Abstract
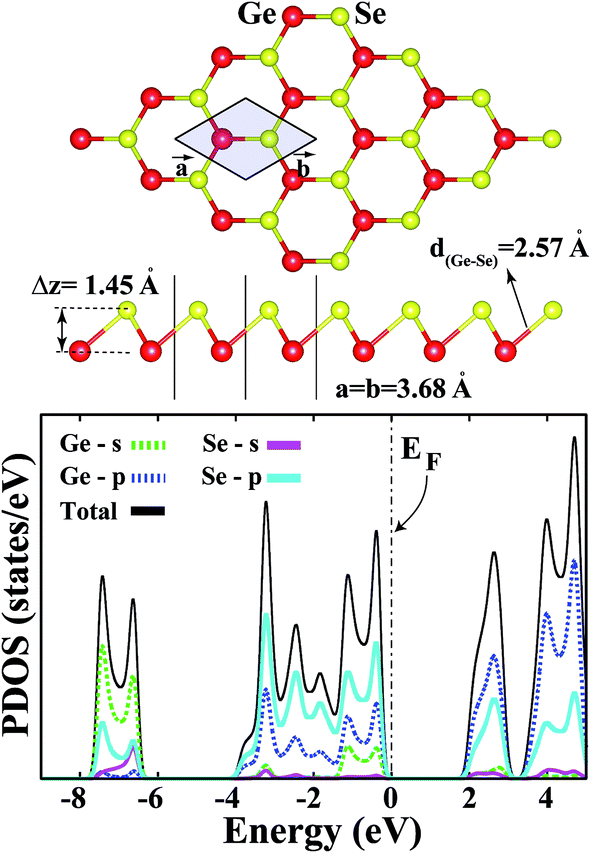
Single-layer GeSe is a new candidate in the two-dimensional family of materials. In our recent study, we showed that GeSe can form a stable buckled honeycomb structure (b-GeSe) and is a semiconductor with a 2.29 eV band gap. This paper investigates the effect of point defects of both hole (Ge, Se) and substitution doping of p-block elements, in single-layer b-GeSe, based on first principles plane wave calculations within spin-polarized density functional theory. In the case of the substitution process, we present an extensive analysis of the effects of substituting atoms (Al, As, Cl, P, C, N, Ge or Se, Si, B, F, Ga and S) on the electronic and magnetic properties of the b-GeSe phase. Our results show that nonmagnetic and semiconducting b-GeSe can be half-metallized by Ge vacancies, while it remains a semiconductor with Se vacancies with a decreasing band gap value. The results of the substitution process can be categorized by the group number in the periodic table. b-GeSe remains a nonmagnetic semiconductor upon the substitution of defects with group IVA and VIA atoms on either the Ge or Se position of the b-GeSe structure. On the other hand, the results show that the influence of group IIIA and VIIA atoms is obvious, as these atoms raise the net magnetic moments (1 μB to 3 μB) of the new b-GeSe system. In particular, the system shows half-metallicity when the Se atoms are replaced with group IIIA atoms. The system has a net magnetic moment when substituting group VA atoms for Se atoms, whereas it does not when substituting them for Ge atoms (except for N). We believe that these results are useful for the further functionalization of b-GeSe with point defects.
Recent experimental and computational studies have demonstrated that the interesting properties of bare SL structures can be modified through adsorption and substitution processes.14,30–33 The band gap of single-layers can be controlled through the substitution of subatoms, and more importantly, it can be tuned for different nanoscale applications.34–37 In this respect, the substitution of atoms in SL-GeSe is highly attractive and gives rise to important changes. Phosphorene-like GeSe and GeS structures (aw-GeS/Se) can exhibit metallic behaviour when substituted with light, non-metallic atoms.38 While aw-GeSe has a 0.99 eV indirect band gap, b-GeSe has a 2.29 eV indirect band gap. This new form of GeSe and its new electronic structure may be useful for future electronic devices. With this motivation, in this manuscript, we investigated the effects of the substitution process on the physical properties of buckled single-layer GeSe. We found that single atom substitution changed the bare single-layer properties significantly. Nonmagnetic b-GeSe gains a net magnetic moment from the substitution of certain atoms. Nonmagnetic and semiconducting b-GeSe can be half-metallized, and it also gains magnetization with Ge vacancies of 2 μB per unit cell, whereas it remains a semiconductor with Se vacancies. In addition, the results of the substitution process can be categorized by the group number of the substituted atoms. We believe that the functionalization of b-GeSe with point defects appears to be a promising way to extend the applications of single-layer GeSe.
