
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe application of a UHPLC system to study the formation of various chemical species by compounds undergoing efficient self-aggregation and to determine the homodimerization constants (KDM) with values in the high range of 106–1010 M−1†
Magdalena Hetmańska*a and
Andrzej Maciejewski *ab
*ab
aPhotochemistry and Spectroscopy Laboratory, Faculty of Chemistry, Adam Mickiewicz University in Poznań, Umultowska 89b, 61-614 Poznań, Poland. E-mail: iwonam@amu.edu.pl; mkiszko@amu.edu.pl; Fax: +48 61 829 15 55; Tel: +48 61 829 15 89
bCentre for Ultrafast Spectroscopy, Adam Mickiewicz University in Poznań, Umultowska 89, 61-614 Poznań, Poland
First published on 20th September 2017
Abstract
This work demonstrates a new concept for the use of UHPLC methodology for identification of the species formed by a self-aggregating compound depending on its concentration and solvent used, as well as to determine very large homodimerization constants (KDM = 106–1010 M−1). It is impossible to obtain such data with traditional UV-VIS and NMR measurements in compounds that undergo easy self-aggregation when their KDM values are very large (≥107 M−1). The application of the UHPLC method in tandem with a UV-VIS photodiode spectrophotometer as a detector, as well as an emission detector allowed us to perform measurements at extremely low dye concentrations (down to 10−8 M in the absorption measurement and down to 10−10 M in the fluorescence measurement). Using the well-known probe 7-aminocoumarin (C120) as a model system, we separated the monomer (M) and dimer (DM) species, and determined their concentrations and individual absorption spectra. The position of the long wavelength band in the monomer absorption spectrum agreed very well with theoretically calculated values of vertical excitation energy to the S1 state of the C120 monomer. To the best of our knowledge, this is the first report on the very efficient self-aggregation of C120 in solution, with KDM = 1.5 × 109 M−1 in polar ACN and KDM = 9 × 109 M−1 in more weakly interacting 1-chlorobutane (ChB).
1. Introduction
If the absorption spectra (AS) and emission spectra (ES) of a compound have a constant shape in the concentration range of 10−5–10−7 M, then that the compound is assumed to exist as single molecules (monomers) in solution. However, many compounds, including bisimide dyes,1 merocyanine dyes,1 squaraine dyes,1 ureido-benzoic acid,2,3 porphyrins,4,5 and phthalocyanines,4,6 and others,1–3,7 that show strong intermolecular interactions resulting from hydrogen bond formation, coordinate bonds, π–π stacking, and dispersive interactions, tend to exist as dimeric or oligomeric species. For such compounds, the homodimerization constant, KDM, is expected to be very high, particularly in weakly interacting solvents, and the dimer species can be efficiently formed in the solution at concentrations as low as 10−6 M.1,4,6,7Equilibrium constants (Keq) for systems containing monomer–dimer, cis–trans isomers, enol–keto tautomers, etc., may be determined by NMR, UV-VIS, IR, emission spectroscopy, CD, etc., when the value of Keq = 1–104,8 with HPLC also being frequently employed for these determinations.8–10 HPLC may be used when the equilibrium is reached rapidly and when the time required to reach equilibrium is negligible compared to the time required to isolate the species.9 However, HPLC cannot determine the individual species within a system. During separation, the analyte behaves as a single compound, exhibiting properties that are a composite of the properties of all the individual analyte species coexisting in the separation system. Only one peak is displayed in the chromatogram. For compounds that produced highly stable species, the equilibrium is attained slowly in comparison with the duration of the separation experiment, resulting in the presence of several peaks in the chromatogram, each corresponding to an individual species. Each species may be separated and determined individually. Thus, the HPLC method can be applied for the determination of KDM values for compounds which easily undergo self-aggregation, when the existing dimers are sufficiently stable.9,10 For the determination of very high homodimerization constants (KDM > 106 M−1), very low concentrations of analyte must be used, e.g., when KDM = 108 M−1, then c = 10−8–10−6 M should be used, so that the concentrations of monomer (M) (cM) and dimer (DM) (cDM) can undergo distinct changes, and the ratio cDM/cM = 1–10. Only UV-VIS absorption spectroscopy (when εmax ∼ 104 M−1 cm−1) and emission spectroscopy (when quantum yield of fluorescence (ΦF) is sufficiently large >10−2) permit measurements at such low concentrations. A very sensitive LC-MS method has enabled the identification of investigated compounds (as well as the formation of sufficiently stable species), but the determination of KDM values has not been possible.
For many heterodimers with very strong noncovalent interactions between monomers from two different compounds (A, B), very high association (heterodimerization) constants (Kass), of the order 106–1012 M−1, have been found by UV-VIS and fluorescence titration methods.1,2,4,5,7,11,12 Unfortunately, these methods cannot be applied to determine homodimerization constants of self-aggregated species. One can expect that KDM values of numerous compounds which easily undergo self-aggregation are very high, similar to the values of Kass.1–4,6,7,13–19 Unfortunately, there is no known experimental method for the determination of KDM > 107 M−1 for noncovalent homodimers. To determine the contents of M and DM species in a sample of known concentration, one must know the value of KDM.
Determination of very high KDM values would allow us to identify the compounds and solvents with the best properties for the formation of systems of practical importance, including supramolecular polymers,2,13,14,20,21 aggregation-induced emission active materials,7,22 optical sensors based on dimer–monomer equilibrium as near-infrared fluorescence sensors for proteins23 to sense pH in a broad range,24 and for nucleic acids determination.25 For the determination of KDM, the monomer, dimer, or larger species formed by the investigated compounds must be identified. NMR is an indispensable method for determining if a studied compound occurs as a monomer, a dimer or a mixture.1–3,26 A comparison of NMR spectra distinguishes between homodimers and heterodimers.1–3,26,27 However, to analyze a sample by NMR methodology, sample concentrations must be ≥10−4 M and thus, NMR spectroscopy is not useful to determine KDM > 106 M−1.
Simple simulations (Table S1†) show that for the compounds whose KDM values are in the range of ≤104 M−1, only monomers are present (cM/cDM = 100) in the solution, if the studies are carried out in the range of low concentrations (≤10−6 M). On the other hand, if KDM = 106–107 M−1, then in the concentration range of 10−6–10−7 M, both monomers (M) and dimers (DM) are present (Table S1†). Thus, the AS and ES represent the combined spectra of M and DM. If the value of KDM ≥ 108 M−1, then even in the range of very low concentrations (10−7–10−8 M), the concentration of dimer is greater than that of monomer (Table S1†). For a compound with KDM ≥ 107 M−1 in the concentration range ≥10−6 M, the AS would represent mostly the absorption of the dimer and not that of the monomer, as commonly assumed. Therefore, the spectrophotometric determination of the AS of the monomer is not applicable to compounds with values of KDM ≥ 107 M−1. To obtain the monomer at concentration 10 times greater than that of dimer, when KDM = 1 × 108 M−1, the concentration of the investigated compound must be less than 1 × 10−9 M (Table S1†). The AS for such low concentrations cannot be measured, even when the values of εmax are very high (∼105 M−1 cm−1) and the cell path length is 10 cm.
For compounds that undergo easy self-aggregation, it is difficult to establish which of the species are present and to determine their concentration, their AS and the ε(λ) values. To obtain these data, it is necessary to determine the KDM value. For the compounds whose monomer and dimer have similar AS and ES,28–33 it is impossible to determine their concentrations (i.e., those of monomer and dimer) and thus their KDM value by measurements of AS on a spectrophotometer and ES on a spectrofluorimeter.
In this work, we demonstrate that by using UHPLC in conjunction with a photodiode UV-VIS spectrophotometer (PDA) and an emission detector, it is possible to determine the species formed in the range of very low concentrations (10−6–10−8 M) after their previous separation. This methodology opens the door to determining very high values of KDM = 106–1010 M−1. The determination of such high KDM values will permit the first ever determination of the type of species (only monomer, only dimer or both) of compounds undergoing efficient self-aggregation in a sample of known concentration.
Coupling the HPLC technique with photodiode UV-VIS spectrophotometers and emission detectors makes a very sensitive method. In an earlier work, we studied compounds with very weak emissions without interference from impurities by using an HPLC system.34 We defined the main assets of using HPLC and UHPLC by studying three tautomers of gossypol34 and two complexes formed by benzopyrantione with water molecules.35 The HPLC method was successfully applied to the separation of monomers, dimers and larger aggregates.36–39 Size exclusion-HPLC has been used to detect and characterize M, DM and higher aggregates of protein39 as well as to study M–DM equilibrium of gramicidin.36 Würthner at al.1 first separated conformationally stable atropo-diastereomers of perylene bisimides, derivatives with a high activation barrier (Ea ≥ 97 kJ mol−1) for racemization. In recent work on 1-butyl-3-methylimidazolium tetrafluoroborate, we showed for the first time that linking together the HPLC setup with a high performance PDA and emission detector enabled separation of the species formed by this ionic liquid from impurities present in the solution and independently measured the absorption and emission spectra and other spectral properties of the species and the impurities.40
7-Aminocoumarins are the derivatives of 1,2-benzopyrone having amino groups at position 7 of the 1,2-benzopyrone moiety. They are often used as fluorescence probes,41–43 chemosensors,44 in biological and biomedical sciences,45,46 and in dye lasers.47 Coumarin-120 (7-amino-4-methyl-1,2-benzopyrone) (Scheme 1), abbreviated as C120, belongs to this group, having a simple NH2 group. The fluorescence quantum yields of this dye in solvents of moderate to higher polarities are very high ΦF ≥ 0.5 (ref. 48 and 49) and the absorption and especially emission maxima, as well as Stokes shifts, were found to be strongly dependent on polarity and hydrogen bonding ability of the solvent. Therefore, C120 has been widely used as a probe for studying solute–solvent interactions and solvatochromic properties of various systems and it has been the focus of intensive studies, both experimental48,49 and theoretical50 ones. C120 is also a well-known laboratory reagent, employed as a fluorogenic probe for detection of enzymatic activity.51 This dye has been used as a fluorescence probe to analyze glycoproteins, monosaccharides and N-linked oligosaccharides via chromatography.52 C120 has also shown effective antitubercular,53 antibacterial and antifungal54 activities. It is known to be non-toxic to humans and animals.
 | ||
| Scheme 1 Chemical structure of the coumarin-120 (C120) molecule. | ||
Spectral and photophysical studies of C120 have usually been performed with dye solutions over the concentration range of 10−4–10−5 M.41–43,48,49 It has always been assumed that within this concentration range, the dye exists in the solution as a monomer and/or possibly as the complex formed by the dye molecule and solvent molecules in hydrogen bonding solvents. However, the formation of stable dimers or even larger aggregates has been recently postulated for several derivatives of aminocoumarins in the concentration range of c = 10−4–10−6 M.55–57 These properties made C120 a good candidate for our investigations using UHPLC in conjunction with a photodiode UV-VIS spectrophotometer and an emission detector (UHPLC-PDA-FL system) for identification of the type of species made by this compound in a wide range of concentrations (c = 10−5–10−8 M), and for determination of its KDM.
2. Experimental section
Coumarin 120 (C120), of purity >99%, (Applied Photophysics Ltd.) was used as received. Acetonitrile (ACN) (Sigma Aldrich, Chromasolv gradient grade for HPLC ≥99.9%) and 1-chlorobutane (ChB) for HPLC (Sigma Aldrich) were used as an eluent and a solvent. The absorption spectra of C120 labeled as conventional were measured using a double beam spectrophotometer type V-650 (Jasco). The emission spectra of C120 labeled as conventional were measured using a Jobin Yvon-Spex Fluorolog3-22 spectrofluorimeter.All chromatographic measurements were performed using a Waters Ultra-High Performance Liquid Chromatography (UHPLC) system with a Photodiode Array Detector (PDA) (flow cell path length of 2.5 cm, volume: 1.25 μl) with linear working range for absorbance near A = 2 and Acquity UPLC fluorescence detector (flow cell of 2 μl volume). An Empower 2 chromatographic interface was used for data collection. The Kinetex Phenyl-Hexyl 150 × 2.10 mm column (Phenomenex) packed with 1.7 μm particles with pore size 100 Å was used. Isocratic elution was carried out with acetonitrile (ACN) or 1-chlorobutane (ChB) at the flow rate of 0.10, 0.25, 0.50 or 1.0 ml min−1. All measurements were carried out at ambient temperature. Isocratic elution mode with ACN or ChB was used and no buffer was added to the eluent. The Core-Shell Technology (a Kinetex type) columns used in this work are much more efficient than traditional fully porous sub-2 μm columns, yielding a remarkable chromatographic resolution, higher peak capacities, and a greater sensitivity because of very narrow chromatographic peaks, ΔtR1/2 = 1–3 s (absorption) and ΔtR1/2 = 3–4.5 s (emission). The use of the UHPLC method allowed a reliable determination of very low concentrations of the separated compound and species by at least one order of magnitude lower than those that could be determined with the HPLC method. All dye concentration data for the UHPLC method measurements are given for injected solutions.
