DOI:
10.1039/C7RA04524G
(Paper)
RSC Adv., 2017,
7, 29752-29761
Synthesis, structure and reactivity of guanidinate rare earth metal bis(o-aminobenzyl) complexes†
Received
22nd April 2017
, Accepted 23rd May 2017
First published on 7th June 2017
Abstract
A series of guanidinate rare-earth metal complexes [(PhCH2)2NC(NC6H4iPr2-2,6)2]RE(CH2C6H4NMe2-o)2 (RE = Y (2a), La (2b), Dy (2c), Lu (2d)) were synthesized by the acid–base reaction of RE(CH2C6H4NMe2-o)3 with (PhCH2)2N[C(NHR)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) (NR)] (R = 2,6-iPr2-C6H3) (1) in THF. Treatment of complexes 2 with two equivalents of carbon dioxide, sulfur and phenyl isothiocyanate gave the corresponding insertion products {[(PhCH2)2NC(NC6H4iPr2-2,6)2]RE(μ-η2:η1-O2CCH2C6H4NMe2-o)(μ-η1:η1-O2CCH2C6H4NMe2-o)}2 (RE = Y (3a), La (3b), Dy (3c), Lu (3d)), {[(PhCH2)2NC(NC6H4iPr2-2,6)2]RE[μ-S(CH2C6H4NMe2-o)]2}2 (RE = Y (4a), La (4b), Dy (4c), Lu (4d)) and {[(PhCH2)2NC(NC6H4iPr2-2,6)2]RE{SC(CH2C6H4NMe2-o)NPh}2 (RE = Y (5a), La (5b), Dy (5c), Lu (5d)) in good yields, respectively. All new complexes were fully characterized by NMR spectroscopy and elemental analysis. The structures of 1, 2d, 3, 4a, 4c–d, 5a, and 5c–d were established by X-ray diffraction studies. Complexes 2 were found to have a high activity and excellent 3,4-selectivity for isoprene polymerization in the presence of [Ph3C][B(C6F5)4].
(NR)] (R = 2,6-iPr2-C6H3) (1) in THF. Treatment of complexes 2 with two equivalents of carbon dioxide, sulfur and phenyl isothiocyanate gave the corresponding insertion products {[(PhCH2)2NC(NC6H4iPr2-2,6)2]RE(μ-η2:η1-O2CCH2C6H4NMe2-o)(μ-η1:η1-O2CCH2C6H4NMe2-o)}2 (RE = Y (3a), La (3b), Dy (3c), Lu (3d)), {[(PhCH2)2NC(NC6H4iPr2-2,6)2]RE[μ-S(CH2C6H4NMe2-o)]2}2 (RE = Y (4a), La (4b), Dy (4c), Lu (4d)) and {[(PhCH2)2NC(NC6H4iPr2-2,6)2]RE{SC(CH2C6H4NMe2-o)NPh}2 (RE = Y (5a), La (5b), Dy (5c), Lu (5d)) in good yields, respectively. All new complexes were fully characterized by NMR spectroscopy and elemental analysis. The structures of 1, 2d, 3, 4a, 4c–d, 5a, and 5c–d were established by X-ray diffraction studies. Complexes 2 were found to have a high activity and excellent 3,4-selectivity for isoprene polymerization in the presence of [Ph3C][B(C6F5)4].
1. Introduction
Since the first structural characterization of a half sandwich rare-earth bis(alkyl) complex was reported by Schaverien in 1989,1 rare-earth dialkyl complexes have demonstrated great potential applications in synthetic chemistry and polymerization.2 The reported results indicated that the proper design of the coligand is of great importance and can not only improve the stability of rare-earth bis(alkyl) complexes but also influence their reactivity.2c,3 Recently, myriads of rare-earth complexes with non-Cp ancillary ligands were explored.3,4 Among various non-Cp ligands, amidinate and guanidinate stand out and are widely employed in lanthanide chemistry because of their tunable steric and electronic properties, rich coordination mode and easy accessibility.5 Guanidinate scaffolds are of tremendous popularity in lanthanide chemistry and numerous rare-earth complexes bearing guanidiante ligands have been reported. However, just a handful of neutral mono(guanidinate) bis(alkyl) rare-earth complexes were synthesized. And neither their reactivity towards small molecules nor their performance as precatalyst towards conjugated diene polymerization have been unveiled so far.6
Recently, our group reported the reactions of amidinate rare-earth dialkyl complexes7 with various small molecules, which not only shed some light on the reaction chemistry of amidiante rare-earth dialkyl complexes, but also provided different options for the synthesis of some organolanthanide derivatives.
In order to further explore the effect of subtle ligand change on the reaction patterns of rare-earth bis(alkyl) complexes, we designed a new bulky tetraalkylated guanidinate with two benzyl groups at the axis N atom. Herein, we report the synthesis of this new guanidine and the corresponding rare-earth bis(alkyl) complexes. The reactions of the dialkyl species with small molecules, such as CO2, S8, and PhNCS are also disclosed. Moreover, the catalytic performance of the mono(guanidiante) rare-earth bis(alkyl) complexes for isoprene polymerization will be discussed as well.
2. Results and discussion
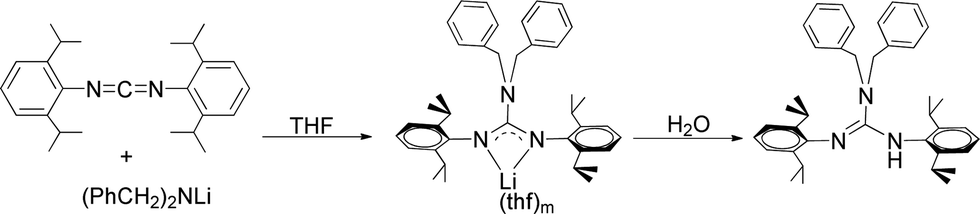
2.1 Synthesis of (PhCH2)2N[C(NHR)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) (NR)] (R = 2,6-iPr2C6H3) (1)
(NR)] (R = 2,6-iPr2C6H3) (1)
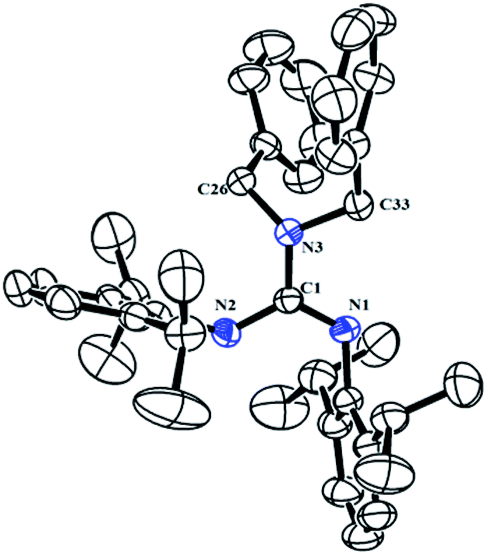
The neutral guanidinate compound (PhCH2)2N[C(NHR)NR] (R = 2,6-iPr2C6H3) (1) was synthesized in good yield by the reaction of lithium dibenzylamido with one equivalent of carbodiimide (Scheme 1). Compound 1 was characterized by NMR spectroscopy and X-ray structural analysis. The X-ray diffraction reveals that the ΔCN parameter of complex 1 is 0.102 Å (Fig. 1), which shows the difference in interatomic distance in the supposed “double” and “single” bonds is very evident. This compound is very easy to dissolve in hexane, toluene and THF. In the 1H NMR spectrum (in CDCl3) of 1, four doublets at δ = 1.34, 1.25, 1.05 and 0.91 ppm can be assigned to methyl protons, and the multiples appear at 3.31–3.20 assignable to methine protons. After scrutiny of the 1H NMR spectrum (in CDCl3), we found that the multiples were formed by two sets of overlapped multiples. And this was further confirmed by the 13C NMR spectrum (in CDCl3): two resonances at δ = 29.0 and 28.4 ppm can be assigned to methine carbons of the –CHMe2 units. Besides, we further collected the NMR spectra of 1 in C6D6, and in the 1H NMR spectrum (in C6D6) of 1, the signals of methine protons in –CHMe2 units appear as multiple peaks at δ = 3.50 and 3.35 ppm. In its 13C NMR spectrum (in C6D6), corresponding carbon resonances appear at δ = 29.3 and 28.6 ppm respectively.
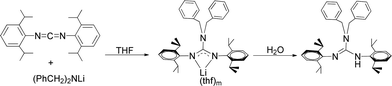 |
| | Scheme 1 | |
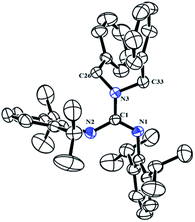 |
| | Fig. 1 Molecular structure of complex 1 with thermal ellipsoids at 30% probability. All of the hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): C(1)–N(1) 1.269(3), C(1)–N(2) 1.371(3), C(1)–N(3) 1.389(3), C(26)–N(3) 1.454(3), C(33)–N(3) 1.474(3), N(2)–C(1) 1.371(3); C(1)–N(1)–C(2) 121.1(2), C(1)–N(2)–C(14) 132.7(2), N(1)–C(1)–N(2) 122.8(2), N(1)–C(1)–N(3) 119.1(2), N(2)–C(1)–N(3) 118.1(2), C(1)–N(3)–C(26) 121.61(19), C(1)–N(3)–C(33) 115.15(18), C(26)–N(3)–C(33) 114.26(18). | |
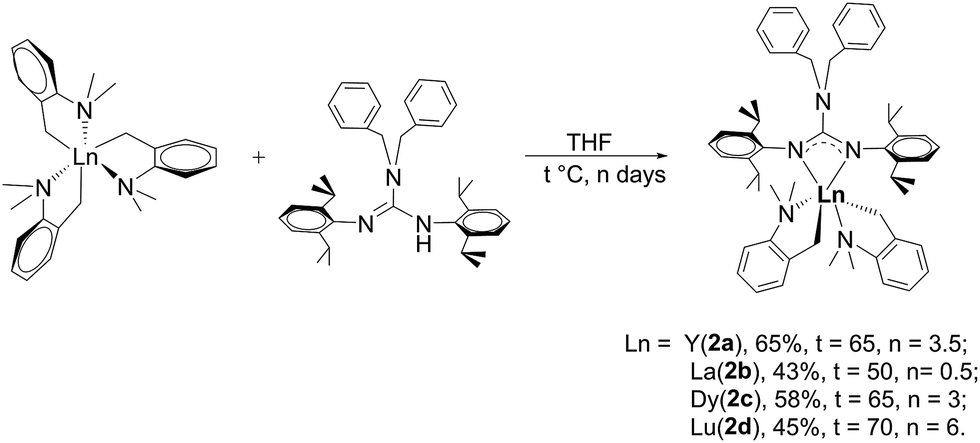
2.2 Synthesis of [(PhCH2)2NC(NC6H4iPr22,6)2]RE(CH2C6H4NMe2-o)2 (RE = Y (2a), La (2b), Dy (2c), Lu (2d))
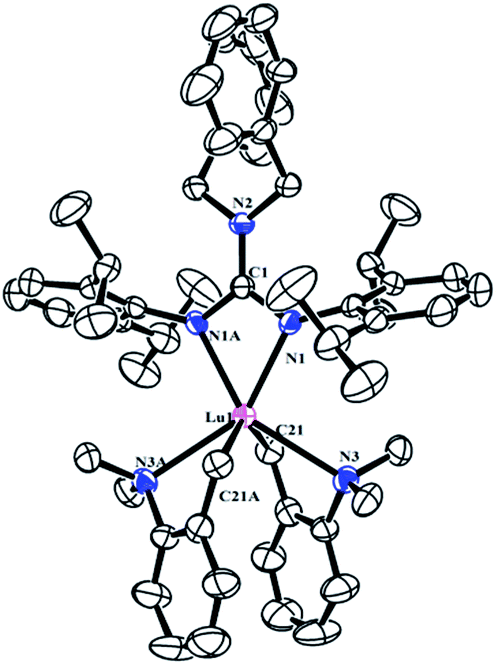
As shown in Scheme 2, treatment of RE(CH2C6H4NMe2-o)3 with 1 afforded the guanidinate-stabilized bis(aminobenzyl) complexes [(PhCH2)2NC(NC6H3iPr2-2,6)2]RE(CH2C6H4NMe2-o)2 (RE = Y (2a), La (2b), Dy (2c), Lu (2d)) in moderate yields. All the new compounds were characterized by elemental analysis, NMR spectroscopy (except for 2c). In 1H NMR spectra of 2, the resonances of the methylene protons in aminobenzyl group appear as sharp singlets at δ = 1.81(2a), 1.84(2b) and 1.80(2d) ppm respectively. And the signals at δ = 48.0(2a), 61.6(2b) and 53.3(2d) in their 13C NMR spectra can be assigned to their corresponding methylene carbons. And the signals of methine protons in the guanidinate ligand of the complexes 2 are observed at δ = 4.12 ppm for 2a, 3.93 ppm for 2b and 4.20 ppm for 2d, while their corresponding carbon resonances almost remain unchanged, the similar phenomenon was observed in [(CH3)2NC(NC6H4iPr2-2,6)2]Y(CH2SiMe3)2 (THF) and the corresponding neutral ligand (CH3)2N[C(NHR)(NR)] (R = 2,6-iPr2C6H3).6c Other characteristic peaks can also be assigned clearly. Single crystal X-ray structural analysis of complex 2d established the distorted-octahedral geometry of its core structure. The structure of 2d is depicted in Fig. 2; selected bond lengths and angles are listed in the caption. The lutetium atom was coordinated by six atoms: two nitrogen atoms from the guanidinate ligand, two carbon atoms and two amino nitrogen atoms. The guanidinate ligand coordinates symmetrically to the Lu atom. Because of the better symmetry of the coordinated guanidinate ligand, the numbers of the methine and methyl resonances of the ligand decrease in the NMR spectra of complexes 2 in comparison with the neutral ligand.6c The bond length of Lu–N (2.318(3) Å) is very close to the Lu–N distance in [CyC(N-2,6-iPr2C6H3)2]Lu(CH2SiMe3)2(THF) (average 2.313 Å)8 and slightly shorter than Y–N distance (2.373 Å) in [PhC(NC6H4iPr2-2,6)2]Y(CH2C6H4NMe2-o)2,7b after taking into consideration of the difference between metal radii.
