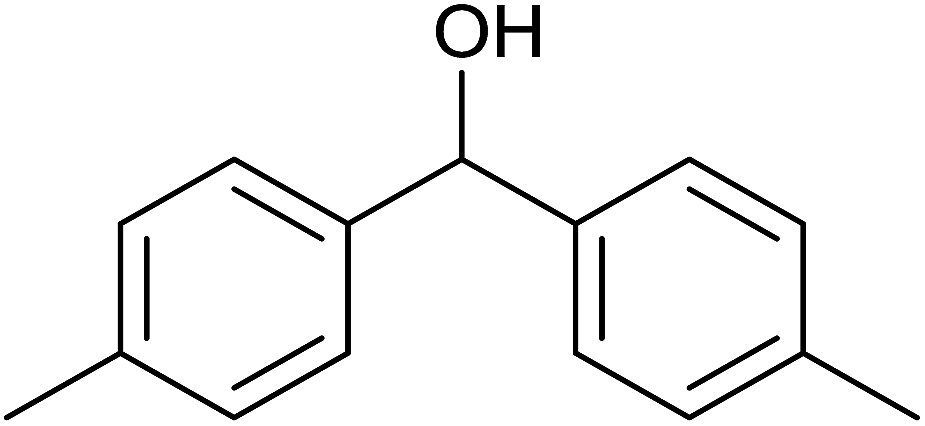
TH of acetophenone was carried out in the presence of 3 under base free condition by varying substrate/catalyst (S/C) ratio and time as shown in Table 3. The best conversion was achieved in the presence of 1 mol% of 3 (entry 3). In the presence of 0.1 mol% of 3, only 71% conversion was achieved after 24 h (entry 6). Following the condition listed in entry 3, various ketones were subjected to TH both in the presence and in the absence of base and the results of this investigation are listed in Table 4. The extent of reduction appears to depend upon neither steric factor nor electronic factor of the substrates (entries 1–7). Sterically less hindered cyclohexanone is reduced efficiently both under basic and base free conditions while sterically more hindered benzophenone and 4,4′-dimethyl benzophenone are reduced with less and no efficiencies respectively (see entries 8–10). The 99% conversion reported for reduction of 2-methyl acetophenone is remarkable as this substrate was not reduced under base free TH in the literature (see entry 1). However, Ru(II) phosphine based catalyst under basic condition was shown to reduce this substrate more efficiently.35 Cyclohexanone was reduced efficiently under base free condition in the present investigation as has been analogously reported for this substrate with Ru(II) complexes.32
| Entry | Substrate | Product | Conversionb (%) | |||
|---|---|---|---|---|---|---|
| In presence of KOH | TON | In absence of KOH | TON | |||
| a Substrate/catalyst/KOH = 100/1/100; iPrOH, 82 °C, 4 h.b Conversion estimated by 1H NMR spectroscopy and reported as an average of two trials. | ||||||
| 1 |  |
 |
35 ± 8 | 35 | 99 ± 1 | 99 |
| 2 |  |
 |
79 ± 2 | 79 | 93 ± 6 | 93 |
| 3 |  |
 |
18 ± 9 | 18 | 61 ± 1 | 61 |
| 4 |  |
 |
97 ± 2 | 97 | 91 ± 3 | 91 |
| 5 | 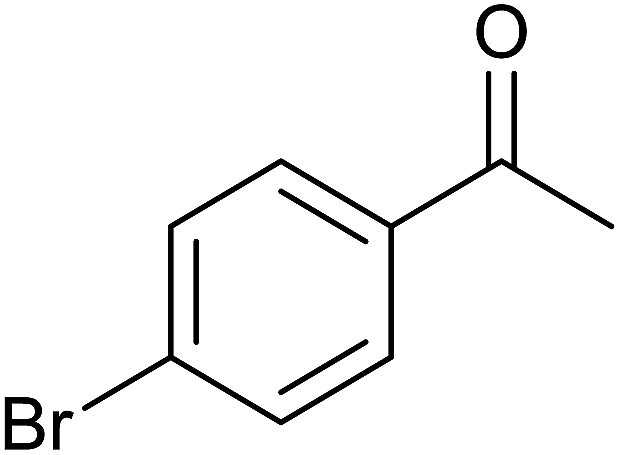 |
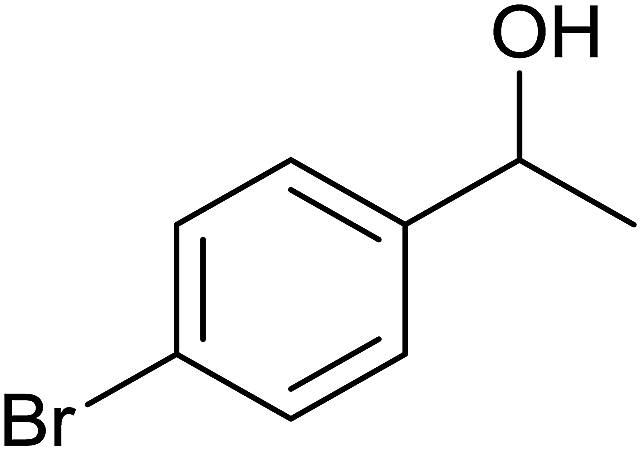 |
84 ± 4 | 84 | 72 ± 8 | 72 |
| 6 | 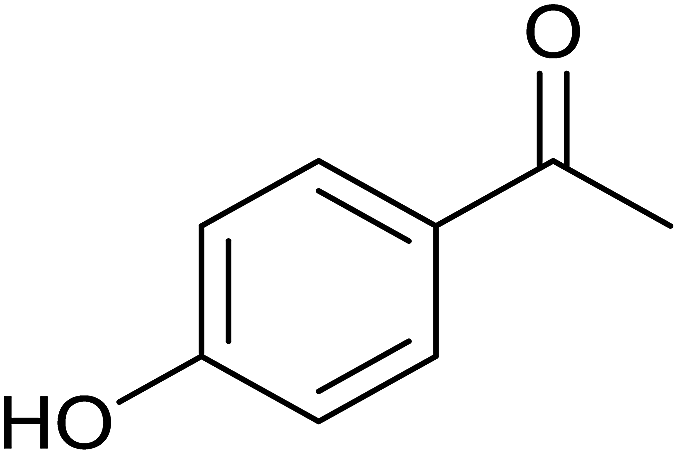 |
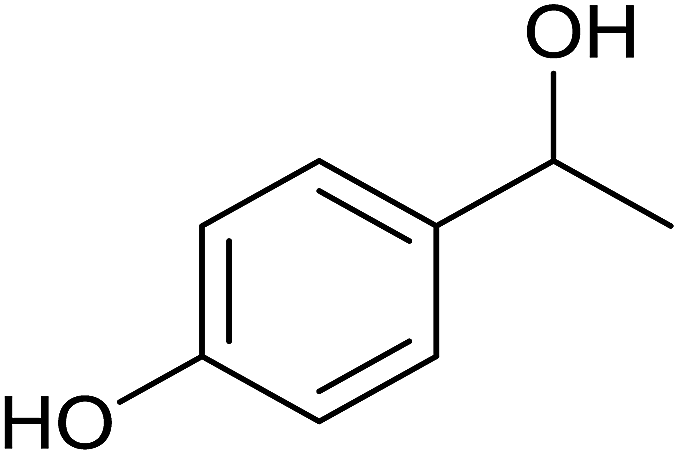 |
<1 | 0 | 7 ± 3 | 7 |
| 7 | 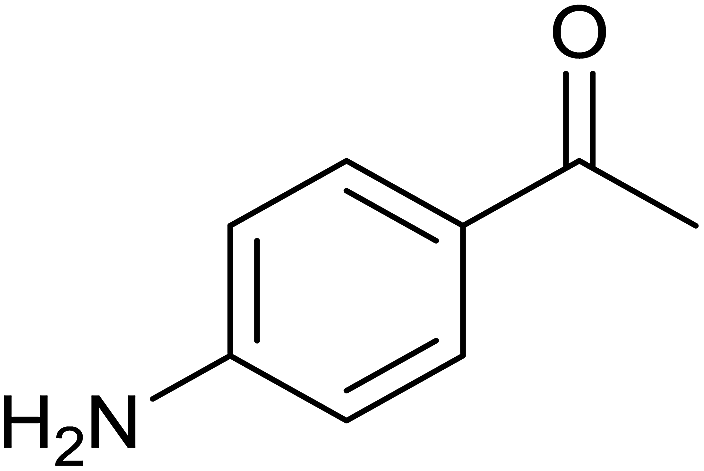 |
 |
0 ± 0 | 0 | <1 | 0 |
| 8 |  |
 |
99 ± 1 | 99 | 97 ± 2 | 97 |
| 9 | 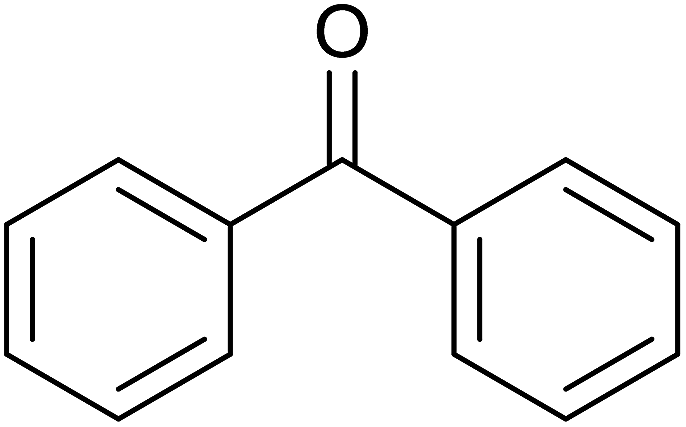 |
 |
39 ± 2 | 39 | 61 ± 2 | 61 |
| 10 | 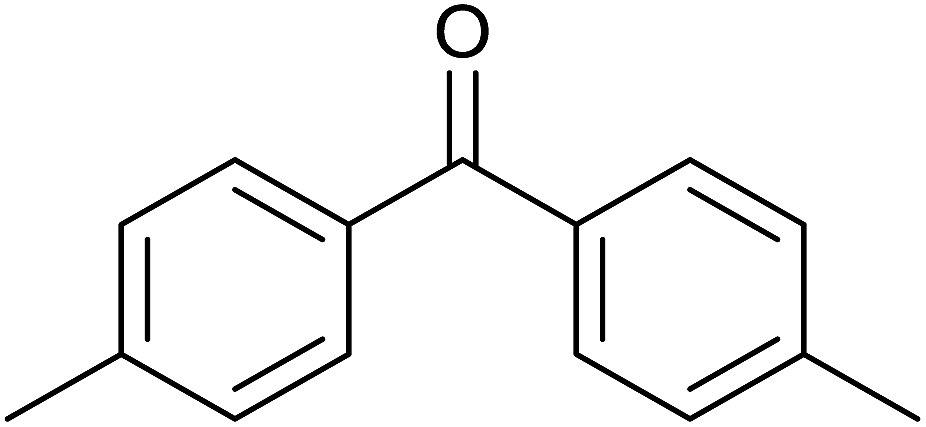 |
 |
<1 | 0 | 20 ± 5 | 20 |
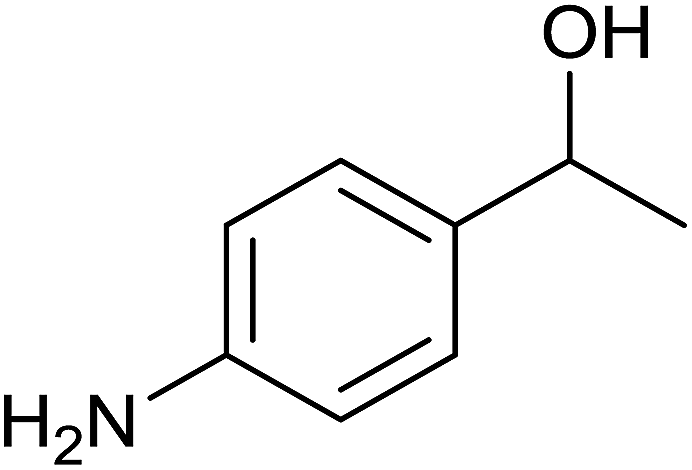
Interestingly, TH of 4-nitroacetophenone in the presence of 1 mol% of 3 under base free condition gave the corresponding alcohol while under basic condition gave 4-aminoacetophenone in >99% yield in both cases (see Scheme 3). The observed chemoselectivity is attributed to two distinct mechanisms operating in TH under basic and base free conditions (see later). The chemoselective reduction of either ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CO group10,12,31a or NO2 group36 in 4-nitroacetophenone in TH catalysed by Ir(III), Ru(II) and Rh(III) metal complexes are known. However, chemoselective reduction of either one functional group in 4-nitroacetophenone by the same catalyst to the best of our knowledge is unprecedented in homogeneous catalysis. Further, percent conversion associated with carbonyl reduction of 4-nitroacetophenone reported herein is comparable with the percent conversion reported for the related Ir(III) amido complex.12 The reduction of nitro group in 4-nitroacetophenone was not reported in the literature under TH condition. TH of 4-nitroacetophenone carried out in the presence of ruthenium(II) carbene complex, [(η6-benzene)RuCl2(Im(Et,CH2CH2OEt))] and KOtBu resulted in no carbonyl reduction37 and this observation indirectly suggests that it is the nature of guanidinato ligand in 3 that is responsible for NO2 reduction. TH of nitrobenzene and 4-nitrotoluene was carried out separately in the presence of 3 under basic condition which gave aniline in 3% yield and a mixture of as yet unidentified products respectively. Further, TH of methyl 4-nitrobenzoate under basic condition afforded insoluble material. The aforementioned three experiments suggest a limited scope of 3 as catalyst in the reduction of nitroarenes.