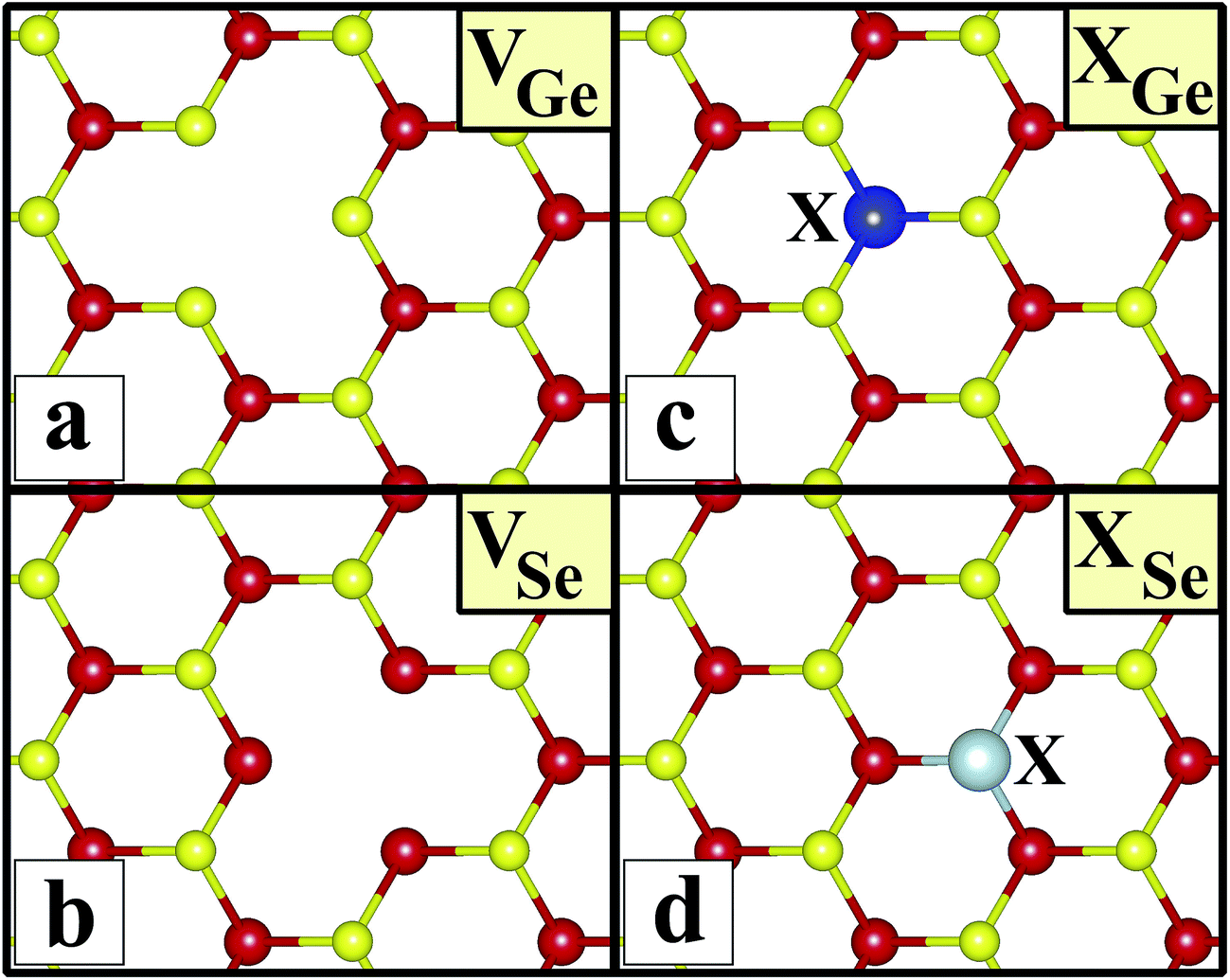
Substitution (extrinsic). An atom of X placed instead of a Ge(Se) atom, labeled as XGe(XSe). The X atoms chosen were Al, As, Cl, P, C, N, Se or Ge, Si, B, F, Ga and S.All atomic positions, including those of the subatom and the remaining substrate atoms, were relaxed to find the ground state energy for the new geometric configuration. After geometrical optimization, we calculated their cohesive energies using the equation above, and the formation energy for the substitution of defect XY, where atom X occupies the site of atom Y, using the equation below:
| | |
Eform = Esubstrate+XY − Esubstrate + EY − EX.
| (2) |
We also used this equation to determine the vacancy formation energy by using the term
Esubstrate–Ge(Se) instead of
Esubstrate+XY and omitting the
EX term.