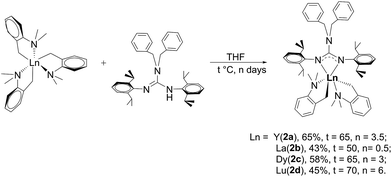 |
| | Scheme 2 | |
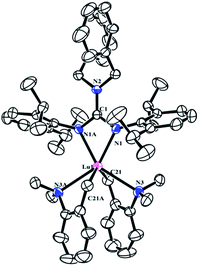 |
| | Fig. 2 Molecular structure of complex 2d with thermal ellipsoids at 30% probability. All of the hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Lu(1)–N(1) 2.318(3), Lu(1)–N(1A) 2.318(3), Lu(1)–N(3) 2.624(3); N(1)–C(1)–N(1A) 109.4(4), N(1)–C(1)–N(2) 125.31(19), N(1A)–C(1)–N(2) 125.31(19), N(1)–Lu(1)–N(1A) 57.07(11). | |
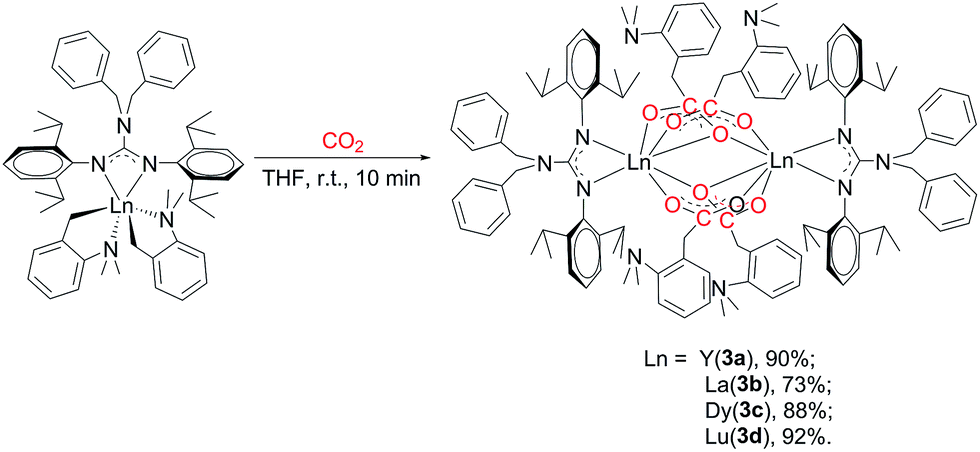
2.3 Reaction of [(PhCH2)2NC(NC6H4iPr2-2,6)2]RE(CH2C6H4NMe2-o)2 (RE = Y (2a), La (2b), Dy (2c), Lu (2d)) with CO2
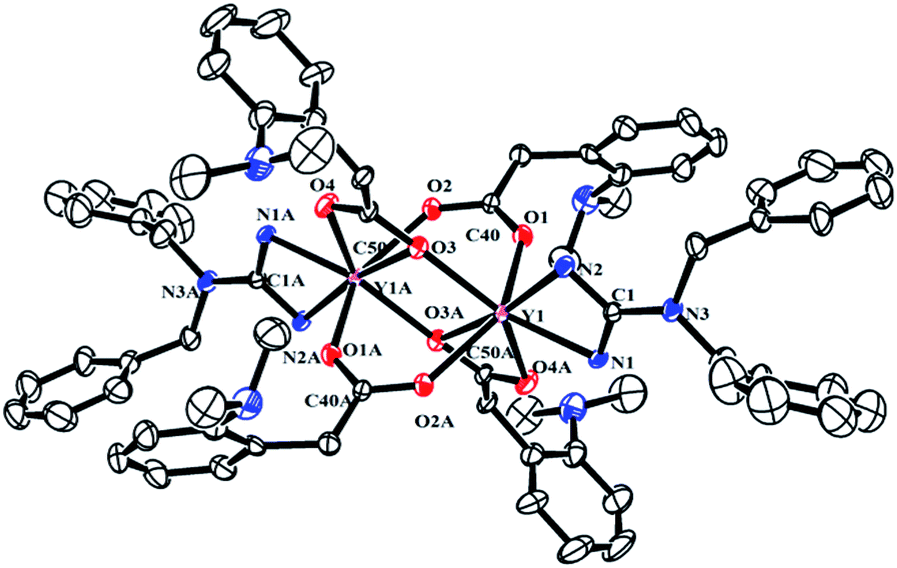
To explore the ligand effect on the structure and reactivity of complexes, reactions of complexes 2 with CO2 were conducted firstly. The THF solution of complexes 2 was stirred under an atmosphere of CO2 (0.1 MPa), an immediate colour change from yellow to colourless was observed for complexes 2b and 2c, while the colour change was not observed in the reaction of 2a and 2d (Scheme 3). Expected insertion products {[(PhCH2)2NC(NC6H4iPr2-2,6)2]RE(μ-η2:η1-O2CCH2C6H4NMe2-o)(μ-η1:η1-O2CCH2C6H4NMe2-o)}2 (RE = Y (3a), La (3b), Dy (3c), Lu (3d)) were obtained in mediate to excellent yields. In their 1H NMR spectra, the broad peak observed at δ = 3.47(3b), 3.50(3d) ppm can be assigned to methylene protons of the aminobenzyl groups. Complex 3b displays a sharp singlet at δ = 2.54 ppm assignable for the methyl protons of –NMe2 groups, while for 3d, the methyl signal turns into a set of broad multiple peaks at δ = 2.52 ppm. However, in the room temperature 1H NMR spectrum of 3a, there is only one broad peak at δ = 2.47 ppm that can be assigned to methylene and methyl protons in the aminobenzyl groups. Compared to their corresponding dialkyl complexes, in complexes 3 (except 3c), the methylene protons of the aminobenzyl groups obviously shift to downfield which indicates the insertion of CO2 molecules. And in their 13C NMR spectra, the resonances at δ = 186.2(3a), 183.6(3b) and 183.7(3d) ppm are assignable to the carbons in carboxyl groups. The structures of complexes 3 were further confirmed by the X-ray single crystal diffraction. The X-ray crystallographic analysis indicates that complexes 3a–d are isostructural, and crystal structure of 3a is presented in Fig. 3. The generated carboxyl units coordinate to lanthanide centres in two different fashions: μ-η2:η1 and μ-η1:η1. And the C–O bond lengths of complexes 3 range from 1.222(9) Å to 1.309(8) Å, which are consistent with typical delocalized carboxylate species.9 The Y–O bond length are generally in the normal range except the Y(1)–O(3A) (2.737(4) Å) bond. The Y(1)–O(3A) bond is remarkably longer than those observed in [(C5Me4)SiMe2(CH2CHCH2)]Y(μ-η2:η1-O2CCH2SiMe3)(μ-η1:η1-O2CCH2SiMe3)}2 (ref. 9b) (2.563(2) Å) and {[PhC(NC6H4iPr2-2,6)2]Y(μ-η2:η1-O2CCH2C6H4NMe2-o)(μ-η1:η1-O2CCH2C6H4NMe2-o)}2 (2.53(1) Å).7b This unusual bond length can be ascribed to the bulky size and electronic effect of the guanidinate ligand. The poor solubility of complexes 3 even in THF at room temperature can provide extra evidence for the sterically crowded coordination sphere around the lanthanide centres. The reactions of complexes 2 with CO2 provide not only an effective way for the activation of CO2, but also a good synthetic method for guanidinate rare-earth derivatives.