CO group10,12,31a or NO2 group36 in 4-nitroacetophenone in TH catalysed by Ir(III), Ru(II) and Rh(III) metal complexes are known. However, chemoselective reduction of either one functional group in 4-nitroacetophenone by the same catalyst to the best of our knowledge is unprecedented in homogeneous catalysis. Further, percent conversion associated with carbonyl reduction of 4-nitroacetophenone reported herein is comparable with the percent conversion reported for the related Ir(III) amido complex.12 The reduction of nitro group in 4-nitroacetophenone was not reported in the literature under TH condition. TH of 4-nitroacetophenone carried out in the presence of ruthenium(II) carbene complex, [(η6-benzene)RuCl2(Im(Et,CH2CH2OEt))] and KOtBu resulted in no carbonyl reduction37 and this observation indirectly suggests that it is the nature of guanidinato ligand in 3 that is responsible for NO2 reduction. TH of nitrobenzene and 4-nitrotoluene was carried out separately in the presence of 3 under basic condition which gave aniline in 3% yield and a mixture of as yet unidentified products respectively. Further, TH of methyl 4-nitrobenzoate under basic condition afforded insoluble material. The aforementioned three experiments suggest a limited scope of 3 as catalyst in the reduction of nitroarenes.
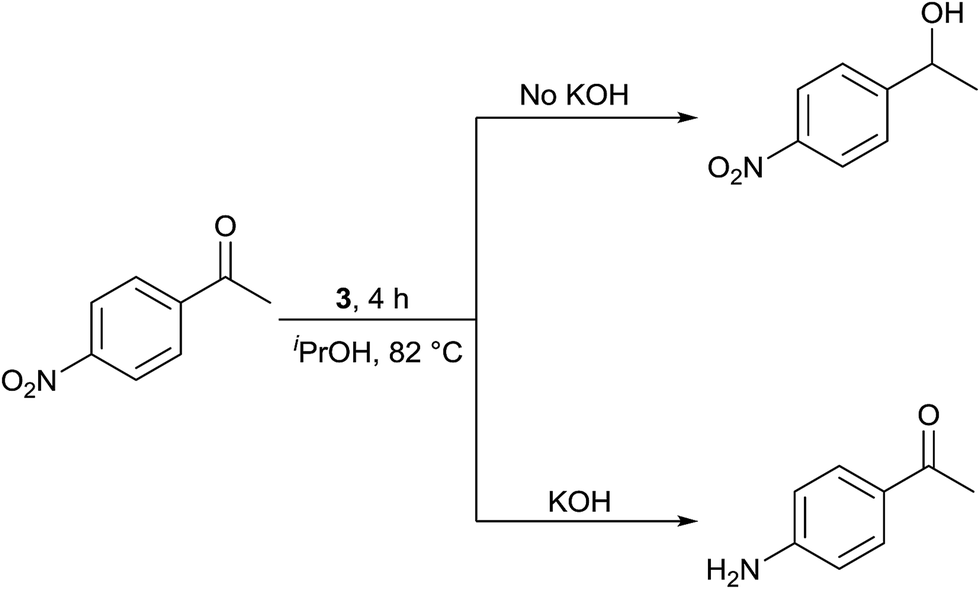 | ||
| Scheme 3 TH of 4-nitroacetophenone under base free and basic conditions. | ||
TH of aromatic aldehydes catalysed by phosphine free Ru(II), Os(II) and Ir(III) complexes under basic11,38,39 and base free30−32 conditions are known. Aldehydes are more challenging substrates than ketones in TH carried out under basic condition due to potential side reactions such as aldol condensation and decarbonylation.40,41 The reaction conditions were optimised for base free TH of benzaldehyde in the presence of 3 in iPrOH by varying S/C ratio, temperature and time and the results of this investigation are listed in Table 5. TH of benzaldehyde is sluggish at RT and the reaction is complete only after 24 h (entries 1–3). TH of benzaldehyde is 88% complete in 1 h at 82 °C and 99% complete in 4 h at the same temperature (entries 4 and 5). When 0.1 mol% of 3 was used at 82 °C for 4 h, only 20% conversion was achieved (entry 6). Thus, the optimised condition listed in entry 5 was applied for TH of other aldehydes outlined below.
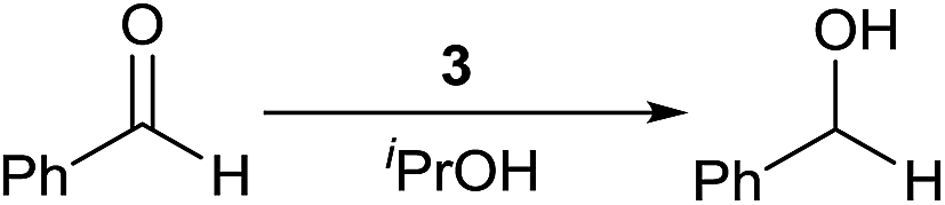
Benzaldehyde, 4-substituted benzaldehydes and furfural were reduced to the corresponding alcohols in >99% yield under base free condition (see Scheme 4). The reactions were carried out in air with aldehydes as received from commercial vendors. The significance of TH of commercial grade aldehydes catalysed by Ru(II) phosphine complexes has been discussed recently.40 The efficiency of 3 in reduction of benzaldehyde is comparable with that of Ir(III) carbene complex.41 TH of 4-nitrobenzaldehyde under basic condition gave a solid which was insoluble in organic solvents thus hampering its characterisation by 1H NMR spectroscopy. This is likely due to the formation of 4-aminobenzaldehyde which subsequently undergoes self-condensation to afford a polymeric product.