Because it was necessary to work at very low concentrations of C120 (cC120 = 10−6–10−9 M), we took special precautions to assure that our data were accurate and reliable. Within one measuring cycle, the measurements were carried out repeatedly with numerous injections. Only reproducible absorption chromatograms and absorption spectra as well as reproducible emission chromatograms were selected for analysis of results. It is worth noting that the signal-to-noise ratios (S/N) for the lowest investigated concentrations (cC120 = 1.9 × 10−8 M) were sufficiently high (S/N ≥ 3 for the peak B and S/N ∼ 25 for the peak A) when absorption chromatograms were obtained in the range of λ = 280–340 nm. Since the measurement error was very small (ΔA = ±2 × 10−5), the value of S/N ≥ 3 was reliable. The peaks A and B were also intensive in emission chromatograms, (particularly the peak A). For low C120 concentration (1.9 × 10−8M), the S/N ratio was greater than 500 for the peak A and S/N ∼ 50 for the peak B, provided that suitable λex and λem were selected. Due to this, accurate measurements of emission chromatograms were possible even for concentrations of 10−9–10−10 M.
To eliminate the effect of impurities in the solvent and the eluent as well the wavelength effect on the contribution from reflected and dispersed light, measurements were carried out for C120 samples in ACN and for ACN alone and also for C120 samples in ChB and for ChB alone. Absorption and emission chromatograms and AS originating solely from the dimer (DM) and the monomer (M) (see below) were obtained by subtracting experimental absorption and emission chromatograms and AS of the solvent (ACN and ChB) from experimental absorption and emission chromatogram and AS of a sample (C120 in ACN and C120 in ChB). For cC120 ≥ 10−6 M, absorption and emission of ACN and ChB alone was so small that they could be neglected.
3. Results and discussion
Absorption and emission spectra of C120 in the concentration range of 10−4–10−6 M (in ACN and other solvents) measured on UV-VIS spectrophotometers and on spectrofluorimeters, both by us and other authors, had the same shape.48,49 In low concentrations (10−4–10−6 M) of C120 applied in these measurements, it was always assumed that the spectra originate from single molecules (monomers) of C120.48,49 To verify this assumption, measurements of absorption chromatograms and AS, as well as of emission chromatograms, for solutions of C120 in ACN in the concentration range of 10−5–10−8 M were performed using the UHPLC-PDA-FL system.The application of this new generation UHPLC instrument and columns of core–shell type produced on absorption chromatograms very narrow peaks (ΔtR1/2 = 1.5–2 s) of separated species and allowed measuring very low absorbancies (A = 10−3–10−4) with a very small error (ΔA = ±2 × 10−5). Due to such instrumental parameters, the separation (at least a partial one) of species with very close retention times (differing by merely 1 second) is possible and measurements can be carried out in the spectral range of λ = 190–800 nm and at very low concentrations (10−6–10−8 M). The complete separation of M (peak B) from DM (peak A), obtained in the absorption chromatograms (Fig. 2, 5 and S1†), was also maintained in the emission chromatograms (Fig. 6, S2 and S3†), although it appeared to be slightly worse. It should be mentioned that due to installing the PDA detector first and placing the emission detector immediately after it, tmaxR of M and DM peaks in the emission chromatograms are by 3–6 s longer depending on the eluent flow rate. Since the cell volume (2 μl) in the emission detector was considerably higher than that (1.25 μl) in the PDA detector and data acquisition in the emission detector is slower (20–50 Hz) than in the PDA detector (80 Hz), the peaks of M and DM in the emission chromatograms were about twice as wide as those in the absorption chromatograms.
3.1. Species formed by C120 in ACN
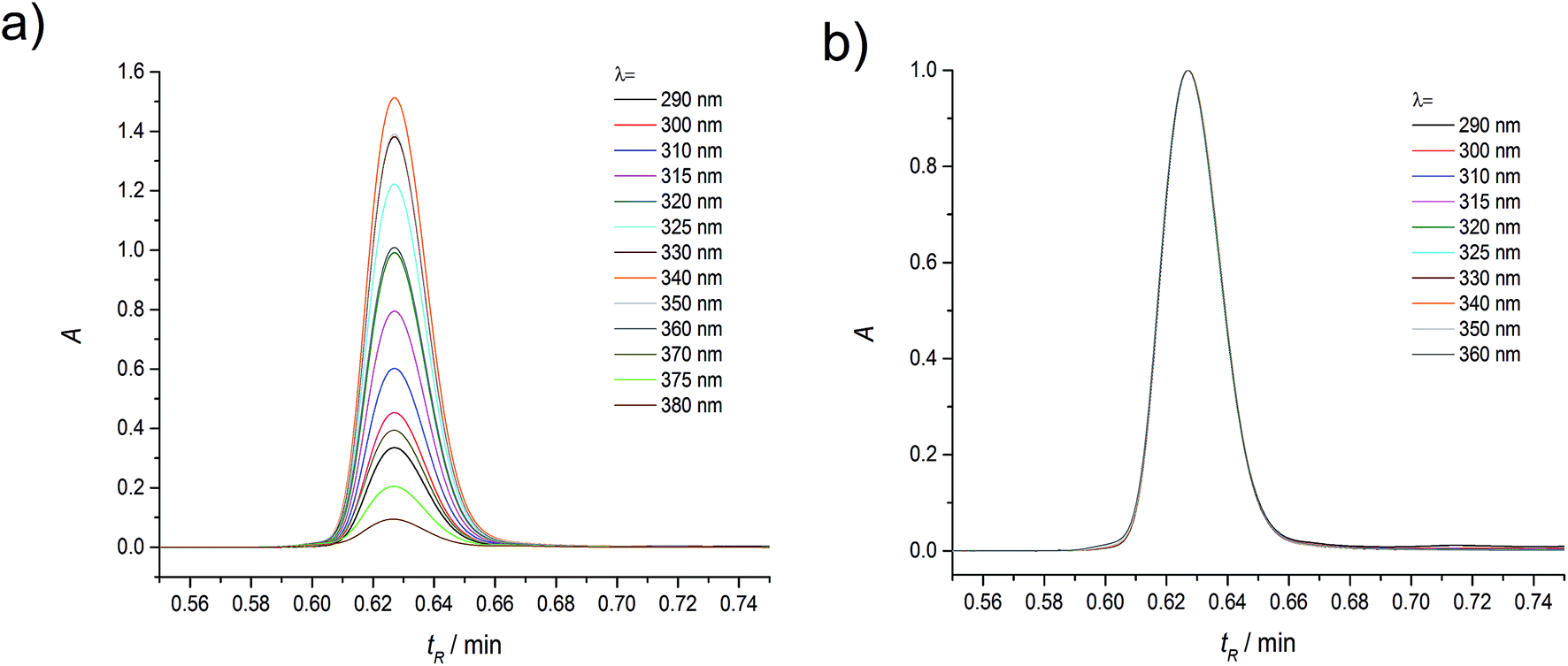 | ||
| Fig. 1 Raw (a) and normalized (b) absorption chromatograms of coumarin-120 (C120) in ACN at the dye concentration of 4.6 × 10−5 M, recorded over a wide range of detection wavelengths (the flow rate was 0.5 ml min−1). | ||
Moreover, the position of the maximum and the shape of the long-wavelength band in the absorption spectrum determined for different retention times did not change and were the same as the absorption spectrum measured using an UV-VIS spectrophotometer for C120 solution (c = 10−4–10−6 M) in ACN (see Fig. 4b). The only peak present in the absorption chromatogram in the range 10−4–10−5 M (Fig. 1) can be ascribed to the monomer of the investigated compound, in compliance with the all previous interpretations. However, the KDM value for the compounds easily undergoing self-aggregation can be large enough to expect that the above peak can originate from the dimer. Therefore, to determine whether the only peak present in the absorption chromatogram should be ascribed to the monomer or the dimer, we performed measurements in a wide enough range of very small concentrations of C120.
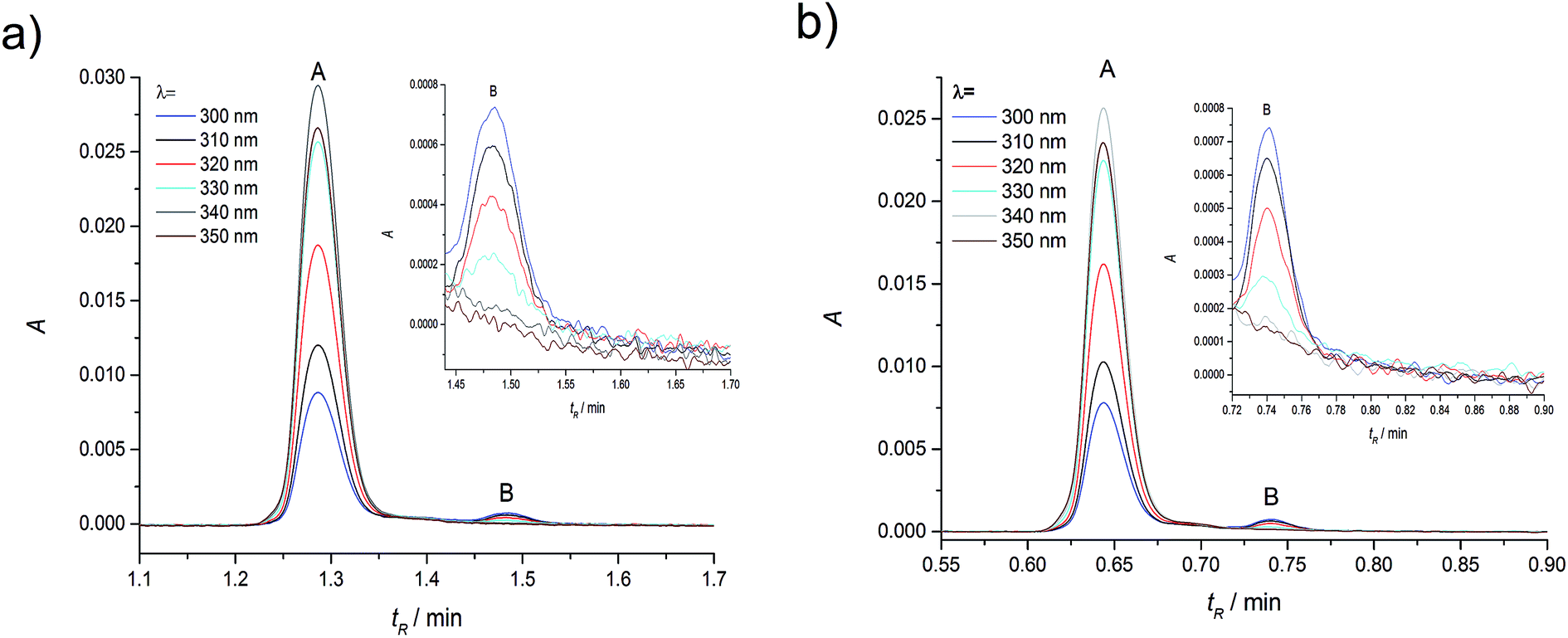 | ||
| Fig. 2 Experimental absorption chromatograms of coumarin 120 (C120) in ACN at the dye concentration of 1.1 × 10−6 M, recorded over a wide range of detection wavelengths. (a) Flow rate of 0.25 ml min−1; (b) flow rate of 0.5 ml min−1. | ||
As in the case of higher dye concentrations, the absorption spectra, in particular the position of the maximum and the shape of the long-wavelength band determined for the retention times within the chromatographic peak A (Fig. 3a), corresponded to the absorption spectrum measured using a Jasco V-650 Spectrophotometer for the C120 solutions in ACN, (Fig. 4b). The absorption spectra recorded for different retention times from the peak B (Fig. 3b) were the same or similar, however, they were totally different from those within the chromatographic peak A. The long-wavelength bands in the AS, determined at the maximum of the peaks A and B and normalized to the same intensity, are presented in Fig. 4. The maximum of the long-wavelength band (λ ∼ 295 nm) in the absorption spectrum recorded at the peak B was clearly shifted towards shorter wavelengths relative to the corresponding band in the spectrum recorded at the peak A (the maximum at 340 nm).