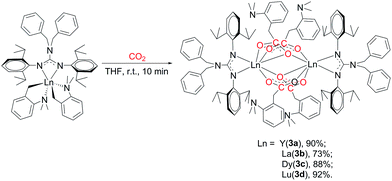 |
| | Scheme 3 | |
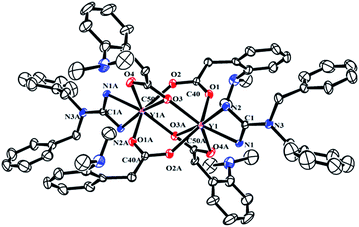 |
| | Fig. 3 Molecular structure of complex 3a with thermal ellipsoids at 30% probability. 2,6-Diisopropylphenyl groups of guanidinate ligand and all of the hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Y(1)–N(1) 2.318(4), Y(1)–N(2) 2.354(5), Y(1)–O(3) 2.254(4), Y(1)–O(1) 2.269(4), Y(1)–O(2A) 2.276(4), Y(1)–O(4A) 2.330(4), Y(1)–O(3A) 2.737(4), O(1)–C(40) 1.259(7), O(2)–C(40) 1.276(6), O(3)–C(50) 1.280(6), O(4)–C(50) 1.257(7); O(3)–Y(1)–O(1) 81.80(15), O(1)–Y(1)–O(4A) 85.33(16), O(3)–Y(1)–O(4A) 135.12(15), O(3)–Y(1)–O(3A) 84.96(14), O(1)–Y(1)–O(2A) 141.14(14), N(1)–C(1)–N(2) 110.5(4), N(1)–Y(1)–N(2) 56.38(15), O(1)–C(40)–O(2) 122.2(5), O(4)–C(50)–O(3) 118.5(5). | |
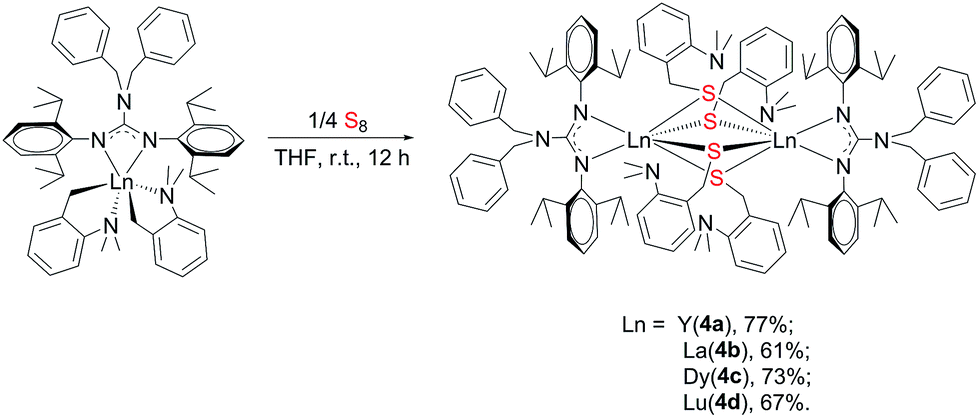
2.4 Reaction of [(PhCH2)2NC(NC6H4iPr2-2,6)2]RE(CH2C6H4NMe2-o)2 (RE = Y (2a), La (2b), Dy (2c), Lu (2d)) with S8
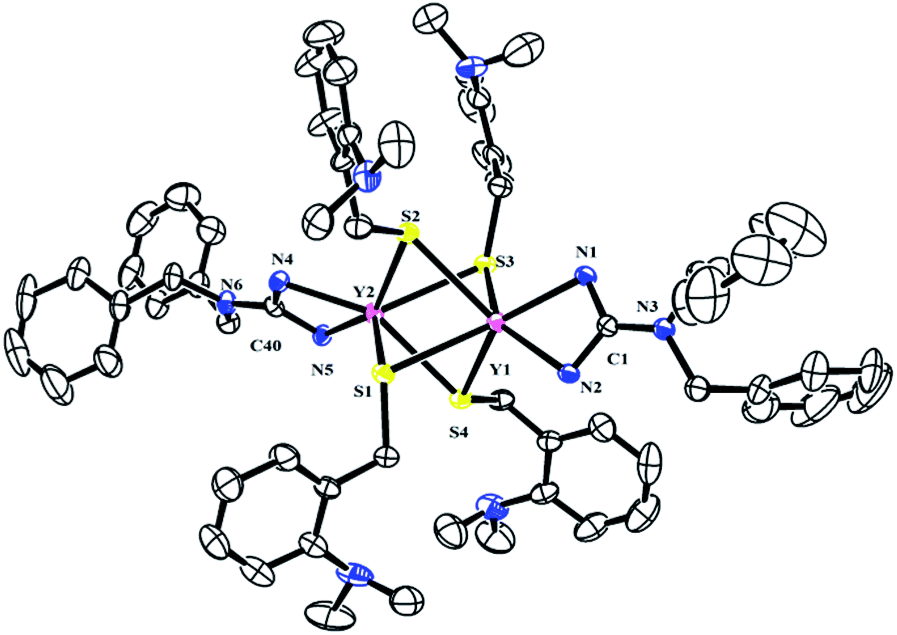
To further study the reactivity of complexes 2, reactions with S8 were also carried out. Different from their amidinate counterparts,7b the reactions of complexes 2 with 1/4 equivalent S8 gave dinuclear lanthanide insertion products {[(PhCH2)2NC(NC6H4iPr2-2,6)2]RE[μ-S(CH2C6H4NMe2-o)]2}2 (RE = Y (4a), La (4b), Dy (4c), Lu (4d)) in good yields (Scheme 4).10d These complexes are easily soluble in toluene and THF, and slightly soluble in hexane. Complexes 4a, 4b and 4d were characterized by the 1H and 13C NMR spectra in C6D6 at 25 °C. The 1H NMR spectra show multiple signals at δ = 3.89(4a), 3.77(4b) and 3.95(4d) ppm which are assignable for the methine protons of the CHMe2 groups. Compared to their corresponding dialkyl complexes, the signals of the methylene protons obviously shift to lower field because of the insertion of the sulfur atoms. For 4b, the signal for the methylene protons of aminobenzyl group appears as a sharp singlet at δ = 4.52 ppm, while for 4a and 4d, it becomes a broad peak at δ = 4.28 and 4.46 ppm respectively. And their corresponding carbon signals show up at δ = 34.6(4a), 33.5(4b) and 34.3(4d) ppm as sharp singlets. The X-ray single crystal diffraction analysis established the bimetallic structure of complexes 4a, 4c and 4d (Fig. 4). Two yttrium ions are connected by four bridging thiolate units which definitely proves the insertion of sulfur atoms into each of Y–C bonds. In lanthanide chemistry, the similar core structure is only observed in complex [{tBuC(NC6H3-2,6-iPr2)2}Yb(μ-SCH2Ph)2]2.11 The lengths of Y–S bonds range from 2.778(1) Å to 2.841(1) Å and are comparable to the previously reported Y–S(μ2-SR) bonds.10 The dihedral angles between the Y1S1Y2 and Y1S3Y2 plane is 179.7°, indicating that the Y1S1Y2S3 is coplanar. Y1S2Y2S4 unit is also coplanar. Besides, the two planes are almost vertical to each other with a dihedral angle of 89.86°.
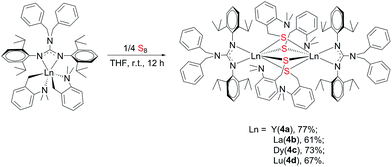 |
| | Scheme 4 | |
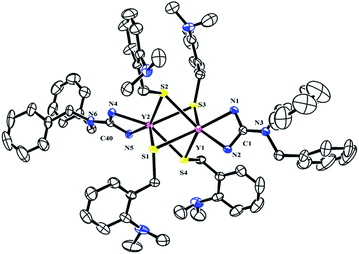 |
| | Fig. 4 Molecular structure of complex 4a with thermal ellipsoids at 30% probability. 2, 6-Diisopropylphenyl groups of guanidinate ligand and all of the hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Y(1)–N(2) 2.320(3), Y(1)–N(1) 2.338(3), Y(1)–S(3) 2.7781(10), Y(1)–S(4) 2.7900(9), Y(1)–S(2) 2.8012(10), Y(1)–S(1) 2.8079(9), Y(2)–N(4) 2.320(3), Y(2)–N(5) 2.323(3), Y(2)–S(1) 2.7947(10), Y(2)–S(2) 2.795(1), Y(2)–S(4) 2.807(1), Y(2)–S(3) 2.8412(9); N(2)–Y(1)–N(1) 56.99(9), S(3)–Y(1)–S(4) 69.31(3), S(4)–Y(1)–S(2) 106.2(1), N(4)–Y(2)–N(5) 57.08(9), S(1)–Y(2)–S(2) 69.28(3), S(2)–Y(2)–S(4) 105.9(1), N(2)–C(1)–N(1) 111.3(3), N(4)–C(40)–N(5) 110.5(3), Y(2)–S(1)–Y(1) 73.84(2), Y(2)–S(2)–Y(1) 73.94(2), Y(1)–S(3)–Y(2) 73.58(2), Y(1)–S(4)–Y(2) 73.93(2). | |
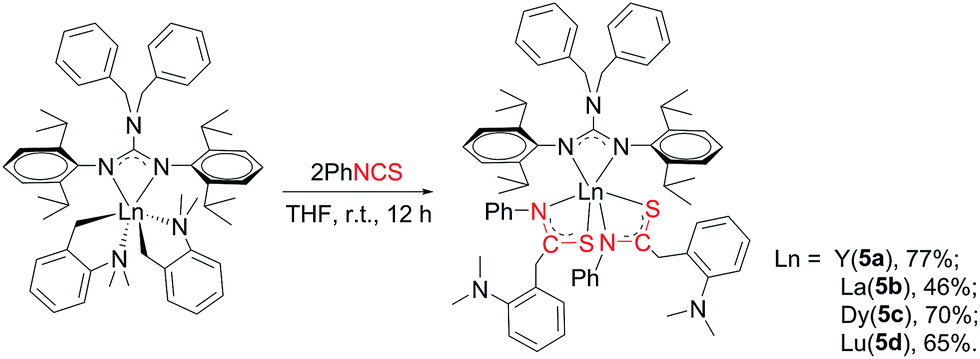
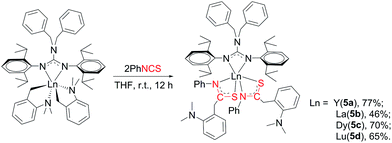
2.5 Reaction of [(PhCH2)2NC(NC6H4iPr2-2,6)2]RE(CH2C6H4NMe2-o)2 (RE = Y (2a), La (2b), Dy (2c), Lu (2d)) with PhNCS
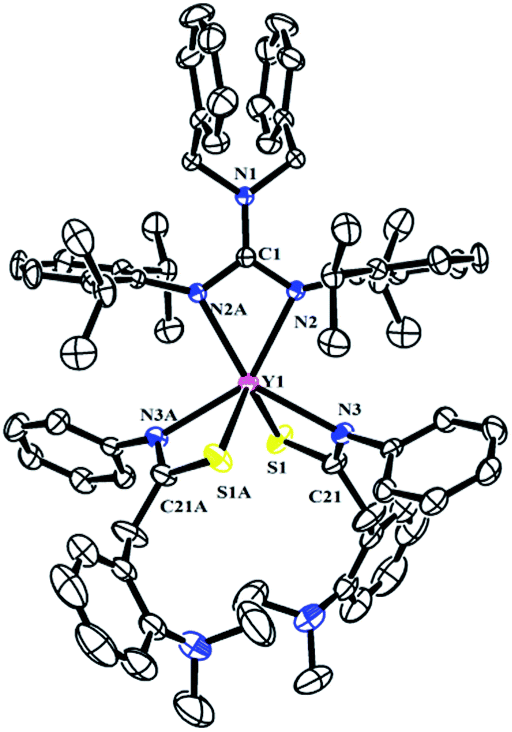
The reactivity of complexes 2 towards phenyl isothiocynate was also studied. These reactions provided insertion products [(PhCH2)2NC(NC6H4iPr2-2,6)2]RE{SC(CH2C6H4NMe2-o)NPh}2 (THF)n (n = 1, RE = La (5b); n = 0, RE = Y (5a), Dy (5c), Lu (5d)) in 46–77% yields (Scheme 5). Compared to the guanidinate dialkyl complexes 2, the resonances of methylene protons in aminobenzyl groups of these insertion products shift from upfield to downfield δ = 4.14 for 5a, 4.17 for 5b and 4.15 for 5d ppm respectively. In their 13C NMR spectra, the corresponding signals were observed at δ = 42.4(5a), 43.2(5b) and 42.6(5d) ppm as singlets. Complex 5b is of high solubility even in hexane while 5a, 5c and 5d are sparingly soluble in hexane. Noticeably, the larger lanthanum bears one coordinated THF molecule, while complexes 5a, 5c and 5d are all solvent free mononuclear complexes. As shown in Fig. 5, the yttrium ion of 5a is surrounded by four nitrogen atoms, two from the guanidinate ligand, two from the NCS fragments and two sulfur atoms from the NCS moieties. The Y–S bond length of 2.7154(9) Å is identical with the corresponding distances in 1,4-C6H4[C(NR)2Y{SC(CH2C6H4NMe2-o)NPh}2]2 (ref. 7a) (2.71 Å) and {(CH3C5H4)2Y[η2-SC(NPh2)NPh]}2 (2.7847(8) Å).12 The S1–C21 and N3–C21 distances, 1.735(3) and 1.287(4) Å, are between the corresponding single and double bond lengths, respectively, indicating that the negative charge is delocalized over the N–C–S moiety.13
 |
| | Scheme 5 | |
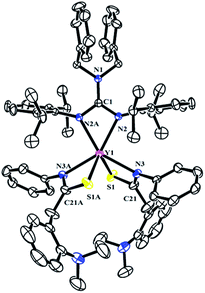 |
| | Fig. 5 Molecular structure of complex 5a with thermal ellipsoids at 30% probability. All of the hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Y(1)–N(2/2A) 2.329(2), Y(1)–N(3/3A) 2.405(3), Y(1)–S(1/1A) 2.7154(9), N(1)–C(1) 1.359(5), N(1)–C(2) 1.468(3), N(2)–C(1) 1.351(3), S(1)–C(21) 1.735(3), N(3)–C(21) 1.287(4); N(2)–Y(1)–N(2A) 56.81(11), N(2)–Y(1)–S(1A) 128.56(6), N(3)–Y(1)–S(1) 60.70(6), N(3)–C(21)–S(1) 118.0(2), N(3A)–Y(1)–N(3) 120.99(12), C(21)–N(3)–Y(1) 99.6(2). | |
2.6 Polymerization of isoprene
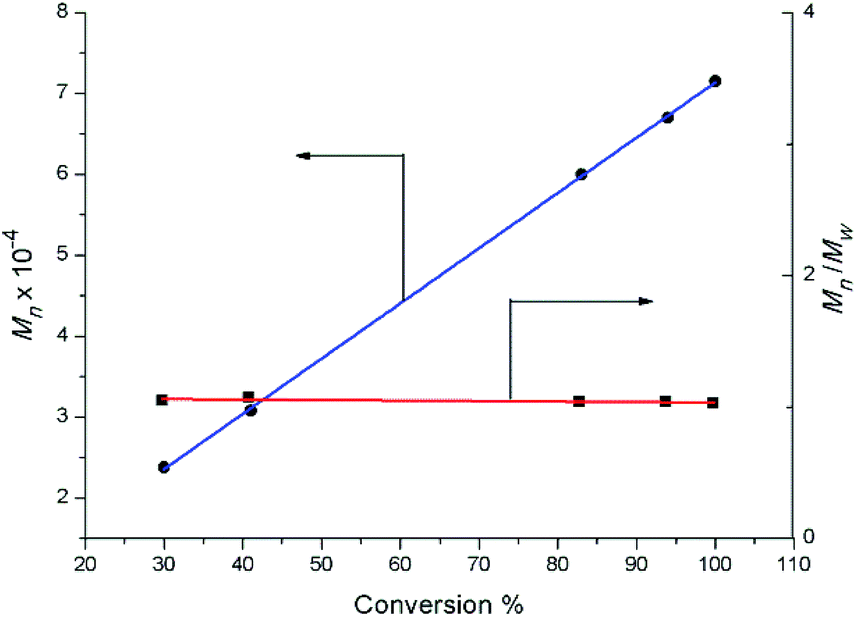
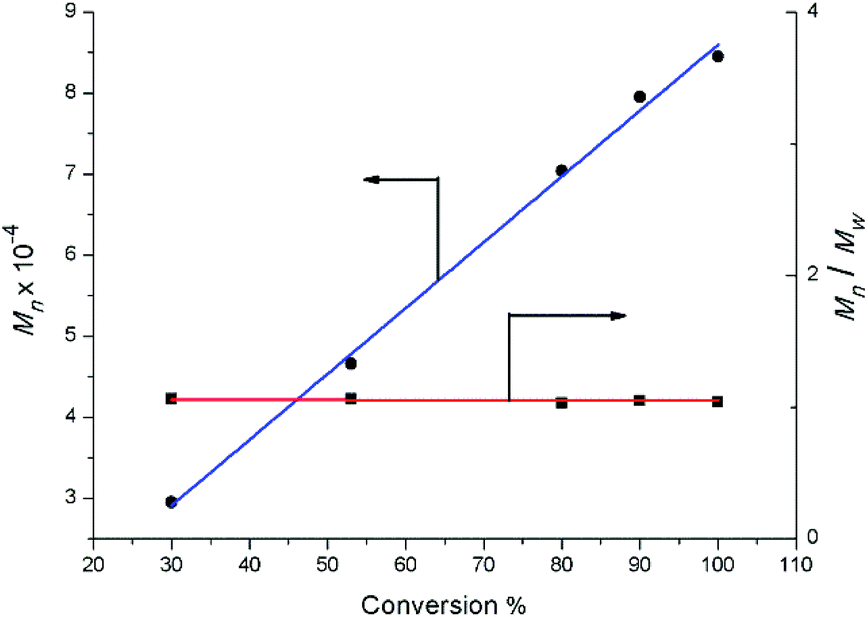