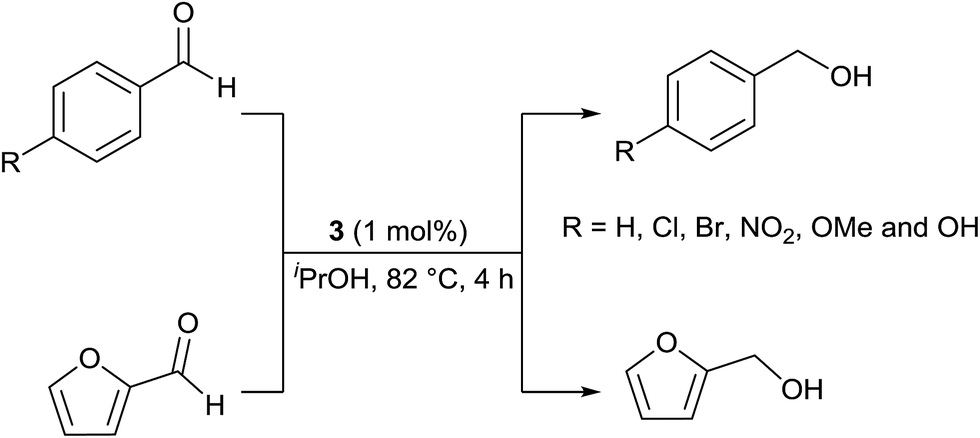 | ||
| Scheme 4 TH of aromatic aldehydes carried out under base free condition. 99 ± 1% conversion was obtained in each case as estimated from 1H NMR spectroscopy and this value is an average of two trials. | ||
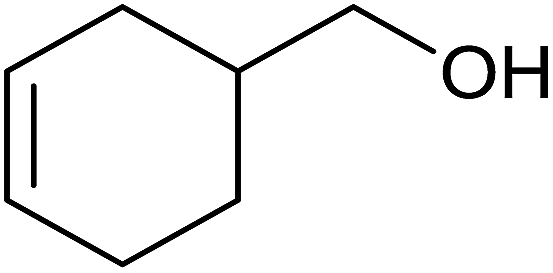
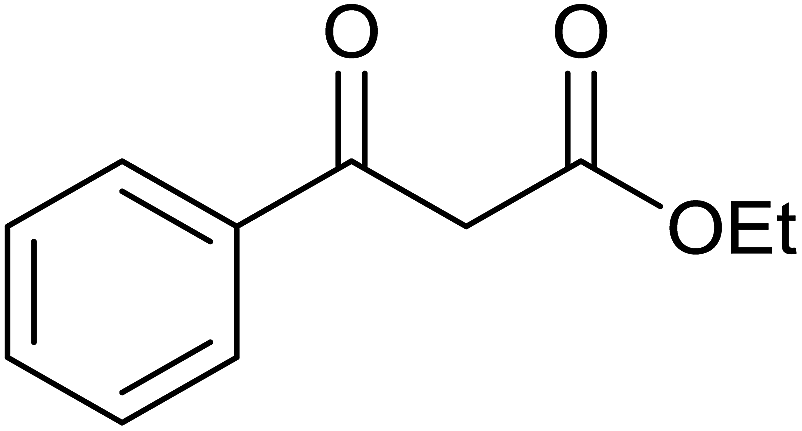
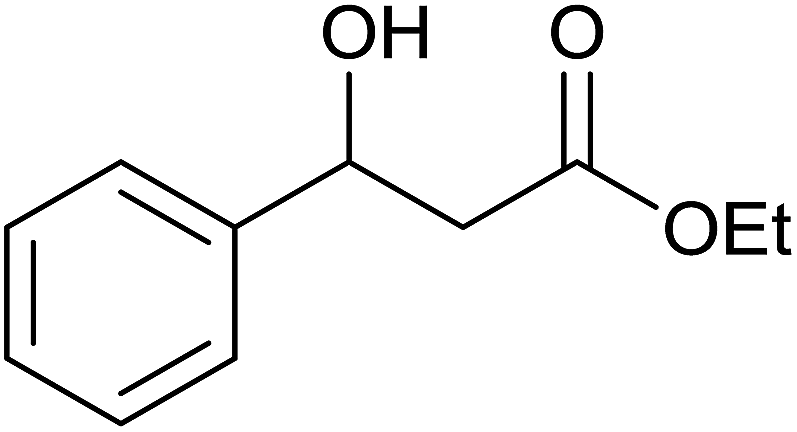
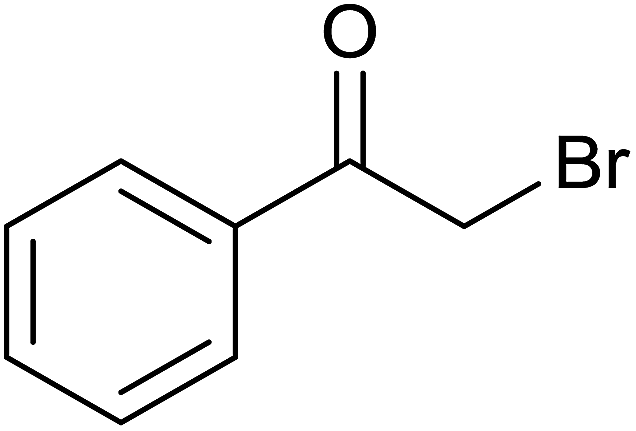
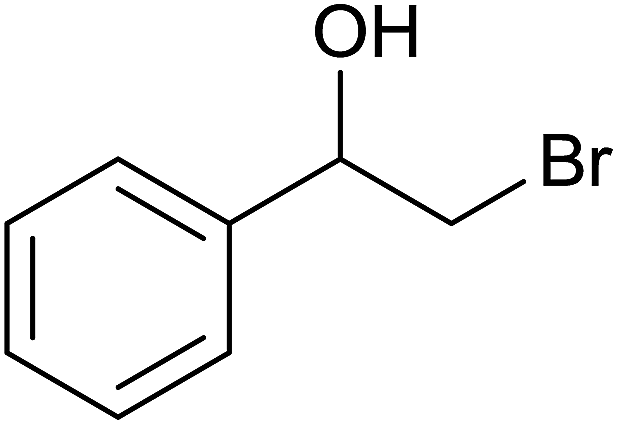
Base sensitive carbonyl compounds were subjected to TH in the presence of 3 under base free condition (Table 6). 3-Cyclohexene-1-carboxaldehyde was reduced to the corresponding alcohol in 96% conversion without reducing the CC![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) unit (entry 1). 2-Phenylacetophenone and ethyl benzoylacetate that contain active methylene group were reduced to the corresponding alcohols in 84% and 93% yields respectively (entries 2 and 3). 2-Bromoacetophenone remained largely unreacted and the reduction product was obtained only in 39%. The lower yield of the reduction product is likely due to steric hindrance caused by the Br in the substrate. When acetylacetone, 4-cyanobenzaldehyde and 4-cyanoacetophenone were subjected to TH, as yet unidentified mixture of products were formed as revealed by 1H NMR spectroscopy.
unit (entry 1). 2-Phenylacetophenone and ethyl benzoylacetate that contain active methylene group were reduced to the corresponding alcohols in 84% and 93% yields respectively (entries 2 and 3). 2-Bromoacetophenone remained largely unreacted and the reduction product was obtained only in 39%. The lower yield of the reduction product is likely due to steric hindrance caused by the Br in the substrate. When acetylacetone, 4-cyanobenzaldehyde and 4-cyanoacetophenone were subjected to TH, as yet unidentified mixture of products were formed as revealed by 1H NMR spectroscopy.
