
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A green approach to the synthesis of Ag doped nano magnetic γ-Fe2O3@SiO2-CD core–shell hollow spheres as an efficient and heterogeneous catalyst for ultrasonic-assisted A3 and KA2 coupling reactions†
S. Sadjadi*a,
M. Malmirb and
M. M. Heravi *b
*b
aGas Conversion Department, Faculty of Petrochemicals, Iran Polymer and Petrochemical Institute, PO Box 14975-112, Tehran, Iran. E-mail: samahesadjadi@yahoo.com
bDepartment of Chemistry, School of Science, Alzahra University, PO Box 1993891176, Vanak, Tehran, Iran. E-mail: mmh1331@yahoo.com; mmheravi@alzahra.ac.ir; Fax: +98 21 88041344; Tel: +98 21 88044051
First published on 25th July 2017
Abstract
Using a green approach and ultrasonic-assisted template-free five-step process, a novel heterogeneous catalyst, γ-Fe2O3@SiO2-CD/Ag hollow spheres (h-Fe2O3@SiO2-CD/Ag), was fabricated. The design of the catalyst was based on the synthesis of core–shell h-Fe2O3@SiO2 followed by amine functionalization, reaction with tosylated cyclodextrin and subsequent Ag(0) doping using hollyhock flowers collected from the Banaruiyeh District, in Larestan, Iran as extract-based reducing agent. The novel catalyst was fully characterized using SEM/EDX, BET, TGA, ICP-AES, FTIR, VSM and XRD techniques. The hybrid catalyst was successfully applied for promoting A3 and KA2 coupling reactions under mild and green conditions, i.e. ultrasonic irradiation at room temperature in aqueous media. Studying the reusability of the catalyst and Ag(0) nanoparticle leaching established that the catalyst can be recovered and reused with preserving its catalytic activity and negligible Ag(0) leaching for five reaction runs. Upon reusing for the sixth run, however, slight leaching of Ag(0) and consequently loss of catalytic activity was observed.
Introduction
Propargylamine derivatives are key intermediates which are broadly applied for the synthesis of a diverse range of natural products and biologically active nitrogen containing chemicals such as herbicides, fungicides, restricted peptide isosteres, oxotremorine analogues, indolizines, pyrroles and quinolines.1–4 Propargylamines can be obtained from three-component coupling reaction of amines, aldehydes and alkynes, referred to as the A3 coupling reaction. Using this valuable one-pot procedure several elements can be introduced into a molecule.5 This reaction can be promoted by using various metal catalysts such as gold, copper, iron, silver and zinc based catalysts3,6,7 that are able to activate the terminal C–H bond of the alkyne. Despite developing some promising methodologies, some of them require a toxic solvent such as toluene, or expensive media, like ionic liquids.8 Therefore, development of a novel, efficient and heterogeneous catalyst for this process is of great interest.Magnetic nanoparticles, MNPs, and its core–shell structures received ongoing interest due to their outstanding properties such as high magnetic susceptibility and coercivity, super paramagnetic behaviour and high surface area.9 This class of materials has been widely used for catalysis, target-drug delivery, hyperthermia treatment of cancer, diagnosis, MRI, enzyme immobilization, magnetic separation and sensors.10–12 Among various uses, catalysis has gained growing interest. Using MNPs diverse range of heterogeneous catalysts with tune-able size and morphologies can be designed and synthesized. Another merit of MNPs is their facile separation from the reaction mixture, which is more efficient and economical than conventional filtration and centrifuge approaches.13–15
Hollow MNPs (h-MNPs), benefit from high surface areas, tiny particle sizes and low densities. However, they may suffer from aggregations. To circumvent this issue, the surface of h-MNPs is usually decorated with stabilizing agents.14