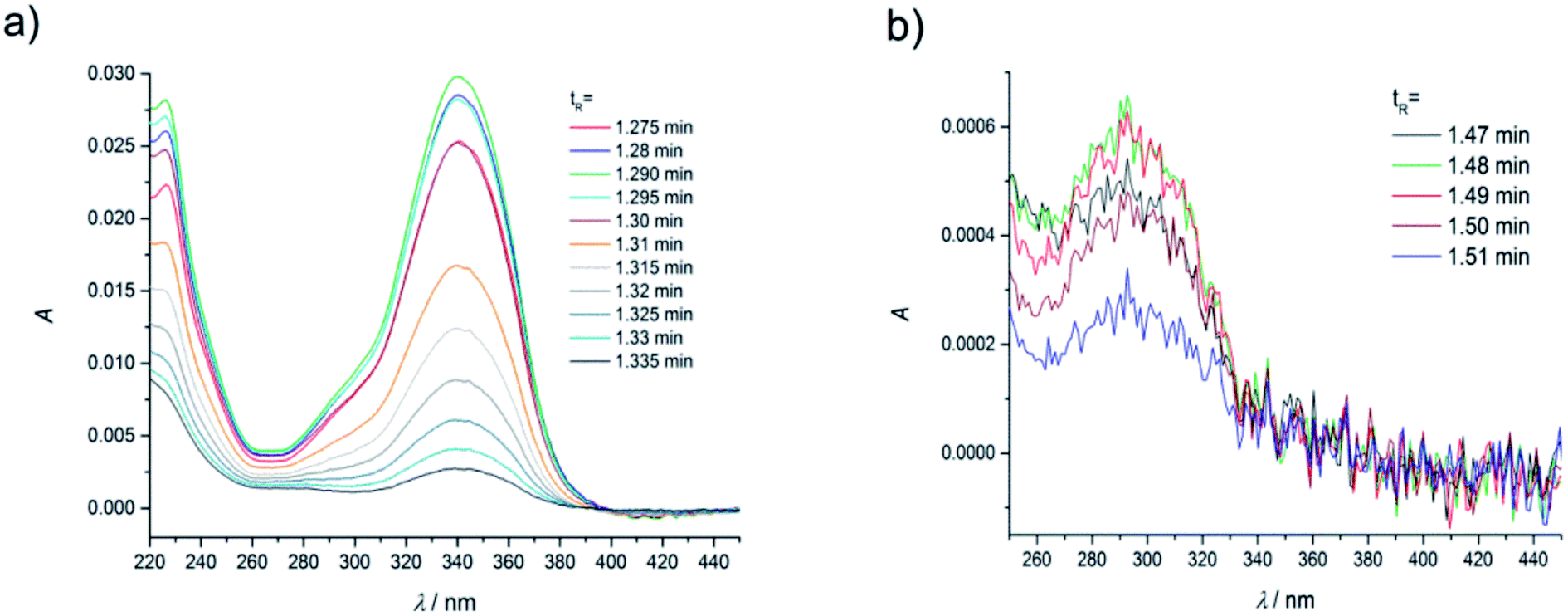 | ||
| Fig. 3 Absorption spectra of coumarin-120 (C120) in ACN at the dye concentration of 1.1 × 10−6 M, recorded (a) in the range of retention times 1.275–1.335 min (peak A) from Fig. 2a and b in the range of retention times 1.47–1.51 min (peak B), from Fig. 2a, flow rate of 0.25 ml min−1. | ||
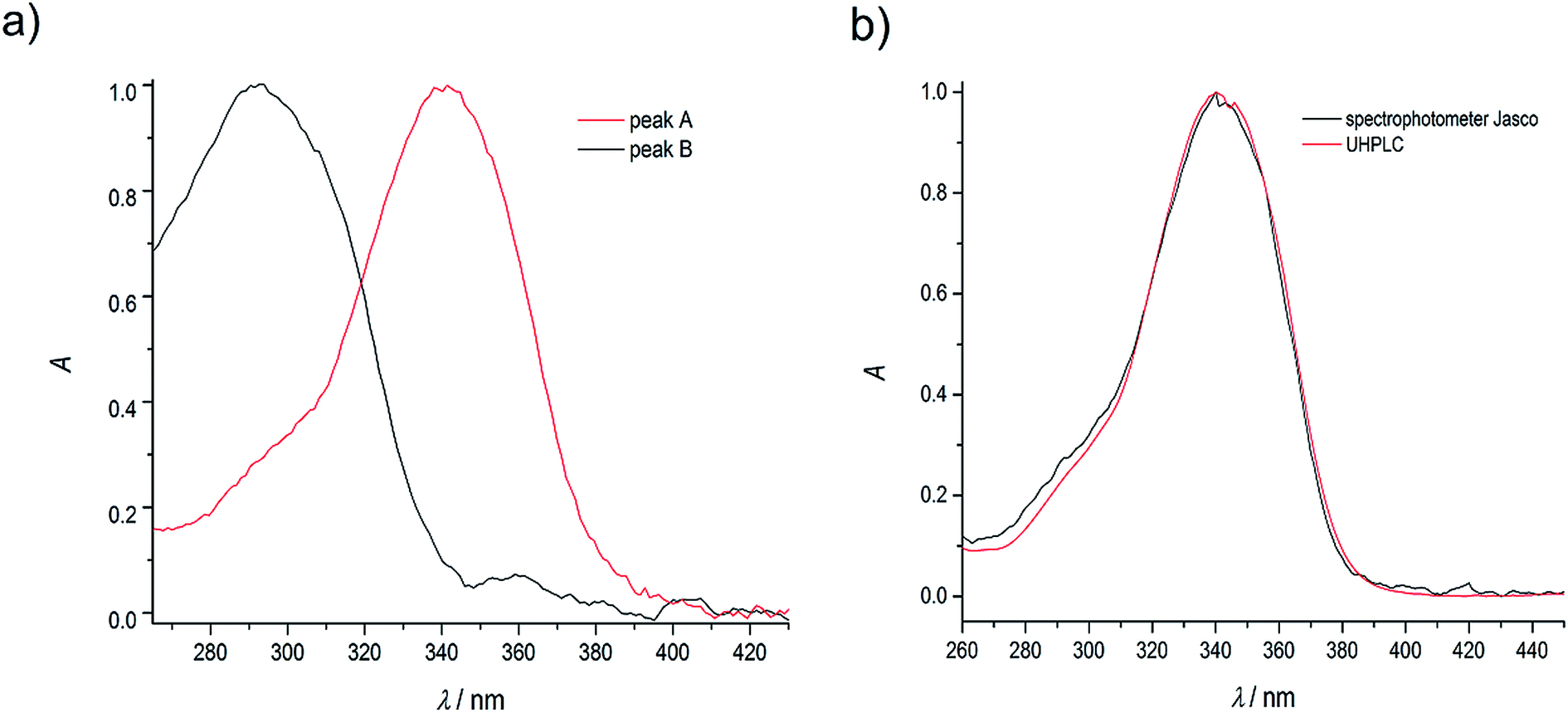 | ||
| Fig. 4 (a) Normalized absorption spectra of coumarin-120 (C120) in ACN at the dye concentration of 1.1 × 10−6 M, recorded at the maxima of chromatographic peaks A and B (see Fig. 2 and 3); (b) a comparison of normalized absorption spectra at the maximum of the peak A (a) and absorption spectrum measured using a Jasco V650 UV-VIS spectrophotometer at the dye concentration of 4.6 × 10−7 M. | ||
The presence of two peaks in the absorption chromatogram and clearly different AS within both peaks suggested the existence of at least two absorbing species in the C120 solutions.
The two peaks present in the chromatogram (Fig. 2) could originate from:
(a) two monomers with considerably different concentrations,
(b) the monomer and an impurity present in it,
(c) the monomer and the dimer.
To distinguish among these possibilities, we conducted measurements over a wide range of concentrations. Since in the case of concentrations ≥10−5 M only the peak A was observed (Fig. 1), we also carried out studies over the range from 1.1 × 10−6 to 1.9 × 10−8 M. Herein, we present the outcomes of studies with three concentrations that show marked changes in the ratio of Amax (peak B)/Amax (peak A) and for the sample of the lowest concentration the above ratio was as high as possible. By evaluating the data in this way for C120 (particularly the interpretation of absorption chromatograms) our conclusions could be maximally reliable.
Measurements of absorption chromatograms by using a photodiode spectrophotometer and absorption spectra taken for chosen tR values are single beam measurements. Studies carried out with samples having the lowest C120 concentrations (c = 10−7–10−8 M) inevitably entailed measuring very low values of absorbance (A = 10−3–10−4). Because we performed our studies under conditions that corrected for any experimental artifacts from the solvents (see Experimental procedures), we were able to obtain reliable absorption chromatograms, even at very small concentrations (Fig. S1†). Therefore, we were confident that the number of peaks in a chromatogram corresponded to the number of species formed by C120.
The presence of two peaks in all absorption chromatograms, characterized by identical profiles, as well as the same tmaxR and ΔtR1/2 (for the same flow rates) indicated that two species formed by C120 existed in the range of cC120 = 10−6–10−8 M.
Results of measurements obtained in compliance with the procedures developed by us, described in the Experimental section are presented in Fig. 5. Shapes of peak A and peak B, determined across a wide range of wavelength, were very similar. Also the positions (tmaxR) and widths of peaks were very similar in the entire range of C120 concentrations. Moreover, the absorption spectra recorded for different retention times in the range of each peak are similar (Fig. 3). The chromatographic peak A, observed in the chromatograms recorded for the whole range of C120 concentrations (cC120 = 1.1 × 10−6–1.9 × 10−8 M) was always characterized by a considerably higher intensity than the peak B.
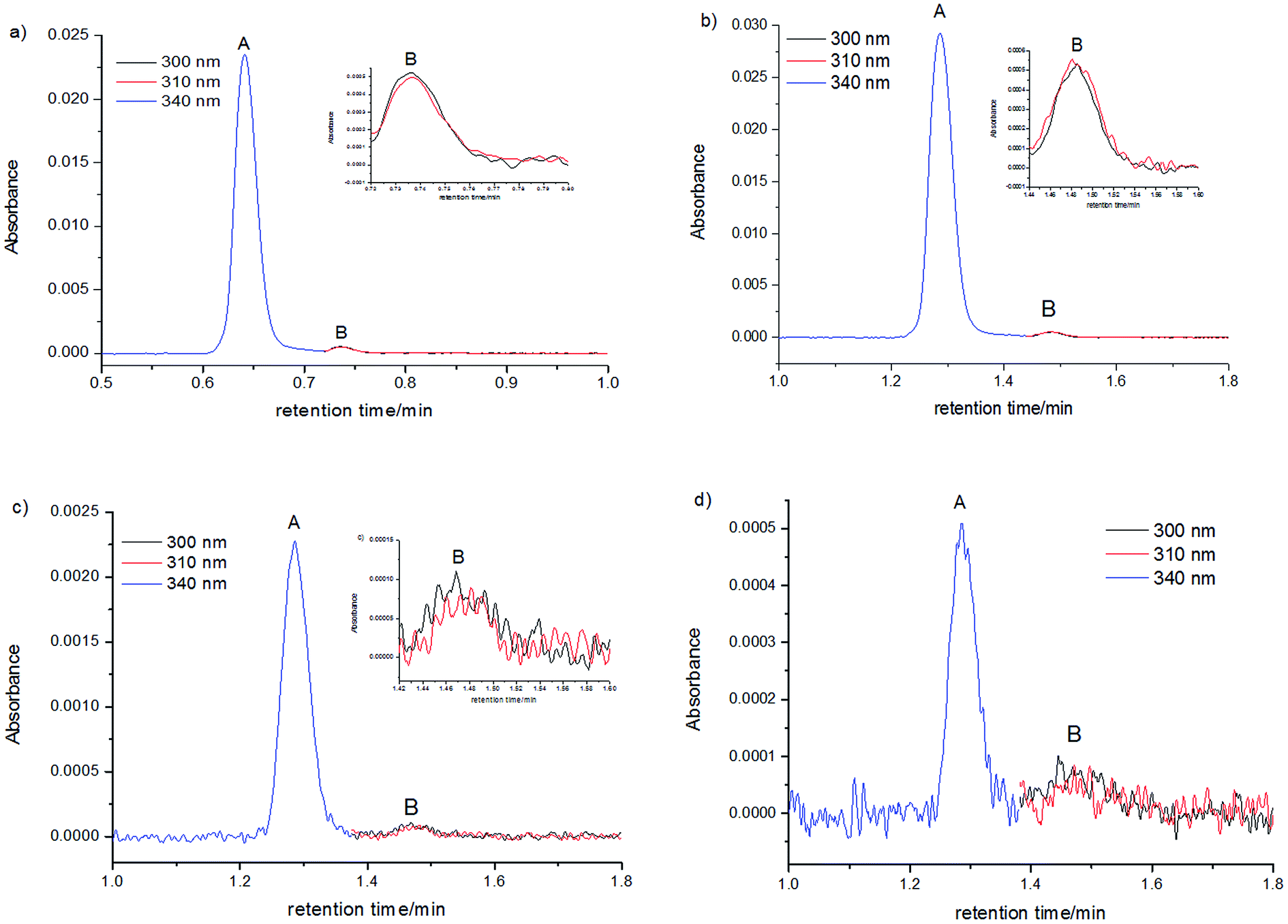 | ||
| Fig. 5 Absorption chromatograms for C120 samples in ACN with concentrations of 1.1 × 10−6 M (a and b) 9.3 × 10−8 M, (c) 1.9 × 10−8 M, (d) measured for the dimer (see below). Peak A, (λ = 340 nm) in the retention time range of 1.0–1.42 min (b–d) and 0.5–0.72 min (a) and for the monomer (see below); peak B, (λ = 300, 310 nm) in the retention time range of 1.42–1.8 min (b–d) and 0.72–1.0 min (a); (a) flow rate 0.5 ml min−1, (b–d) flow rate 0.25 ml min−1. | ||
A comparison of peaks A and B in samples differing significantly in their concentrations (Table 1) indicated that absorbance of the peak A was reduced as the C120 concentration decreased. On the other hand, absorbance of the peak B decreased much less. With the reduction in concentration from 1.1 × 10−6 M to 1.9 × 10−8, absorbance of the peak A diminished ∼61-fold, whereas that of the peak B decreased only ∼7-fold. Thus, the peak A and the peak B could originate neither from two monomers of C120 nor from C120 monomer plus an impurity. Indeed, the absorbances of both peaks decreased as the concentration of C120 decreased, but to a different extent. The clear increase in the ratio of Aλ=300 (peak B)/Aλ=340 (peak A) with the decrease in C120 concentration (Table 1) suggests that the peak A originates from the dimer formed by two molecules of C120, whereas the peak B arises from a single molecule of C120 (the monomer).