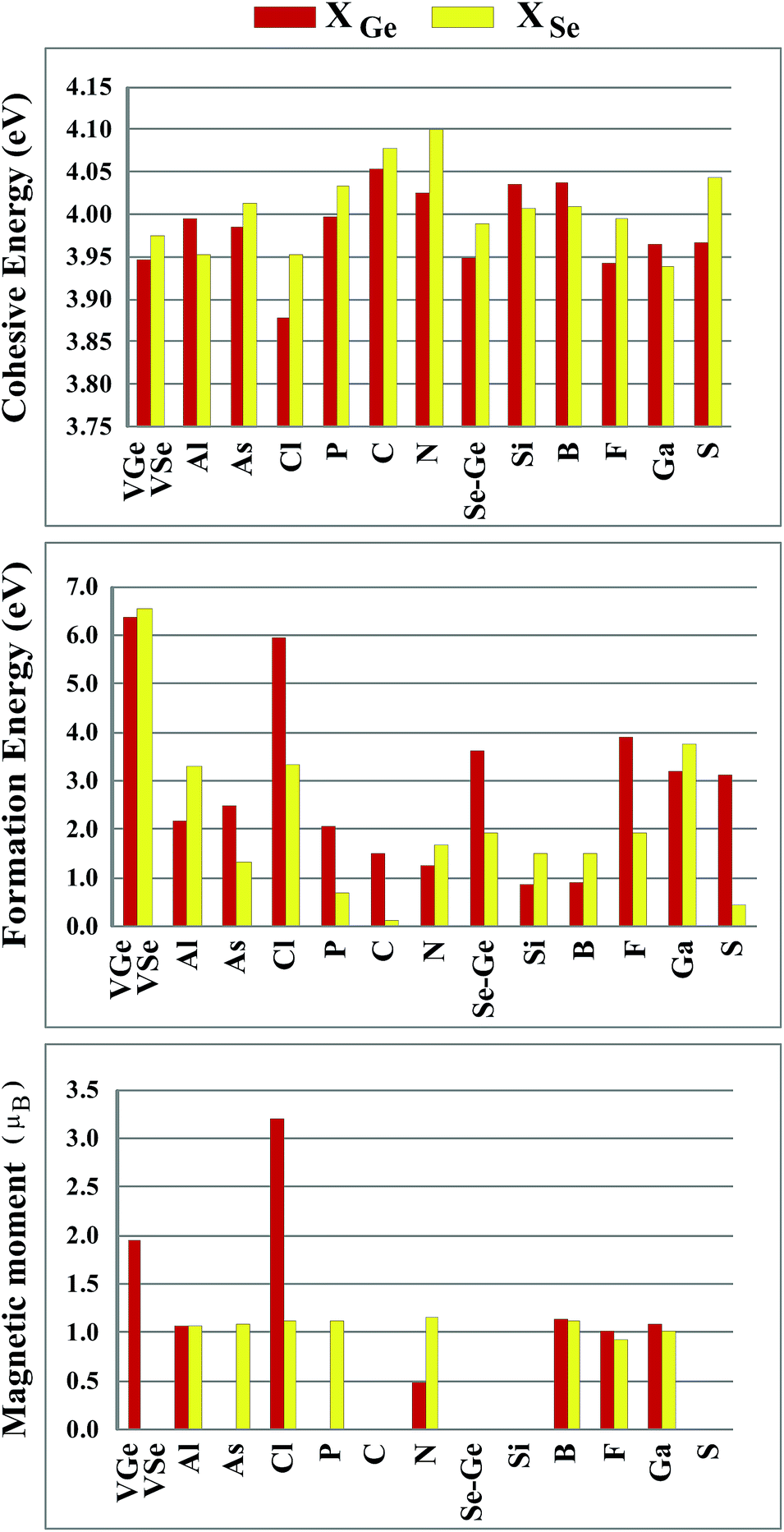
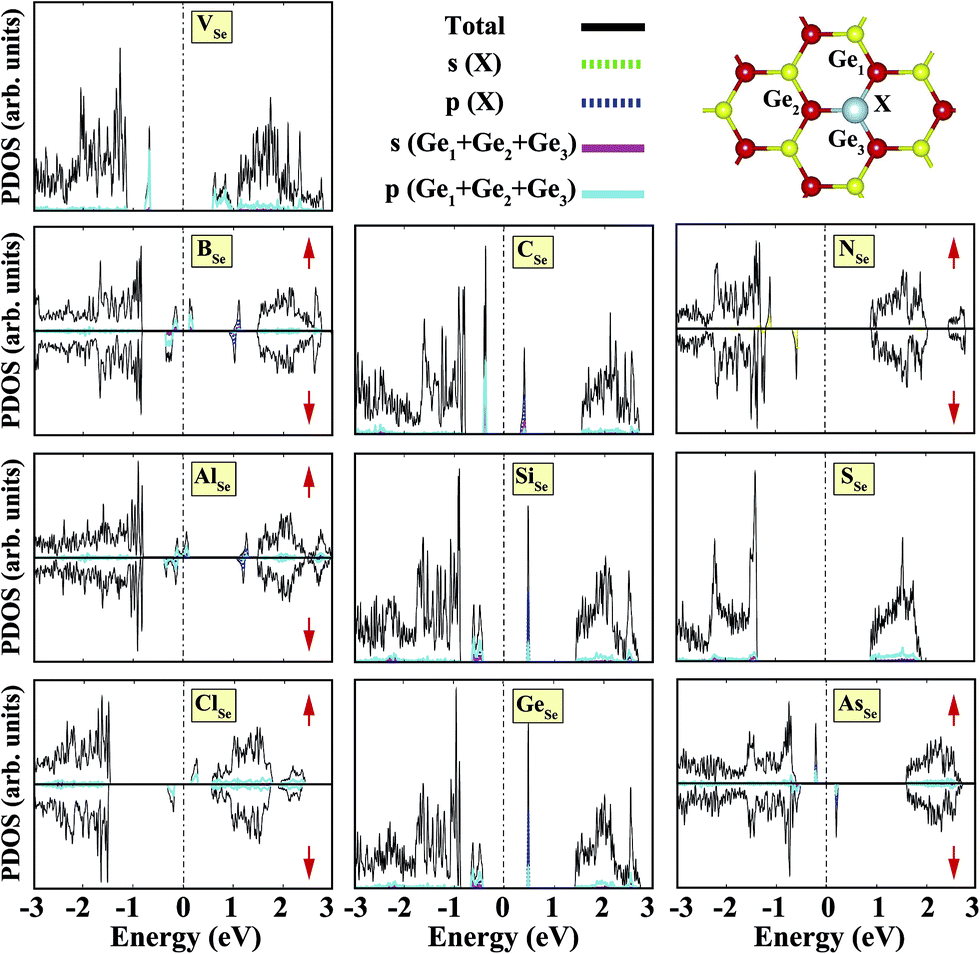
Sketches for each system considered in this study are given in Fig. 2(a)–(d) in detail. The charge at the substituted atom (X), ρL, was obtained by Löwdin charge analysis. The excess charge of atom X was calculated as ρ* = ZA − ρL. Here, ZA is the valance charge of atom X and ρL is the charge at atom X. In Tables 1 and 2, we present the relevant structural parameters, such as the length of the X–Ge and X–Se bonds and the lattice constant, and energetic values, such as the cohesive energy and defect formation energy, magnetic moments and excess charge on the subatom, for the defected structures built by substitution in the Ge and Se positions of the SL-GeSe network, respectively. The calculated cohesive energies of the intrinsic and extrinsic point defects are almost equal and interchangeable in the small range of 3.9 eV to 4.1 eV, while the formation energies for the vacancies and ClGe are two to three times higher than those for the other atom substitutions. Based on the formation energy equation without van der Waals (vdW) correction, we obtained values of 6.354 eV and 6.543 eV for the required energy to form VGe and VSe, respectively, indicating that the Ge vacancy is more easily formed in SL-GeSe, while an anion (S or Se) vacancy is more favorable for SL-GaS and -GaSe.52,53 The Ge vacancy does not change the supercell lattice constant, while the Se vacancy causes a small structural shrink. Upon Ge vacancy formation, three dangling bonds remain surrounding the vacancy, with 0.07 Å smaller Ge–Se bond lengths than those in the bare single-layer. Because of these dangling bonds, three unpaired electrons exist in the atoms surrounding the vacancy, and they form one bonding and one antibonding orbital. Due to spin-polarization, unoccupied antibonding states occur above the Fermi level, as illustrated in Fig. 4. The new system has a local magnetic moment of 2 μB, in addition to ferromagnetism, and half-metallicity occurs upon Ge vacancy formation. In contrast to the Ge vacancy, after relaxation, the GeSe lattice constant of the supercell becomes 0.14 Å smaller than that of the bare one. This atomic replacement is called the pseudo Jahn–Teller effect,54 and it induces a nonmagnetic system, whereas the system has a magnetic moment before relaxation according to Liebs theorem. Another way to tune the electronic and magnetic properties of the system is to change the carrier density by creating impurities in the bare system. For this investigation we chose p-block elements. These atoms can be categorized for the Ge(Se) host atom as follows: (1) fewer p electrons than the Ge(Se) atom, (2) isovalency with Ge(Se) and (3) more p electrons than Ge(Se).