Ultrasonic irradiation is a useful tool for developing green and clean procedures for the synthesis of organic compounds. The notable advantageous of ultrasonic-assisted approaches are accelerated reaction rates, waste minimizing, and simplicity of the procedure, high yields and purities of products.16,17 The effect of ultrasonic irradiation can be defined on the base of cavitation effect. The ultrasonic irradiation can cause high local temperatures and pressure inside the bubbles and accelerates mass transfer and turbulent flow in the liquid.17
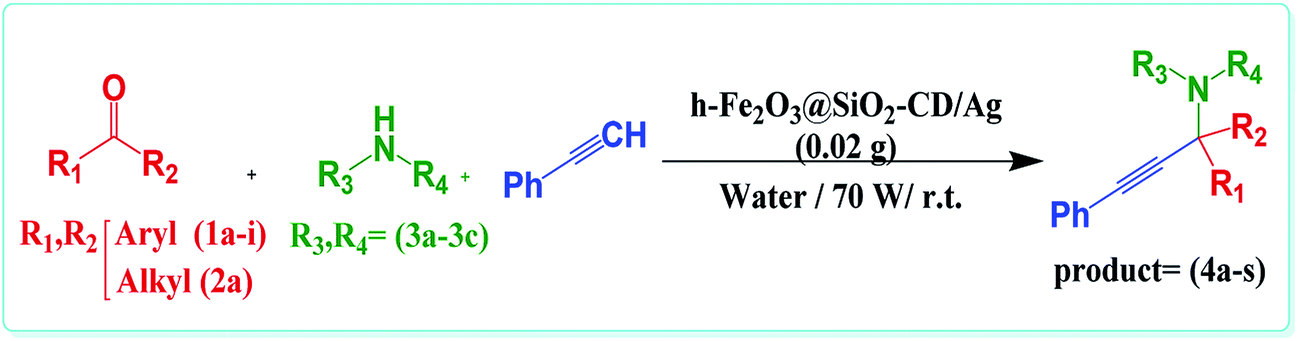
In last decade, we devoted our research to develop new heterogeneous and reusable catalysts for promoting organic transformations.14,18–22 In continuation of our attempts, herein, we wish to disclose a novel efficient hybrid catalyst for promoting A3 and KA2 coupling reactions under mild and green reaction condition. The design of the catalyst is based on fabrication of γ-Fe2O3 hollow spheres, which we disclosed in our previous report,14 followed by formation of SiO2 shell. The obtained h-Fe2O3@SiO2 was subsequently amine functionalized with 3-N-(2-(trimethoxysilyl)ethyl)methanediamine and reacted with tosylated cyclodextrin to form h-Fe2O3@SiO2-CD. The latter was then doped with Ag(0) NPs by using hollyhock flower extract as a extract-based reducing agent (Scheme 1). The reusability and Ag(0) leaching were also studied. Moreover, the catalytic performance of the catalyst was compared with some of previously reported catalytic procedures.
 | ||
| Scheme 1 The possible formation process of h-Fe2O3@SiO2-CD/Ag hollow spheres. | ||
Results and discussion
Catalyst characterization
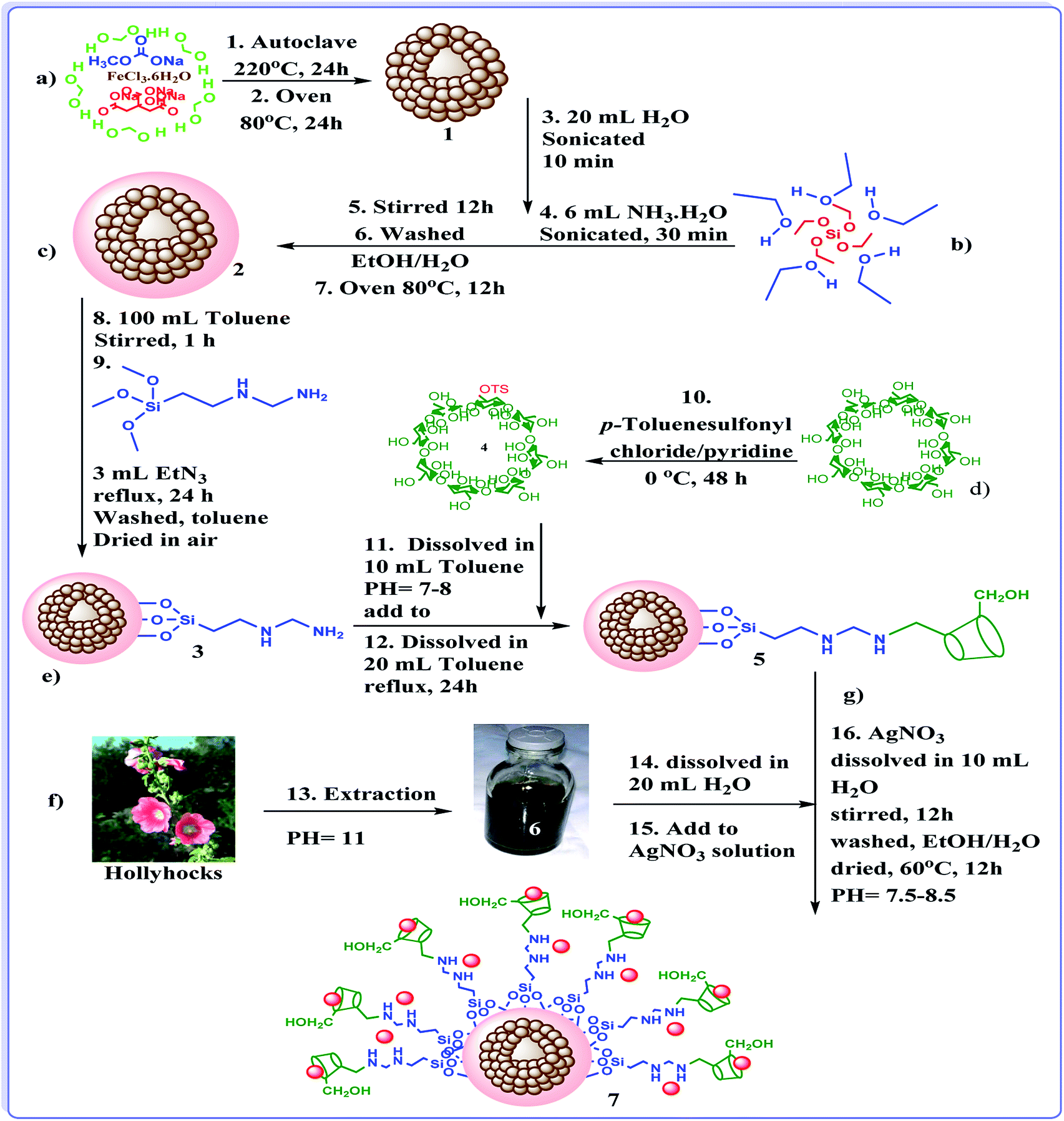
To confirm the formation of h-Fe2O3@SiO2-CD/Ag, the FTIR spectrum of the catalyst was obtained and compared with h-Fe2O3@SiO2-CD, Fig. 1. The strong absorption bands observed at 470–590 cm−1 can be assigned to Fe–O stretching vibration and confirm the formation of magnetic NPs.14 Both spectra exhibited the characteristic band of Si–O stretching at 1082 cm−1 indicating the successful coverage of SiO2 shell. The absorption peaks at 980, 810 and 470 cm−1 were in good agreement with the bending vibrations of the Si–O–Si bond.9 The bands at 2925 cm−1 and 3493 cm−1 can be attributed to the –CH2 stretching and –NH functionality and prove the conjugation of 3-N-(2-(trimethoxysilyl)ethyl)methanediamine.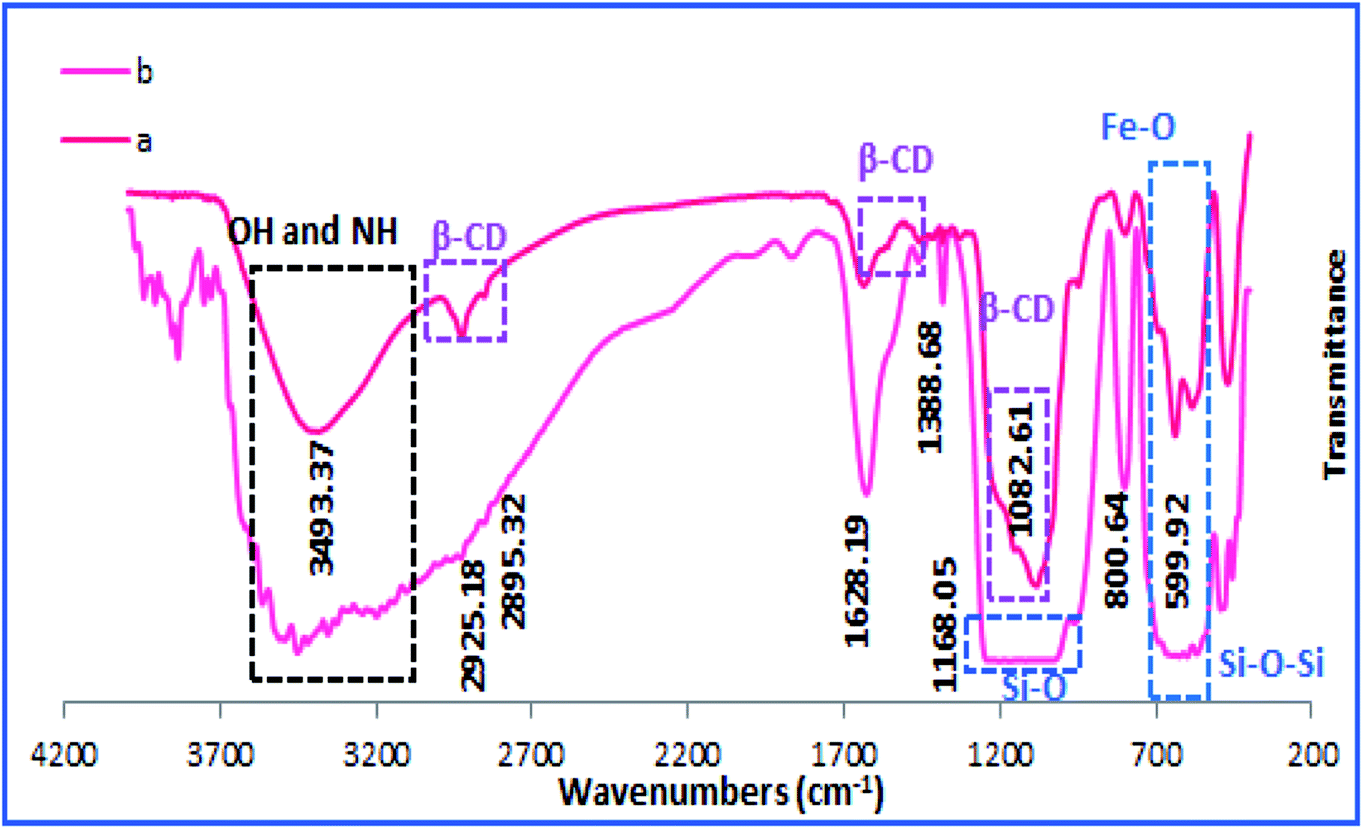 | ||
| Fig. 1 The FT-IR spectra of (a) h-Fe2O3@SiO2-CD and (b) h-Fe2O3@SiO2-CD/Ag. | ||
These bands can also represent the –OH and –CH2 groups in CD. Comparing the FTIR spectra of h-Fe2O3@SiO2-CD and h-Fe2O3@SiO2-CD/Ag reveals the broadening and sharpening of the bands upon Ag incorporation. According to the previous reports,23 biological extracts contain several functional groups that can be clearly detected in FTIR analysis. Hence the broadening and sharpening of the bands in and h-Fe2O3@SiO2-CD/Ag can be assigned to the presence of some functionalities in hollyhock flower extract such as –OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, N–H, C–H etc.
N, N–H, C–H etc.
The X-ray diffraction pattern, XRD, was exploited for investigation of the phase and composition of h-Fe2O3@SiO2-CD/Ag, Fig. 2. According to the literature,24 the broad halo observed at 2θ = 3–20 (labelled as *) can be assigned to the amorphous silica and CD. Moreover, the observed peaks at 30.3, 35.6, 43.2, 54.0, 57.3, 63.0 and 74.6 (labelled as F) can be attributed to the {220}, {311}, {400}, {422}, {511}, {440} and {533} planes of the typical cubic structure hematite (γ-Fe2O3) (JCPDS card no. 39-1346).14 The peaks labelled as A are characteristic peaks of Ag(0) NPs (JCPDS card no. 04-0783). Using Debye–Scherrer equation, the average particle size of Ag(0) NPs was estimated to be 37 nm.