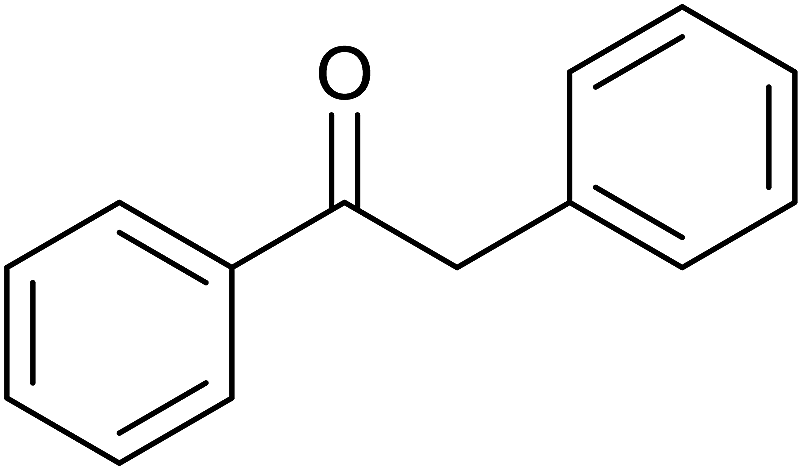
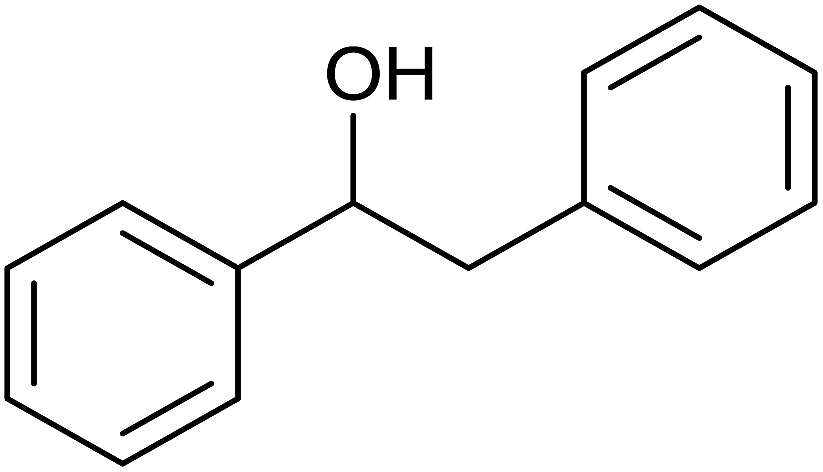
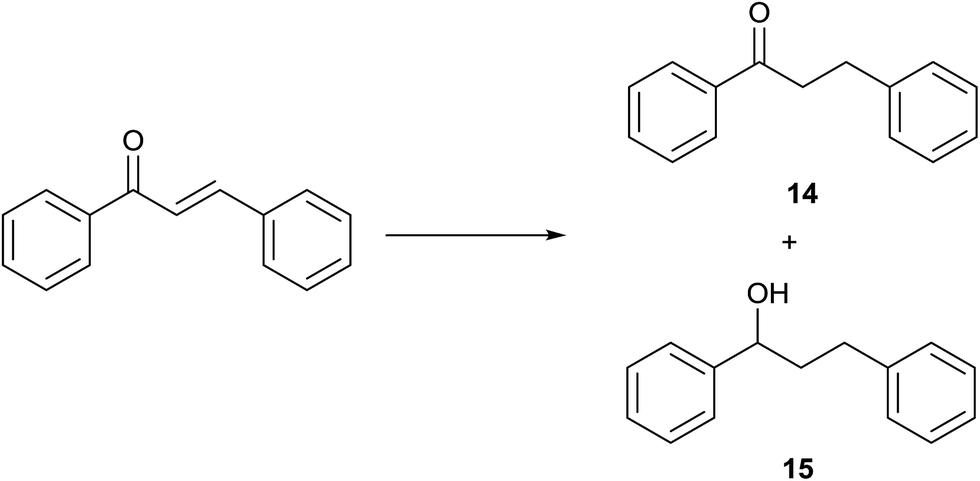
(E)-Chalcone was subjected to TH in the presence of 3 under base free condition and the progress of the reaction was monitored by 1H NMR spectroscopy (Table 7). In 30 min, 40% of (E)-chalcone was converted to 1,3-diphenylpropan-1-one, 14. In 60 min, (E)-chalcone completely disappeared with the concomitant formation of products 14 and 1,3-diphenylpropan-1-ol, 15 in 85% and 15% conversions respectively. The proportion of the product 15 increased with concomitant decrease in the proportion of 14 at 90 min and 240 min and thereafter the proportion of 14 and 15 remains unchanged. Thus, (E)-chalcone is completely consumed at 240 min to afford products 14 and 15 in 60:40 chemoselectivity (entries 4 and 5). Prior to this work, TH of α,β-unsaturated ketones was studied in the presence of Ir,42 and Rh43 complexes only under basic condition. The formation of 14 in the present investigation can be explained possibly through 1,4-addition pathway as previously shown for CC reduction of 3-buten-2-one in the presence of [(η5-C5H5)Rh{κ2(N,N′)(NHCH2CH2NH2)}H] by Deng and co-workers via DFT calculations.43
Methyl cinnamate was subjected to TH in the presence of 3 under base free condition. Only the CC bond was reduced to afford 16 in 29, 66 and 81% conversions after 4, 12 and 24 h respectively while the ester group remained unaltered (see Scheme 5). The faster reduction of the CC bond in methyl cinnamate was observed in TH catalysed by Pd(OAc)2 in the presence of ionic liquid.44 The reduction of methyl acrylate in TH catalysed by 3 under base free condition was not clean.
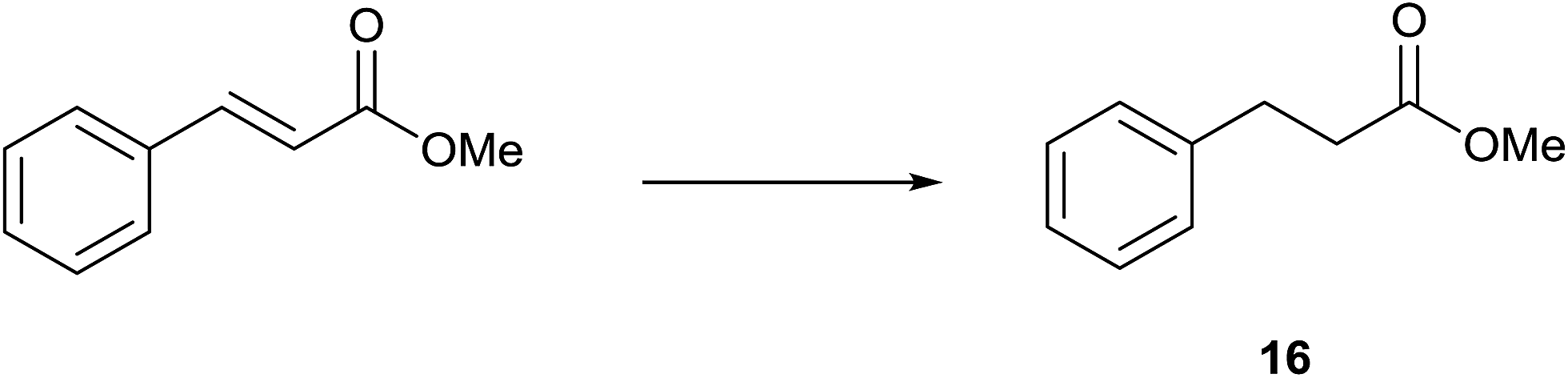 | ||
| Scheme 5 TH of methyl cinnamate with 3 as a catalyst under base free condition (reaction condition: substrate/catalyst = 100, iPrOH, 82 °C). | ||
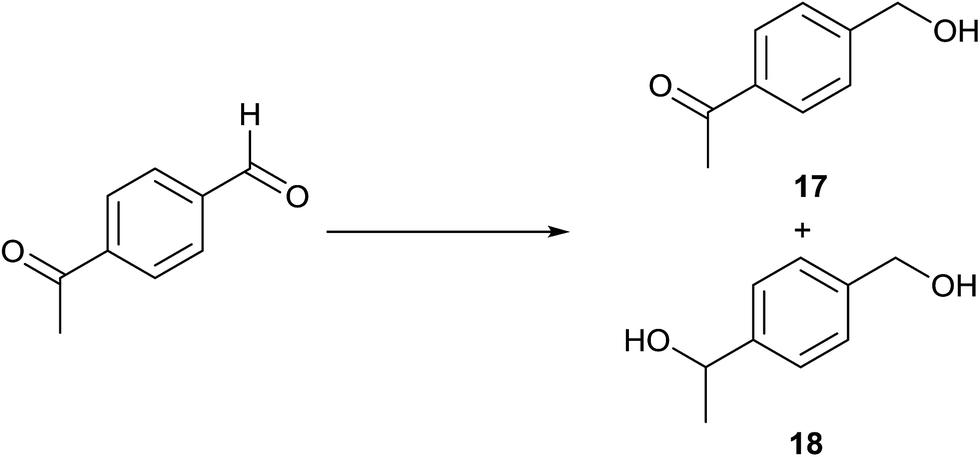
TH of 4-acetylbenzaldehyde in the presence of 3 under base free condition gave products 17 and 18 and their ratio is time dependent (Table 8). In 5 min, only the formyl group of the substrate is reduced to afford 17 in 50% conversion. Upon extending the reaction period to 10 min, the substrate is completely consumed leading to the formation of products 17 and 18 in 99:1 ratio. Upon extending the time, the concentration of 18 gradually increased at the expense of 17. In 500 min, the product 18 was exclusively present in the reaction mixture. This observation indicates that more reactive formyl group in the substrate is reduced first followed by reduction of the acetyl group. The reaction was truncated after 10 and 500 min which enabled us to isolate 17 in 91% yield and 18 in 98% yield respectively after column chromatographic workup (see the ESI†). 4-Acetylbenzaldehyde was subjected to TH in the presence of 0.1 mol% of [{(NHC)(linker)(NHC)}IrI2(κ2O,O′-OAc)] and 0.1 mol% of K2CO3 in iPrOH to afford a mixture of 17 and 18 in 95:5 chemoselectively.45 When TH of 4-acetylbenzaldehyde was carried out in presence of 3 under basic condition, a precipitate was formed, which is insoluble in CDCl3 and DMSO-d6, presumably due to the formation of a polymer formed through base catalysed self-aldol condensation.