Complexes 2 were also evaluated as precatalysts for the polymerization of isoprene, and the results are presented in Table 1. Complexes 2 were inert towards the polymerization of isoprene. The binary system comprised of 2a (2c or 2d)/[Ph3C][B(C6F5)4] (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) displayed a high catalytic activity and predominant 3,4-regioselectivity for isoprene polymerization at room temperature (entries 1 to 4). But significant catalytic activity drop was observed for the lanthanum binary system (entry 2), probably due to the lower Lewis acidity compared to Y, Dy and Lu ions. Molecular weight distributions of all the polymers obtained from these binary systems are very narrow and unimodal (1.03–1.05) (entries 1 to 8). Further kinetics study on yttrium binary system was carried out and displayed in Fig. 6. It is noteworthy that the number-average molecular weight (Mn) of the yielded polymer samples was linearly relative to the conversion. In the meanwhile, the molecular weight distribution (Mw/Mn) was almost constant (1.03 to 1.07). The kinetics study indicated that the binary system 2a/[Ph3C][B(C6F5)4] demonstrated characteristics of living polymerization. To the best of our knowledge, examples of isoprene living polymerization with high 3,4-regioselectivity remain relatively rare.14 Moreover, the reaction temperature also influenced the polymerization performance. When the polymerization was carried out at 0 °C (entry 9), the yielded polymer showed the higher proportion of 3,4 units (95%). At −20 °C (entries 10–14), although the activity apparently dropped, it took 24 hours for 2a/[Ph3C][B(C6F5)4] system to get 100% conversion, the higher 3,4-regioselectivity (99%) was achieved and the yttrium binary system still demonstrates characteristics of living polymerization (Fig. 7). Interestingly, in contrast to the binary system comprised of 2a/[Ph3C][B(C6F5)4], it was found that the ternary system 2a/[Ph3C][B(C6F5)4]/AlMe3 (1:1:5) showed a high cis-1,4-regioselectivity (entry 15).15 However, the introduction of AliBu3 to the 2a/[Ph3C][B(C6F5)4] system led to the formation of polymer blend (entry 16).
:1) displayed a high catalytic activity and predominant 3,4-regioselectivity for isoprene polymerization at room temperature (entries 1 to 4). But significant catalytic activity drop was observed for the lanthanum binary system (entry 2), probably due to the lower Lewis acidity compared to Y, Dy and Lu ions. Molecular weight distributions of all the polymers obtained from these binary systems are very narrow and unimodal (1.03–1.05) (entries 1 to 8). Further kinetics study on yttrium binary system was carried out and displayed in Fig. 6. It is noteworthy that the number-average molecular weight (Mn) of the yielded polymer samples was linearly relative to the conversion. In the meanwhile, the molecular weight distribution (Mw/Mn) was almost constant (1.03 to 1.07). The kinetics study indicated that the binary system 2a/[Ph3C][B(C6F5)4] demonstrated characteristics of living polymerization. To the best of our knowledge, examples of isoprene living polymerization with high 3,4-regioselectivity remain relatively rare.14 Moreover, the reaction temperature also influenced the polymerization performance. When the polymerization was carried out at 0 °C (entry 9), the yielded polymer showed the higher proportion of 3,4 units (95%). At −20 °C (entries 10–14), although the activity apparently dropped, it took 24 hours for 2a/[Ph3C][B(C6F5)4] system to get 100% conversion, the higher 3,4-regioselectivity (99%) was achieved and the yttrium binary system still demonstrates characteristics of living polymerization (Fig. 7). Interestingly, in contrast to the binary system comprised of 2a/[Ph3C][B(C6F5)4], it was found that the ternary system 2a/[Ph3C][B(C6F5)4]/AlMe3 (1:1:5) showed a high cis-1,4-regioselectivity (entry 15).15 However, the introduction of AliBu3 to the 2a/[Ph3C][B(C6F5)4] system led to the formation of polymer blend (entry 16).
Table 1 Polymerization of isoprene catalyzed by complexes 2a
| Entry |
Precat. |
Al reagent ([Al]o/[RE]o) |
t [min] |
Tp [°C] |
Conversion [%] |
Microstructuresb [%] |
Mn (×104)c |
Mw/Mnc |
| 3,4 |
cis-1,4 |
trans-1,4 |
| Conditions: 2 20 μmol, [Ph3C][B(C6F5)4] 20 μmol, [IP]0/[RE]0 750, chlorobenzene 10 mL. Determined by 1H NMR and 13C NMR spectroscopy in CDCl3. Determined by GPC in THF at 40 °C against polystyrene standard. |
| 1 |
2a |
— |
10 |
25 |
100 |
91 |
2.3 |
6.7 |
7.15 |
1.03 |
| 2 |
2b |
— |
300 |
25 |
100 |
70 |
11.8 |
18.2 |
7.50 |
1.04 |
| 3 |
2c |
— |
12 |
25 |
100 |
83 |
3.9 |
13.1 |
9.04 |
1.03 |
| 4 |
2d |
— |
35 |
25 |
100 |
80 |
2.3 |
17.2 |
7.13 |
1.05 |
| 5 |
2a |
— |
8 |
25 |
94 |
91 |
2.3 |
6.7 |
6.70 |
1.04 |
| 6 |
2a |
— |
5 |
25 |
83 |
91 |
2.3 |
6.7 |
6.00 |
1.04 |
| 7 |
2a |
— |
3 |
25 |
41 |
91 |
2.3 |
6.7 |
3.08 |
1.07 |
| 8 |
2a |
— |
2 |
25 |
30 |
91 |
2.3 |
6.7 |
2.38 |
1.05 |
| 9 |
2a |
— |
180 |
0 |
100 |
95 |
0.6 |
4.4 |
7.10 |
1.03 |
| 10 |
2a |
— |
1440 |
−20 |
100 |
99 |
0.6 |
0.3 |
8.43 |
1.06 |
| 11 |
2a |
— |
1080 |
−20 |
90 |
99 |
0.6 |
0.3 |
7.95 |
1.05 |
| 12 |
2a |
— |
720 |
−20 |
80 |
99 |
0.6 |
0.3 |
7.04 |
1.03 |
| 13 |
2a |
— |
360 |
−20 |
53 |
99 |
0.6 |
0.3 |
4.66 |
1.06 |
| 14 |
2a |
— |
180 |
−20 |
30 |
99 |
0.6 |
0.3 |
2.95 |
1.06 |
| 15 |
2a |
AlMe3(5) |
40 |
25 |
100 |
7 |
87.5 |
5.5 |
17.90 |
1.93 |
| 16 |
2a |
AliBu3(5) |
40 |
25 |
100 |
53 |
42 |
5 |
9.9/3.36 |
3.1/1.16 |
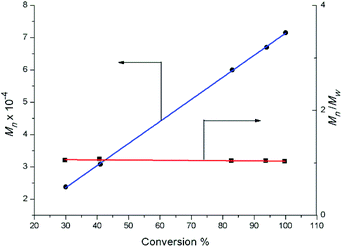 |
| | Fig. 6 Polymerization of isoprene with 2a/[Ph3C][B(C6F5)4]: molecular weight vs. conversion ([Y]0 = 2.0 μmol mL−1, [IP]0/[Y]0 = 750, chlorobenzene, 25 °C). | |
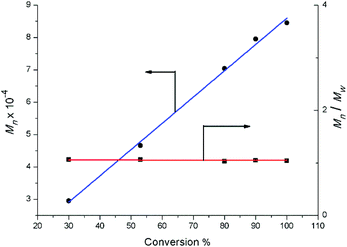 |
| | Fig. 7 Polymerization of isoprene with 2a/[Ph3C][B(C6F5)4]: molecular weight vs. conversion ([Y]0 = 2.0 μmol mL−1, [IP]0/[Y]0 = 750, chlorobenzene, −20 °C). | |
3. Conclusions
In summary, a series of neutral mono(guanidinate) rare-earth bis(alkyl) complexes have been synthesized via the protonolysis reaction of homoleptic rare-earth alkyl complexes with one equimolar amount of a new bulky guanidine. The reactions of bis(aminobenzyl) complexes with CO2, S8 and PhNCS provide some new options for effective synthetic routes for guanidinate lanthanide derivatives. Moreover, upon activation of an organoborate, such as [Ph3C][B(C6F5)4], complexes 2 show excellent activity and predominant 3,4-selectivity towards isoprene polymerization in a living fashion, whereas the ternary system 2a/[Ph3C][B(C6F5)4]/AlMe3 (1:1:5) showed a high cis-1,4-regioselectivity.