 | ||
| Fig. 2 The XRD pattern of h-Fe2O3@SiO2-CD/Ag. | ||
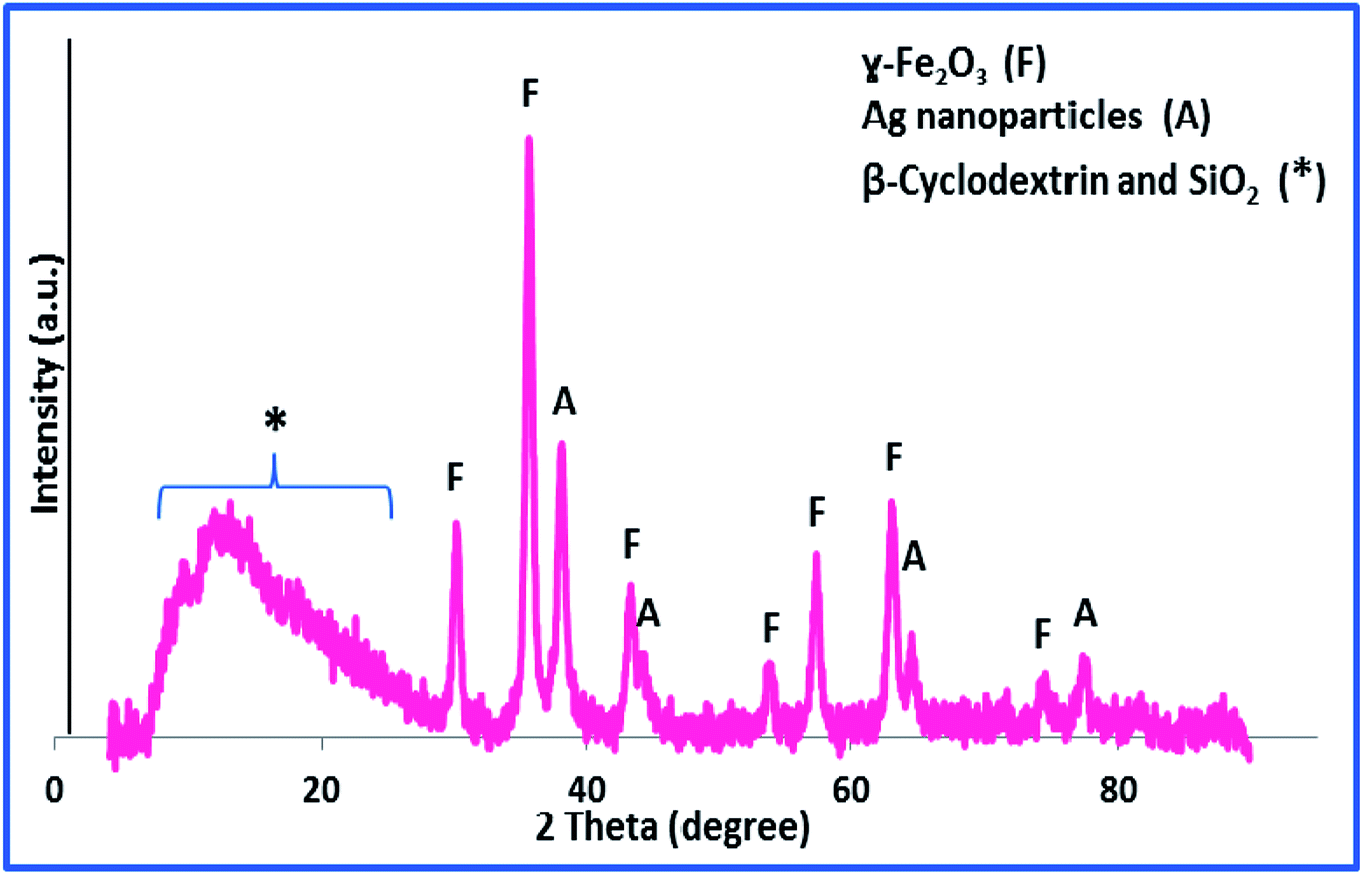
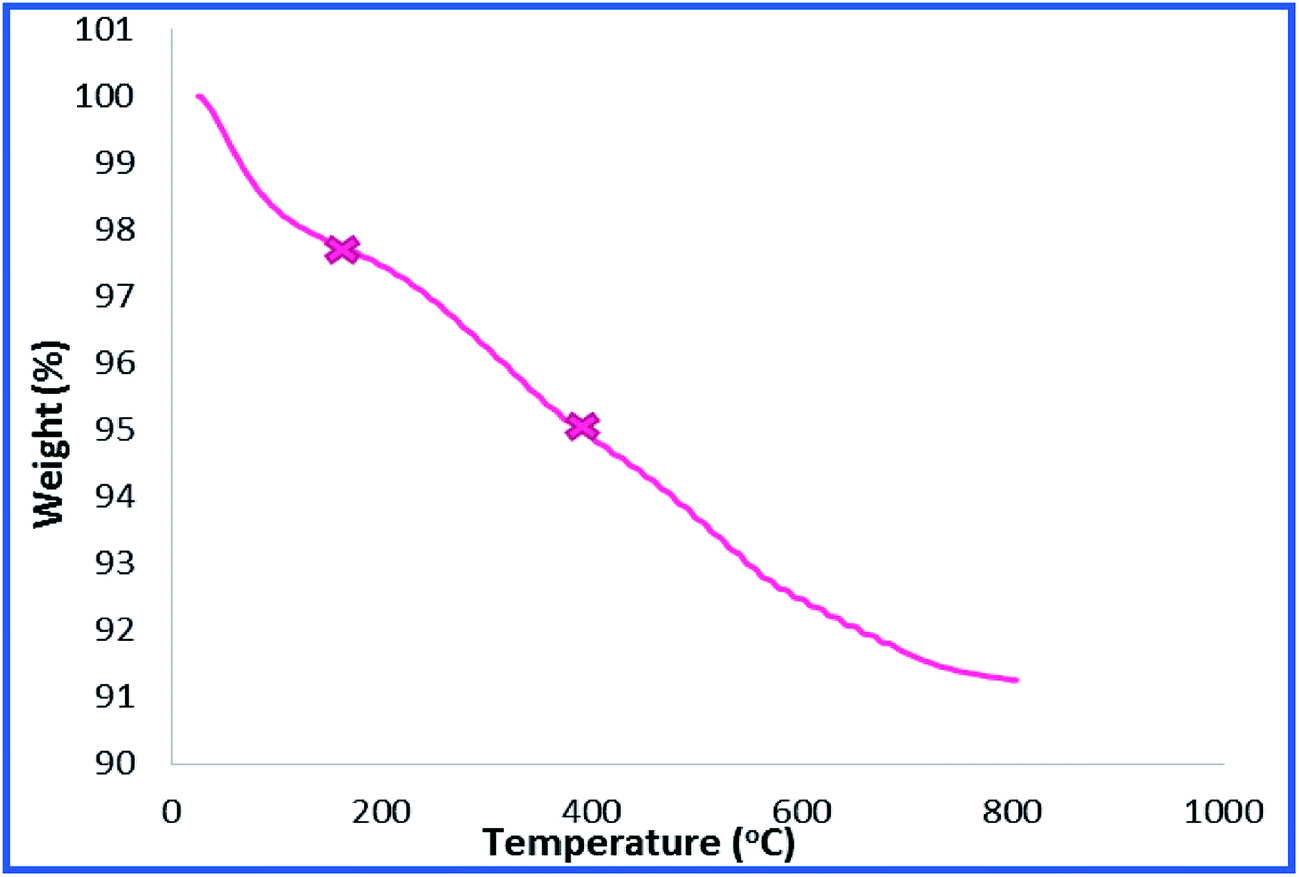
To provide more insight into the properties of the catalyst, thermo gravimetric analysis (TGA) of h-Fe2O3@SiO2-CD/Ag was carried out, Fig. 3. Two degradation steps can be detected for h-Fe2O3@SiO2-CD/Ag over the range of 40–800 °C. The first weight loss, observed at about 180 °C, is due to loss of adsorbed water molecules and/or surface hydroxyl groups. The second weight loss can be assigned to the degradation of organic moiety. Using TGA analysis, the content of organic moiety in h-Fe2O3@SiO2-CD/Ag was estimated to be about 4 w/w%. To study the morphology of the catalyst and investigating the effect of attachment of 3-N-(2-(trimethoxysilyl)ethyl)methanediamine, CD and embedding Ag(0) NPs on the morphology, the FE-SEM analysis of h-Fe2O3@SiO2 and SEM analyses of h-Fe2O3@SiO2-CD and h-Fe2O3@SiO2-CD/Ag were obtained, Fig. 4. As expected, the h-Fe2O3@SiO2 possessed a spherical morphology. Both h-Fe2O3@SiO2-CD and h-Fe2O3@SiO2-CD/Ag exhibited spherical morphologies, indicating that functionalization of h-Fe2O3@SiO2 with CD and incorporation of Ag(0) NPs did not alter the morphology dramatically.
 | ||
| Fig. 3 The TGA analysis of the h-Fe2O3@SiO2-CD/Ag. | ||
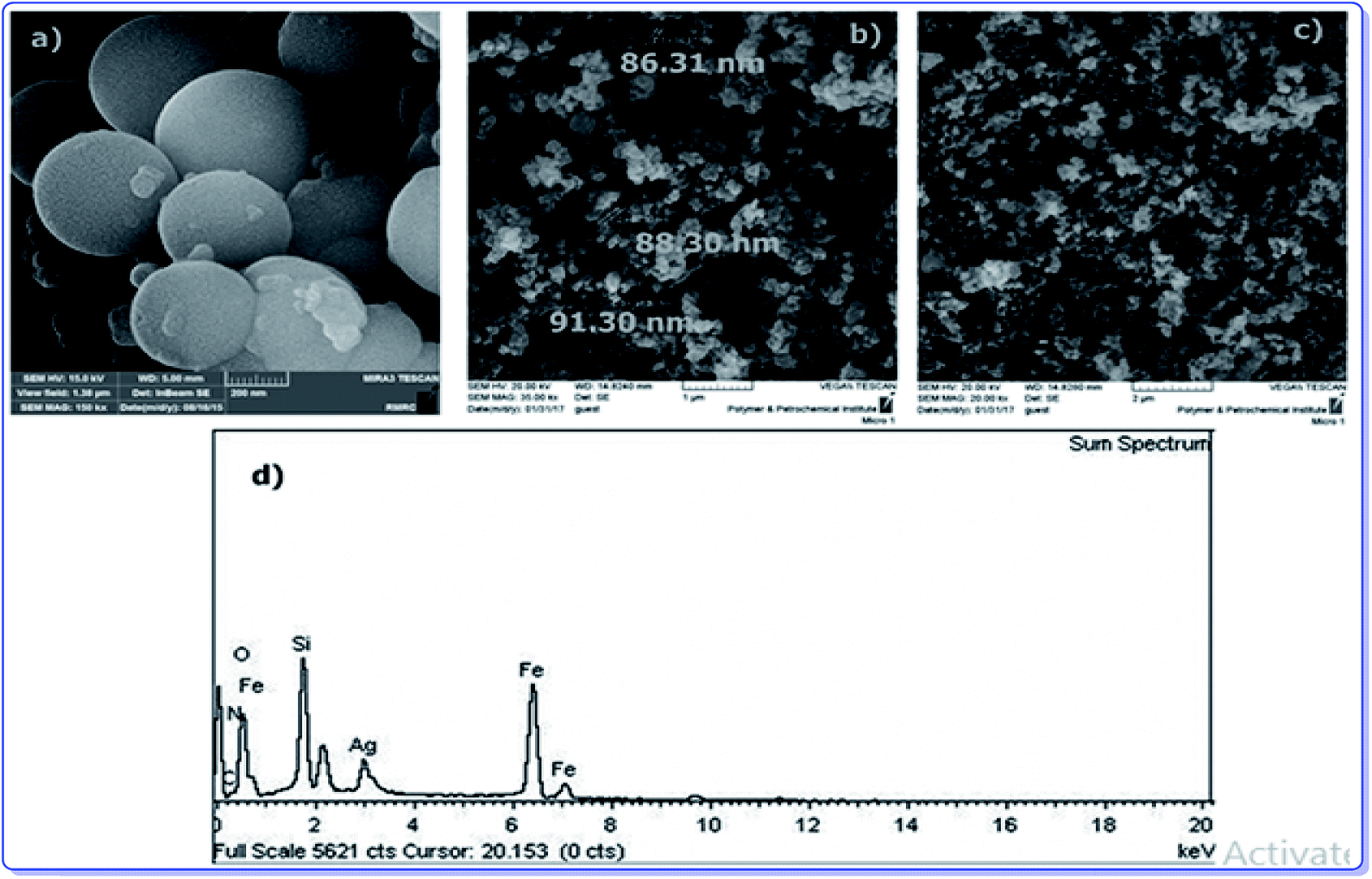 | ||
| Fig. 4 The FE-SEM analysis of (a) h-Fe2O3@SiO2 and SEM analyses of (b) h-Fe2O3@SiO2-CD, (c) h-Fe2O3@SiO2-CD/Ag and (d) the EDX analysis of h-Fe2O3@SiO2-CD/Ag. | ||
The EDX analysis of h-Fe2O3@SiO2-CD/Ag is shown in Fig. 4d. The presence of the Fe and O atoms can represent the γ-Fe2O3 magnetic NPs. Moreover, observation of the Si and O atoms can prove the existence of SiO2 shell. The conjugation of 3-N-(2-(trimethoxysilyl)ethyl)methanediamine and CD can be confirmed by the presence of N, Si, C and C and O elements, respectively. The existence of Ag atom in EDX analysis can indicate the incorporation of Ag.