| cC120 [M] | cC120(rel) | N | Aλ=340a | Aλ=340(rel)a | Aλ=300b | Aλ=300(rel)b | Aλ=300/Aλ=340 | cM/cDM |
|---|---|---|---|---|---|---|---|---|
| a Peak A, the retention time tmaxR = 1.29 min.b Peak B, the retention time tmaxR = 1.48 min; Aλ=300 and Aλ=340 are mean values determined from several independent measurements of absorption chromatograms, N – the number of measurements of absorption chromatograms used for the determination of mean values of Aλ=300 for the monomer and Aλ=340 for the dimer. | ||||||||
| 1.1 × 10−6 | 57.9 | 3 | 0.0293 | 61.0 | 0.00053 | 7.4 | 0.018 | 0.038 |
| 9.3 × 10−8 | 4.9 | 6 | 0.0023 | 4.8 | 0.00012 | 1.7 | 0.052 | 0.11 |
| 1.9 × 10−8 | 1.0 | 8 | 0.00048 | 1.0 | 0.00007 | 1.0 | 0.15 | 0.32 |
Two molecules of C120 can form two identical hydrogen bonds between each other, between the oxygen atom of the carbonyl group of one molecule and the N–H bond of another molecule. Moreover, π–π interactions, as well as electrostatic and dispersion interactions can substantially contribute to the dimer formation. There are no literature reports presenting results or discussing formation of a homodimer by C120 molecules. A very efficient formation of dimers with various structures have been recently calculated for 7-hydroxycoumarin57 and the dimerization energy for the most stable dimer was very similar to that obtained by us for C120 (7-aminocoumarin).58
To confirm whether the peak A in the chromatograms originates from the dimer and peak B from the monomer (Fig. 2, 5 and S1†) we have calculated concentrations of the monomer (cM) and dimer (cDM) using the eqn (1):
| KDM = (cDM)/(cM)2 | (1) |
To directly benefit from the calculated cM and cDM, the calculations were performed for samples with the same concentrations as those used in experiments on C120 in ACN, knowing that cC120 = cM + 2cDM.
Since the concentration of the monomer cM (peak B) is considerably lower than the dimer concentration cDM (peak A), even for the lowest concentration (cC120 = 1.9 × 10−8 M) (Fig. 5d), the KDM value for C120 in ACN must be very high. Therefore, we have performed calculations for very high values of KDM = 105–1010 M−1, covering a wide concentration range to obtain values of cM and cDM (Table S2†). To employ the results of calculations (Table S2†) for the confirmation that the peak A originates from dimer and the peak B from the monomer (Fig. 2, 5 and S1†), we have calculated relative concentrations of the monomer cM(rel) and dimer cDM(rel), Table 2, using the values of cM and cDM presented in Table S2.† As the concentration of C120 increased, relative changes of cDM(rel) were considerably greater than relative changes of cM(rel), calculated over the whole range of KDM values. This means that the peak A observed in absorption chromatograms (Fig. 5), for which relative changes of Aλ=340DM(rel) are large (Table 1), should be assigned to the dimer and the peak B, for which changes of Aλ=300M(rel) were small, should correspond to the monomer.
| cC120 [M] | cC120(rel) | KDM = 1 × 105 [M−1] | KDM = 1 × 106 [M−1] | KDM = 1 × 107 [M−1] | KDM = 1 × 108 [M−1] | KDM = 1 × 109 [M−1] | KDM = 1 × 1010 [M−1] | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cDM(rel) | cM(rel) | cDM(rel) | cM(rel) | cDM(rel) | cM(rel) | cDM(rel) | cM(rel) | cDM(rel) | cM(rel) | cDM (rel) | cM(rel) | ||
| 1.1 × 10−6 | 57.9 | 2457 | 49.6 | 838 | 29 | 205.6 | 14.35 | 90 | 9.5 | 66.7 | 8.07 | 60.7 | 7.79 |
| 9.3 × 10−8 | 4.9 | 23.9 | 4.89 | 18.8 | 4.35 | 10.5 | 3.24 | 6.47 | 2.54 | 5.3 | 2.31 | 5.04 | 2.24 |
| 1.9 × 10−8 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
Alternatively, the peak B may originate from dimer, while the peak A may arise from a larger aggregate, such as trimer. However, it is unlikely for the monomer content to be so small at very low C120 concentrations (cC120 = 10−7–10−8 M) that its peak is undetectable in absorption chromatogram. In such a case, the KDM value should be much greater than 1010 M−1, which seems improbable. Also the trimerization constant (KTM) would be unbelievably high.
3.2. Dimerization constant, KDM, determination for C120 in acetonitrile and 1-chlorobutane
Knowledge of KDM value permits the investigator to determine the optimal concentration and solvent to be used with a compound. Depending on the dimer stability, KDM values can vary widely and cover many orders of magnitude. Our calculations presented in Table S1† confirm that the contents of the monomer and the dimer depend very strongly on the concentration of a studied compound and the KDM value and thus, the monomer-to-dimer concentration ratio for a given concentration of a compound cannot be predicted if the KDM value is unknown. Concentration of a studied compound that is necessary for the presence of monomer only is also unknown.As emphasized earlier, large KDM values can be determined with sensitive UV-VIS or fluorescence emission spectroscopy methods. However, the spectra obtained in these studies are the summary absorption and emission of all species that are present in the sample. Therefore, when determining KDM on the basis of changes occurring in a UV-VIS absorption spectrum with a change in concentration, it is assumed that only one monomer and one dimer are present in the whole concentration range of the compound studied.1–3 Moreover, it is assumed that the presence of isosbestic points indicates the existence of equilibria between the two species. The determination of KDM = 106–107 M−1 from UV-VIS spectroscopic measurements is also subject to large errors, because despite using low concentrations (usually 10−5–10−6 M), the dimer is the predominant species in the sample, cDM/cM > 3. The very small changes in the absorption spectrum across the whole concentration range make it difficult to establish the presence of an isosbestic point.1,2 It should be noted that the presence of isosbestic point in absorption spectra of samples with different concentrations of a compound studied does not ensure that only one monomer and one dimer are present.59 However, such an assumption has to be made to enable the determination of KDM value from measurements of UV-VIS spectra of the samples on a spectrophotometer. Moreover, when the absorption spectra of monomer and dimer are similar, the determination of KDM by UV-VIS methods is very difficult and not very accurate, especially if the absorption spectra are very similar.15,37–41
If the dimerization constant KDM of a compound is very large (108–109 M−1), then even for a very low sample concentration of about 10−6 M, the content of dimer species in solution will always be greater than that of monomer species (Table S1†). If the KDM is 1.5 × 109 M−1 (data obtained for C120 in ACN), the concentration of C120 should be less than 10−9 M (Table S1†), to have monomer species content significantly greater than that of dimer species. However, under such conditions, the absorbance of the two species would be too small to measure, even if their molar absorption coefficients εmax were fairly large (of the order of 104 mol−1 dm3 cm−1) and quantum yield of fluorescence ΦF < 10−2.
As indicated below, when applying our proposed UHPLC method in tandem with UV-VIS photodiode spectrometry and independently, additionally with an emission detector, the peaks assigned to dimer (A) and monomer (B) species are well resolved and intense enough to be measured with high (DM) and satisfactory (M) accuracy in the absorption chromatograms (see Fig. 5 and S1†) and in the emission chromatograms (Fig. 6 and S2†) and we were able to calculate KDM values. Under such conditions, concentrations of the monomer and dimer species can be determined independently using a UHPLC-PDA system (absorbance measurements) or UHPLC-PDA-FL system (absorbance and emission measurements).
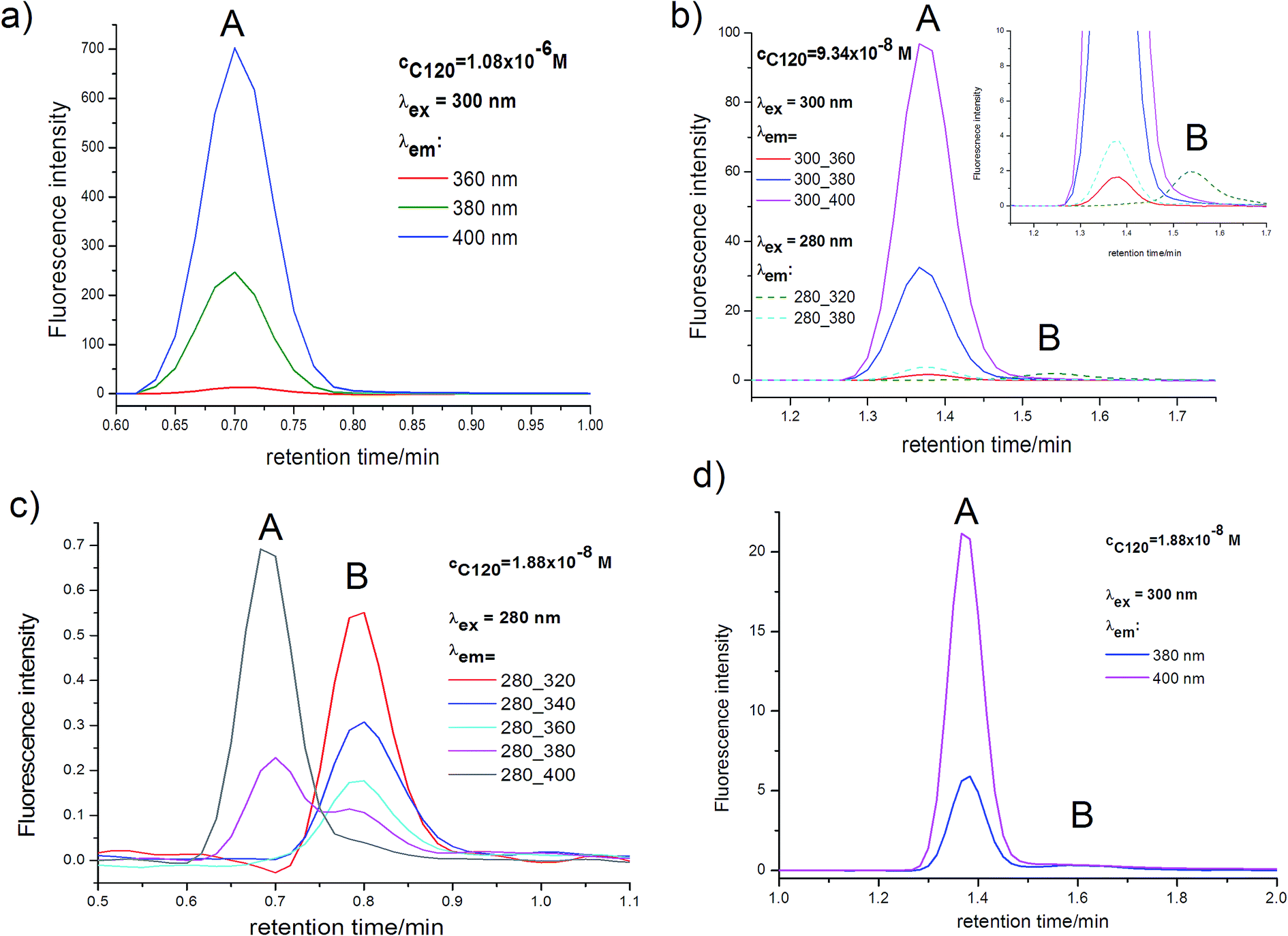 | ||
| Fig. 6 Emission chromatograms for C120 samples in ACN with concentrations of 1.9 × 10−8 M (a), 9.3 × 10−8 M (b), 1.1 × 10−6 M (c). Peak A – dimer, peak B – monomer; (a and c) flow rate 0.25 ml min−1, (b) flow rate 0.25 ml min−1. | ||
The value of KDM can also be determined by comparing relative absorbancies at the monomer peak maximum Aλ=300M(rel) (peak B) (Table 1), with theoretically calculated values of cM(rel) (Table 2) and relative absorbancies at the dimer peak maximum Aλ=340DM(rel) (peak A) (Table 1), with theoretically calculated values of cDM(rel) (Table 2). The values of Aλ=300M(rel) for the monomer and Aλ=340DM(rel) for the dimer were determined at one wavelength therefore, in compliance with the Lambert–Beer law, they depend only on the monomer and dimer concentrations. Thus, these values can be directly compared with the calculated relative concentration of the monomer (cM(rel)) and of the dimer (cDM(rel)) (Table 2), for the same C120 concentration. As results from Table 2, the cM(rel) and cDM(rel) values strongly depended on C120 concentration and changed considerably with KDM value. Having the values of Aλ=300M(rel) (the monomer peak) and Aλ=340DM(rel) (the dimer peak), determined from absorption chromatograms in the range cC120 = 1.1 × 10−6–1.9 × 10−8 M (Fig. 5 and Table 1), as well as calculated values of cM(rel) and cDM(rel) (Table 2) in the samples of the same concentrations of C120 (which were investigated experimentally), it was possible to determine the most reliable KDM values. Both Aλ=300M(rel) and cM(rel) values for the monomer and also Aλ=340DM(rel) and cDM(rel) values for the dimer (Table 1) were similar to calculated cM(rel) and cDM(rel) values, when KDM was very large and fell within the range 1 × 109–1 × 1010 M−1 (Table 2). Thereby, KDM values, estimated on the basis of two different relationships and results of the measurements of absorption chromatograms, were very large and similar.