Mechanistic hypotheses
A plausible mechanism of TH of carbonyl compounds in the presence of KOH is illustrated in Scheme 6. The reaction of 1–11 with KOH can afford 16e complex, D wherein the guanidinato ligand is present in the dianionic form. The formation of species D in the catalytic cycle is conceivable as [(η5-Cp*)Ir{κ2(N,N)[(ArN)3C]}] (Ar = 4-MeC6H4) has been isolated46 and the related 16e complexes have been structurally characterised.5a,13 TH of ketones catalysed by M/NH bifunctional catalysts were shown to proceed via a six-membered pericyclic transition state (see Chart 6).4a,b,14 In 2013, Ikaria and co-workers suggested the formation of contact ion pair intermediate rather than six-membered pericyclic transition state in asymmetric TH of ketones.47,48 One of the coordinated amide N atoms of the guanidinate(2−) ligand in D can be protonated by iPrOH to form contact ion pair intermediate, E. Subsequently, the intermediate, E could transform to the Rh–H species F via 1,2-elimination of hydride from iPrO− in the former while simultaneously releasing acetone. The species F upon reaction with the substrate, RC(O)R′ afforded another contact ion pair intermediate, G before releasing the alcohol and regenerating the species D. Unlike in the species D, the guanidinato ligand in species E–G is present in the monoanionic form.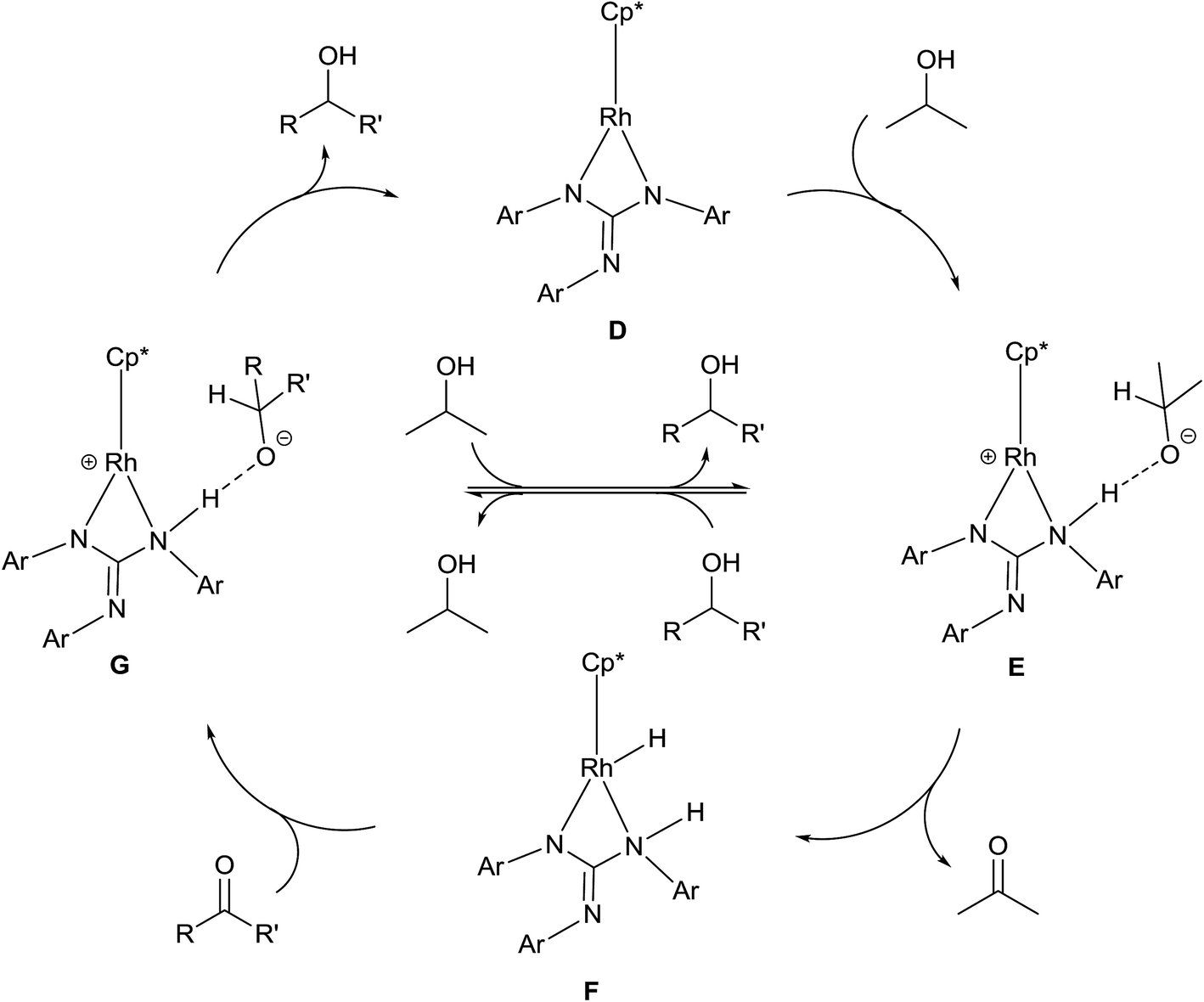 | ||
| Scheme 6 Plausible mechanism of TH of carbonyl compounds with 1–11 as catalysts under basic condition. | ||
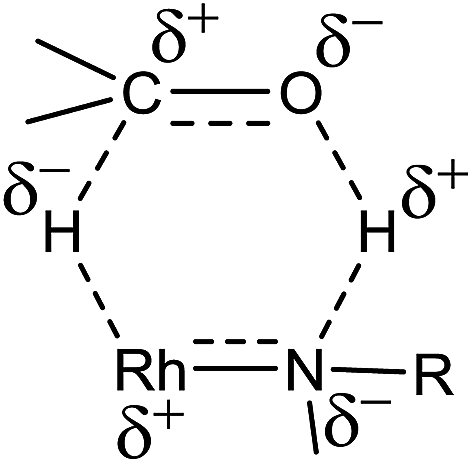 | ||
| Chart 6 | ||
The plausible mechanism of base free TH of carbonyl compounds is illustrated in Scheme 7. This mechanism is based on (i) products analyses discussed in the preceding section and (ii) a mechanism proposed by Sarkar and co-workers for related ruthenium(II) azocarboxamide complexes under base free TH.31b In the presence of excess of iPrOH, 1–11 could ionise to possibly form a cationic complex H which upon reaction with iPrOH can give rise to an intermediate, I. The species I transforms to Rh–H intermediate, J while simultaneously releasing acetone. The species J upon reaction with the substrate, RC(O)R′ regenerates species H via the species K while simultaneously releasing the reduction product, RCH(OH)R′. The guanidine unit in species H is monoanionic while that in species I–K is neutral. The formation of acetophenone reduction product in 50% yield when 8 was used as a catalyst in the absence of base can be ascribed to the difficulty in the formation of species H due to the presence of a coordinated MeCN in the former. In TH of carbonyl compounds catalysed by Ru(II) acetamido complex, the carbonyl reduction was shown to occur via iminol-to-amide tautomerisation of the acetamido ligand thereby allowing the formation of a Ru–H intermediate.49
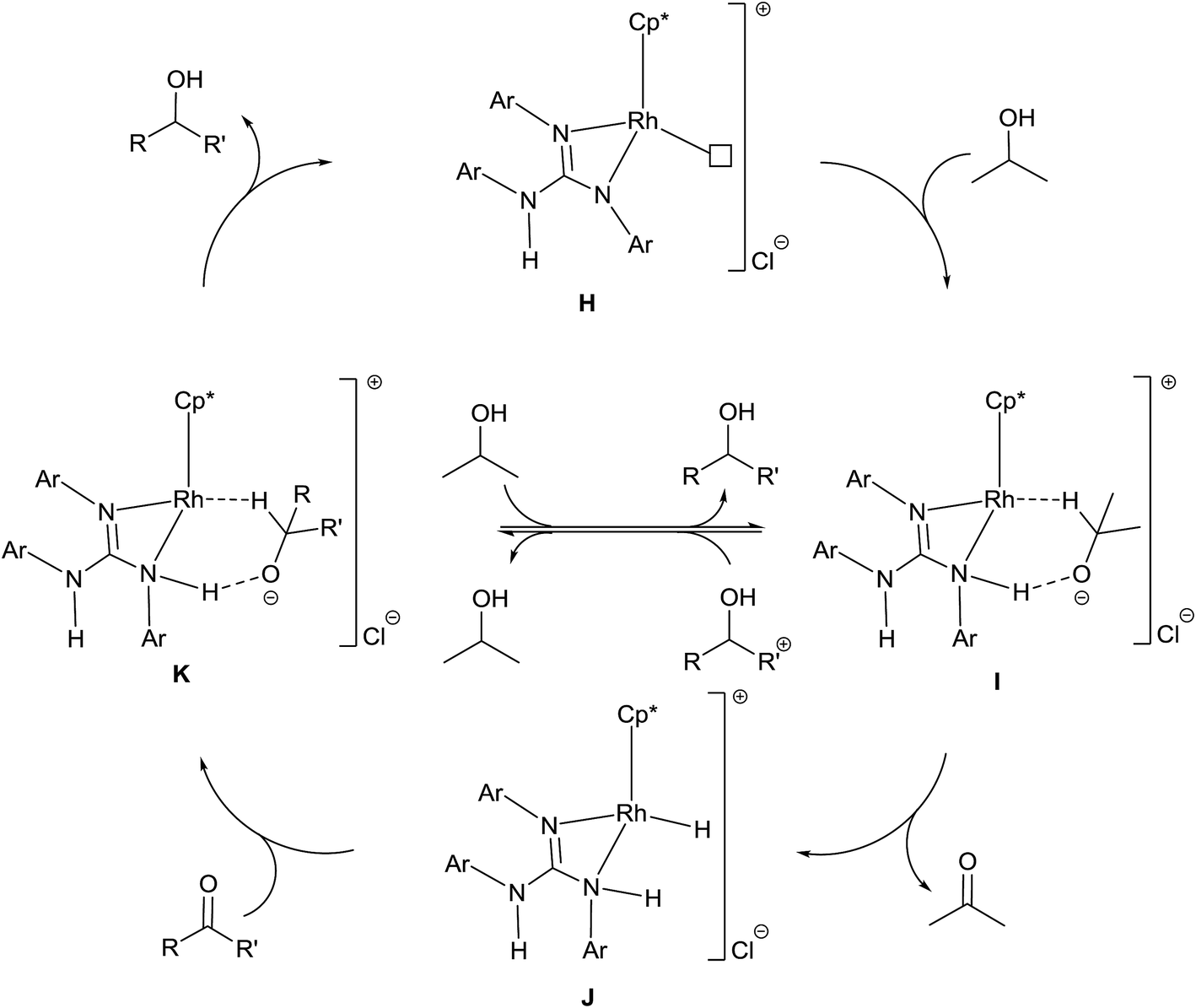 | ||
| Scheme 7 Plausible mechanism of TH of carbonyl compounds with 1–11 as catalysts under base free condition. The symbol □ in H refers to a vacant site. | ||
There have been several attempts to understand the nature of catalytically active species formed during TH of carbonyl compounds mediated by Ru(II) and M(III) (M = Rh and Ir) complexes with iPrOH as solvent and as a hydride source.13,30,31b,32,37,49,50 To shed light on the nature of catalytically active species formed during TH of carbonyl compounds, 3 was treated with two equiv. of 4-methoxybenzaldehyde in CD3OD in the presence of excess of iPrOH and the 1H NMR spectrum was recorded for the reaction mixture. No 1H NMR signals characteristic of species J could be detected although formation of acetone (δ 2.15 ppm) and the 4-methoxybenzyl alcohol were detected. On the other hand, the 1H NMR spectrum of 3 in CD3OD in the presence of excess of iPrOH revealed the presence of a large amount of unreacted 3, acetone and a new species in about 5%. The new species revealed a triplet at δ −8.74 ppm (JRh2H 26 Hz) and a singlet at δ 1.88 ppm assignable to Rh–H and CH3 protons of Cp* ring respectively. These two 1H NMR signals closely matched with those published for the related complex, L (δ −8.59 ppm (t, JRh2H 27.4 Hz)) and 1.94 ppm (s, CH3, Cp*) (see Chart 7).6 From this study it appears that the species J is formed transiently in the presence of the 4-methoxybenzaldehyde resulting in the formation of 4-methoxybenzyl alcohol while in the absence of 4-methoxybenzaldehyde and in the presence of 3, species J deactivates to afford M as revealed by 1H NMR spectroscopy (see Chart 7).