The elemental mapping analysis of h-Fe2O3@SiO2-CD/Ag is illustrated in Fig. 5. As obvious, the N has been distributed uniformly in the catalyst, indicating that 3-N-(2-(trimethoxysilyl)ethyl)methanediamine decorated the whole surface of the h-Fe2O3@SiO2–N2 surface evenly. The good dispersion of Ag NPs also established that Ag(0) NPs were dispersed all around the catalyst.
 | ||
| Fig. 5 The elemental mapping analysis of h-Fe2O3@SiO2-CD/Ag. | ||
To disclose the textural properties of the catalyst, the nitrogen adsorption–desorption isotherm of the h-Fe2O3@SiO2-CD/Ag was obtained, Fig. 6. The shape of the isotherm confirmed the porous structure and according to IUPAC classification exhibits type II nitrogen adsorption–desorption isotherms with H3 hysteresis loops.25 The textural properties including specific surface area, total pore volume and average pore diameter of h-Fe2O3@SiO2-CD/Ag are reported in Table 1. Additionally, these parameters were compared with those of h-Fe2O3@SiO2-CD to investigate the influence of incorporation of Ag(0) doping on the textural properties of final catalyst. Comparison of these parameters established that doping of Ag(0) NPs increased the specific surface area and total pore volume while decreased average pore diameter. According to the previous reports, the increase in specific surface area can emerge from the presence of Ag(0) NPs on h-Fe2O3@SiO2-CD.26
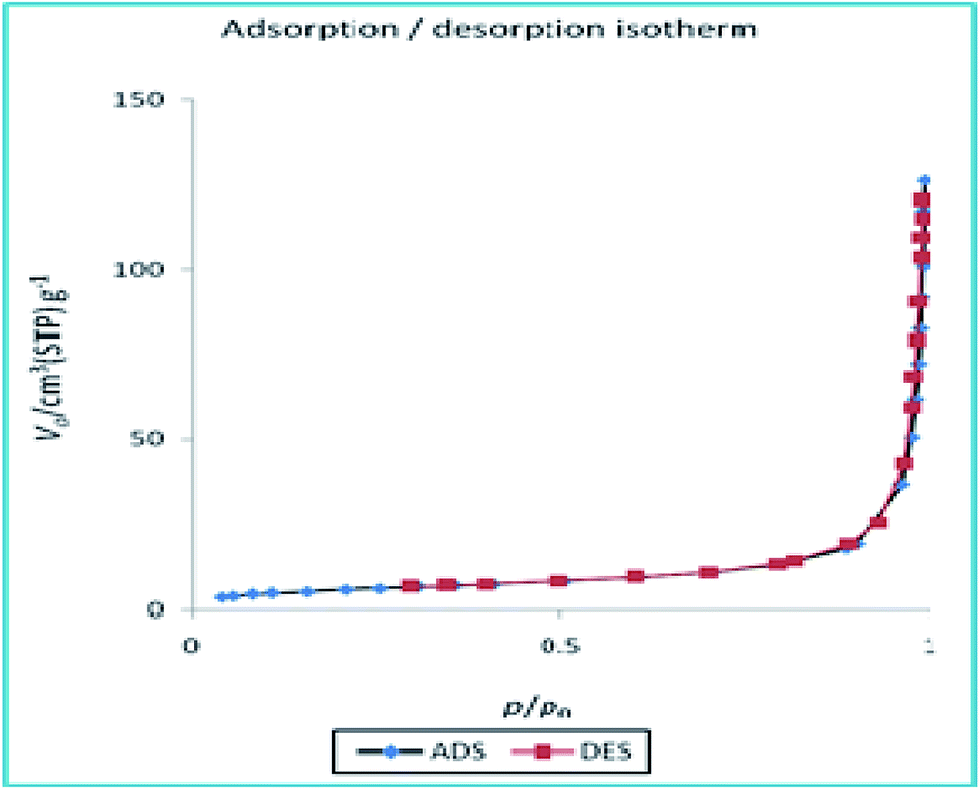 | ||
| Fig. 6 N2 adsorption–desorption isotherms of the catalyst. | ||
| Sample | SBET (m2 g−1) | Total pore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|
| h-Fe2O3@SiO2-CD/Ag | 21.067 | 0.1834 | 34.822 |
| h-Fe2O3@SiO2-CD | 12.139 | 0.1143 | 37.679 |
Using room temperature vibrating sample magnetometer (VSM), the magnetic property of h-Fe2O3@SiO2-CD/Ag was studied and compared with that of h-Fe2O3 Fig. 7. As depicted, the maximum saturation magnetization (Ms) value of h-Fe2O3@SiO2-CD/Ag was calculated as 41.15 emu g−1 that is slightly lower than Ms of h-Fe2O3 (45.28 emu g−1).
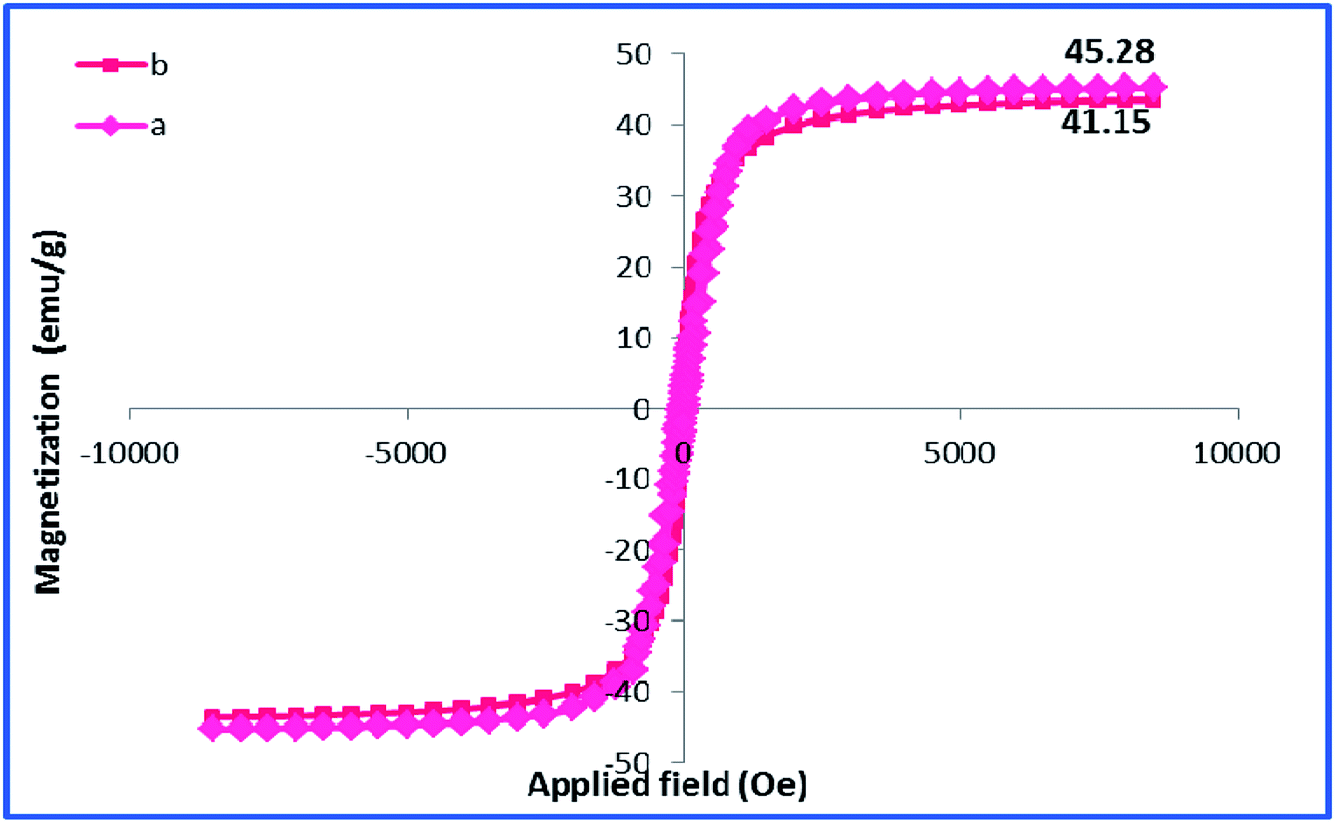 | ||
| Fig. 7 VSM analyses of (a) h-Fe2O3 and (b) h-Fe2O3@SiO2-CD/Ag. | ||
This result implies that the functionalization of h-Fe2O3 and subsequent attachment of CD and incorporation of Ag(0) NPs did not affect the magnetic property of h-Fe2O3 dramatically. Furthermore, it can be found that there are no evident changes in the coercivity from the enlarged view of the central loop of the h-Fe2O3 and h-Fe2O3@SiO2-CD/Ag. The content of silver in h-Fe2O3@SiO2-CD/Ag was also estimated by using ICP-AES analysis. Initially, the definite amount of catalyst was digested in concentrated hydrochloric and nitric acids solution. The resulting extract was then analyzed by ICP-AES. The Ag content was calculated as 0.4 w/w%.