The most sensitive method of accurate determination of very low concentrations is emission spectroscopy, provided that the emission quantum yield (ΦF) is large. The fluorescence of C120 in practically all solvents is very intense,48,49 and only in saturated hydrocarbons is somewhat less intense.48 In the case of C120 (cC120 ∼ 10−5 M) in ACN ΦF = 0.63.48 Due to this fact, the determination of M and DM content on the basis of measurements of emission chromatograms, even at very low C120 concentrations (cC120 = 10−8–10−10 M) is possible. KDM values can be estimated using results of the measurements of emission chromatograms (Fig. 6, Tables 6 and 7). Very good quality of these results, particularly for the DM peak, made it possible to precisely determine very large KDM values including KDM = 108–1010 M−1. The results obtained from emission chromatograms will be presented in the further part of this paper.
The results of measurements of absorption chromatograms. The determination of concentrations of monomer and dimer in solution requires the knowledge of molar absorption coefficients of both species. The value of molar absorption coefficient for the dimer at the maximum of its long-wavelength absorption band (λmax = 340 nm) was determined to be εmaxDM = 32
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 mol−1 dm3 cm−1. It was determined directly from the absorption spectra measured using a Jasco V-650 spectrophotometer for C120 solutions in ACN at concentrations in the range 2 × 10−5–5 × 10−5 M, when the dimer species are exclusively present in the sample (cDM/cM ≥ 100) (we verified that the AS measured with UV-VIS spectrophotometer was the same as the AS of the dimer measured from the peak A (Fig. 4b)). On the other hand, for the monomer species it was assumed on the basis of the exciton theory60 that at the maximum of the long-wavelength absorption band (λmax = 295 nm), the molar absorption coefficient εmaxM was half that for the dimer species, i.e., εmaxM = 16300 mol−1 dm3 cm−1.
600 mol−1 dm3 cm−1. It was determined directly from the absorption spectra measured using a Jasco V-650 spectrophotometer for C120 solutions in ACN at concentrations in the range 2 × 10−5–5 × 10−5 M, when the dimer species are exclusively present in the sample (cDM/cM ≥ 100) (we verified that the AS measured with UV-VIS spectrophotometer was the same as the AS of the dimer measured from the peak A (Fig. 4b)). On the other hand, for the monomer species it was assumed on the basis of the exciton theory60 that at the maximum of the long-wavelength absorption band (λmax = 295 nm), the molar absorption coefficient εmaxM was half that for the dimer species, i.e., εmaxM = 16300 mol−1 dm3 cm−1.The dimerization constant KDM for C120 in ACN was determined from the equation:1,61,62
| KDM = (1 − α)/(2α2cC120) | (2) |
The concentration of the dimer (cDM) was calculated from the area under the chromatographic peak A (PDM), taking into account the value of ε(λ) for λ at which the dimer peak was measured in the absorption chromatogram. The concentration of the monomer was calculated from the area under the chromatographic peak B (PM), taking into account the value of ε(λ) for λ at which the monomer peak was measured in the absorption chromatogram.
In the studies carried out at very low C120 concentrations, while calculating the dimer concentration, the value of PDM and molar absorption coefficient of the dimer (εmaxDM) were determined at λ = 340 nm. It is worth mentioning that the same concentrations of the dimer were obtained when PDM and εDM were determined at λ = 330 and 350 nm. On the other hand, to calculate the monomer concentration, PM and εM were determined at λ = 300 nm and 310 nm.
The dimerization constant KDM, determined using the absorption chromatograms for C120 solutions in ACN in the concentrations range from 1.1 × 10−6 to 9.3 × 10−8 M, was found to be very large and covered a relatively narrow range (KDM = 0.9–2.6 × 109 M−1, Table 3). The mean value of KDM = 1.5 × 109 M−1 was calculated for two concentrations (cC120 = 1.1 × 10−6 M and cC120 = 9.3 × 10−8 M) for which the error of the determined KDM is the smallest. The obtained results show that KDM does not depend on C120 concentration and wavelength at which the monomer concentration was derived from absorption chromatograms (Table 3). KDM also was independent of conditions used in absorption chromatogram measurements (i.e., flow rate).
| cC120 [M] | P340DM | P300M | P310M | α300 [×10−2] | α310 [×10−2] | K300DM [×109] [M−1] | K310DM [×109] [M−1] |
|---|---|---|---|---|---|---|---|
| a Flow rate – 0.25 ml min−1.b Flow rate – 0.5 ml min−1; P300M and P310M – area under chromatographic peak of the monomer (peak B), P340DM – area under chromatographic peak of the dimer (peak A). | |||||||
| 1.1 × 10−6a | 1.47 × 10−3 | 2.7 × 10−5 | 3.3 × 10−5 | 1.8 | 2.2 | 1.4 | 0.9 |
| 1.1 × 10−6b | 6.29 × 10−4 | 1.4 × 10−5 | 1.5 × 10−5 | 2.2 | 2.3 | 0.9 | 0.9 |
| 9.3 × 10−8a | 1.13 × 10−4 | 6.1 × 10−6 | 5.2 × 10−6 | 5.1 | 4.4 | 2.0 | 2.6 |
| 1.9 × 10−8a | 2.6 × 10−5 | 4.5 × 10−6 | 4.4 × 10−6 | 17 | 14 | 0.8 | 1.0 |
The KDM values determined at each concentration (Table 3) are mean values calculated from several independent and reproducible absorption chromatograms (see Fig. S1†). The mean value of KDM determined for the sample with the concentration of 1.1 × 10−6 M was very similar to KDM values determined for single measurements. In the case of the aforementioned sample, S/N = 1500 for the dimer peak, while for the monomer peak, S/N = 25. With a relatively large measurement error of PM (for the monomer peak S/N = 3) and PDM (for the dimer peak S/N = 25), determined from eight absorption chromatograms measured for the sample with cC120 = 1.9 × 10−8 M, values of KDM covered a relatively wide range: KDM = (0.4–3.0) × 109 M−1. Although the mean value of KDM determined from eight measurements was close to the mean value of KDM determined for samples with greater concentrations, the lower reliability of KDM value determined for the sample with c = 1.9 × 10−8 M led us to omit this value from the determination of the final value of KDM = 1.5 × 109 M−1.
The necessity of conducting research on extremely low concentrations of C120 inevitably entailed measurements of absorption chromatograms in which the dimer peak, and particularly the monomer peak, had exceptionally low intensity. At cC120 = 1.9 × 10−8 M, Aλ=300M < 1 × 10−4 for the monomer peak, and Aλ=340DM ∼ 5 × 10−4 for the dimer peak. To check whether the above values agreed with the expected ones, they were compared with calculated Aλ=340DM for DM and Aλ=300M for M. For samples with experimentally studied concentrations of C120 and for the determined value of KDM = 1.5 × 109 M−1, calculations were performed of cM and cDM values. Since we knew that εmaxDM = 32600 M−1 cm−1 for dimer and it was assumed on the basis of the exciton theory,60 that ε300M = 15500 M−1 cm−1 for monomer, as well as l = 2.5 cm (in the PDA detector), we were able to calculate Aλ=340DM and Aλ=300M on the basis of the Lambert–Beer law. Both the shape of the monomer and dimer peaks, as well as their width in absorption chromatograms were very similar, at least for the range of C120 concentrations cC120 = 1.0 × 10−6–9.3 × 10−8 M (Fig. 5), allowing us to directly compare experimentally obtained Aλ=300M and Aλ=340DM with those calculated theoretically. A very good agreement was obtained between experimentally determined and calculated Aλ=340DM values for all cC120 concentrations (Table 4). Also for the sample with the highest concentration, cC120 = 1.1 × 10−6 M, there is a very good agreement between the experimental Aλ=300M = 5.3 × 10−4 (S/N > 20) and calculated Aλ=300M = 5.0 × 10−4 values (Table 4) (the latter was calculated assuming that ε300M = 15500 M−1 cm−1). This agreement proves that the assumption that εmaxM = 0.5εmaxDM, made on the basis of the exciton model,60 is correct. Therefore, one can expect that the value of KDM = 1.5 × 109 M−1, determined from absorption chromatograms using the above assumption, is also correct. Moreover very important is the fact that, an independent confirmation was provided by results of the measurements of emission chromatograms (see below).
| cC120 [M] | Aλ=340DM cal | Aλ=340DM exp | Aλ=300M cal | Aλ=300M exp | Aλ=300M/Aλ=340DM cal | Aλ=300M/Aλ=340DM exp |
|---|---|---|---|---|---|---|
| 1.1 × 10−6 | 2.92 × 10−2 | 2.93 × 10−2 | 5.0 × 10−4 | 5.3 × 10−4 | 0.017 | 0.018 |
| 9.3 × 10−8 | 2.38 × 10−3 | 2.30 × 10−3 | 1.4 × 10−4 | 1.2 × 10−4 | 0.059 | 0.052 |
| 1.9 × 10−8 | 4.51 × 10−4 | 4.8 × 10−4 | 6.1 × 10−5 | 7.0 × 10−5 | 0.135 | 0.150 |
Since UHPLC system enables to perform measurements of absorption chromatograms for very low concentrations (10−7–10−8 M), if ε(λ) ∼ 104 M−1 cm−1, therefore the monomer content in samples with cC120 = 1.9 × 10−8 M and cC120 = 9.3 × 10−8 M was relatively large, in spite of exceptionally high value of KDM. For the sample with the lowest concentration, the monomer content was about 30% (Table 5). Due to this fact, the cM/cDM ratio for the sample with the concentration of 1.9 × 10−8 M was relatively large and 7 times greater than that for the sample with the concentration of 1.1 × 10−6 M, in which the monomer content was about 4% (Table 5).
| cC120 [M] | cDM [M] cal | cDM [M] exp | cM [M] cal | cM [M] exp | cDM [%] cal | cDM [%] exp | cM [%] cal | cM [%] exp | cM/cDM cal | cM/cDM exp |
|---|---|---|---|---|---|---|---|---|---|---|
| a cM (exp) was determined from the monomer peak area (P300M); cDM (exp) was determined from the dimer peak area (P340DM). | ||||||||||
| 1.1 × 10−6 | 5.4 × 10−7 | 5.4 × 10−7 | 1.9 × 10−8 | 2.2 × 10−8 | 96.6 | 96.1 | 3.4 | 3.9 | 0.04 | 0.04 |
| 9.3 × 10−8 | 4.4 × 10−8 | 4.4 × 10−8 | 5.4 × 10−9 | 5.2 × 10−9 | 89.0 | 89.4 | 11.0 | 10.6 | 0.12 | 0.12 |
| 1.9 × 10−8 | 8.3 × 10−9 | 8.1 × 10−9 | 2.4 × 10−9 | 2.8 × 10−9 | 77.9 | 74.0 | 22.1 | 26.0 | 0.29 | 0.35 |
The values of cM, cDM and cM/cDM determined from absorption chromatograms and calculated by taking the value of KDM = 1.5 × 109 M−1 closely agreed with each other for C120 in the concentration range of 10−6–10−8 M (Table 5). This means that both the measured absorption chromatograms (Fig. 5) and cM and cDM values calculated on their basis indicate that the monomer–dimer model assumed for C120 in ACN and the determined very high KDM value are correct.