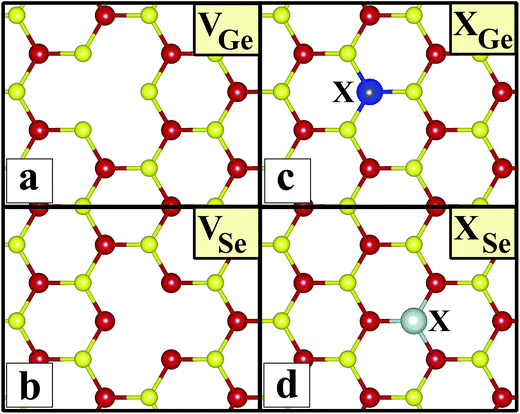 |
| | Fig. 2 Sketches of the systems considered in this study in the (4 × 4) supercell of the b-GeSe phase. (a) Ge vacancy, VGe. (b) Se vacancy, VSe. (c) Substitution of atom X in the Ge position, XGe, where atom X is Al, As, Cl, P, C, N, Se, Si, B, F, Ga or S. (d) Substitution of atom X in the Se position, XSe, where atom X is Al, As, Cl, P, C, N, Ge, Si, B, F, Ga or S. | |
Table 1 Calculated parameters for atom substitutions in the (4 × 4) supercells of GeSe in the Ge position. Ecoh is the cohesive energy; Eform is the formation energy; a is the lattice constant (NC indicates that the lattice constant is not changed), which is the same as in the bare single-layer; d(X–Ge) and d(X–Se) are the bond distances between the substituted (X) and Ge and Se atoms in b-GeSe, respectively (for VGe, the bond distances are given for Se–Ge and Se–Se); μ is the local magnetic moment (NM indicates a nonmagnetic system); ρ* is the excess charge on the substituted atom, obtained by subtracting the charge on the substituted atom from its valence charge (a negative sign indicates excess electrons). For VGe, the excess charges on the atoms around the vacancy are given in the table
| System |
Ecoh (eV) |
Eform (eV) |
a (Å) |
d(X–Ge) (Å) |
d(X–Se) (Å) |
μ (μB) |
ρ* (e) |
| VGe |
3.946 |
6.354 |
NC |
2.50 |
3.67 |
2.00 |
−0.05 |
| BGe |
4.037 |
0.895 |
14.62 |
3.83 |
1.99 |
1.00 |
−0.37 |
| AlGe |
3.995 |
2.170 |
NC |
3.68 |
2.45 |
1.00 |
0.22 |
| GaGe |
3.965 |
3.211 |
NC |
3.74 |
2.51 |
1.00 |
0.04 |
| CGe |
4.053 |
1.496 |
14.52 |
3.52 |
2.08 |
NM |
−0.58 |
| SiGe |
4.036 |
0.856 |
14.67 |
3.65 |
2.47 |
NM |
0.10 |
| NGe |
4.026 |
1.268 |
14.58 |
3.58 |
2.07 |
0.48 |
−0.57 |
| PGe |
3.998 |
2.073 |
NC |
3.63 |
2.39 |
NM |
0.07 |
| AsGe |
3.985 |
2.497 |
NC |
3.64 |
2.54 |
NM |
0.13 |
| SGe |
3.967 |
3.127 |
14.65 |
3.36 |
2.26 |
NM |
−0.04 |
| SeGe |
3.949 |
3.632 |
NC |
3.44 |
2.40 |
NM |
0.08 |
| FGe |
3.943 |
3.903 |
14.66 |
3.14 |
1.84 |
1.00 |
−0.41 |
| ClGe |
3.879 |
5.934 |
NC |
3.68 |
3.01 |
3.00 |
−0.33 |