Catalytic activity of nanomagnetic h-Fe2O3@SiO2-CD/Ag
Considering the importance of the A3 and KA2 coupling reactions, the catalytic activity of the h-Fe2O3@SiO2-CD/Ag was studied for promoting these reactions. Initially, reaction of benzaldehyde, phenyl acetylene and morpholine was selected as a model reaction and carried out in the presence of catalytic amount of the catalyst (20 mg) under conventional reflux condition in aqueous media.The results established high catalytic activity of the h-Fe2O3@SiO2-CD/Ag (90%). Motivated by this result and in attempt to achieve a green and rapid procedure, the model reaction was performed under ultrasonic irradiation. Gratifyingly, the desired product was furnished in high yield (97%) in a very short reaction time. According to the previous reports,16,27 we define the efficiency of ultrasound irradiation on the base of cavitation phenomena. That is, formation, growth and collapse of the cavities which result in generation of high local temperatures and pressures. These can concentrate huge amount of energy from the conversion of kinetic energy of liquid motion into heating of the contents of the cavities. This can be considered as a driving force for promoting the organic transformation.27 It is proved that the ultrasonic irradiation not only can accelerate the reaction rate, but also affect the reaction through altering the reaction pathway and enhancing chemical reactivity. Subsequently, to elucidate whether the extract-based reduction of the silver species can affect the catalytic activity, the catalytic activity of h-Fe2O3@SiO2-CD/Ag was compared with that of h-Fe2O3@SiO2-CD/Ag–H (in which the silver was reduced by hydrazine). Interestingly, h-Fe2O3@SiO2-CD/Ag–H exhibited comparative catalytic activity. This result established that both chemical reduction and extract-based reduction can lead to the catalysts with high catalytic activity. Hence, both preparation procedures can be chosen. When the green chemistry is of focus, the green-synthesis approach is privileged while using commercially available hydrazine as the reduction agent can render chemical reduction more feasible.
According to the literature, the Ag(0) NPs is the dominant catalytic active site for promoting A3 coupling reaction.28–31 Ag(0) NPs can activate the substrate (phenyl acetylene) and promote formation of intermediates which then transformed into the desired product (see Reaction mechanism section). Hence, it can be concluded that the content of Ag(0) NPs can influence the catalytic activity. To confirm this postulation, h-Fe2O3@SiO2-CD/Ag with various content of Ag(0) (0.2–0.8 w/w%) were synthesized and their catalytic activities were compared. It was found that small amount of silver can catalyse the reaction efficiently. Therefore, the need for scant amount of silver can be considered as the merit of this protocol. Moreover, it was found that increasing the amount of silver content from 0.2 to 0.4 w/w% can improve the catalytic activity. This can be attributed to the increase of the catalytic active sites. However, this trend was not sustainable and further increase of silver content not only did not improve the catalytic activity but also had a detrimental effect on the catalytic performance. This observation can be attributed to the aggregation of the Ag(0) NPs aggregation.
To explore the role of CD in the catalytic activity, the catalytic activity of h-Fe2O3@SiO2-CD/Ag was compared with that of Fe2O3@SiO2–N2/Ag. The result clearly demonstrated the superior catalytic activity of the former, indicating the effective role of CD in the catalytic activity. According to the literature, this observation can be attributed to the NPs and CD.32 Moreover, it is postulated that the presence of the CD can stabilize the NPs.33
Then, the reaction variables including ultrasonic irradiation, solvent and catalyst amount were optimized by altering the reaction variables and studying their effects on the yield of the model product (Fig. 8). As shown in Fig. 8, increasing the amount of catalyst from 10 mg to 20 mg resulted in higher yields. However, dependent on the reaction condition, more increase in the catalyst amount (to 30 mg) had different effects on the yield of the product. As depicted, in toluene, acetonitrile and EtOH, this had a detrimental effect on the yield of the reaction and no product was obtained using 30 mg catalyst. This can be assigned to the formation of various by-products. In the case of water as a solvent, the effect of the catalyst amount is influenced by the power of the used ultrasonic irradiation. Using 30 mg catalyst under low (50 W) or high (100 W) power of ultrasonic irradiation resulted in increase of the yield of the product while using medium (70 W) ultrasonic power reduced the yield slightly. The optimum reaction variables were found to be using 20 mg catalyst under ultrasonic irradiation of power 70 W in aqueous media and ambient temperature.
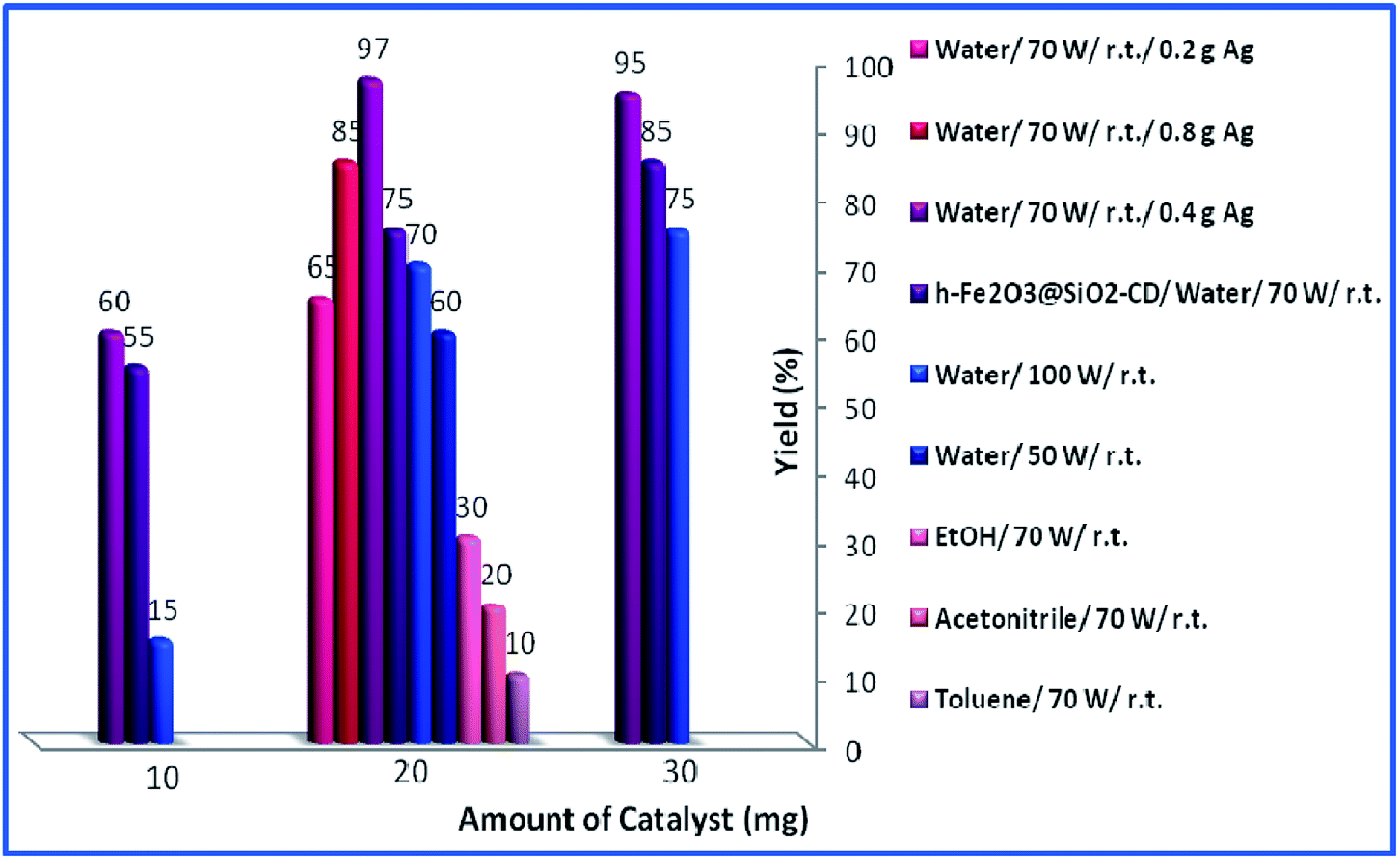 | ||
| Fig. 8 Effects of loading of catalyst, duration of reaction and solvent for the synthesis of propargylamine. | ||
Having the optimum reaction condition in hand, the generality of this protocol was examined by using various amines and aldehydes with different electron densities (Table 2). As shown in this table, aldehydes with both electron donating and electron withdrawing functional groups could furnish the corresponding products in high yields. However, using heterocyclic aldehyde, furfural, led to slightly lower yield. These results established the reliability of this protocol. Encouraged by these results, we studied the possibility of replacing aldehydes with ketones, KA2 coupling reaction (Table 2, products 4r and 4s). Interestingly, h-Fe2O3@SiO2-CD/Ag could also catalyse this reaction to furnish the corresponding products in high yields. Finally, the efficiency of this protocol and the catalytic performance of h-Fe2O3@SiO2-CD/Ag for catalysing the model reaction were compared with those of some previously reported catalytic methodologies to disclose the merits of this procedure (Table 3). As obvious, h-Fe2O3@SiO2-CD/Ag resulted in desired product in higher or comparative yields. However, compared to all cases, h-Fe2O3@SiO2-CD/Ag led to the product in shorter reaction time. Moreover, the presented protocol does not required any harsh reaction condition, inert atmosphere or toxic solvent.