4. Experiment section
4.1 Materials and general procedures
All manipulations involving air- and moisture sensitive compounds were performed under an inert atmosphere of purified nitrogen with rigorous exclusion of air and moisture using standard Schlenk techniques and a nitrogen filled glove box operating at less than 1 ppm oxygen and 1 ppm moisture. Solvents (toluene, hexane, and THF) were distilled from sodium/benzophenone ketyl, and dried over fresh Na chips in the glove box. Bis(2,6-diisopropylphenyl)carbodiimide was obtained from Tokyo Chemical Industry Co., Ltd and used without purification. CH3C6H4NMe2-o was purchased from Acros and used without purification. nBuLi (2.5 mol L−1 in hexane), AlMe3 (1 mol L−1 in hexane) and AliBu3 (1 mol L−1 in hexane) were purchased from J&K and used without purification. Phenyl isothiocyanate were purchased from Dar Rui and distilled from CaH2 before being used. Highly pure CO2 gas (99.99%) was purchased from Pujiang Gas and dried by passing through activated 4 Å molecular sieves. Isoprene was obtained from Tokyo Chemical Industry Co., Ltd and purified by distillation over CaH2 under a nitrogen atmosphere. C6D6 and CDCl3 was obtained from Cambridge Isotope and dried by sodium chips. RE(CH2C6H4NMe2-o)3 (ref. 16) were prepared according to the literature procedures. 1H NMR and 13C NMR spectra were recorded on a JEOL ECA-400 NMR spectrometer (FT, 400 MHz for 1H; 100 MHz for 13C) in C6D6 at room temperature. GPC data were collected on a Waters 1515 Breeze GPC system using a polystyrene standard in THF.
4.2 X-ray crystallographic analysis
Suitable crystals were sealed in thin-wall glass capillaries under a microscope in the glove box. Data collections were performed on a Bruker SMART APEX diffractometer with a CCD area detector using graphite-monochromated MoKα radiation (λ = 0.71073 Å). The determination of crystal class and unit cell was carried out by the SMART program package. The raw frame data were processed using SAINT17 and SADABS18 to yield the reflection data file. The structure was solved by using SHELXTL program.19 Refinement was performed on F2 anisotropically by the full-matrix least-squares method for all the non-hydrogen atoms. The analytical scattering factors for neutral atoms were used throughout the analysis. Except for the hydrogen atoms on bridging-carbons, hydrogen atoms were placed at the calculated positions and included in the structure calculation without further refinement of the parameters. The hydrogen atoms on bridging carbons were located by difference Fourier syntheses and their coordinates and isotropic parameters were refined. The disordered toluene and THF molecules within the crystal lattice are not crystallographically well defined and are squeezed by the PLATON program. Details of this SQUEEZE are given in the cif files. Residual electron densities were of no chemical significance. Crystal data, data collection, and processing parameters for complexes 2b, 3a, 4a and 5a are summarized in Table 2. CCDC – 1542242 (1), 893571 (2d), 1541968 (3a), 1541967 (3b), 1541964 (3c), 1541970 (3d), 1541962 (4a), 1541966 (4c), 1541963 (4d), 1541969 (5a), 1541965 (5c), 1541961 (5d) contain the supplementary crystallographic data for this paper.†
Table 2 Crystallographic data for complexes 2d, 3a, 4a, 5a
| |
2d |
3a |
4a |
5a |
| Formula |
C57H72LuN5 |
C59H72N5O4Y |
C114H144N10S4Y2 |
C71H82N7S2Y |
| Formula weight |
1002.17 |
1004.13 |
1960.44 |
1186.47 |
| Temperature (K) |
293(2) |
296(2) |
273(2) |
296(2) |
| Wavelength (Å) |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| Crystal system |
Rhombohedral |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
C2/c |
| a (Å) |
24.986(7) |
13.041(5) |
15.488(2) |
13.050(2) |
| b (Å) |
24.986(7) |
16.053(6) |
23.909(3) |
26.302(5) |
| c (Å) |
47.359(9) |
16.979(11) |
31.418(4) |
19.537(4) |
| α (deg) |
90 |
103.789(10) |
90 |
90 |
| β (deg) |
90 |
104.711(10) |
98.781(2) |
104.170(3) |
| γ (deg) |
120 |
112.479(7) |
90 |
90 |
| V (Å3) |
13025(9) |
2945(3) |
11497(3) |
6502(2) |
| Z |
18 |
2 |
4 |
4 |
| μ (mm−1) |
1.771 |
1.036 |
1.125 |
1.007 |
| F(000) |
9360 |
1064 |
4160 |
2512 |
| θ range (°) |
1.63 to 25.50 |
1.48 to 25.05 |
1.075 to 25.050 |
1.79 to 25.04 |
| h, k, l range |
−24 ≤ h ≤ 30, |
−15 ≤ h ≤ 15 |
−18 ≤ h ≤ 18 |
−15 ≤ h ≤ 14 |
| |
−29 ≤ k ≤ 30 |
−19 ≤ k ≤ 18 |
−28 ≤ k ≤ 24 |
−29 ≤ k ≤ 31 |
| |
−57 ≤ l ≤ 56 |
−14 ≤ l ≤ 20 |
−28 ≤ l ≤ 37 |
−23 ≤ l ≤ 23 |
| Reflections collected |
27469 |
16741 |
56587 |
19415 |
| Reflections unique |
5296[R(int) = 0.0484] |
10159[R(int) = 0.0594] |
20283[R(int) = 0.0538] |
5746[R(int) = 0.0538] |
| Goodness-of-fit on F2 |
1.002 |
1.044 |
1.042 |
1.032 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0600, wR2 = 0.1539 |
R1 = 0.0819, wR2 = 0.2077 |
R1 = 0.0491, wR2 = 0.1116 |
R1 = 0.0440, wR2 = 0.1154 |
| R indices (all data) |
R1 = 0.1189, wR2 = 0.1834 |
R1 = 0.1219, wR2 = 0.2282 |
R1 = 0.0915, wR2 = 0.1212 |
R1 = 0.0702, wR2 = 0.1348 |
4.3 Synthesis of (PhCH2)2N[C(NHR)(NR)] (R = 2,6-iPr2-C6H3) (1)
A nBuLi solution (10.0 mmol, 4 mL, 2.5 M in hexane) was added slowly to a stirred solution of (PhCH2)2NH (1.97 g, 10.0 mmol) in THF (30 mL) at room temperature, then stirred for 2 h. Subsequently, the solution of the in situ (PhCH2)2NLi was added to a stirred THF solution of bis(2,6-diisopropylphenyl)carbodiimide (3.63 g, 10.0 mmol) at room temperature, the mixture was stirred for 4 h and added slowly the distilled water to afford a clear orange solution. All volatiles were removed under vacuum. The residue was washed with water, and the product was extracted with hexane (3 × 25 mL), all volatiles were removed under vacuum, and the yellowish powder was recrystallized in hexane at −35 °C to give colourless crystals (4.53 g, 81%). 1H NMR (400 MHz, CDCl3, 25 °C): 7.25–6.97 (m, 16H, Ar), 5.17 (s, 1H, –NH–), 4.29 (s, 4H, –CH2Ph), 3.31–3.20 (m, 4H, –CHMe2), 1.34 (d, J = 8 Hz, 6H, –CHMe2), 1.25 (d, J = 8 Hz, 6H, –CHMe2), 1.03 (d, J = 8 Hz, 6H, –CHMe2), 0.91 (d, J = 8 Hz, 6H, –CHMe2), 13C NMR (100 MHz, CDCl3, 25 °C): δ = 149.8 (NCN), 146.0 (Ar), 144.7 (Ar), 140.2 (Ar), 138.6 (Ar), 128.7 (Ar), 128.2 (Ar), 127.3 (Ar), 123.0 (Ar), 122.5 (Ar), 51.0 (–CH2Ph), 29.0 (–CHMe2), 28.4 (–CHMe2), 25.6 (–CHMe2), 24.6 (–CHMe2), 22.6 (–CHMe2), 21.9 (–CHMe2); 1H NMR (400 MHz, C6D6, 25 °C): 7.28 (s, 1H, Ar), 7.26 (s, 1H, Ar), 7.14–7.09 (m, 9H, Ar), 7.06–7.03 (m, 3H, Ar), 6.95 (s, 1H, Ar), 6,94 (s, 1H, Ar), 5.40 (s, 1H, –NH–), 4.43 (s, 4H, –CH2Ph), 3.50 (m, 2H, –CHMe2), 3.35 (m, 2H, –CHMe2), 1.42 (d, J = 4 Hz, 6H, –CHMe2), 1.38 (d, J = 8 Hz, 6H, –CHMe2), 1.03 (d, J = 4 Hz, 6H, –CHMe2), 0.83 (d, J = 4 Hz, 6H, –CHMe2), 13C NMR (100 MHz, C6D6, 25 °C): δ = 150.1 (NCN), 146.1 (Ar), 145.1 (Ar), 140.3 (Ar), 138.9 (Ar), 135.5 (Ar), 128.8 (Ar), 128.5 (Ar), 127.2 (Ar), 124.1 (Ar), 123.5 (Ar), 123.4 (Ar), 51.5 (–CH2Ph), 29.3 (–CHMe2), 28.6 (–CHMe2), 25.6 (–CHMe2), 24.9 (–CHMe2), 22.7 (–CHMe2), 22.0 (–CHMe2). Calcd for C39H49N3 (%): C, 83.67; H, 8.82; N, 7.51; found: C, 83.96; H, 8.77; N, 7.48.