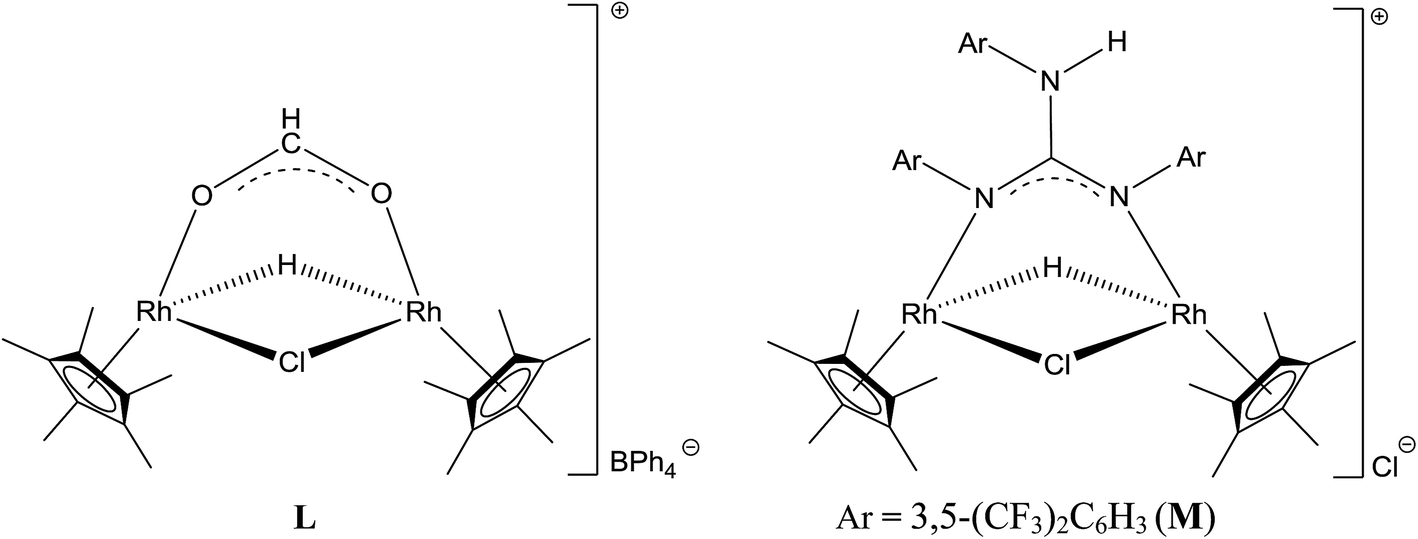 | ||
| Chart 7 Hydrido bridged Cp*Rh(III) formato complex found in the literature (L) and the related Cp*Rh(III) guanidinato complex detected in the present investigation (M). | ||
The Rh–H Intermediate F can react with potassium enolate of 4-nitroacetophenone51 to afford an intermediate N (see Scheme S1 in the ESI†). This species upon reaction with iPrOH can regenerate the intermediate F via Rh(III) isopropoxide intermediate O while simultaneously releasing the reduction product, 4-aminoacetophenone.
TH–etherification tandem catalysis
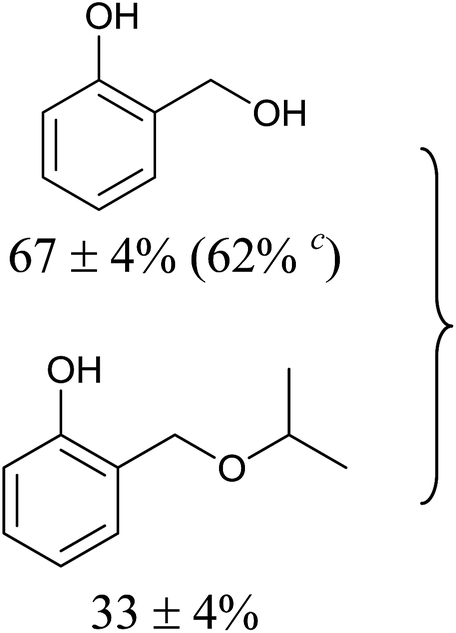
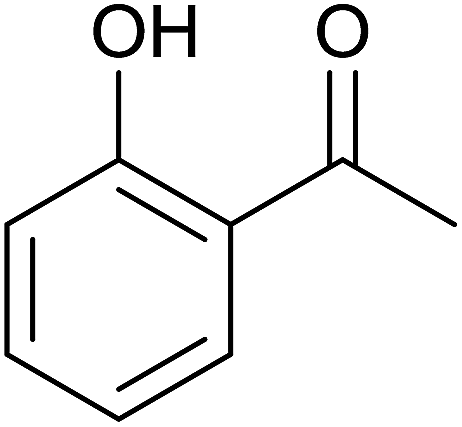
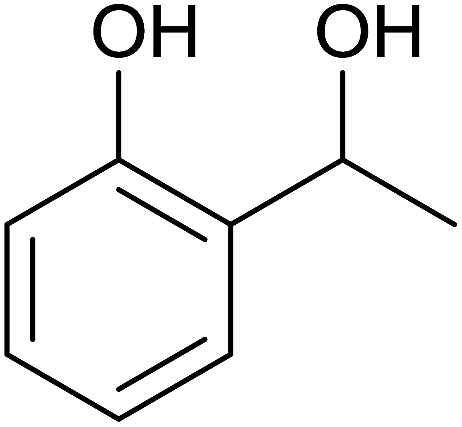
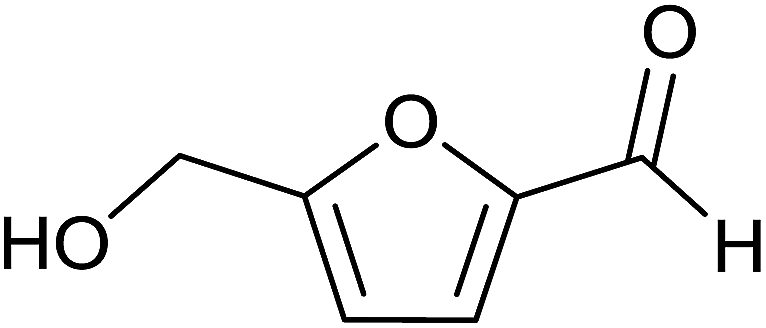
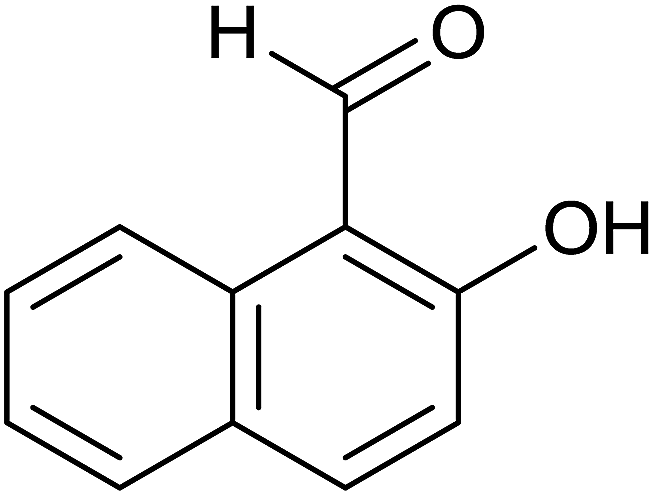
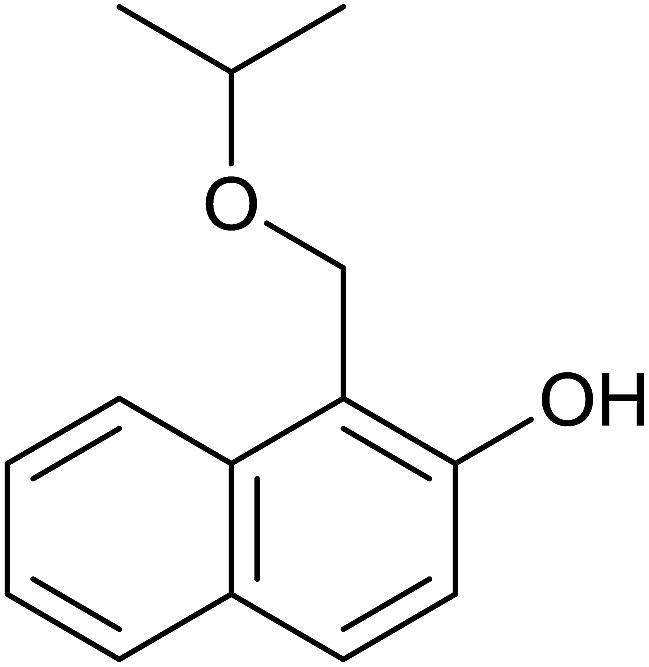
Aromatic carbonyl compounds that also bear OH group were subjected to TH in the presence of 3 under base free condition and the results of this study are listed in Table 9. Salicylaldehyde upon TH afforded 2-hydroxy benzyl alcohol and 2-(isopropoxymethyl)phenol in 67% and 33% yields respectively with overall conversion being 99% (entry 1). There have been some difficulties encountered in TH of salicylaldehyde50 but in the present investigation the substrate is not only reduced but also partially etherified. Interestingly, 2-hydroxy acetophenone upon TH under base free condition afforded the corresponding alcohol without etherifying the resulting –CMe(H)(OH) unit (entry 2). The contrasting behaviour of salicylaldehyde and 2-hydroxy acetophenone in TH in the present investigation is ascribed to sterically less hindered nature of the carbonyl group in the former. On the other hand, 5-(hydroxymethyl) furfural (HMF) upon TH under the base free condition afforded (5-(isopropoxymethyl)furan-2-yl)methanol and 2,5-diisopropoxymethyl-furan in 90% and 10% yields respectively with the overall conversion being 99% (entry 3). HMF is a biomass derived aldehyde, extensively studied in relation to the formation of mono- and di-etherification products catalysed by heterogeneous Lewis acid catalysts.52,53 Interestingly, 2-hydroxy-1-naphthaldehyde upon TH under base free condition afforded 1-(isopropoxymethyl)naphthalene-2-ol as the only product in >99% yield (entry 4). Further, the aforementioned transformation can be achieved in ≥95% yields even when the reaction was carried out in the presence of 1 mol% of 1, 2, 4, 6, 9 and 10 under base free condition (see Table S1 in the ESI†). Hydrogenation–etherification tandem catalysis of benzaldehyde, acetophenone and cinnamaldehyde was effected in the presence of 5 mol% of anionic Os(III)–H complex at 130 °C using Dean–Stark apparatus for water removal.54 Thus, TH–etherification tandem catalysis reported in Table 9 for three substrates to the best of our knowledge is unprecedented in the literature.

Mechanistic hypothesis of etherification
The isolation of 2-hydroxy benzyl alcohol in TH of salicylaldehyde led us to conclude that TH occurs first followed by etherification. A plausible mechanism of etherification of 1-(hydroxymethyl)naphthalen-2-ol is illustrated in Scheme 8. Complex 3 upon reaction with the substrate can lead to the formation of an intermediate P, the formation of which is driven not only by the ability of the substrate to chelate the Rh but also its ability to form intramolecular hydrogen bond involving the benzylic OH hydrogen of the substrate and the imine nitrogen atom of the guanidinato ligand. The intermediate P upon reaction with iPrOH can afford 1-(isopropoxymethyl)naphthalen-2-ol with regeneration of 3 and simultaneous elimination of water.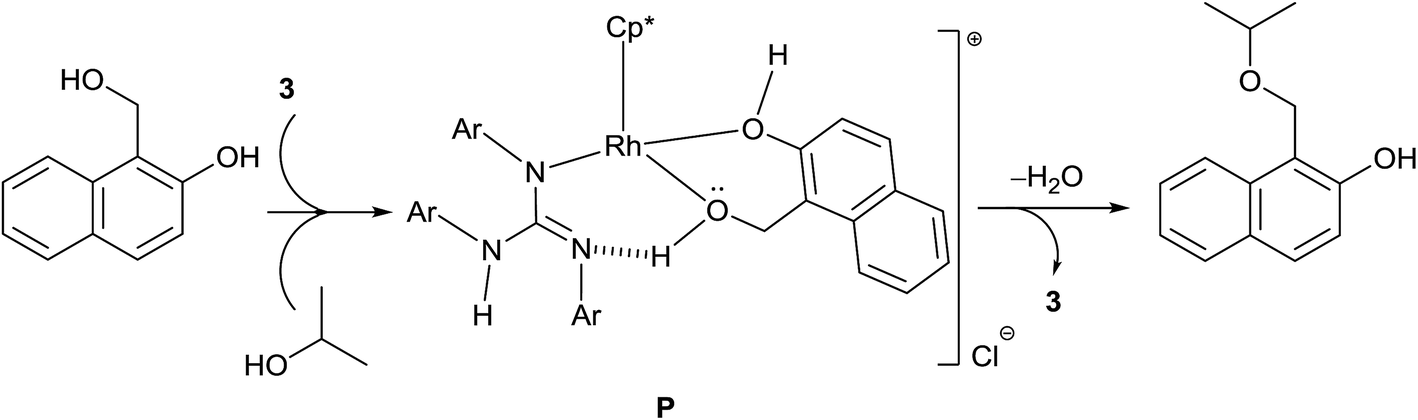 | ||
| Scheme 8 Plausible mechanism of etherification of 1-(hydroxymethyl)naphthalene-2-ol. | ||