Although all results presented thus far are in agreement with the assumed model indicating the presence monomer and dimer, it is possible that trimer and not the dimer is present in addition to the monomer. To establish whether it is possible in the case of C120 in ACN, the value of trimerization constant (KTM) was determined for a sample with the concentration of 1.1 × 10−6 M (Fig. 5a and b) for which the monomer concentration (peak B) and the trimer concentration (peak A) would be the same as the concentrations determined from intensities of peaks A and B in the absorption chromatogram (assuming εmaxTM = εmaxDM). The determined KTM value must be equal to 1 × 1017 M−2. For the above value of KTM, the calculated concentration ratios cM/cTM for the samples with two lower concentrations (cC120 = 1.9 × 10−8 M and 9.3 × 10−8 M) clearly differed from the values obtained experimentally (Table S3†), which were in a very good agreement with experimentally determined cM/cDM ratios (Table 5). This outcome directly showed that the peak A cannot originate from trimer.
The results of measurements of emission chromatograms. Emission spectra of C120 (cC120 = 10−4–10−6 M) in solvents of different properties, including ACN, have been published many times,48,49 likewise ΦF values48 and lifetimes in the S1 state. The same ES and the value of ΦF = 0.63, as those reported earlier,48 we have obtained when measuring ES of C120 (cC120 ∼ 10−5–10−6 M) in ACN on a spectrofluorimeter. Taking into consideration C120 concentrations used in our studies, one can assume, on the basis of the above presented absorption chromatograms (Fig. 5) and determined KDM = 1.5 × 109 M−1, that in the emission measurements performed by us in this study and earlier,48,49 cDM ≥ 100cM. Moreover, if the position of the long-wavelength band in absorption spectra of M and DM (Fig. 3 and 4) is taken into consideration, it becomes obvious that the emission observed hitherto for λex ∼ 340 nm originates from DM and not, as was believed, from M! Although emission properties of M are unknown, one can expect that its ES is short-wavelength shifted relative to ES of DM, analogously to their long-wavelength band in AS. Also ΦF value of the monomer should be large.
Emission chromatograms, measured on the UHPLC-PDA-FL system immediately after absorption chromatograms, are shown in Fig. 6. Due to relatively high concentration of DM and its ΦF = 0.63, only a DM-originated peak is visible in the long-wavelength range (λem ≥ 380 nm) in the emission chromatogram of the sample with the concentration of cC120 = 1.1 × 10−6 M (Fig. 6a). The mentioned peak is also visible for the same λem in the case of samples with lower concentrations (Fig. 6b and c), however, its intensity is considerably lower in accordance with the relation IF ∼ ΦF(1–10−Aλex). In Fig. 6b and c visible is also another peak originating from M, whose intensity strongly depends on λem. The peak's intensity increases for shorter and shorter λem (Fig. 6c) and is the highest for λem = 320 nm (Fig. 6b and c). The retention time of M and DM in emission chromatograms is the same as that of M and DM in absorption chromatograms (after elimination of the difference in tmaxR values, originating from the configuration of the UHPLC-PDA-FL system, in both chromatograms). Unfortunately, ES measurements are not possible using emission detector. However, on the basis of the obtained emission chromatograms (Fig. 6b and c) one can assume that maximum in the emission spectrum of M is located near λmaxF ∼ 320 nm, so it is strongly short-wavelength shifted relative to that in the fluorescence spectrum of DM (λmaxF ∼ 410 nm), analogously as in AS. The initial part of the emission spectrum of DM at the short-wavelength side is located at λF ≥ 350 nm.48 This is why the DM peak is not visible in emission chromatograms for λem ≤ 340 nm (Fig. 6b and c), despite relatively high concentration of the dimer.
Although the M peak is well visible in emission chromatograms for very low concentrations cC120 ≤ 9.3 × 10−8 M (Fig. 6b and c), however, only a very intense DM peak has been employed for the determination of KDM values from emission chromatograms. For the latter peak and the concentration cC120 = 1.9 × 10−8 M (Fig. 6d), for λem = 380 nm, the ratio of S/N = 600 and for λem = 400 and 420 nm the S/N ratio is even considerably greater. Therefore, one can expect that DM peak areas (PDM) determined from emission chromatograms and calculated PDM(rel), as well as KDM values determined on their basis will be reliable and encumbered with small error. PDM values were measured for λex (300, 340 nm) and λem (380, 400, 420 nm) chosen in such a way that the DM peak was intense even for the lowest C120 concentrations (Fig. 6). The values of PDM(rel) and PDM, given for the concentration cC120 = 1.9 × 10−8 M were presented in Table 6. The PDM(rel) values are similar for all emission chromatograms. The experimentally determined PDM correspond directly to DM concentrations (cDM), (Table S4†) and PDM(rel) correspond to cDM(rel) values. To determine KDM values for measured emission chromatograms we have calculated cDM(rel) values for samples with the same concentrations of the C120 as those studied experimentally (Table 7). The calculations were performed for KDM in the range of (0.5–3.0) × 109 M−1. This range was chosen in such a way that it included KDM = 1.5 × 109 M−1 for C120 in ACN, determined from absorption chromatograms (Fig. 5 and Table 3). In Table 6 presented are KDM values determined by comparing cDM(rel) obtained from emission chromatograms (Fig. 6) with cDM(rel) calculated theoretically (Table 7).
| cC120 [M] | cC120(rel) | PDM(rel) | Pavr.DM(rel) | KDM [M−1] | ||
|---|---|---|---|---|---|---|
| 300/380 | 300/400 | 340/420 | ||||
| a DM peak area (PDM) is given in parentheses.b See Table 7. | ||||||
| 1.08 × 10−6 | 57.4 | 64.3 | 65.9 | 65.0 | 65.1 | 1.1 × 109 b |
| 9.34 × 10−8 | 4.97 | 5.25 | 5.38 | 5.14 | 5.26 | 2.5 × 109 b |
| 1.88 × 10−8 | 1.0 | 1.0 (3.03 × 105)a | 1.0 (10.3 × 105)a | 1.0 (9.2 × 105)a | 1.0 | |
| cC120 [M] | cC120(rel) | cDM(rel) | |||||
|---|---|---|---|---|---|---|---|
| KDM [M−1] | |||||||
| 0.5 × 109 | 1.0 × 109 | 1.5 × 109 | 2.0 × 109 | 2.5 × 109 | 3.0 × 109 | ||
| 1.08 × 10−6 | 57.4 | 70.0 | 66.0 | 64.5 | 62.8 | 62.8 | 62.4 |
| 9.34 × 10−8 | 4.97 | 5.636 | 5.425 | 5.346 | 5.29 | 5.26 | 5.23 |
| 1.88 × 10−8 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
The KDM values determined for C120 in ACN on the basis of the measured absorption chromatograms (Fig. 5) and emission chromatograms (Fig. 6) are given in Table 8. All KDM values calculated from eqn (2) or obtained from cDM(rel) are similar (Table 8). The mean value of KDM is 1.5 × 109 M−1 and does not depend on C120 concentration.
| cC120 (M) | KDM × 109 (M−1) | ||
|---|---|---|---|
| a Obtained from absorption chromatograms and eqn (2), see Table 3.b Obtained using the cDM(rel) values determined from the absorption chromatograms, see Table 5; for details see text.c Obtained from the cDM(rel) values determined from the emission chromatograms, see Table 6; for details see text. | |||
| 1.1 × 10−6 | 1.2a | 1.0b | 1.1c |
| 9.3 × 10−8 | 2.3a | 1.0b | 2.5c |
| 1.9 × 10−8 | 0.9a | — | — |
The study of the solvent effect on KDM value was carried out for many compounds using NMR and UV-VIS techniques. The KDM value increases, as expected, with a decrease in the energy of interactions with a solvent.1–3,6,16 However, sometimes the observed effect of a solvent on KDM value is surprising. In our work, in addition to the determination of KDM in a polar solvent such as ACN (f(ε,n2) = 0.306), we present results of KDM determination in a less polar solvent such as 1-chlorobutane (ChB) (f(ε,n2) = 0.204). This permits to expect in the case of the latter solvent even a higher KDM value than in that of ACN. In emission chromatogram, likewise in absorption chromatogram, there are two peaks originating from M and DM (not shown) and, contrary to the measurement in ACN, the retention time (tmaxR) of M is somewhat shorter (∼6 s for flow rate 0.25 ml min−1), than tmaxR of DM. Since a higher KDM value was expected in ChB than in ACN, we have used even lower C120 concentrations than those in ACN to get a sufficiently large cM/cDM ratio in the sample with the lowest concentration (cC120 = 6.82 × 10−9 M). The PDM values were measured for λex (300, 340 nm) and λem (380, 400, 420 nm) chosen in such a way that the DM peak was intense even for the lowest C120 concentrations. The results obtained for DM are presented in Table 9 in which experimental PDM values obtained for the lowest C120 concentration and calculated PDM(rel) values are given for all concentrations. The PDM(rel) values are similar for all emission chromatograms. By using calculated cDM values (Table S5†) and calculated cDMrel values (Table S6†), for samples with the same C120 concentrations as those investigated experimentally, we have determined the value of KDM = 9 × 109 M−1. A clearly higher KDM value determined in ChB compared to that in ACN is in agreement with the higher value of EDM = 7300 cm−1 in ChB58 than in ACN, EDM = 7000 cm−1.58
| cC120 [M] | cC120(rel) | PDM(rel) | Pavr.DM(rel) | KDM (M−1) | ||
|---|---|---|---|---|---|---|
| 300/380 | 300/400 | 340/420 | ||||
| a DM peak area is given in parentheses.b See Table S6. | ||||||
| 8.52 × 10−7 | 124.9 | 137.4 | 134.6 | 132.7 | 134.9 | 10.5 × 109b |
| 3.40 × 10−8 | 4.99 | 5.19 | 5.35 | 5.30 | 5.28 | 7.5 × 109b |
| 6.82 × 10−9 | 1.0 | 1.0 (3.543 × 105)a | 1.0 (1.987 × 105)a | 1.0 (1.276 × 105)a | 1.0 | |
Conclusions on the determination of very large KDM values on the basis of measurements of absorption chromatograms and emission chromatograms using a UHPLC-PDA-FL system. This is the first reported application of the UHPLC-PDA-FL method for the determination of a KDM value. This method permits the separation of M from DM and provides a higher sensitivity than any other method used thus far. With the use of a UHPLC-PDA-FL system experimental data needed for the determination of KDM are obtained directly from absorption and emission chromatograms. Isocratic elution mode with ACN and ChB was used and no buffer was added to the eluent so that the system peaks (“ghost peaks”) were avoided.63–68 By carefully controlling the number of peaks in the absorption and emission chromatograms, their intensities and shapes, measured in a wide range of wavelengths (λ) (absorption chromatogram) or wavelengths of the excitation (λex) and emission (λem), (emission chromatogram), we were confident that in the results presented in this work all chromatographic peaks came only from the species formed by C120. In all chromatograms of C120 samples in ACN and in ChB with the concentration of 10−6–10−8 M, the same two peaks with very similar retention times, shapes and absorption spectra were always present.
The results obtained for C120 in ACN and ChB confirm that by using the UHPLC-PDA-FL one can determine very high KDM values, even of order 1010 M−1. For such high KDM values, the maximum monomer content will be no more than 10%, however, this is enough for the determination of a reliable KDM value, particularly when making use of the emission chromatograms of compounds with a large ΦF. A good illustration is provided by the emission chromatograms obtained for C120 in ACN (Fig. 6). The peak of M, and particularly that of DM, are characterized by a relatively high intensity, even for very low concentration, cC120 ∼ 1.9 × 10−8 M. This is a consequence of a very high value of ΦF = 0.63 for DM, as well as high ΦF for M. Taking into account good quality and reproducibility of the results presented and the way we conducted the experiments, we are confident that the KDM value of 1.5 × 109 M−1 for C120 in ACN and KDM = 9 × 109 M−1 in ChB are correct.
The presented absorption chromatograms, particularly that measured for the lowest concentration of C120 in ACN (cC120 = 1.9 × 10−8 M), where the monomer peak is unusually small, suggest that obtaining such data is extremely difficult, if possible at all. Therefore, the proposed method for the determination of very high KDM values, although theoretically correct, is not useful in practice. However, if this method is applied to the determination of KDM of order 107–108 M−1, then due to significantly lower KDM value, the monomer concentration will be considerably higher and consequently, the absorbance for the monomer peak will be considerably greater (Tables S7 and S8†). As a result, measurements of absorption chromatograms will be easier and error of determined KDM values will be considerably smaller than those obtained in the case of KDM ∼ 109 M−1, as observed with C120 in ACN. Moreover, when KDM = 1 × 107–1 × 108 M−1, then an ample value of Aλ=300M/Aλ=340DM ratio is also obtained; as noted with the concentration of 10−7 M, a relatively large value of cM/cDM ratio was obtained. Thus, for the determination of a reliable KDM it would not be necessary to carry our measurements for concentrations of order of 10−8 M. For the concentration of 10−7 M, the measurement error of Aλ=300M would be much smaller. Thereby, when KDM = 107–108 M−1, large values of S/N ratio will be obtained from absorption chromatograms not only for DM, but also for M (Tables S7 and S8†).