4.4 Synthesis of [(PhCH2)2NC(NC6H4iPr2-2,6)2]Y(CH2C6H4NMe2-o)2 (2a)
A THF solution (10 mL) of Y(CH2C6H4NMe2-o)3 (0.25 g, 0.5 mmol) was added into a stirred solution (20 mL) of (PhCH2)2N[C(NHR)(NR)] (R = 2,6-iPr2-C6H3) (1) (0.28 g, 0.5 mmol) in THF. The reaction solution was left to stir for 3.5 days at 65 °C and all volatiles were removed under vacuum. The oily residue was washed with cold hexane and pale yellow powder was obtained by filtration. The pale yellow powder was recrystallized in toluene at −35 °C for 2 days to give white powder of 2a 0.30 g (65%). 1H NMR (400 MHz, C6D6, 25 °C): δ = 7.15–7.13 (m, 4H, Ar), 7.02–6.86 (m, 12H, Ar), 6.68 (d, J = 8 Hz, 4H, Ar), 6.59 (d, J = 12 Hz, 2H, Ar), 6.95–6.87 (m, 10H, Ar), 6.75–6.73 (m, 4H, Ar), 6.68 (d, J = 8 Hz, 4H, Ar), 6.52 (m, 2H, Ar), 4.28 (br s, 4H, –CH2Ph), 4.12 (m, 4H, –CHMe2), 2.19 (s, 12H, –NMe2), 1.81 (s, 4H, –CH2C6H4NMe2-o), 1.50 (d, J = 6 Hz, 12H, –CHMe2), 1.45 (d, J = 4 Hz, 12H, –CHMe2), 13C NMR (100 MHz, C6D6, 25 °C): δ = 161.3 (NCN), 145.4 (d, J = 26 Hz, Ar), 144.2 (s, Ar), 141.8 (s, Ar), 136.0 (s, Ar), 129.9 (s, Ar), 127.4 (s, Ar), 126.7 (s, Ar), 124.4 (s, Ar), 123.8 (s, Ar), 120.8 (s, Ar), 118.2 (s, Ar), 53.2 (s, –CH2Ph), 48.0 (d, J = 20 Hz, –CH2C6H4NMe2-o), 46.6 (br s, –NMe2), 29.1 (s, –CHMe2), 24.7 (s, –CHMe2), 24.2 (br s, –CHMe2). Calcd for C57H72N5Y (%): C, 74.73; H, 7.92; N, 7.64; found: C, 75.25; H, 7.76; N, 7.20.
4.5 Synthesis of [(PhCH2)2NC(NC6H4iPr2-2,6)2]La(CH2C6H4NMe2-o)2 (2b)
A THF solution (10 mL) of La(CH2C6H4NMe2-o)3 (0.27 g, 0.5 mmol) was added into a stirred THF solution (20 mL) of (PhCH2)2N[C(NHR)(NR)] (R = 2,6-iPr2-C6H3) (1) (0.280 g, 0.5 mmol). The reaction solution was left to stir for 12 hours at 50 °C and all volatiles were removed under vacuum. The oily residue was washed with cold hexane and yellow powder was obtained by filtration. The powder was recrystallized in toluene at −35 °C for 3 days to give yellow powder of 2b 0.21 g (43%). 1H NMR (400 MHz, C6D6, 25 °C): δ = 7.14 (s, 2H, Ar), 7.12 (s, 2H, Ar), 6.99 (s, 1H, Ar), 6.97 (s, 1H, Ar), 6.95–6.87 (m, 10H, Ar), 6.75–6.73 (m, 4H, Ar), 6.68 (s, 1H, Ar), 6.66 (s, 1H, Ar), 6.61–6.58 (m, 2H, Ar), 4.18 (s, 4H, –CH2Ph), 3.93 (m, 4H, –CHMe2), 2.06 (s, 12H, –NMe2), 1.84 (s, 4H, –CH2C6H4NMe2-o), 1.42 (d, J = 8 Hz, 12H, –CHMe2), 1.35 (d, J = 8 Hz, 12H, –CHMe2), 13C NMR (100 MHz, C6D6, 25 °C): δ = 158.4 (NCN), 145.1 (s, Ar), 143.8 (s, Ar), 141.1 (s, Ar), 141.0 (s, Ar), 136.4 (s, Ar), 130.0 (s, Ar), 129.3 (s, Ar), 128.6 (s, Ar), 128.0 (s, Ar), 127.6 (s, Ar), 127.4 (s, Ar), 124.2 (s, Ar), 122.9 (s, Ar), 119.7 (s, Ar), 119.3 (s, Ar), 61.6 (s, –CH2C6H4NMe2-o), 52.4 (s, –CH2Ph), 44.8 (s, –NMe2), 29.1 (s, –CHMe2), 24.6 (s, –CHMe2), 24.1 (s, –CHMe2). Calcd for C57H72N5La (%): C, 70.86; H, 7.51; N, 7.25; found: C, 70.71; H, 7.43; N, 6.89.
4.6 Synthesis of [(PhCH2)2NC(NC6H4iPr2-2,6)2]Dy(CH2C6H4NMe2-o)2 (2c)
A THF solution (10 mL) of Dy(CH2C6H4NMe2-o)3 (0.28 g, 0.5 mmol) was added into a stirred solution (20 mL) of (PhCH2)2N[C(NHR)(NR)] (R = 2,6-iPr2-C6H3) (1) (0.28 g, 0.5 mmol) in THF. The reaction solution was left to stir for 3 days at 65 °C, and all volatiles were removed under vacuum. The oily residue was washed with cold hexane and pale yellow powder was obtained by filtration. The powder was recrystallized in toluene at −35 °C for 2 days to give pale yellow powder of 2c 0.29 g (58%). Calcd for C57H72N5Dy (%): C, 69.17; H, 7.33; N, 7.08; found: C, 69.25; H, 7.83; N, 6.71.
4.7 Synthesis of [(PhCH2)2NC(NC6H4iPr2-2,6)2]Lu(CH2C6H4NMe2-o)2 (2d)
A THF solution (10 mL) of Lu(CH2C6H4NMe2-o)3 (0.29 g, 0.5 mmol) was added into a stirred THF solution (20 mL) of (PhCH2)2N[C(NHR)(NR)] (R = 2,6-iPr2-C6H3) (1) (0.28 g, 0.5 mmol). The reaction solution was left to stir for 3.5 days at 65 °C and all volatiles were removed under vacuum. The oily residue was washed with cold hexane and white powder was obtained by filtration. The white powder was recrystallized in toluene at −35 °C for 2 days to give a colourless crystalline complex 2d 0.23 g (45%). 1H NMR (400 MHz, C6D6, 25 °C): δ = 7.14–7.12 (m, 4H, Ar), 7.00–6.96 (m, 8H, Ar), 6.90–6.84 (m, 8H, Ar), 6.97 (s, 1H, Ar), 6.95–6.87 (m, 10H, Ar), 6.75–6.73 (m, 4H, Ar), 6.68 (d, J = 8 Hz, 4H, Ar), 6.52 (m, 4H, Ar), 4.31 (br s, 4H, –CH2Ph), 4.20 (m, 4H,–CHMe2), 2.21 (s, 12H, –NMe2), 1.80 (s, 4H, –CH2C6H4NMe2-o), 1.53 (d, J = 12 Hz, 12H, –CHMe2), 1.47 (d, J = 8 Hz, 12H, –CHMe2), 13C NMR (100 MHz, C6D6, 25 °C): δ = 161.8 (NCN), 145.7 (s, Ar), 145.5 (s, Ar), 142.3 (s, Ar), 136.0 (s, Ar), 129.9 (s, Ar), 127.4 (s, Ar), 126.3 (s, Ar), 124.5 (s, Ar), 124.2 (s, Ar), 121.2 (s, Ar), 117.8 (s, Ar), 53.4 (s, –CH2Ph), 53.3 (s, –CH2C6H4NMe2-o), 47.2 (s, –NMe2), 28.9 (s, –CHMe2), 25.1 (s, –CHMe2), 24.4 (s, –CHMe2). Calcd for C57H72LuN5 (%): C, 68.31; H, 7.24; N, 6.99; found: C, 69.01; H, 7.19; N, 6.72.
4.8 Synthesis of {[(PhCH2)2NC(NC6H4iPr2-2,6)2]Y(μ-η2:η1-O2CCH2C6H4NMe2-o)(μ-η1:η1-O2CCH2C6H4NMe2-o)}2 (3a)
A THF (10 mL) solution of complex 2a (0.46 g, 0.5 mmol) was placed in a tube with a Teflon stopcock and degassed by a freeze pump thaw cycle. One atmospheres of CO2 was introduced into the tube and the solution was concentrated to saturation after 10 minutes. Colourless crystals of 3a (0.45 g, 90%) were harvested after the solution stood at ambient temperature for 3 days. 1H NMR (400 MHz, C6D6, 25 °C): δ = 7.18–7.12 (m, 8H, Ar), 6.98 (br s, 14H, Ar), 6.94–6.87 (m, 18H, Ar), 6.71–6.69 (m, 8H, Ar), 4.14 (s, 8H, –CH2Ph), 3.88 (m, 8H,–CHMe2), 2.47 (br s, 32H, –OOCCH2C6H4NMe2-o), 1.28 (t, J = 8 Hz, 48H, –CHMe2), 13C NMR (100 MHz, C6D6, 25 °C): δ = 186.2 (s, OCO), 165.1 (s, NCN), 152.7 (s, Ar), 143.9 (s, Ar), 142.5 (s, Ar), 136.6 (s, Ar), 131.1 (s, Ar), 130.6 (s, Ar), 129.0 (s, Ar), 127.0 (s, Ar), 126.6 (s, Ar), 123.6 (s, Ar), 123.4 (s, Ar), 123.3 (s, Ar), 123.1 (s, Ar), 122.9 (s, Ar), 119.3 (s, Ar), 52.2 (s, –CH2Ph), 45.1 (br s, –OOCCH2C6H4NMe2-o), 27.9 (s, –CHMe2), 25.6 (s, –CHMe2), 23.9 (s, –CHMe2). Calcd for C118H144N10O8Y2 (%): C, 70.57; H, 7.23; N, 6.97; found: C, 70.52; H, 7.33; N, 6.74.
4.9 Synthesis of {[(PhCH2)2NC(NC6H4iPr2-2,6)2]La(μ-η2:η1-O2CCH2C6H4NMe2-o)(μ-η1:η1-O2CCH2C6H4NMe2-o)}2 (3b)
Complex 3b was obtained as a colourless crystalline product (0.38 g, 73%), similarly to the preparation of 3a described above. 1H NMR (400 MHz, C6D6, 25 °C): δ = 7.35–7.29 (m, 4H, Ar), 7.22–7.18 (m, 5H, Ar), 7.04–7.00 (m, 12H, Ar), 6.91–6.88 (m, 10H, Ar), 6.86–6.82 (m, 4H, Ar), 6.68 (d, J = 4 Hz, 8H, Ar), 4.08 (s, 8H, –CH2Ph), 3.80 (m, 8H, –CHMe2), 3.47 (br s, 8H, –OOCCH2C6H4NMe2-o), 2.54 (s, 24H, –OOCCH2C6H4NMe2-o), 1.29 (d, J = 4 Hz, 24H, –CHMe2), 1.25 (d, J = 4 Hz, 24H, –CHMe2), 13C NMR (100 MHz, C6D6, 25 °C): δ = 183.6 (s, OCO), 163.4 (s, NCN), 153.3 (s, Ar), 143.8 (s, Ar), 142.3 (s, Ar), 131.5 (s, Ar), 129.3 (s, Ar), 128.8 (s, Ar), 127.5 (s, Ar), 127.0 (s, Ar), 123.6 (s, Ar), 123.6 (s, Ar), 123.4 (s, Ar), 119.8 (s, Ar), 52.0 (s, –CH2Ph), 45.6 (s, –OOCCH2C6H4NMe2-o), 38.6 (s, –OOCCH2C6H4NMe2-o), 28.4 (s, –CHMe2), 26.6 (s, –CHMe2), 23.7 (s, –CHMe2). Calcd for C118H144N10O8La2 (%): C, 67.22; H, 6.88; N, 6.64; found: C, 66.70; H, 7.45; N, 5.97.
4.10 Synthesis of {[(PhCH2)2NC(NC6H4iPr2-2,6)2]Dy(μ-η2:η1-O2CCH2C6H4NMe2-o)(μ-η1:η1-O2CCH2C6H4NMe2-o)}2 (3c)
Complex 3c was obtained as a colourless crystalline product (0.47 g, 88%), similarly to the preparation of 3a described above. Calcd for C118H144N10O8Dy2 (%): C, 65.75; H, 6.73; N, 6.50; found: C, 66.16; H, 6.92; N, 6.56.