|
||||||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | Product | Time (min) | Yieldb (%) |
| a Reaction conditions: aliphatic ketone 2a (1.0 equiv.) or arylaldehydes 1a (1.0 equiv.), amines 3a (1.0 equiv.), alkynes (1.1 equiv.), H2O (20 mL) and h-Fe2O3@SiO2-CD/Ag (0.02 g) under ultrasonic irradiation (70 W) at room temperature.b Isolated yield. | ||||||
| 1a: C6H5 | H | 3a: –(CH2)5– | 4a | 7 | 97 | |
| 1b: p-Cl–C6H4 | H | 3a | 4b | 10 | 95 | |
| 1c: p-NO2–C6H4 | H | 3a | 4c | 12 | 90 | |
| 1d: p-Me–C6H4 | H | 3a | 4d | 15 | 90 | |
| 1e: p-MeO–C6H4 | H | 3a | 4e | 8 | 91 | |
| 1f: o-OH–C6H4 | H | 3a | 4f | 10 | 94 | |
| 1g: Furfuryl | H | 3a | 4g | 12 | 85 | |
| 1h: p-OHCC6H4 | H | 3a | 4h | 10 | 90 | |
| 1a | H | 3b: –(CH2)2–O–(CH2)2– | 4i | 8 | 97 | |
| 1b | H | 3b | 4j | 8 | 90 | |
| 1c | H | 3b | 4k | 12 | 85 | |
| 1d | H | 3b | 4l | 15 | 90 | |
| 1e | H | 3b | 4m | 10 | 92 | |
| 1f | H | 3b | 4n | 12 | 96 | |
| 1g | H | 3b | 4o | 20 | 90 | |
| 1i: m-NO2–C6H4 | H | 3b | 4p | 20 | 90 | |
| 1a | H | 3c: C2H5 | C2H5 | 4q | 15 | 88 |
| 2a: –(CH)5– | 3a | 4r | 20 | 89 | ||
| 2a | 3b | 4s | 15 | 90 | ||
| Catalyst (amount) | Reaction conditions | Time (min) | Catalyst amount | Yield (%) | Ref. |
|---|---|---|---|---|---|
| CuI | H2O/r.t./U.S | 45 | 15 mg | 98 | 37 |
| h-Fe2O3@SiO2-CD/Ag | H2O/r.t./70 W | 7 | 20 mg | 97 | This work |
| h-Fe2O3@DA/Ag | S.F./90 °C | 60 | 10 mg | 96 | 14 |
| Gold nanocrystals stabilized on montmorillonite | Reflux/toluene | 180 | 20 mg | 91 | 38 |
| ZnO nanoparticles | Stirrer/90 °C | 120 | 10 mol% | 89 | 5 |
| Immobilized silver on surface-modified ZnONPs | Reflux/H2O | 240 | 10 mol% | 89 | 30 |
| Cu2O–ZnO | S.F./100 °C | 60 | 10 mg | 95 | 35 |
| CuCN | [bmim]PF6/S.F./120 °C | 120 | 2 mol% | 95 | 39 |
| CuNPs/TiO2 | Neat/70 °C | 420 | 0.5 mol% | 91 | 1 |
| Nafion-NR50 | CH3CN/70–80 °C/N2 atm | 300 | 350 mg | 85 | 40 |
| ZnS | Reflux/CH3CN | 270 | 10 mol% | 89 | 41 |
Reaction mechanism
Considering the previous reports,14,34 the plausible mechanism for the formation of propargylamines includes activation of terminal C–H bond of phenylacetylene by Ag NPs of the catalyst and formation of an intermediate, Ag–acetylide I. In this regard the suggested mechanism involves the activation of the C–H bond of alkyne via interaction by the metal nanoparticles (M). Thus, terminal alkyne is converted into the M⋯acetylide intermediate (I). The nucleophilic addition of the amine to the already activated aldehyde by Ag leads to generation of iminium ion (II). Next, the M⋯acetylide intermediate attacks the iminium ion simultaneously to provide the corresponding propargylamine (III). In this process one molecule of water can be formed from hydroxyl group and hydrogen of terminal alkyne, Scheme 2.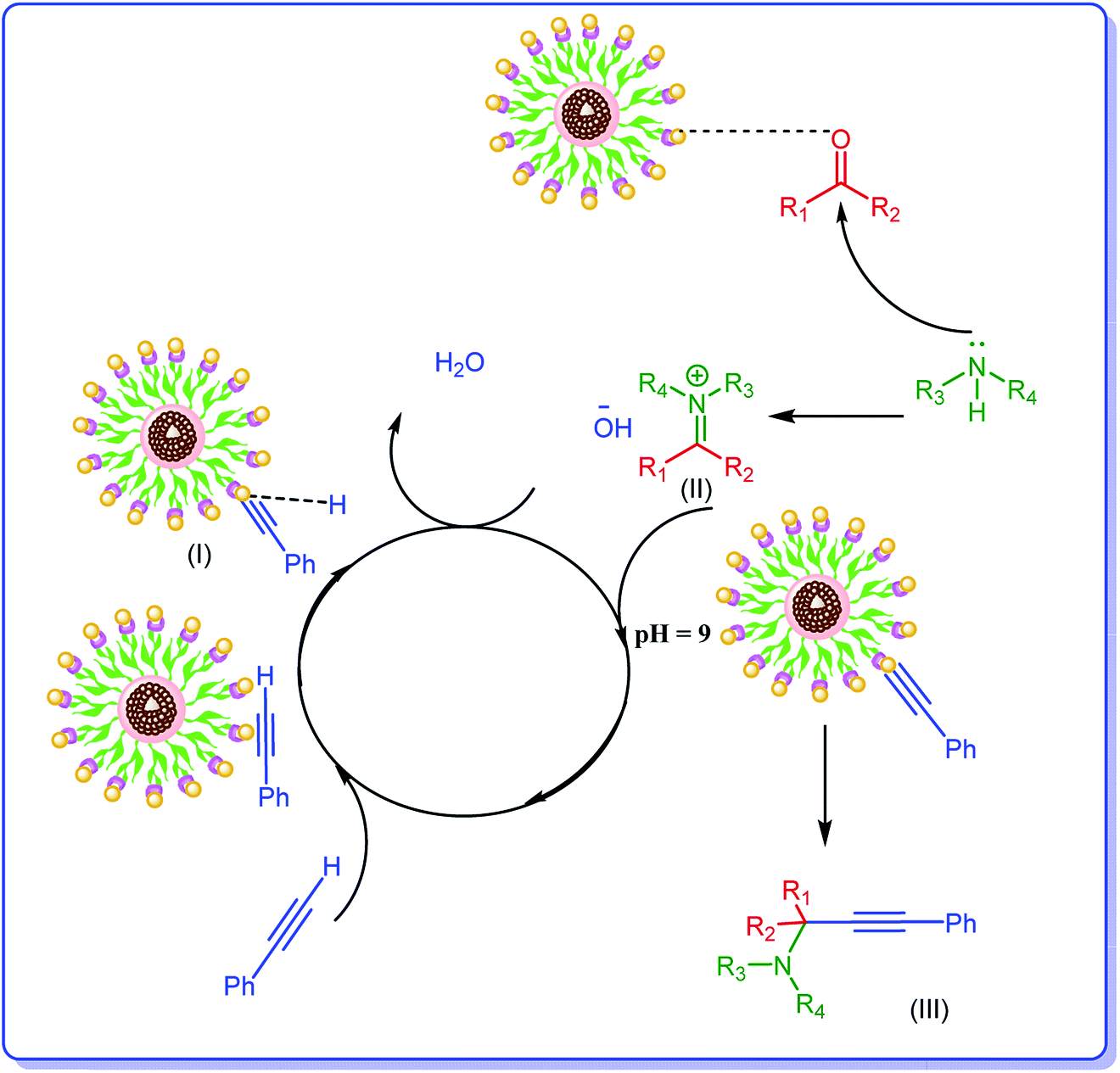 | ||
| Scheme 2 Plausible mechanism for the synthesis of propargylamine catalyzed by h-Fe2O3@SiO2-CD/Ag. | ||
Catalyst reusability
The reusability of a heterogeneous catalyst is an important feature that makes it green and economically-efficient. To investigate the possibility of reusing h-Fe2O3@SiO2-CD/Ag, the yields of the model reaction in the presence of fresh and reused catalysts in various time intervals were measured and compared, Fig. 9.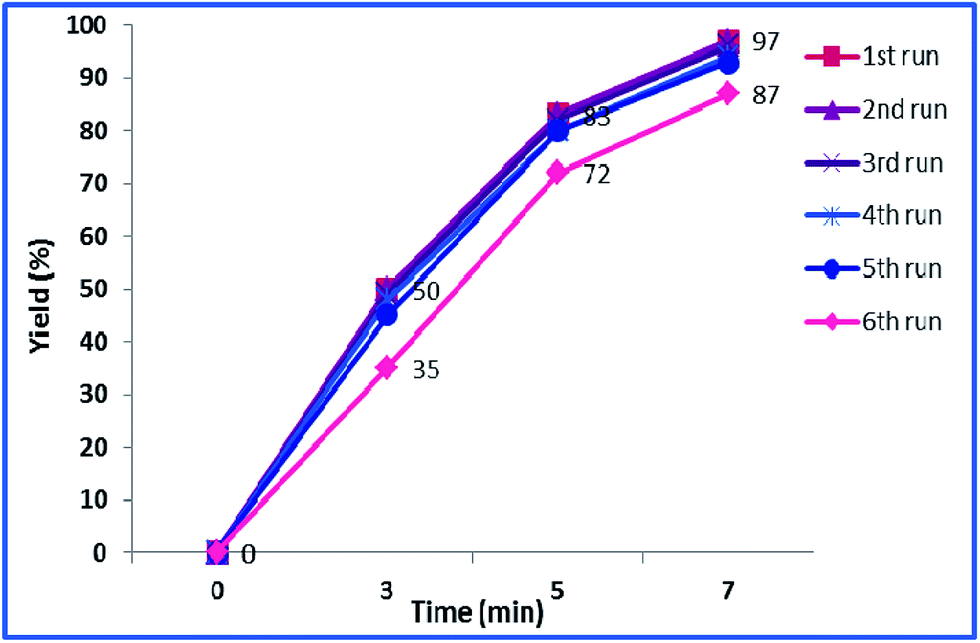 | ||
| Fig. 9 Reusability of the h-Fe2O3@SiO2-CD/Ag catalyst in A3 coupling. | ||
As shown in this figure, the catalyst can be recovered and reused for four times without any loss of catalytic activity. Reusing the catalyst for the fifth run, however, led to slight loss of catalytic activity. Upon reusing the catalyst for the sixth run, remarkable loss of catalytic activity (10%) was observed and the desired product was furnished in lower yield. To justify this behaviour, the leaching of Ag(0) NPs of reused catalysts were studied using hot filtration method and ICP-AES analysis. The results demonstrated zero leaching of Ag(0) NPs for the catalysts reused for four times. For the catalyst reused for fifth run only negligible loss of Ag(0) NPs was observed. This can justify the slightly lower catalytic activity of this catalyst. The leaching of Ag(0) NPs for the catalyst reused for sixth run however, were detectable. This finding can prove that the Ag(0) NPs are the catalytically active species. Moreover, the nature of the catalysis is heterogeneous.