It is worth noting that conditions for measurements by the UHPLC-PDA-FL method do not perturb the equilibrium between monomer and dimer present in ACN. If such a perturbation occurred, then the determined KDM would differ from the true KDM for C120 solution in ACN in the concentration range of 1.1 × 10−6–1.9 × 10−8 M. During the chromatographic separation of monomer from dimer an injected C120 sample undergoes a dilution with the eluent. Thereby, during the chromatographic separation, a decrease in the concentration of dimers could occur. If a sample dilution with an eluent or possibly interactions of dimers with the column caused a cleavage of dimers into monomers on the column, then the KDM determined by the UHPLC-PDA-FL method could be reduced, as compared to the true KDM. This reduction would be greater as the dimer concentration became smaller. The dimer concentration in samples with very different concentrations of C120 (cC120 = 1.1 × 10−6–1.9 × 10−8 M) and identical all the other measurement conditions (volume of injected sample, eluent flow rate) differed considerably. In the sample with the smallest C120 concentration cC120 = 1.9 × 10−8 M, the dimer concentration was about 5 times lower than in the sample with the concentration of 9.3 × 10−8 M and as much as about 65 times lower than in the sample with the concentration of 1.1 × 10−6 M (Table 5). The same determined value of KDM (within the margin of error) (Table 3), irrespective of C120 concentration, permitted us to assume that the determined very high KDM is correct and that the UHPLC-PDA-FL method employed for its determination had no effect on KDM value.
An independent confirmation comes from the comparison of emission chromatograms measured for C120 in ACN. In studies with the same concentration of C120 (cC120 = 3 × 10−8 M), but with different injection volumes (v = 3, 5 and 10 μl) (Table S9†), the corrected peak areas for the dimer in the emission chromatograms were the same when corrected for the effects of volume differences (Fig. S3†). Therefore, irrespective of the extent of sample dilution with the eluent, no cleavage of the dimer into two monomers occurred during the chromatographic process for any of injected samples. Moreover, the same concentration of the dimer, thereby the same KDM value, was obtained from absorption chromatograms for which dimer retention times differed by a factor of two (Fig. 5a and b and Table 3) due to differences in eluent flow rates. In accordance with the ref. 9 and 10, the absence of the effect of chromatographic separation conditions on concentrations of monomer and dimer (and consequently on the value of dimerization constant) is the result of very high stability of C120 dimer in ACN (EDM ≈ 7000 cm−1).58
Finally we would like to point out the problems encountered while determining very high KDM values during measurements of absorption and emission chromatograms of C120 in ACN, and particularly in ChB, carried out using a UHPLC-PDA-FL system. We have noticed that the most important and at the same time very difficult experimental problem is the presence of impurities (Imp) in a solvent-eluent. To ensure M content of at least a few percent in a sample, it is necessary to conduct measurements of a studied compound (SC) at very low concentrations. For instance, if KDM = 1 × 109 M−1, then cSC should be of order 10−8 M. In such conditions, most frequently cImp ≫ cSC even in the solvents of the highest purity. If Imp forms hydrogen or coordinate bonds with M and DM of SC, the fact that cImp ≫ cSC causes that instead of DM formation, the complexes M⋯Imp(solv) and DM⋯Imp(solv) are formed with a high yield from M and DM of a studied compound and impurity molecules. In such cases the determined KDM values can considerably differ from true KDM values. It should be emphasized that the above-described problem is not a consequence of the UHPLC-PDA-FL method applied, but to the unavoidable presence of impurities in solvents.
4. Conclusions
The most important achievement of this work is a new concept for the application of the UHPLC method with UV-VIS photodiode absorption detector (PDA) and emission detector. It was illustrated by the example of measurements of absorption and emission chromatograms and AS for C120 in ACN and in ChB that a UHPLC-PDA-FL apparatus can be applied to measurements in the range of very low concentrations (10−6–10−9 M), provided that the detection is carried out with a very sensitive PDA spectrophotometer and emission detector enabling the measurement of both reliable absorption and emission chromatograms and AS, with an error as small as ΔA = ±2 × 10−5 and ΔIF = ±1 × 10−2. Due to the application of the UHPLC-PDA-FL system and core–shell type columns for the separation, one can obtain very narrow (ΔtR = 1–2 s in absorption chromatograms and 2–4 s in emission chromatograms) and clearly separated peaks originating from species formed by the studied compound (and impurities). An important result obtained for C120 in ACN and in ChB was establishing, on the basis of the presence of only two peaks in its absorption and emission chromatograms, that C120 exists only as monomer and dimer species, and not as larger aggregates, within the dye concentration range of 10−5–10−9 M.Taking advantage of the unique properties of the UHPLC-PDA-FL system, we have proposed in this work a method for the determination of very large homodimerization constant values of KDM = 106–1010 M−1, regardless of the similarity or difference between AS of monomer and the dimer. This method enabled us to determine very high values of KDM = 1.5 × 109 M−1 for C120 in ACN and KDM = 9 × 109 M−1 in ChB. The same KDM value (within the limits of error) determined from emission chromatograms and absorption chromatograms proves that the two procedures for the determination of very large KDM values are correct.
The KDM value is especially important in the case of compounds that can easily undergo self-aggregation due to formation of strong hydrogen bonds or coordination bonds as well as due to π–π, dipole–dipole or dispersion interactions, e.g., porphyrins and phthalocyanines, bisimide, merocyanine and squaraine dyes and donor–acceptor compounds. For these compounds, absorption spectra measured using a spectrophotometer usually have very similar shapes in the range of very small concentrations (10−5–10−7 M). To determine whether a measured AS (as well as ES) comes from M or DM (as is the case for C120) or from both of them, it is necessary to know the KDM value.
Our demonstration that AS and ES spectra of C120 (which have been published many times48,49) in each case originated from the dimer, and not from the monomer has a number of important consequences. The values of vertical excitation energy of C120 monomer to the S1 state, which has been calculated many times using ab initio and TD-DFT methods,50 were always considerably greater (by 3000–5000 cm−1) than the experimental values determined using the position of long-wavelength band in AS of C120. However, the present study shows that all AS of C120 measured hitherto originated from dimers. The application of the UHPLC-PDA method has enabled us to record AS of the monomer for the first time. Fortunately, the position of the band corresponding to the transition to the S1 state of the monomer (Fig. 3b) agreed very well with theoretically calculated values of vertical excitation energy to the S1 state of C120 monomer.50
The coexistence of C120 monomer and dimer species, along with their distinct spectral properties (in particular, the location of the long wavelength band in the absorption and emission spectra), opens the possibility to use C120 as a sensitive absorption and fluorescence probe. For example, C120 as a coupled monomer–dimer pair can be used to control the release of the C120 monomer species from N-linked oligosaccharides and the formation of the C120 dimer species.69
For a number of compounds, very high energies of dimerization have been calculated, EDM > 5000 cm−1,1–3,57,70 sometimes for more than one dimer structure.1,57,70 For such compounds, the value of KDM would be very high. It would be interesting to use the proposed UHPLC-PDA method to determine these values.
Conflicts of interest
There are no conflicts to declare.Abbreviations
| AS | Absorption spectra |
| ES | Emission spectra |
| KDM | Homo-dimerization constant (dimerization constant) |
| A, B | Different chemical compounds |
| Keq | Equilibrium constant |
| Kass | Association constant |
| cM | Monomer concentration |
| cDM | Dimer concentration |
| M | Monomer |
| DM | Dimer |
| ε(λ) | Molar absorption coefficient |
| ΦE | Quantum yield of emission |
| ΦF | Quantum yield of fluorescence |
| λem | Emission wavelength |
| λex | Excitation wavelength |
| εmax | Molar absorption coefficient at the maximum |
| UHPLC | Ultra-high performance liquid chromatography |
| HPLC | High performance liquid chromatography |
| UHPLC-PDA-FL | Ultra-high performance liquid chromatography apparatus in conjunction with a photodiode UV-VIS spectrophotometer and an emission detector |
| tR | Retention time |
| ΔtR1/2 | Peak width at half maximum of intensity |
| A | Absorbance |
| ΔA | Error of the absorbance measurement |
| tmaxR | Retention time at the maximum |
| ACN | Acetonitrile |
| ChB | 1-Chlorobutane |
| S/N | Signal/noise ratio |
| εmaxM | Molar absorption coefficient of the monomer at the maximum |
| εmaxDM | Molar absorption coefficient of the dimer at the maximum |
| cC120 | Concentration of C120 |
| cC120(rel) | Relative concentration of C120 |
| Aλ=300M | Absorbance of monomer at λ = 300 nm |
| Aλ=340DM | Absorbance of dimer at λ = 340 nm |
| cM(rel) | Relative concentration of monomer |
| cDM(rel) | Relative concentration of dimer |
| ε300M | Molar absorption coefficient of monomer at 300 nm |
| ε340DM | Molar absorption coefficient of dimer at 340 nm (at the maximum) |
| Aλ=300M(rel) | Relative absorbance of monomer at λ = 300 nm |
| Aλ=340DM(rel) | Relative absorbance of dimer at λ = 340 nm |
| λmax | Wavelength at the maximum |
| α | Mole fraction of the monomer species |
| PM | Area of dimer peak |
| PDM | Area of monomer peak |
| P340DM | Area of dimer peak at λ = 340 nm |
| P300M | Area of monomer peak λ = 300 nm |
| PM(rel) | Relative area of monomer peak |
| PDM(rel) | Relative area of dimer peak |
| PDM(rel)avr | Average value of relative area of dimer peak |
| K300DM | Dimerization constant, where PM was measured at λ = 300 nm |
| K310DM | Dimerization constant, where PM was measured at λ = 310 nm |
| EDM | Dimerization energy |
| εmaxTM | Molar absorption coefficient of trimer at the maximum |
| KTM | Trimerization constant |
| cTM | Trimer concentration |
Acknowledgements
We thank Julia Józkowiak, MSc, for the technical support. The authors are very grateful to Professors Ryszard Fiedorow and Jacek Kubicki for help in manuscript preparation. A part of chromatographic measurements were performed at the Centre for Ultrafast Laser Spectroscopy at the A. Mickiewicz University in Poznań, Poland.References
- (a) P. Osswald and F. Würthner, J. Am. Chem. Soc., 2007, 129, 14319–14326 CrossRef CAS PubMed; (b) Z. Xie and F. Würthner, Org. Lett., 2010, 12, 3204–3207 CrossRef CAS PubMed; (c) U. Mayerhöffer and F. Würthner, Chem. Sci., 2012, 3, 1215–1220 RSC; (d) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed; (e) Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- (a) S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493 CrossRef; (b) F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS; (c) J. H. K. Ky Hirschberg, F. H. Beijer, H. A. van Aert, P. C. M. M. Magusin, R. P. Sijbesma and E. W. Meijer, Macromolecules, 1999, 32, 2696–2705 CrossRef CAS; (d) T. F. A. de Greef, M. M. L. Nieuwenhuizen, P. J. M. Stals, C. F. C. Fitie, A. R. A. Palmans, R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2008, 4306–4308 RSC; (e) T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- (a) P. S. Corbin and S. C. Zimmerman, J. Am. Chem. Soc., 1998, 120, 9710–9711 CrossRef CAS; (b) D. W. Kuykendall, C. A. Anderson and S. Zimmerman, Org. Lett., 2009, 11, 61–64 CrossRef CAS PubMed; (c) E. M. Todd, J. R. Qinn, T. Park and S. C. Zimmerman, Isr. J. Chem., 2005, 45, 381–389 CrossRef CAS.
- (a) C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS PubMed; (b) B. A. Blight, C. A. Hunter, D. A. Leigh, H. McNab and P. I. T. Thomson, Nat. Chem., 2011, 3, 244–248 CrossRef CAS PubMed; (c) C. A. Hunter and L. D. Sarson, Angew. Chem., Int. Ed. Engl., 1994, 33, 2313–2316 CrossRef.
- (a) D. A. Leigh, C. C. Robertson, A. M. Z. Slawin and P. I. T. Thomson, J. Am. Chem. Soc., 2013, 135, 9939–9943 CrossRef CAS PubMed; (b) S. Djurdjevic, D. A. Leigh, H. McNab, S. Parsons, G. Teobaldi and F. Zerbetto, J. Am. Chem. Soc., 2007, 129, 476–477 CrossRef CAS PubMed.