4.11 Synthesis of {[(PhCH2)2NC(NC6H4iPr2-2,6)2]Lu(μ-η2:η1-O2CCH2C6H4NMe2-o)(μ-η1:η1-O2CCH2C6H4NMe2-o)}2 (3d)
Complex 3d was obtained as a colourless crystalline product (0.50 g, 92%), similarly to the preparation of 3a described above. 1H NMR (400 MHz, C6D6, 25 °C): δ = 7.36–7.29 (m, 10H, Ar), 7.01–6.87 (m, 30H, Ar), 6.70 (d, J = 4 Hz, 8H, Ar), 4.14 (s, 8H, –CH2Ph), 3.91 (m, 8H, –CHMe2), 3.50 (br s, 8H, –OOCCH2C6H4NMe2-o), 2.62–2.40 (m, 24H, –OOCCH2C6H4NMe2-o), 1.31–1.27 (m, 48H, –CHMe2), 13C NMR (100 MHz, C6D6, 25 °C): δ = 183.7 (s, OCO), 164.7 (s, NCN), 153.0 (s, Ar), 144.2 (s, Ar), 143.2 (s, Ar), 136.9 (s, Ar), 131.4 (br s, Ar), 127.3 (br s, Ar), 127.0 (s, Ar), 124.0 (s, Ar), 123.8 (s, Ar), 123.7 (s, Ar), 119.4 (br s, Ar), 52.6 (s, –CH2Ph), 45.4 (s, –OOCCH2C6H4NMe2-o), 38.0 (br s, –OOCCH2C6H4NMe2-o), 28.3 (s, –CHMe2), 26.0 (s, –CHMe2), 24.4 (s, –CHMe2). Calcd for C118H144N10O8Lu2 (%): C, 65.00; H, 6.66; N, 6.50; found: C, 65.68; H, 6.39; N, 6.05.
4.12 Synthesis of {[(PhCH2)2NC(NC6H4iPr2-2,6)2]Y[μ-S(CH2C6H4NMe2-o)]2}2 (4a)
To a stirred THF (20 mL) solution of complex 2a (0.46 g, 0.5 mmol) was added slowly a THF (10 mL) solution of S8 (0.032 g, 0.125 mmol). The reaction solution was left to stir for 3.5 days at 65 °C, and all volatiles were removed under vacuum. After being washed with cold hexane, the oily residue turned to white powder which then was collected by filtration. The white powder was recrystallized in toluene at room temperature for 2 days to give a colourless crystalline complex 4a 0.38 g (77%). 1H NMR (400 MHz, C6D6, 25 °C): δ = 7.76–7.73 (m, 3H, Ar), 7.15–7.15 (m, 11H, Ar), 7.01–6.95 (m, 12H, Ar), 6.88–6.81 (m, 15H, Ar), 6.51–6.49 (m, 7H, Ar), 4.28 (br s, 8H, –SCH2C6H4NMe2-o), 4.12 (s, 8H, –CH2Ph), 3.89 (m, 8H, –CHMe2), 2.58 (s, 24H, –NMe2), 1.43 (br s, 24H, –CHMe2), 1.27 (d, J = 4.0 Hz, 24H, –CHMe2), 13C NMR (100 MHz, C6D6, 25 °C): δ = 166.09 (s, NCN), 152.6 (s, Ar), 143.3 (s, Ar), 142.7 (s, Ar), 137.8 (s, Ar), 135.9 (s, Ar), 131.3 (s, Ar), 129.4 (s, Ar), 127.2 (s, Ar), 126.5 (s, Ar), 124.6 (s, Ar), 124.2 (s, Ar), 123.0 (s, Ar), 118.3 (s, Ar), 52.5 (s, –CH2Ph), 45.2 (s, –NMe2), 34.6 (s, –SCH2C6H4NMe2-o), 29.0 (s, –CHMe2), 26.2 (s, –CHMe2), 24.2 (s, –CHMe2). Calcd for C114H144N10S4Y2 (%): C, 69.84; H, 7.40; N, 7.14; found: C, 69.40; H, 7.44; N, 6.73.
4.13 Synthesis of {[(PhCH2)2NC(NC6H4iPr2-2,6)2]La[μ-S(CH2C6H4NMe2-o)]2}2 (4b)
To a stirred THF (20 mL) solution of complex 2b (0.48 g, 0.5 mmol) was added slowly a THF (10 mL) solution of S8 (0.032 g, 0.125 mmol). After it was stirred at room temperature for 12 h, all volatiles were removed under vacuum, and washed with cold hexane, the oily residue turn to white powder which then was collected by filtration. The white powder was recrystallized in toluene at −35 °C for 3 days to give a colourless crystalline complex 4b 0.31 g (61%). 1H NMR (400 MHz, C6D6, 25 °C): δ = 7.62 (d, J = 4.0 Hz, 3H, Ar), 7.17–7.14 (m, 7H, Ar), 7.10–7.05 (m, 12H, Ar), 6.98–6.92 (m, 8H, Ar), 6.89–6.86 (m, 11H, Ar), 6.61–6.60 (m, 7H, Ar), 4.52 (br s, 8H, –SCH2C6H4NMe2–o), 4.04 (s, 8H, –CH2Ph), 3.77 (m, 8H, –CHMe2), 2.61 (s, 24H, –NMe2), 1.48 (d, J = 4.0 Hz, 24H, –CHMe2), 1.22 (d, J = 4.0 Hz, 24H, –CHMe2), 13C NMR (100 MHz, C6D6, 25 °C): δ = 164.4 (s, NCN), 152.5 (s, Ar), 142.7 (s, Ar), 142.6 (s, Ar), 138.7 (s, Ar), 136.4 (s, Ar), 131.2 (s, Ar), 129.2 (s, Ar), 128.8 (s, Ar), 127.1 (s, Ar), 126.4 (s, Ar), 124.2 (s, Ar), 124.1 (s, Ar), 123.3 (s, Ar), 118.7 (s, Ar), 51.8 (s, –CH2Ph), 45.3 (s, –NMe2), 33.5 (s, –SCH2C6H4NMe2-o), 28.8 (s, –CHMe2), 27.6 (s, –CHMe2), 23.6 (s, –CHMe2). Calcd for C114H144N10S4La2 (%): C, 66.45; H, 7.04; N, 6.80; found: C, 66.07; H, 6.91; N, 6.60.
4.14 Synthesis of {[(PhCH2)2NC(NC6H4iPr2-2,6)2]Dy[μ-S(CH2C6H4NMe2-o)]2}2 (4c)
Complex 4c was obtained as a colourless crystalline product (0.38 g, 73%), similarly to the preparation of 4a described above. Calcd for C114H144N10S4Dy2 (%): C, 64.96; H, 6.89; N, 6.65; found: C, 64.84; H, 6.83; N, 6.73.
4.15 Synthesis of {[(PhCH2)2NC(NC6H4iPr2-2,6)2]Lu[μ-S(CH2C6H4NMe2-o)]2}2 (4d)
Complex 4d was obtained as a colourless crystalline product (0.36 g, 67%), similarly to the preparation of 4a described above. 1H NMR (400 MHz, C6D6, 25 °C): δ = 7.83–7.80 (m, 3H, Ar), 7.18–7.16 (m, 14H, Ar), 6.87–6.81 (m, 12H, Ar), 6.48 (d, J = 4.0 Hz, 6H, Ar), 4.46 (br s, 8H, –SCH2C6H4NMe2-o), 4.13 (s, 8H, –CH2Ph), 3.95 (m, 8H, –CHMe2), 2.57 (s, 24H, –NMe2), 1.41 (d, J = 8.0 Hz, 24H, –CHMe2), 1.30 (d, J = 4.0 Hz, 24H, –CHMe2), 13C NMR (100 MHz, C6D6, 25 °C): δ = 165.3 (s, NCN), 152.3 (s, Ar), 142.8 (s, Ar), 142.6 (s, Ar), 137.3 (s, Ar), 135.4 (s, Ar), 130.8 (s, Ar), 129.0 (s, Ar), 126.7 (s, Ar), 126.1 (s, Ar), 124.4 (s, Ar), 123.7 (s, Ar), 122.5 (s, Ar), 117.9 (s, Ar), 52.4 (s, –CH2Ph), 44.7 (s, –NMe2), 34.3 (s, –SCH2C6H4NMe2-o), 28.6 (s, –CHMe2), 25.6 (s, –CHMe2), 23.8 (s, –CHMe2). Calcd for C114H144N10S4Lu2 (%): C, 64.20; H, 6.81; N, 6.57; found: C, 64.58; H, 7.51; N, 6.18.
4.16 Synthesis of [(PhCH2)2NC(NC6H4iPr2-2,6)2]Y{SC(CH2C6H4NMe2-o)NPh}2 (5a)
A THF (10 mL) solution of PhNCS (0.108 mL, 1 mmol) was added slowly to a stirred THF (20 mL) solution of complex 2a (0.46 g, 0.5 mmol). The reaction solution was left to stir at room temperature for 12 h and all volatiles were removed under vacuum. After being washed with cold hexane, the oily residue turned to white powder which then was collected by filtration and dissolved in toluene. The solution was concentrated to saturation and stood at ambient temperature. Colourless crystals of 5a (0.46 g, 77%) were obtained after 2 days. 1H NMR (400 MHz, C6D6, 25 °C): δ = 7.74–7.71 (m, 2H, Ar), 7.19–7.14 (m, 4H, Ar), 7.04 (br s, 6H, Ar), 6.95–6.92 (m, 2H, Ar), 6.95–6.92 (m, 2H, Ar), 6.89–6.83 (m, 10H, Ar), 6.80–6.76 (m, 2H, Ar), 6.66–6.65 (m, 4H, Ar), 6.39 (d, J = 8.0 Hz, 4H, Ar), 4.20 (s, 4H, –CH2Ph), 4.14 (s, 4H, –CH2C6H4NMe2-o), 3.94 (m, 4H, –CHMe2), 2.37 (s, 12H, –NMe2), 1.27 (d, J = 8.0 Hz, 12H, –CHMe2), 1.23 (d, J = 4.0 Hz, 12H, –CHMe2), 13C NMR (100 MHz, C6D6, 25 °C): δ = 199.8 (s, NCS), 165.2 (s, NCN), 153.4 (s, Ar), 147.7 (s, Ar), 143.6 (s, Ar), 143.1 (s, Ar), 136.1 (s, Ar), 133.0 (s, Ar), 131.0 (s, Ar), 129.5 (s, Ar), 129.0 (s, Ar), 127.2 (s, Ar), 124.6 (s, Ar), 124.4 (s, Ar), 124.1 (s, Ar), 123.8 (s, Ar), 123.3 (s, Ar), 120.2 (s, Ar), 52.4 (s, –CH2Ph), 44.9 (s, –NMe2), 42.4 (s, –CH2C6H4NMe2-o), 28.7 (s, –CHMe2), 25.7 (s, –CHMe2), 24.2 (s, –CHMe2). Calcd for C71H82N7S2Y (%): C, 71.87; H, 6.97; N, 8.26; found: C, 70.97; H, 7.34; N, 8.08.