Table 2 Calculated parameters for atom substitutions in the (4 × 4) supercells of GeSe in the Se position. Ecoh is the cohesive energy; Eform is the formation energy; a is the lattice constant (NC indicates that the lattice constant is not changed), which is the same as in the bare single-layer; d(X–Ge) and d(X–Se) are the bond distances between the substituted (X) and Ge and Se atoms in b-GeSe, respectively (for VSe, the bond distances are given for Ge–Ge and Se–Ge); μ is the local magnetic moment (NM indicates a nonmagnetic system); ρ* is the excess charge on the substituted atom, obtained by subtracting the charge on the substituted atom from its valence charge (a negative sign indicates excess electrons). For VSe, the excess charges on the atoms around the vacancy are given in the table
| System |
Ecoh (eV) |
Eform (eV) |
a (Å) |
d(X–Ge) (Å) |
d(X–Se) (Å) |
μ (μB) |
ρ* (e) |
| VSe |
3.975 |
6.543 |
14.58 |
2.94 |
2.59 |
NM |
0.08 |
| BSe |
4.010 |
1.458 |
14.65 |
2.10 |
3.66 |
1.00 |
−0.36 |
| AlSe |
3.912 |
3.284 |
NC |
2.59 |
3.84 |
1.00 |
0.28 |
| GaSe |
3.939 |
3.743 |
NC |
2.60 |
3.86 |
1.00 |
0.09 |
| CSe |
4.078 |
0.136 |
14.62 |
2.03 |
3.66 |
NM |
−0.46 |
| SiSe |
4.007 |
1.519 |
NC |
2.45 |
3.77 |
NM |
0.19 |
| GeSe |
3.990 |
1.927 |
NC |
2.52 |
3.79 |
NM |
0.14 |
| NSe |
4.099 |
1.676 |
14.60 |
2.02 |
3.66 |
1.00 |
−0.58 |
| PSe |
4.033 |
0.696 |
NC |
2.42 |
3.70 |
1.00 |
−0.03 |
| AsSe |
4.013 |
1.335 |
NC |
2.52 |
3.72 |
1.00 |
0.04 |
| SSe |
4.043 |
0.439 |
14.65 |
2.45 |
3.64 |
NM |
−0.15 |
| FSe |
3.996 |
1.939 |
14.64 |
2.37 |
3.61 |
1.00 |
−0.44 |
| ClSe |
3.953 |
3.325 |
NC |
2.49 |
3.56 |
1.00 |
−0.16 |
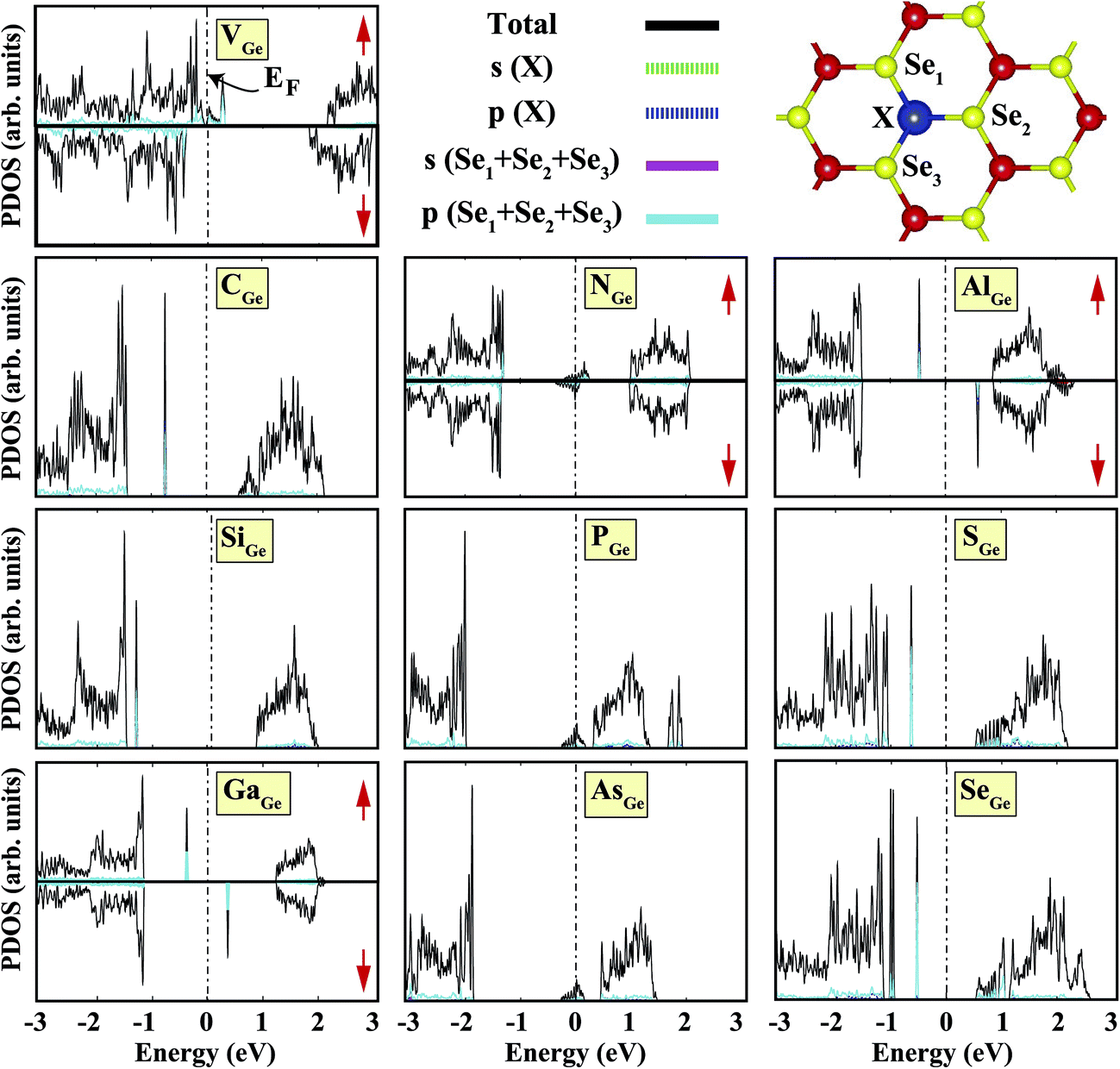
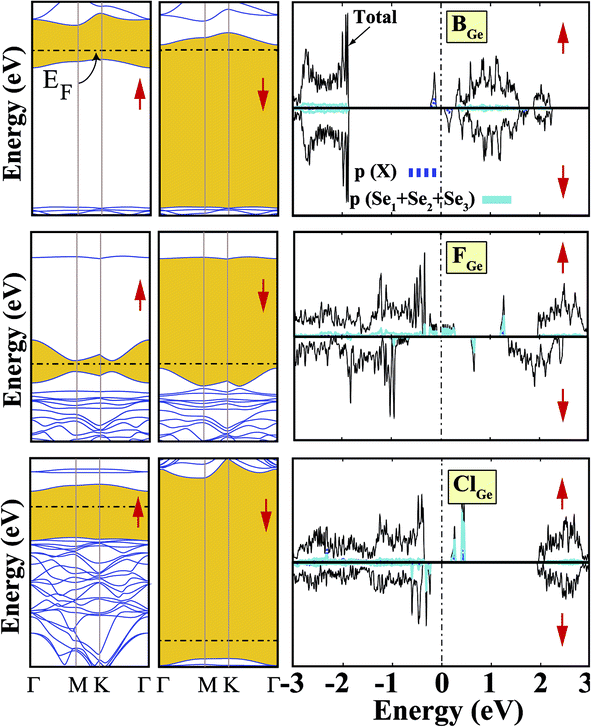
We started our investigation on substituted atoms in the Ge position. After optimization following substitutional doping, local defects and reconstructions occur in single-layer b-GeSe. Some of the substituted atoms change the lattice constant and create new bond lengths around the defects, and these values are given in Table 1. The cohesive energies and formation energies for all of the considered systems are given in Fig. 3. As evident in Fig. 3 and Table 1, the cohesive energies decrease with an increase in period for the same group number, while the formation energies increase. These results are as expected, as with an increasing atomic radius and decreasing electronegativity of the dopant atoms, the doping feasibility becomes harder with the increasing period, and binding will be weaker and the cohesive energy