
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A visible-light-induced photoelectrochemical water-splitting system featuring an organo-photocathode along with a tungsten oxide photoanode†
Yuto Kawaia,
Keiji Nagaib and
Toshiyuki Abe *a
*a
aDepartment of Frontier Materials Chemistry, Graduate School of Science and Technology, Hirosaki University, 3 Bunkyo-cho, Hirosaki 036-8561, Japan. E-mail: tabe@hirosaki-u.ac.jp
bLaboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, Suzukake-dai, Midori-ku, Yokohama 226-8503, Japan
First published on 11th July 2017
Abstract
A photoelectrochemical water-splitting system featuring an organo-photocathode of a p–n bilayer was studied, where WO3 was simultaneously utilized as a photoanode. Stoichiometric formation of H2 and O2 was found to occur due to the decomposition of water. In the reference system of a WO3 photoanode and Pt counter electrode, bias voltages more than 0.4 V were needed to be applied for water splitting; however, the present system successfully led to water decomposition by applying only a low voltage of 0.1 V to the system. In the present water-splitting system, oxidizing and reducing powers can be separately generated at the WO3 photoanode and organo-photocathode, respectively, which is distinct from the reference system. Furthermore, electron transfer from WO3 (conduction band) to the hole-retained p-type layer (valence band) in the organo-photocathode can efficiently occur for completing the photoelectrochemical process, thus, resulting in a high concentration of holes available for rate-limiting O2 evolution at WO3 on the basis of efficient charge separation.
Introduction
Solar hydrogen has attracted attention as a potential clean energy for establishing a sustainable society. The most ideal and ultimate method for acquiring solar hydrogen is water photolysis. Hence, the photoelectrochemical and photocatalytic decomposition of water has been actively investigated.1–10 The construction of an efficient water-splitting system under solar irradiation is the most central issue. Tungsten(VI) oxide (WO3) is a photocatalyst material capable of water oxidation to O2; here, the metal oxide has been realized as a photoanode for O2 evolution.10–17 WO3 cannot solely lead to the overall decomposition of water because the reducing power generated at its conduction band edge is insufficient for the evolution of H2 from H+.18–21 In other words, some of the metal oxides (such as WO3, Fe2O3, etc.) can respond to visible-light energy; however, their conduction band edge of the d-orbital is forced on a downward shift, making it impossible for the reduction of H+.18–21 With respect to the position of valence band edge corresponding to oxidizing power, WO3 is similar to TiO2.20–23 Therefore, making an effective use of the oxidizing power of WO3 can help in developing a visible-light-induced water-splitting system. For instance, when employing a system featuring an organo-photocathode for photoelectrochemical water splitting (cf. Scheme 1), TiO2 photoanode was utilized in the overall splitting of water, particularly under bias-free condition.4 As reported by Honda and Fujishima,24 the water-splitting system, comprising TiO2 photoanode and Pt counter, is a typical instance, but it required chemical or electric bias for completing the water splitting process.25–29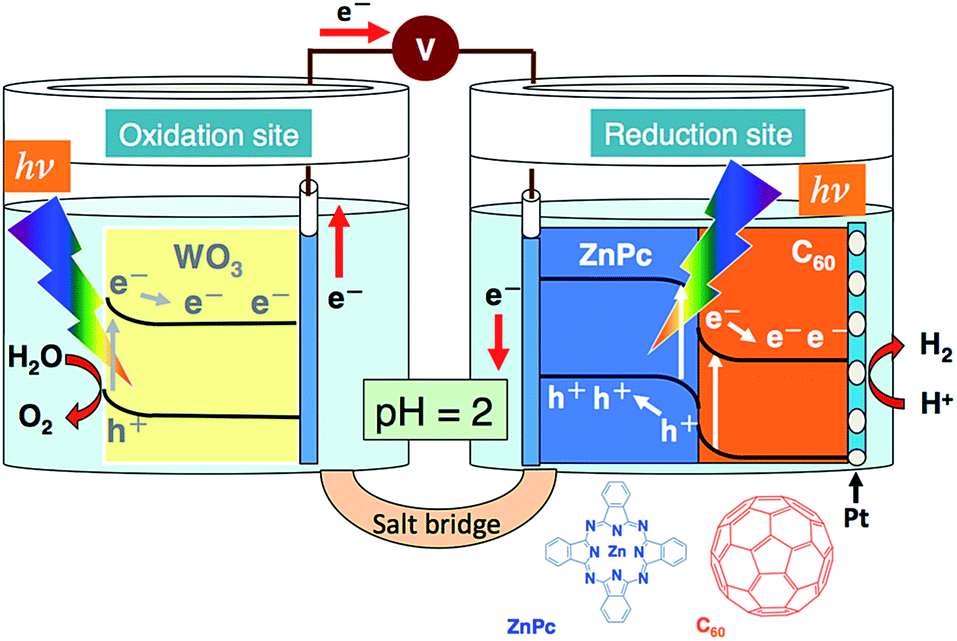 | ||
| Scheme 1 An illustration of the system employed for water splitting. | ||
Photoelectrochemical and photocatalytic systems based on the utilization of both organic semiconductors and p–n bilayer have been studied by our group.30–40 For example, an organic p–n bilayer of phthalocyanine [MPc (M = H2 or Zn), p-type] and fullerene (C60, n-type) can function as a photocathode, where a reducing power can be generated at the C60/water interface through a series of photophysical events within the bilayer.38–40 Particularly, in terms of molecular hydrogen evolution, when loading Pt on the C60 surface in the MPc/C60 organo-photocathode, it induced the reduction of H+ at applied potentials that are more positive than the formal potential of H+/H2.39,40