4.17 Synthesis of [(PhCH2)2NC(NC6H4iPr2-2,6)2]La{SC(CH2C6H4NMe2-o)NPh}2(THF) (5b)
A THF (10 mL) solution of PhNCS (0.108 mL, 1 mmol) was added slowly to a stirred THF (20 mL) solution of complex 2b (0.48 g, 0.5 mmol). The reaction solution was left to stir at room temperature for 12 h and all volatiles were removed under vacuum. After being washed with cold hexane, the oily residue turned to white powder which then was collected by filtration and dissolved in toluene. The solution was concentrated to saturation and layered with 2 mL hexane solvent. White participate 5b was obtained after the solution stood at −35 °C for 3 days. Yield (0.28 g, 46%). 1H NMR (400 MHz, C6D6, 25 °C): δ = 7.72–7.70 (m, 2H, Ar), 7.13–7.08 (m, 8H, Ar), 7.03–6.82 (m, 22H, Ar), 6.37 (d, J = 8 Hz, 4H, Ar), 4.17 (s, 4H, –CH2C6H4NMe2-o), 4.12 (s, 4H, –CH2Ph), 3.98 (m, 4H, –CHMe2), 3.54 (m, 4H, THF), 2.36 (s, 12H, –NMe2), 1.36 (d, J = 8 Hz, 12H, –CHMe2), 1.22 (d & m, J = 8 Hz, 24H, –CHMe2 & THF & Hex), 13C NMR (100 MHz, C6D6, 25 °C): δ = 198.9 (s, NCS), 163.1 (s, NCN), 153.2 (s, Ar), 148.9 (s, Ar), 142.6 (s, Ar), 137.2 (s, Ar), 131.3 (s, Ar), 129.5 (s, Ar), 128.9 (s, Ar), 128.8 (s, Ar), 128.3 (s, Ar), 127.5 (s, Ar), 127.2 (s, Ar), 124.4 (s, Ar), 123.5 (s, Ar), 123.2 (s, Ar), 119.8 (s, Ar), 69.1 (s, THF), 51.7 (s, –CH2Ph), 45.0 (s, –NMe2), 43.2 (s, –CH2C6H4NMe2-o), 28.5 (s, –CHMe2), 27.0 (s, –CHMe2), 25.3 (s, THF), 24.1 (s, –CHMe2). Calcd for C75H90N7S2OLa (%): C, 68.84; H, 6.93; N, 7.49; found: C, 68.70; H, 7.06; N, 7.73.
4.18 Synthesis of [(PhCH2)2NC(NC6H4iPr2-2,6)2]Dy{SC(CH2C6H4NMe2-o)NPh}2 (5c)
Complex 5c was obtained as a colourless crystalline product (0.44 g, 70%), similarly to the preparation of 5a described above. Calcd for C71H82N7S2Dy (%): C, 67.67; H, 6.56; N, 7.78; found C, 67.59; H, 6.87; N, 7.80.
4.19 Synthesis of [(PhCH2)2NC(NC6H4iPr2-2,6)2]Lu{SC(CH2C6H4NMe2-o)NPh}2 (5d)
Complex 5d was obtained as a colourless crystalline product (0.41 g, 65%), similarly to the preparation of 5a described above. 1H NMR (400 MHz, C6D6, 25 °C): δ = 7.79–7.76 (m, 2H, Ar), 7.20–7.14 (m, 5H, Ar), 7.05 (br s, 6H, Ar), 6.95–6.84 (m, 9H, Ar), 6.78–6.75 (m, 2H, Ar), 6.65 (br s, 4H, Ar), 6.38 (d, J = 8.0 Hz, 4H, Ar), 4.25 (br s, 4H, –CH2Ph), 4.15 (s, 4H, –CH2C6H4NMe2-o), 4.02 (m, 4H, –CHMe2), 2.36 (s, 12H, –NMe2), 1.29 (d, J = 8.0 Hz, 12H, –CHMe2), 1.23 (br s, 12H, –CHMe2), 13C NMR (100 MHz, C6D6, 25 °C): δ = 200.2 (s, NCS), 165.1 (s, NCN), 153.4 (s, Ar), 147.4 (s, Ar), 143.6 (s, Ar), 143.5 (s, Ar), 136.0 (s, Ar), 133.0 (s, Ar), 130.9 (s, Ar), 129.6 (s, Ar), 128.9 (s, Ar), 127.2 (s, Ar), 124.8 (s, Ar), 124.4 (s, Ar), 124.3 (s, Ar), 123.8 (s, Ar), 123.4 (s, Ar), 120.2 (s, Ar), 52.6 (s, –CH2Ph), 45.0 (s, –NMe2), 42.6 (s, –CH2C6H4NMe2-o), 28.6 (s, –CHMe2), 25.7 (s, –CHMe2), 24.6 (s, –CHMe2). Calcd for C71H82N7S2Lu (%): C, 67.01; H, 6.49; N, 7.70; found: C, 66.80; H, 6.81; N, 7.60.
4.20 Typical procedure for polymerization of isoprene
The procedures for isoprene polymerization were similar, thus take complex 2a as an example and corresponding polymerization procedure is given below. For 2a/[Ph3C][B(C6F5)4] binary system: in a glovebox, a magnetic stir bar was placed in a 100 mL flask, to which a dropping funnel was attached. Isoprene (1.022 g, 15 mmol), 2a (0.018 g, 0.020 mmol) and C6H5Cl (8 mL) were charged into the flask. A C6H5Cl solution (2 mL) of [Ph3C][B(C6F5)4] (0.0185 g, 0.020 mmol) was charged to the dropping funnel. The reaction apparatus was moved outside and placed in a water bath (25 °C). After 10 min, the C6H5Cl solution of [Ph3C][B(C6F5)4] was dropped into the mixture of 2a and isoprene under rapid stirring. After the mixture was stirred at 25 °C for 10 min, methanol was injected to terminate the polymerization. The reaction mixture was poured into a large quantity (200 mL) of methanol containing a small amount of hydrochloric acid and butylhydroxytoluene (BHT) as a stabilizing agent under stirring. The precipitated polymer was isolated by decantation, washed with methanol, and then dried under vacuum at 60 °C to a constant weight to afford 1.02 g of 3,4-rich polyisoprene (∼100% yield). For 2a/[Ph3C][B(C6F5)4]/AlR3 ternary systems: isoprene (1.022 g, 15 mmol), 2a (0.018 g, 0.020 mmol), AlR3 (100 μL, 1 mol L−1 in hexane) and C6H5Cl (8 mL) were charged into the flask and other operations are same as above-mentioned binary system.
Acknowledgements
This work was supported by the NSFC, The National Basic Research Program (973 Program, No. 2012CB821604).
Notes and references
- H. van der Heijden, C. J. Schaverien and A. G. Orpen, Organometallics, 1989, 8, 255 CrossRef CAS.
-
(a) M. Nishiura and Z. Hou, Nat. Chem., 2010, 2, 257 CrossRef CAS PubMed;
(b) M. Nishiura, F. Guo and Z. Hou, Acc. Chem. Res., 2015, 48, 2209 CrossRef CAS PubMed;
(c) A. A. Trifonov and D. M. Lyubov, Coord. Chem. Rev., 2017, 340, 10 CrossRef CAS.
- W. E. Piers and D. J. H. Emslie, Coord. Chem. Rev., 2002, 233(234), 131 CrossRef.
-
(a) G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428 CrossRef CAS;
(b) F. T. Edelmann, D. M. M. Freckmann and H. Schumann, Chem. Rev., 2002, 102, 1851 CrossRef CAS PubMed;
(c) M. C. Baier, M. A. Zuideveld and S. Mecking, Angew. Chem., Int. Ed., 2014, 53, 9722 CrossRef CAS PubMed.
-
(a) F. T. Edelmann, Chem. Soc. Rev., 2009, 38, 2253 RSC;
(b) A. A. Trifonov, Coord. Chem. Rev., 2010, 254, 1327 CrossRef CAS;
(c) F. T. Edelmann, Chem. Soc. Rev., 2012, 41, 7657 RSC;
(d) F. T. Edelmann, Adv. Organomet. Chem., 2013, 61, 55 CrossRef CAS.
-
(a) A. A. Trifonov, D. M. Lyubov, G. K. Fukin, E. V. Baranov and Y. A. Kurskii, Organometallics, 2006, 25, 3935 CrossRef CAS;
(b) D. M. Lyubov, G. K. Fukin and A. A. Trifonov, Inorg. Chem., 2007, 46, 11450 CrossRef CAS PubMed;
(c) S. Ge, A. Meetsma and B. Hessen, Organometallics, 2008, 27, 3131 CrossRef CAS.
-
(a) M. Li, J. Hong, Z. Chen, X. Zhou and L. Zhang, Dalton Trans., 2013, 42, 8288 RSC;
(b) J. Hong, L. Zhang, K. Wang, Z. Chen, L. Wu and X. Zhou, Organometallics, 2013, 32, 7312 CrossRef CAS.
- Y. J. Luo, X. L. Wang, J. Chen, C. C. Luo, Y. Zhang and Y. M. Yao, J. Organomet. Chem., 2009, 694, 1289 CrossRef CAS.
-
(a) W. J. Evans, C. A. Seibel, J. W. Ziller and R. J. Doedens, Organometallics, 1998, 17, 2103 CrossRef CAS;
(b) W. J. Evans, J. C. Brady and J. W. Ziller, J. Am. Chem. Soc., 2001, 123, 7711 CrossRef CAS PubMed;
(c) D. Cui, M. Nishiura and Z. Hou, Macromolecules, 2005, 38, 4089 CrossRef CAS;
(d) W. J. Evans, J. M. Perotti and J. W. Ziller, J. Am. Chem. Soc., 2005, 127, 3894 CrossRef CAS PubMed;
(e) D. Cui, M. Nishiura, O. Tardif and Z. Hou, Organometallics, 2008, 27, 2428 CrossRef CAS.
-
(a) Y. Li, C. Pi, J. Zhang, X. Zhou, Z. Chen and L. Weng, Organometallics, 2005, 24, 1982 CrossRef CAS;
(b) W. J. Evans, B. M. Schmiege, S. E. Lorenz, K. A. Miller, T. M. Champagne, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, J. Am. Chem. Soc., 2008, 130, 8555 CrossRef CAS PubMed;
(c) J. Zhang, L. Ma, Y. Han, F. Han, Z. Zhang, R. Cai, Z. Chen and X. Zhou, Dalton Trans., 2009, 3298 RSC;
(d) Z. Zhang, L. Zhang, Y. Li, L. Hong, Z. Chen and X. Zhou, Inorg. Chem., 2010, 49, 5715 CrossRef CAS PubMed;
(e) W. Yi, J. Zhang, S. Huang, L. Weng and X. Zhou, Chem.–Eur. J., 2014, 20, 867 CrossRef CAS PubMed;
(f) D. M. Lyubov, L. Luconi, A. Rossin, G. Tuci, A. V. Cherkasov, G. K. Fukin, G. Giambastiani and A. A. Trifonov, Chem.–Eur. J., 2014, 20, 3487 CrossRef CAS PubMed.
- I. V. Basalov, D. M. Lyubov, G. K. Fukin, A. V. Cherkasov and A. A. Trifonov, Organometallics, 2013, 32, 1507 CrossRef CAS.
- H. Li, Y. Yao, Q. Shen and L. Weng, Organometallics, 2002, 21, 2529 CrossRef CAS.
-
(a) G. Hafelinger, Chem. Ber., 1970, 103, 2920 Search PubMed;
(b) F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
-
(a) B. Wang, K. Lv and D. Cui, Macromolecules, 2008, 41, 1983 CrossRef CAS;
(b) G. Du, Y. Wei, L. Ai, Y. Chen, Q. Xu, X. Liu, S. Zhang, Z. Hou and X. Li, Organometallics, 2011, 30, 160 CrossRef CAS;
(c) C. Yao, D. Liu, P. Li, C. Wu, S. Li, B. Liu and D. Cui, Organometallics, 2014, 33, 684 CrossRef CAS.
- L. Zhang, M. Nishiura, M. Yuki, Y. Luo and Z. Hou, Angew. Chem., Int. Ed., 2008, 47, 2642 CrossRef CAS PubMed.
- W. Zhang, M. Nishiura, T. Mashiko and Z. Hou, Chem.–Eur. J., 2008, 14, 2167 CrossRef CAS PubMed.
- SAINTPlus Data Reduction and Correction Program v. 6.02 a, Bruker AXS, Madison, WI, 2000 Search PubMed.
- G. M. Heldrick, A. ADABS, Program for Empirical Absorption Correction, University of Göttingen, Göttingen, Germany, 1998 Search PubMed.
-
(a) G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Meng Li
,
Meng Li