will decrease. In accordance with these results, the formation energies of defects with atoms B, C, N and Si in the Ge position in the network are lower than those of the other atoms and vacancies. These differences can indicate that these point defects may be more dominant defects during the fabrication of single-layer b-GeSe. To investigate the change in the electronic structure of the new systems, we calculated their electronic partial density of states (PDOS) and their band structures, as illustrated in Fig. 4 and 5. According to our extensive calculations, the substitution of group IIIA atoms on Ge sites yields a magnetic semiconductor material (μ = 1 μB), as seen in Fig. 4 and 5. By increasing the atomic radius, impurity states in the spin-up channel slip below the Fermi level, and also the conduction bands move to the upper energy levels. As seen in the figures, the impurity states are composed of the p-orbitals of the dopants (B, Al, Ga, etc.) and the p-orbitals of the Se atoms, which are around the defects. All group IIIA dopant systems show n-type semiconductor properties for the spin-down channels, therefore charge transfer becomes easier than in the bare GeSe state. When C or Si are substituted on a Ge site, as seen in Table 1, the cohesive energies of the system are closest to the cohesive energy of bare GeSe, because they have the same number of valence electrons. If we compare the total density of states of GeSe with that of CGe and SiGe, we can see similarities in the general trends of the DOS, and also their band structures are the same except for the impurity states. Again, with an increase in the atomic radius, the impurity states go to deeper energy levels. Although the atomic radii are changing, the nonmagnetic semiconductor properties are conserved because C and Si possess the same number of valence electrons as the Ge atom. For example, when the dopants have three more electrons from the host Ge atom, such as in group VIIA, the system attains a magnetic moment and GeSe becomes a magnetic semiconductor. As seen in the FGe part of Fig. 5, there is one shallow state in both of the spin-up and down states, which lies near the conduction band minimum (CBM). For FGe and ClGe, there are donor bands instead of donor states in the spin-up states in the vicinity of the Fermi level. If the first Bohr radius is large, these kinds of band occur due to the donor orbital overlapping, even though the doping ratio is