Experimental
Materials and instruments
All chemicals and reagents, including trisodium citrate dihydrate, sodium acetate trihydrate, FeCl3·6H2O, TEOS, β-cyclodextrin, N-(2-(trimethoxysilyl)ethyl)methanediamine, toluene, triethylamine, acetone, ethanol, ethylene glycol (EG), urea, AgNO3, NH3·H2O and hydrazine hydrate were analytical grade reagents, purchased from Sigma-Aldrich, and used without further purification. The progress of the organic reactions were monitored by TLC on commercial aluminum-backed plates of silica gel 60 F254, visualized, using ultraviolet light. Melting points were determined in open capillaries using an Electrothermal 9100 without further corrections. 1H NMR and 13C NMR spectra were recorded on Bruker DRX-400 spectrometer at 400 and 100 MHz respectively.The catalyst characterization was performed by using various characterization techniques including XRD, FTIR, BET, SEM/EDX, TGA, and ICP-AES. FTIR spectra were obtained by using PERKIN-ELMER-Spectrum 65 instrument. The BET analyses were carried out using BELSORP Mini II instrument. Prior to BET analyses, the samples were degassed at 423 K for 3 h. SEM/EDX images were recorded by employing a Tescan instrument, using Au-coated samples and acceleration voltage of 20 kV. Room temperature powder X-ray diffraction patterns were obtained by using a Siemens, D5000. CuKα radiation from a sealed tube.
All compounds were known and were identified by comparison of their physical and spectroscopic data with those of authentic compounds and were found identical. However, we compared all compounds by their melting points. 1H and 13C NMR spectra of selected compounds are reported and given as ESI.†
Synthesis of nanomagnetic Fe2O3 hollow sphere (a)
Nanomagnetic Fe2O3 hollow sphere were prepared through solvothermal method.14 Initially, FeCl3·6H2O (5 mmol) was dissolved in 70 mL of ethylene glycol in a flask. Next, 30 mmol of sodium acetate trihydrate, 1.5 mmol of trisodium citrate dehydrate and urea (17 mmol) were added to the aforementioned solution. The resulting mixture was stirred vigorously for 60 minutes and then, transferred to the Teflon-lined stainless-steel autoclave (150 mL capacity) and kept at 220 °C for 24 h. Upon completion of the process, the reactor was cooled to room temperature and the resulting brown product 1 was washed for three times with EtOH/H2O and dried at 80 °C for 24 h in oven.Synthesis of h-Fe2O3@SiO2 core–shell NPs (b)
The h-Fe2O3@SiO2 core–shell NPs was prepared according to a previously reported method with little modification.42 Briefly, h-Fe2O3 (1 g) was dispersed in a mixture of 20 mL of deionized water, 50 mL of ethanol and 6 mL of NH3. The pH value of the suspension was kept at ca. 11 and then, the mixture was sonicated for 30 min at room temperature. After that, TEOS (2 g) was added into the mixture, which was continuously stirred for 12 h at room temperature in air. Finally, the obtained h-Fe2O3@SiO2 core–shell, 2, was collected by an external magnet, washed three times with EtOH/H2O to remove blank silica, and dried at 80 °C for 12 h in oven.Synthesis of h-Fe2O3@SiO2–N2 core–shell NPs (c)
For amine-functionalization of the surface of h-Fe2O3@SiO2core–shell (h-Fe2O3@SiO2) (3 g), was dispersed by sonication in 100 mL dry toluene for 1 h. Subsequently, 3-N-(2-(trimethoxysilyl)ethyl)methanediamine (3 mL) as functionalization group and Et3N (3 mL) as a catalyst were added and the reaction mixture was refluxed overnight. At the end of the reaction, the mixture was cooled to room temperature and the amine-functionalized h-Fe2O3@SiO2core–shell, 3, was collected by using an external magnet and washed with toluene, and then dried under air for 12 h.Synthesis of β-cyclodextrin-OTs (d)
Tosylation of CD with p-toluenesulfonyl chloride was performed according to literature procedure.43 Typically, p-toluenesulfonylchloride (7.9 mmol) was added to a solution of pyridine containing 15.86 mmol of CD. The mixture was cooled to 0 °C and kept for 48 h. Upon completion, distilled water was added to the mixture and the solvent was evaporated to afford an oily product. The latter transformed into a white precipitate upon addition of cold water. The final product was achieved by filtration, recrystallization from water and drying.Synthesis of h-Fe2O3@SiO2-CD (e)
To synthesize the h-Fe2O3@SiO2-CD, β-cyclodextrin-OTs (0.5 g) was dispersed in dry toluene (20 mL) under stirring condition. The pH value of the suspension was kept at ca. 7–8. Then, the resulting mixture was added to the suspension of h-Fe2O3@SiO2–N2 core–shell 3 (1 g) in 20 mL dry toluene. After that, the suspension was heated under reflux condition overnight. Upon completion of the reaction, the brown product 5 was magnetically collected, washed with dry toluene for three times and dried in oven at 80 °C for 12 h.Preparations of hollyhocks flower extract (f)
Fresh leaves of hollyhock flower were collected around Banaruiyeh District, in Larestan, Iran. First, the collected hollyhock flowers (2 g) were crushed in porcelain mortar. Then, the resulting powder was mixed thoroughly with 100 mL deionized water and boiled for 60 min at 80 °C. The extract 6 was obtained by cooling the mixture and simple filtration.Extract-based synthesis of Ag NPs and their embedding into h-Fe2O3@SiO2-CD: synthesis of h-Fe2O3@SiO2-CD/Ag (g)
h-Fe2O3@SiO2-CD (1 g) was dispersed into a solution of AgNO3 (0.1 g) in 20 mL deionized H2O under stirring condition at room temperature for 30 minutes. The Ag+ ions were adsorbed onto the surfaces of h-Fe2O3@SiO2-CD via the electrostatic attraction. After that the fresh extract 6 (1 mL) as a reducing agent was added into the suspension. Upon addition of the extract 6 the colour of the mixture was changed to black which indicate the reduction of silver to Ag(0). To immobilize Ag(0) NPs the into the h-Fe2O3@SiO2-CD, the mixture was stirred for 12 h. Finally, the product 7 was successfully collected by magnetic separation, washed three times with EtOH/H2O and dried at 60 °C for 12 h. The schematic processes of synthesis of the catalyst are depicted in Fig. 1. Notably, to compare the catalytic activity of the catalyst achieved from the extract-based approach with the one obtained from conventional chemical reduction of silver species, the immobilization of Ag(0) nanoparticles on h-Fe2O3@SiO2-CD was also performed by reduction of silver precursor with hydrazine. To this purpose, the same procedure reported above was used except that the reduction was carried out by using 2 mL hydrazine hydrate. This catalyst was named as h-Fe2O3@SiO2-CD/Ag–H. h-Fe2O3@SiO2–N2/Ag was also prepared through synthesis of h-Fe2O3@SiO2–N2 (c) and subsequent immobilization of Ag(0) NPs via extract-based approach similar to the one reported for the synthesis of h-Fe2O3@SiO2-CD/Ag except that h-Fe2O3@SiO2–N2 was used as the starting material.General procedure for A3 and KA2 coupling reactions
To a mixture of phenyl acetylene (1.1 equiv.), amine (1.0 equiv.), aldehyde or ketone (1.0 equiv.) in H2O (10 mL) as a green solvent, nanomagnetic h-Fe2O3@SiO2-CD/Ag (20 mg) was added. The mixture was subjected to ultrasonic irradiation of power of 70 W. The progress of the reaction was monitored by TLC. Upon completion of the reaction, the reaction mixture was cooled to ambient temperature and diluted with hot ethanol (10 mL). The catalyst was separated by an external magnet from the cooled mixture, washed with ethanol, dried in air and re-used for a consecutive reaction run under the same reaction conditions. The resulting residue was purified by recrystallization (hot ethanol) to obtain the excellent yield of product.Spectral data of some selected compounds
Conclusions
In summary, hollyhock flower extract was used as a reducing agent for the synthesis of Ag(0) NPs on cyclodextrin decorated h-Fe2O3@SiO2 core–shell, obtained through a green facile ultrasonic-assisted and template-free process, to furnish a novel and heterogeneous catalyst (h-Fe2O3@SiO2-CD/Ag). The utility of the novel catalyst for catalyzingA3 and KA2 coupling reactions under mild and green condition, i.e. ultrasonic irradiation at room temperature in aqueous media was proved. Hot filtration experiments ruled out the remarkable Ag(0) leaching up to five reaction runs and the reused catalyst can be reused without negligible loss of catalytic activity.Acknowledgements
The authors are thankful to Alzahra University Research Council S. Sadjadi and M. M. Heravi are also thankful to Iran National Science Foundation (INSF) for supporting this research under contract no. 95834576.Notes and references
- M. J. Albaladejo, F. Alonso, Y. Moglie and M. Yus, Eur. J. Org. Chem., 2012, 2012, 3093–3104 CrossRef CAS.