The formation of unsymmetrical ethers from the reaction of benzyl alcohol with various other alcohols catalysed by [(η5-Cp*)IrCl2(NHC)] was proposed to occur through IrV–H intermediate.55 In the present investigation, we do not believe the formation of RhV–H species as an intermediate in the ether formation but we do believe the formation of P as an intermediate for two reasons. The imine N atom of the guanidinato ligand in P can act as an internal base in abstracting the benzylic OH proton while the guanidinate ligand simultaneously releases the NH proton in the formation of water. Thus, the guanidinate ligand in the catalysts acts as a proton shuttle between the substrate and iPrOH for ether formation. Further, we did not observe the formation of ether in TH of various aldehydes discussed in Scheme 4. Thus, the guanidinato ligands in the catalyst have to partially decoordinate to accommodate the chelating substrate such as 1-(hydroxymethyl)naphthalen-2-ol prior to the formation of the intermediate P.
Conclusions
Eight half sandwich rhodium(III)/iridium(III) guanidinato complexes were isolated in moderate to good yields. Molecular structures of eleven compounds were determined by SCXRD. The solution behaviour of representative complexes was studied by a detailed NMR experiments. The presence of more than one solution species of Cp*Rh(III) guanidinato complexes was ascribed to arise from either due to unsymmetrical substitution pattern of aryl ring or due to the presence of hydrogen bond donor unit, –N(H)Ar in the guanidinate ligands. Fourteen complexes including those related known complexes in the literature were screened as catalysts for TH of acetophenone under basic and base free conditions and from the screening, 3 emerged as the preferred catalyst. Complex 3 was used as the catalyst for TH of various ketones under basic and base free conditions. The efficiency of reductions varied depending upon the substrates and reaction conditions.The chemoselective reduction of nitro and carbonyl groups in 4-nitroacetophenone was achieved in the presence of 3 as the catalyst under basic and base free conditions respectively and to the best of our knowledge, this is the first such report in homogeneous catalysis. Complex 3 acts as an excellent catalyst in TH of aldehydes even in the absence of base. TH of base sensitive carbonyl compounds was also achieved in good yields except one substrate for steric reason.
Time dependent chemoselective TH of (E)-chalcone and 4-acetylbenzaldehyde were carried out in the presence of 3 under base free condition which gave a mixture of products or only one product depending upon the substrates. Complex 3 reduces CC group under TH condition when this group is in conjugation with the carbonyl group. Salicylaldehyde, 2-hydroxy naphthaldehyde and HMF upon TH in the presence of 3 under base free condition gave the corresponding alcohols with the phenolic OH group in the substrates remaining intact while newly formed –CH2OH group was either partially or fully etherified depending upon substrates. Plausible mechanisms of TH of carbonyl compounds under basic and base free conditions and etherification were proposed. The guanidinate ligands in the new complexes acts as a proton shuttle between iPrOH and the carbonyl substrates in both base assisted and base free TH. The presence of an in-built amido N atom in the guanidinato ligand of 3 does not necessitate the use of external base for successful base free TH. The dual nature of the guanidinato ligand in 3 as an acid and as a base in water elimination was ascribed for its successful role as catalyst in etherification of the substrate such as 1-(hydroxymethyl)naphthalene-2-ol.
Acknowledgements
We acknowledge the Department of Science and Technology, Delhi for financial support (Grant No: EMR/2014/000698) and University Grant Commission for a fellowship (R. K.). We also acknowledge University Science Instrumentation Centre, University of Delhi, Delhi 110 007 for infrastructural facilities and NMR Research Centre, Indian Institute of Science, Bangalore 560 012 for VT NMR measurements. The authors also thank Prof. R. Murugavel, Indian Institute of Technology, Bombay for the use of his single crystal X-ray diffraction facility for the structure determination of 8 established through a DAE-SRC outstanding investigator award. Sophisticated Test and Instrumentation Centre, Cochin University of Science and Technology, Cochin 682 022 is also acknowledged for elemental analysis data for some compounds.References
- R. Krämer, M. Maurus, K. Polborn, K. Sünkel, C. Robl and W. Beck, Chem.–Eur. J., 1996, 2, 1518 CrossRef.