low. Different to the other mentioned dopants, GeSe can show metallic behavior with a doped atom in the Ge position which has one more valence electron than the Ge atom, such as atoms from group VA. By substituting N, P or As atoms into a Ge site, impurity bands occur in the Fermi level due to the p-orbitals of the dopants and Se atoms around the defect. In addition, N doping on a Ge site raises the magnetic moment of the system by 0.5 μB.
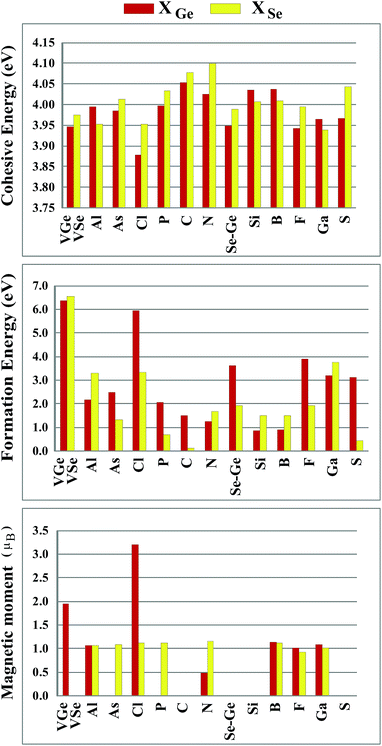 |
| | Fig. 3 Cohesive energy, defect formation energy and magnetic moments for the vacancy systems (VGe and VSe) and X atom substituted systems (X = Al, As, Cl, P, C, N, Se or Ge, Si, B, F, Ga and S) in the Ge position and Se position in the (4 × 4) supercell of the b-GeSe phase. | |
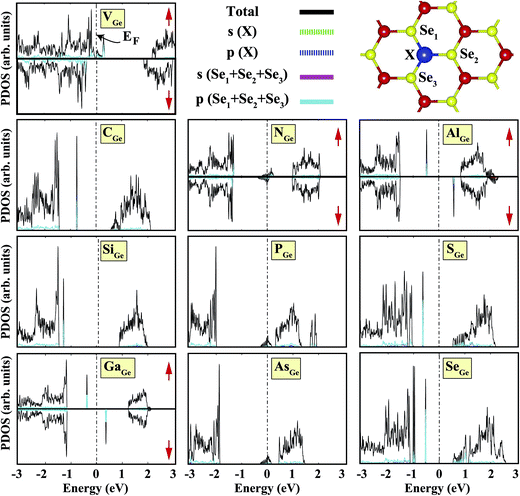 |
| | Fig. 4 Total (shown by black lines), subatom and atoms around the defect projected density of states for a single atom, substituted in the Ge position, in the (4 × 4) supercell of the b-GeSe phase. Zero energy is set to the Fermi level, EF, shown by dashed dotted lines. | |
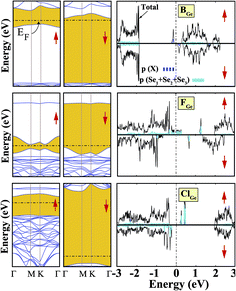 |
| | Fig. 5 Total (shown by black lines), subatom and atoms around the defect projected density of states for a single atom, substituted in the Ge position, in the (4 × 4) supercell of the b-GeSe phase. Band structures are also illustrated in the vicinity of the Fermi level. Zero energy is set to the Fermi level, EF, shown by dashed dotted lines. | |
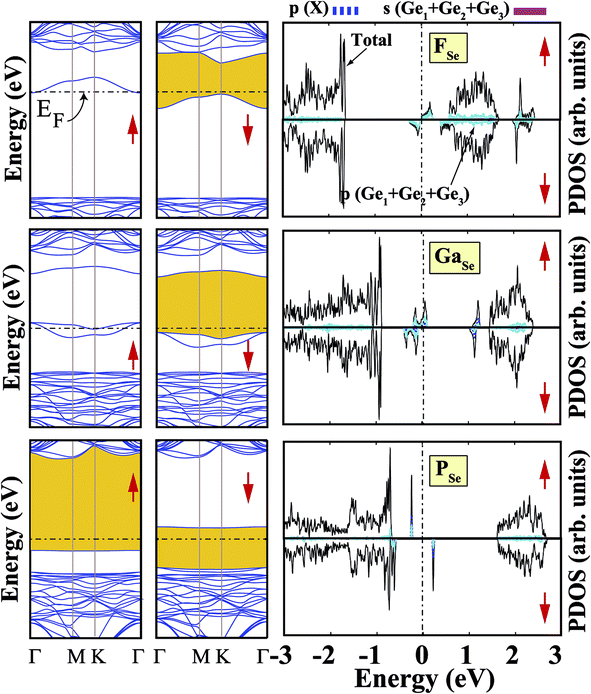
We also investigated the substitution of the same atoms in the Se position. As seen in Fig. 3, similar trends for the formation (cohesive) energy values are valid for the XSe structures. Their values increase (decrease) with an increasing period. The most dominant defect is the CSe defect, and the substitution of an S atom instead of a Se atom is another favorable defect type with a lower formation energy. Similar to the XGe defect, some of the XSe structures have smaller lattice constants than that of the bare GeSe structure. We did not encounter a structure that was larger than bare GeSe. This situation can be attributed to our selection of dopant atoms, which are all light elements. We suppose that the GeSe structure will be enlarged with heavy elements. Similar to XGe, substituted atoms in the Se position change the electronic structure and cause localized states in the fundamental band gap or resonance states in the band continua