- G. Villaverde, A. Corma, M. Iglesias and F. Sanchez, ACS Catal., 2012, 2, 399–406 CrossRef CAS.
- S. V. Katkara and R. V. Jayaram, RSC Adv., 2014, 4, 47958–47964 RSC.
- N. Salam, S. K. Kundu, A. Singha Roy, P. Mondal, S. Roy, A. Bhaumik and S. Manirul Islam, Catal. Sci. Technol., 2013, 3, 3303–3316 CAS.
- K. V. V. Satyanarayana, P. Atchuta Ramaiah, Y. L. N. Murty, M. R. Chandra and S. V. N. Pammi, Catal. Commun., 2012, 25, 50–53 CrossRef CAS.
- B. J. Borah, S. J. Borah, K. l. Saikia and D. K. Dutta, Catal. Sci. Technol., 2014, 4, 4001–4009 CAS.
- T. Zeng, W.-W. Chen, C. M. Cirtiu, A. Moores, G. Song and C.-J. Li, Green Chem., 2010, 12, 570–573 RSC.
- G.-P. Yong, D. Tian, H.-W. Tong and S.-M. Liu, J. Mol. Catal. A: Chem., 2010, 323, 40–44 CrossRef CAS.
- X. Zhang, Y. Niu, Y. Yang, Y. Li and J. Zhao, New J. Chem., 2014, 38, 4351–4356 RSC.
- N. E. A. El-Gamel, L. Wortmann, K. Arroubb and S. Mathur, Chem. Commun., 2011, 47, 10076–10078 RSC.
- T. Georgelin, V. Maurice, B. Malezieux, J.-M. Siaugue and V. Cabuil, J. Nanopart. Res., 2010, 12, 675–680 CrossRef CAS.
- M. Shao-cong, W. Hong, Q. Ming, Q. Cheng-wu, Y. Yong and L. Yong-wang, J. Fuel Chem. Technol., 2015, 43, 692–700 CrossRef.
- J. Zhu, Y. Shen, C. Yao and A. Xie, Colloid J., 2016, 78, 156–163 CrossRef CAS.
- A. Elhampour, M. Malmir, E. Kowsari, F. A. Boorboor and F. Nemati, RSC Adv., 2016, 6, 96623–96634 RSC.
- Q. He, Z. Wu and C. Huang, J. Nanosci. Nanotechnol., 2012, 12, 2943–2954 CrossRef CAS PubMed.
- H. Naeimi and R. Shaabani, Ultrason. Sonochem., 2017, 34, 246–254 CrossRef CAS PubMed.
- M. Navid and S. Rad, Ultrason. Sonochem., 2017, 34, 865–872 CrossRef PubMed.
- S. Sadjadi, M. M. Heravi and M. Daraie, J. Mol. Liq., 2017, 231, 98–105 CrossRef CAS.
- S. Sadjad, M. M. Heravi and M. Daraie, Res. Chem. Intermed., 2017, 43, 843–857 CrossRef.
- M. M. Heravi, E. Hashemi, Y. Shirazi Beheshtiha, S. Ahmadi and T. Hosseinnejad, J. Mol. Catal. A: Chem., 2014, 394, 74–82 CrossRef CAS.
- M. M. Heravi, F. Mousavizadeh, N. Ghobadi and M. Tajbakhsh, Tetrahedron Lett., 2014, 55, 1226–1228 CrossRef CAS.
- S. Sadjadi, M. M. Heravi and M. Daraie, Res. Chem. Intermed., 2017, 43, 2201–2214 CrossRef CAS.
- F. Benakashani, A. R. Allafchian and S. A. H. Jalali, Int. J. Mod. Eng. Sci., 2016, 2, 251–258 Search PubMed.
- C. Cannas, A. Musinu, G. Navarra and G. Piccaluga, Phys. Chem. Chem. Phys., 2004, 006, 3530–3534 RSC.
- P. Yuan, P. D. Southon, Z. Liu, M. E. R. Green, J. M. Hook, S. J. Antill and C. J. Kepert, J. Phys. Chem. C, 2008, 112, 15742–15751 CAS.
- S. Kumar-Krishnan, A. Hernandez-Rangel, U. Pal, O. Ceballos-Sanchez, F. J. Flores-Ruiz, E. Prokhorov, O. Arias de Fuentes, R. Esparza and M. Meyyappan, J. Mater. Chem. B, 2016, 4, 2553–2560 RSC.
- S. Sadjadi and M. Eskandari, Ultrason. Sonochem., 2013, 20, 640–643 CrossRef CAS PubMed.
- N. Salam, S. K. Kundu, R. A. Molla, P. Mondal, A. Bhaumik and S. M. Islam, RSC Adv., 2014, 4, 47593–47604 RSC.
- M. Trose, M. Dell'Acqua, T. Pedrazzini, V. Pirovano, E. Gallo, E. Rossi, A. Caselli and G. Abbiati, J. Org. Chem., 2014, 79, 7311–7320 CrossRef CAS PubMed.
- F. Movahedia, H. Masrouria and M. Z. Kassaee, J. Mol. Catal. A: Chem., 2014, 395, 52–57 CrossRef.
- M. Jeganathan, A. Dhakshinamoorthy and K. Pitchumani, Sustainable Dev. Chem. Eng., 2014, 2, 781–787 CrossRef CAS.
- H. Wu, H. Liu, W. Yang and D. He, Catal. Sci. Technol., 2016, 6, 5631–5646 CAS.
- C. Putta, V. Sharavath, S. Sarkar and S. Ghosh, RSC Adv., 2015, 5, 6652–6660 RSC.
- L. Shi, Y.-Q. Tu, M. Wang, F.-M. Zhang and C.-A. Fan, Org. Lett., 2004, 6, 1001–1003 CrossRef CAS PubMed.
- M. S. Hosseini and F. Moeini, New J. Chem., 2014, 38, 624–635 RSC.
- M. Tajbaksh, M. Farhang, H. R. Mardani, R. Hosseinzadeh and Y. Sarrafi, Chin. J. Catal., 2013, 34, 2217–2222 CrossRef CAS.
- B. Sreedhar, P. R. Surendra, B. P. Veda and A. Ravindra, Tetrahedron Lett., 2005, 46, 7019–7022 CrossRef CAS.
- B. Jyoti Borah, S. Jyoti Borah, K. Saikia and D. Kumar Dutta, Catal. Sci. Technol., 2014, 44, 4001–4009 Search PubMed.
- S. B. Park and H. Alper, Chem. Commun., 2005, 1315–1317, 10.1039/b416268d.
- M. Kidwai and A. Jahan, J. Iran. Chem. Soc., 2011, 8, 462–469 CrossRef CAS.
- N. P. Eagalapati, A. Rajack and Y. L. N. Murthy, J. Mol. Catal. A: Chem., 2014, 381, 126–131 CrossRef CAS.
- T. K. H. Ta, M.-T. Trinh, N. V. Long, T. T. M. Nguyen, T. L. T. Nguyen, T. L. Thuoc, B. T. Phan, D. Mott, S. Maenosono, H. Tran-Van and V. H. Le, Colloids Surf., A, 2016, 504, 1–8 CrossRef.
- B. Martel, Y. Leckchiri, Y. Alain Pollet and M. Morcellet, Eur. Polym. J., 1995, 31, 1083–1088 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04635a |
| This journal is © The Royal Society of Chemistry 2017 |