- (a) A. Satake and Y. Kobuke, Tetrahedron, 2005, 61, 13–41 CrossRef CAS; (b) Y. Kobuke and H. Miyaji, J. Am. Chem. Soc., 1994, 116, 4111–4112 CrossRef CAS.
- S. Schlund, C. Schmuck and B. Engels, J. Am. Chem. Soc., 2005, 127, 11115–11124 CrossRef CAS PubMed.
- C. Maeda, T. Kamada, N. Aratani, T. Sasamori, N. Tokitoh and A. Osuka, Chem.–Eur. J., 2009, 15, 9681–9684 CrossRef CAS PubMed.
- A. Shao, Z. Guo, S. Zhu, P. Tian, H. Shi and W. Zhu, Chem. Sci., 2014, 5, 1383–1389 RSC.
- S. Gadde, E. K. Batchelor, J. P. Weiss, Y. Ling and A. E. Kaifel, J. Am. Chem. Soc., 2008, 130, 17114–17119 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- M. Kubista, R. Sjöback and B. Albinsson, Anal. Chem., 1993, 65, 994–998 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. Ky Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- (a) G. B. W. L. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 810–811 CrossRef CAS PubMed; (b) T. F. A. de Greef, M. M. L. Nieuwenhuizen, R. P. Sijbesma and E. W. Meijer, J. Org. Chem., 2010, 75, 598–610 CrossRef CAS PubMed.
- M. Mizumura, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 5378–5381 CrossRef CAS PubMed.
- (a) M. M. Safont-Sempere, P. Ossswald, K. Radacki and F. Würthner, Chem.–Eur. J., 2010, 16, 7380–7384 CrossRef CAS PubMed; (b) S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Chem.–Eur. J., 2008, 14, 11343–11357 CrossRef CAS PubMed.
- (a) S. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 15959–15967 CrossRef CAS PubMed; (b) A. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831–4835 CrossRef CAS PubMed.
- Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem.–Eur. J., 2007, 13, 450–465 CrossRef CAS PubMed.
- (a) F. C. Spano, J. Am. Chem. Soc., 2009, 131, 4267–4278 CrossRef CAS PubMed; (b) F. C. Spano, S. C. J. Meskers, E. Hennebicq and D. Beljonne, J. Chem. Phys., 2008, 129, 024704 CrossRef PubMed.
- L. Brusveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- (a) Y. Kang and M. E. Meyerhoff, Anal. Chim. Acta, 2006, 565, 1–9 CrossRef CAS; (b) Y. Kang, J. W. Kampf and M. E. Meyerhoff, Anal. Chim. Acta, 2007, 598, 295–303 CrossRef CAS PubMed.
- Y. Xu, Z. Li, A. Malkovskiy, S. Sun and Y. Pang, J. Phys. Chem. B, 2010, 114, 8574–8580 CrossRef CAS PubMed.
- S. Chen, et al., Chem. Sci., 2012, 3, 1804–1809 RSC.
- H.-M. Zhang, X.-Q. Guo, Y.-B. Zhao, D.-Y. Wang and J.-G. Xu, Anal. Chim. Acta, 1998, 361, 9–17 CrossRef CAS.
- T. F. A. de Greef, G. Ercolani, G. B. W. L. Ligthart, E. W. Meijer and R. P. Sijbesma, J. Am. Chem. Soc., 2008, 130, 13755–13764 CrossRef CAS PubMed.
- X.-Z. Wang, X.-Q. Li, X.-B. Shao, X. Zhao, P. Deng, X.-K. Jiang, Z.-T. Li and Y.-Q. Chen, Chem.–Eur. J., 2003, 9, 2904–2913 CrossRef CAS PubMed.
- F. Bergström, I. Mikhalyov, P. Hägglöf, R. Wortmann, T. Ny and L. B.-Å. Johansson, J. Am. Chem. Soc., 2002, 124, 196–204 CrossRef.
- K. A. Kistler, C. M. Pochas, H. Yamagata, S. Matsika and F. C. Spano, J. Phys. Chem. B, 2012, 116, 77–86 CrossRef CAS PubMed.
- T. Kamada, N. Aratani, T. Ikeda, N. Shibata, Y. Higuchi, A. Wakamiya, S. Yamaguchi, K. Suk Kim, Z. Yoon, D. Kim and A. Osuka, J. Am. Chem. Soc., 2006, 128, 7670–7678 CrossRef CAS PubMed.
- A. Niazi, A. Yazdanipour, J. Ghasemi and M. Kubista, Spectrochim. Acta, Part A, 2006, 65, 73–78 CrossRef PubMed.
- S. Nagao, H. Ishikawa, T. Yamada, Y. Mizutani and S. Hirota, J. Biol. Inorg Chem., 2015, 20, 523–530 CrossRef CAS PubMed.
- G. Ercolani, Chem. Commun., 2001, 1416–1417 RSC.
- (a) B. Ciesielska, A. Łukaszewicz, L. Celewicz, A. Maciejewski and J. Kubicki, Appl. Spectrosc., 2007, 61, 102–109 CrossRef CAS PubMed; (b) B. Ciesielska, J. Kubicki, A. Maciejewski and S. Paszyc, Chem. Phys. Lett., 2002, 353, 69–76 CrossRef CAS.
- E. Krystkowiak, R. A. Bachorz and A. Maciejewski, Phys. Chem. Chem. Phys., 2016, 18, 492–502 RSC.
- (a) L. Braco, C. Bano, F. Chillaron and C. Abad, J. Biol. Macromol., 1988, 10, 343–348 CrossRef CAS; (b) D. Salom, M. C. Bañó, L. Braco and C. Abad, Anal. Chim. Acta, 1997, 352, 309–317 CrossRef CAS.
- (a) F. Zeng, S. C. Zimmerman, S. V. Kolotuchin, D. E. C. Reichert and Y. Ma, Tetrahedron, 2002, 58, 825–843 CrossRef CAS; (b) J. Yang, E. Fan, S. J. Geib and A. D. Hamilton, J. Am. Chem. Soc., 1993, 115, 5314–5315 CrossRef CAS.
- (a) R. A. Haycock, C. A. Hunter, D. A. James, U. Michelsen and L. R. Sutton, Org. Lett., 2000, 2, 2435–2438 CrossRef CAS PubMed; (b) G. M. Whitesides, E. E. Simanek, J. P. Mathias, C. T. Seto, D. N. Chin, M. Mammen and D. M. Gordon, Acc. Chem. Res., 1995, 28, 37–44 CrossRef CAS; (c) K. J. Lampi, J. T. Oxford, H. P. Bachinger, T. R. Shearer, L. L. David and D. Kapfer, Exp. Eye Res., 2001, 72, 279–288 CrossRef CAS PubMed.
- (a) H. Ye, Anal. Biochem., 2006, 356, 76–85 CrossRef CAS PubMed; (b) M. D. Bond, M. E. Panek, Z. Zhang, D. Wang, P. Mehndiratta, H. Zhao, K. Gunton, A. Ni, M. L. Nedved, S. Burman and D. B. Volkin, J. Pharm. Sci., 2010, 99, 2582–2597 CrossRef CAS PubMed; (c) K. Ahrer, A. Buchacher, G. Iberer, D. Josic and A. Jungbauer, J. Chromatogr. A, 2003, 1009, 89–96 CrossRef CAS PubMed; (d) G. B. Irvine, Anal. Chim. Acta, 1997, 352, 387–397 CrossRef CAS.
- (a) N. Gutowska and A. Maciejewski, RSC Adv., 2014, 4, 31775–31781 RSC; (b) N. Gutowska and A. Maciejewski, J. Photochem. Photobiol., A, 2016, 29, 1–8 CrossRef.
- N. Nandi, K. Bhattacharyya and B. Bagchi, Chem. Rev., 2000, 100, 2013–2046 CrossRef CAS PubMed.
- B. D. Wagner, Molecules, 2009, 14, 210–237 CrossRef CAS PubMed.
- S. George, M. Kumbhakar, P. K. Singh, R. Ganguly, S. Nath and H. Pal, J. Phys. Chem. B, 2009, 113, 5117–5127 CrossRef CAS PubMed.
- M. Grazula and E. Budzisz, Coord. Chem. Rev., 2009, 253, 2588–2598 CrossRef.
- M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641–2684 CrossRef CAS PubMed.
- M. V. Kulkarni, G. M. Kulkarni, C.-H. Lin and C.-M. Sun, Curr. Med. Chem., 2006, 13, 2795–2818 CrossRef CAS PubMed.
- S. S. Anufrik and V. V. Tarkovsky, J. Appl. Spectrosc., 2010, 77, 640–647 CrossRef CAS.
- H. Pal, S. Nad and M. Kumbhakar, J. Chem. Phys., 2003, 119, 443–452 CrossRef CAS.
- K. Rechthaler and G. Kohler, Chem. Phys., 1994, 189, 99–116 CrossRef CAS.
- (a) W. Zhao, L. Pan, W. Bian and J. Wang, ChemPhysChem, 2008, 9, 1593–1602 CrossRef CAS PubMed; (b) P. Zhou, P. Song, J. Liu, K. Han and G. He, Phys. Chem. Chem. Phys., 2009, 11, 9440–9449 RSC; (c) T. Sakata, Y. Kawashima and H. Nakano, J. Phys. Chem. A, 2010, 114, 12363–12368 CrossRef CAS PubMed; (d) R. J. Cave, K. Burke and E. W. Castner, J. Phys. Chem. A, 2002, 106, 9294–9305 CrossRef CAS.
- M.-G. Choi, E. Lee, H.-S. Chung, S.-H. Jang and C. W. Lee, BMB Rep., 2011, 44, 458–461 CrossRef CAS PubMed.
- M. Yodoshi, A. Tani, Y. Ohta and S. Suzuki, J. Chromatogr. A, 2008, 1203, 137–145 CrossRef CAS PubMed.
- R. Tandon, et al., J. Antimicrob. Chemother., 2011, 66, 2543–2555 CrossRef CAS PubMed.
- X. Liu, M. Dong, X. Chen, M. Jiang, X. Lv and J. Zhou, Appl. Microbiol. Biotechnol., 2008, 78, 241–247 CrossRef CAS PubMed.
- (a) X. Liu, J. M. Cole and K. S. Low, J. Phys. Chem. C, 2013, 117, 14723–14730 CrossRef CAS; (b) X. Liu, J. M. Cole, P. C. Y. Cow, L. Zhang, Y. Tan and T. Zhao, J. Phys. Chem. C, 2014, 118, 13042–13051 CrossRef CAS.
- (a) P. Verma and H. Pal, J. Phys. Chem. A, 2012, 116, 4473–4484 CrossRef CAS PubMed; (b) P. Verma and H. Pal, J. Phys. Chem. A, 2014, 118, 6950–6964 CrossRef CAS PubMed.
- (a) M. Cigan, J. Donovalova, V. Szöcs, J. Gaspar, K. Jakusowa and A. Gaplovsky, J. Phys. Chem. A, 2013, 117, 4870–4883 CrossRef CAS PubMed; (b) A. Stamm, K. Schwing and M. Gerdhardsm, J. Chem. Phys., 2014, 141, 194304 CrossRef CAS PubMed.
- J. Koput, M. Hetmańska and A. Maciejewski, to be published.
- J. Kozłowski, A. Maciejewski, M. Milewski and W. Urjasz, J. Phys. Org. Chem., 1992, 12, 47–52 CrossRef.
- (a) M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS; (b) E. G. McRae and M. Kasha, J. Chem. Phys., 1958, 28, 721–722 CrossRef CAS; (c) A. S. Davydov, Phys.-Usp., 1964, 7, 145–178 Search PubMed.
- J. C. Dearden, Can. J. Chem., 1963, 41, 2683–2691 CrossRef CAS.
- M. Ito, J. Mol. Spectrosc., 1960, 4, 125–143 CrossRef CAS.
- F. Gritti and G. Guiochon, J. Chromatogr. A, 2004, 1028, 197–210 CrossRef CAS PubMed.
- S. Levin and E. Grushka, Anal. Chem., 1986, 58, 1602–1607 CrossRef CAS.
- D. V. McCalley, Anal. Chem., 2003, 75, 3404–3410 CrossRef CAS PubMed.
- J. Srbek, P. Coufal, Z. Bosáková and E. Tesařová, J. Sep. Sci., 2005, 28, 1263–1270 CrossRef CAS PubMed.
- S. Williams, J. Chromatogr. A, 2004, 1052, 1–11 CrossRef CAS PubMed.
- S. Levin and E. Grushka, Anal. Chem., 1987, 59, 1157–1164 CrossRef CAS.
- M. Yodoshi, A. Tani, Y. Ohta and S. Suzuki, J. Chromatogr. A, 2008, 1203, 137–145 CrossRef CAS PubMed.
- M. Bayda, F. Dumoulin, G. L. Hug, J. Koput, R. Gorniak and A. Wójcik, Dalton Trans., 2017, 46, 1914–1926 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05051h |
| This journal is © The Royal Society of Chemistry 2017 |