- R. D. Simpson and W. J. Marshall, Organometallics, 1997, 16, 3719 CrossRef CAS.
- K. Murata and T. Ikariya, J. Org. Chem., 1999, 64, 2186 CrossRef CAS.
- (a) T. Ikariya, K. Murata and R. Noyori, Org. Biomol. Chem., 2006, 4, 393 RSC; (b) T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300 CrossRef CAS PubMed; (c) X. Wu, C. Wang and J. Xiao, Platinum Met. Rev., 2010, 54, 3 CrossRef CAS; (d) F. Foubelo, C. Nájera and M. Yus, Tetrahedron: Asymmetry, 2015, 26, 769 CrossRef CAS.
- (a) A. J. Blacker, E. Clot, S. B. Duckett, O. Eisenstein, J. Grace, A. Nova, R. N. Perutz, D. J. Taylor and A. C. Whitwood, Chem. Commun., 2009, 6801 RSC; (b) A. Nova, D. J. Taylor, A. J. Blacker, S. B. Duckett, R. N. Perutz and O. Eisenstein, Organometallics, 2014, 33, 3433 CrossRef CAS.
- A. J. Blacker, S. B. Duckett, J. Grace, R. N. Perutz and A. C. Whitwood, Organometallics, 2009, 28, 1435 CrossRef CAS.
- M. Yadav, A. K. Singh and D. S. Pandey, Organometallics, 2009, 28, 4713 CrossRef CAS.
- A. Zamorano, N. Rendón, J. E. V. Valpuesta, E. Álvarez and E. Carmona, Inorg. Chem., 2015, 54, 6573 CrossRef CAS PubMed.
- L. Casarrubios, M. A. Esteruelas, C. Larramona, J. G. Muntaner, M. Oliván, E. Oñate and M. A. Sierra, Organometallics, 2014, 33, 1820 CrossRef CAS.
- A. L. Müller, T. Bleith, T. Roth, H. Wadepohl and L. H. Gade, Organometallics, 2015, 34, 2326 CrossRef.
- A. H. Ngo, M. Ibañez and L. H. Do, ACS Catal., 2016, 6, 2637 CrossRef CAS.
- A. Ruff, C. Kirby, B. C. Chan and A. R. O'Connor, Organometallics, 2016, 35, 327 CrossRef CAS.
- K.-J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285 CrossRef CAS.
- R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931 CrossRef CAS PubMed.
- B. Zhao, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744 CrossRef CAS PubMed.
- M. K. T. Tin, G. P. A. Yap and D. S. Richeson, Inorg. Chem., 1998, 37, 6728 CrossRef CAS PubMed.
- (a) P. J. Bailey and S. Pace, Coord. Chem. Rev., 2001, 214, 91 CrossRef CAS; (b) F. T. Edelmann, Adv. Organomet. Chem., 2013, 61, 55 CrossRef CAS.
- T. Singh, R. Kishan, M. Nethaji and N. Thirupathi, Inorg. Chem., 2012, 51, 157 CrossRef CAS PubMed.
- R. Kishan, R. Kumar, S. Baskaran, C. Sivasankar and N. Thirupathi, Eur. J. Inorg. Chem., 2015, 3182 CrossRef CAS.
- P. J. Bailey, L. A. Mitchell and S. Parsons, J. Chem. Soc., Dalton Trans., 1996, 2839 RSC.
- M. B. Dinger, W. Henderson and B. K. Nicholson, J. Organomet. Chem., 1998, 556, 75 CrossRef CAS.
- S. Aharonovich, M. Kapon, M. Botoshanski and M. S. Eisen, Organometallics, 2008, 27, 1869 CrossRef CAS.
- G. Ciancaleoni, C. Zuccaccia, D. Zuccaccia, E. Clot and A. Macchioni, Organometallics, 2009, 28, 960 CrossRef CAS.
- (a) X. Wu and J. Xiao, in Comprehensive Organic Synthesis, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, 2nd edn, 2014, vol. 8, p. 198 Search PubMed; (b) R. H. Morris, Chem. Rec., 2016, 16, 2644 CrossRef PubMed.
- (a) J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237 RSC; (b) W. Baratta and P. Rigo, Eur. J. Inorg. Chem., 2008, 4041 CrossRef CAS.
- M. Nordin, R.-Z. Liao, K. Ahlford, H. Adolfsson and F. Himo, ChemCatChem, 2012, 4, 1095 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621 CrossRef CAS PubMed.
- H. G. Nedden, A. Zanotti-Gerosa and M. Wills, Chem. Rec., 2016, 16, 2623 CrossRef PubMed.
- S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562 CrossRef CAS.
- R. Castarlenas, M. A. Esteruelas and E. Oñate, Organometallics, 2008, 27, 3240 CrossRef CAS.
- (a) M. Kumar, J. DePasquale, N. J. White, M. Zeller and E. T. Papish, Organometallics, 2013, 32, 2135 CrossRef CAS; (b) M. G. Sommer, S. Marinova, M. J. Krafft, D. Urankar, D. Schweinfurth, M. Bubrin, J. Košmrlj and B. Sarkar, Organometallics, 2016, 35, 2840 CrossRef CAS.
- M. C. Carrión, F. Sepúlveda, F. A. Jalón, B. R. Manzano and A. M. Rodríguez, Organometallics, 2009, 28, 3822 CrossRef.
- R. Kishan, PhD Thesis, University of Delhi, 2015.
- R. Kumar, PhD Thesis, University of Delhi, 2017.
- K. Li, J.-L. Niu, M.-Z. Yang, Z. Li, L.-Y. Wu, X.-Q. Hao and M.-P. Song, Organometallics, 2015, 34, 1170 CrossRef CAS.
- Y. Wei, J. Wu, D. Xue, C. Wang, Z. Liu, Z. Zhang, G. Chen and J. Xiao, Synlett, 2014, 25, 1295 CrossRef.
- J. DePasquale, M. Kumar, M. Zeller and E. T. Papish, Organometallics, 2013, 32, 966 CrossRef CAS.
- A. Bolje, S. Hohloch, J. Košmrlj and B. Sarkar, Dalton Trans., 2016, 45, 15983 RSC.
- (a) J. R. Miecznikowski and R. H. Crabtree, Organometallics, 2004, 23, 629 CrossRef CAS; (b) X. Wu, J. Liu, X. Li, A. Zanotti-Gerosa, F. Hancock, D. Vinci, J. Ruan and J. Xiao, Angew. Chem., Int. Ed., 2006, 45, 6718 CrossRef CAS PubMed.
- S. Baldino, S. Facchetti, A. Zanotti-Gerosa, H. G. Nedden and W. Baratta, ChemCatChem, 2016, 8, 2279 CrossRef CAS.
- R. Corberán and E. Peris, Organometallics, 2008, 27, 1954 CrossRef.
- (a) S.-J. Chen, G.-P. Lu and C. Cai, RSC Adv., 2015, 5, 13208 RSC; (b) J. L. Gomez-Lopez, D. Chávez, M. Parra-Hake, A. T. Royappa, A. L. Rheingold, D. B. Grotjahn and V. Miranda-Soto, Organometallics, 2016, 35, 3148 CrossRef CAS.
- X. Li, L. Li, Y. Tang, L. Zhong, L. Cun, J. Zhu, J. Liao and J. Deng, J. Org. Chem., 2010, 75, 2981 CrossRef CAS PubMed.
- Z. Baán, Z. Finta, G. Keglevich and I. Hermecz, Tetrahedron Lett., 2005, 46, 6203 CrossRef.
- J. R. Miecznikowski and R. H. Crabtree, Polyhedron, 2004, 23, 2857 CrossRef CAS.
- A. W. Holland and R. G. Bergman, J. Am. Chem. Soc., 2002, 124, 9010 CrossRef CAS PubMed.
- P. A. Dub and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 2604 CrossRef CAS PubMed.
- P. A. Dub and J. C. Gordon, Dalton Trans., 2016, 45, 6756 RSC.
- C. S. Yi, Z. He and I. A. Guzei, Organometallics, 2001, 20, 3641 CrossRef CAS.
- P. Pelagatti, M. Carcelli, F. Calbiani, C. Cassi, L. Elviri, C. Pelizzi, U. Rizzotti and D. Rogolino, Organometallics, 2005, 24, 5836 CrossRef CAS.
- L. R. Domingo and J. Andrés, The Chemistry of Metal Enolates, Part 1, ed. J. Zabicky, John Wiley & Sons Ltd, West Sussex, 2009, ch. 1, p. 1 Search PubMed.
- J. Jae, E. Mahmoud, R. F. Lobo and D. G. Vlachos, ChemCatChem, 2014, 6, 508 CrossRef CAS.
- J. D. Lewis, S. Van de Vyver, A. J. Crisci, W. R. Gunther, V. K. Michaelis, R. G. Griffin and Y. Román-Leshkov, ChemSusChem, 2014, 7, 2255 CrossRef CAS PubMed.
- M. A. Esteruelas, C. García-Yebra and E. Oñate, Organometallics, 2008, 27, 3029 CrossRef CAS.
- A. Prades, R. Corberán, M. Poyatos and E. Peris, Chem.–Eur. J., 2008, 14, 11474 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data (CIF files for L5, L6, L9 and 1–8), general considerations including syntheses and characterisation of all new compounds and their precursors. CCDC 1538611–1538621. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra04152g |
| This journal is © The Royal Society of Chemistry 2017 |