of b-GeSe. However, different to XGe, there are many half metallic systems for the XSe defect type. Group IIIA doping gives rise to localized states in the fundamental band gap of GeSe. B, Al and Ga doping yields similar band structures with different band gap values, so we only give the GaSe band structure (see Fig. 6). As seen in Fig. 7, there are half-occupied acceptor bands in the Fermi level, and one unoccupied acceptor state near the VBM for the spin-up channel. These acceptor bands and state become nearly 0.1 eV lower in energy, and the system shows semiconductor properties for the spin-down channel. C, Si or Ge doping in the Se position gives similar electronic structures to each other, and the new systems display p-type nonmagnetic semiconductor properties due to the shallow states near the CBM. When we substituted group VA atoms in the Se position, we attained magnetic (μ = 1 μB) semiconductor materials resulting from the one extra hole in the crystals. PDOS are given in Fig. 6, and we also plot the band structure of PSe and illustrate it in Fig. 7. NSe and AsSe also have similar bands. As is evident in Fig. 7, there is one occupied and unoccupied impurity state in the spin-up and spin-down channels, respectively. All group VA doped systems are p-type semiconductors for the spin-down state. Replacing Se with S atoms almost does not change the electronic structure of GeSe, and only the degeneracy decreases due to a very small structural shrink. If we add dopants which have one more electron than Se, such as F and Cl, the systems have a 1 μB magnetic moment and show half-metallicity properties for F doping, while remaining a semiconductor for Cl doping.
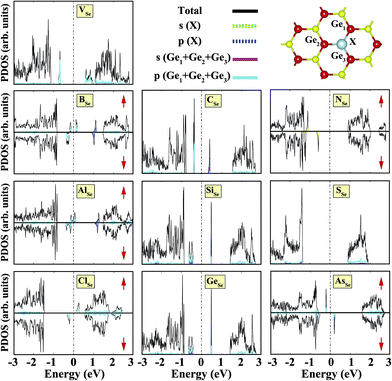 |
| | Fig. 6 Total (shown by black lines), subatom and atoms around the defect projected density of states for a single atom, substituted in the Se position, in the (4 × 4) supercell of the b-GeSe phase. Zero energy is set to the Fermi level, EF, shown by dashed dotted lines. | |
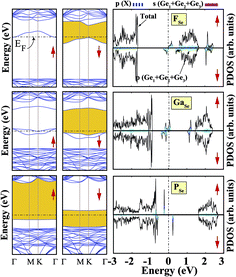 |
| | Fig. 7 Total (shown by black lines), subatom and atoms around the defect projected density of states for a single atom, substituted in the Se position, in the (4 × 4) supercell of the b-GeSe phase. Band structures are also illustrated in the vicinity of the Fermi level. Zero energy is set to the Fermi level, EF, shown by dashed dotted lines. | |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ac
*ac