In the present study, a photoelectrochemical water-splitting system was investigated, where WO3 (photoanode) and Pt-loaded ZnPc/C60 bilayer (photocathode, vide supra) were simultaneously employed in the system depicted in Scheme 1. The stoichiometric decomposition of water into H2 and O2 occurred on applying a bias voltage less than the theoretical voltage of 1.23 V. It is noted that the present system successfully led to water decomposition under a low bias condition (i.e. 0.1 V), where a reference system of WO3 photoanode and Pt counter cannot induce the redox reaction of water. Details of the results are discussed in the following sections.
Experimental
The ZnPc/C60 bilayer was prepared by vapor deposition (pressure, <1.0 × 10−3 Pa; deposition speed, ca. 0.03 nm s−1) using an indium-tin-oxide (ITO)-coated glass plate as the base material.40 ZnPc was first coated on ITO, followed by the coating of C60 on top of the ZnPc layer; moreover, a Pt co-catalyst was photocathodically deposited onto the C60 surface of the bilayer.39,40 The resulting photocathode is abbreviated as ITO/ZnPc/C60–Pt. A WO3 film coated on an F-doped tin oxide (FTO) was prepared according to the following procedures. WO3 (72 g, Kojundo Chemical Laboratory) and acetylacetone (0.8 mL, Wako Chemical) were mixed well in a mortar, and then, pure water (24 mL) was slowly added during 2 h. Subsequently, Triton X-100 detergent (1 mL, Aldrich) was added and mixed well with the resulting slurry of WO3, following which the mixture was subject to ultrasonic irradiation. Furthermore, the sample was centrifuged to eliminate large-sized particles of WO3 (1000 rpm, 5 min). This centrifugation process was repeated 3 times. The resulting suspension of WO3 (800 μL) was spin-coated (2000 rpm, 1 min) on an FTO electrode (4 × 4 cm), followed by drying at 373 K for 30 min. The processes of both spin-coating and drying were repeated 3 times. Prior to use, the resulting film was calcined in an electric furnace [at 823 K for 2 h (rate of temperature rising: 2 °C min−1)]. The SEM image of the WO3 film is shown in Fig. S1 (in the ESI†). The photoanode of WO3 is denoted as FTO/WO3. FTO is usually utilized in preparing photoelectrodes at a high temperature, in order to avoid an unfavorable increase of sheet resistance.41 The preparation of FTO/WO3 was conducted by modifying a method for FTO/TiO2.41 The aforementioned conditions of FTO/WO3 preparation were optimal (cf. thickness of WO3, ca. 1 μm). The effective area of all the photoelectrodes employed was 1 cm × 1 cm. A cell made up of twin compartments separated by a salt bridge was utilized for the water-splitting studies (see Scheme 1). All studies were performed under an Ar atmosphere in an aqueous H3PO4 solution (pH = 2). Other experimental details are provided in the ESI.†Results and discussion
Cyclic voltammogram (CV) was measured at FTO/WO3 photoanode in an H3PO4 solution (Fig. S2 in the ESI†), where a three-electrode system was employed for the voltammetric measurement (see Scheme S1 in the ESI†). CV measured at ITO/ZnPc/C60–Pt photocathode is also depicted in Fig. S2.†40 Considering the formal potentials for H+ reduction (−0.32 V vs. Ag/AgCl (sat.) for H+/H2 couple) and water oxidation (+0.91 V vs. Ag/AgCl (sat.) for O2/H2O couple) at pH = 2, the voltammograms indicated that the photo-induced reactions for H2 evolution and O2 evolution can efficiently occur at both photoelectrodes. In other words, a photoelectrochemical water splitting can be expected to take place under the condition of a low bias voltage, on the basis of the evidence that both photoanodic and photocathodic currents occurred at close potentials.In addition, action spectrum for the photocurrents generated at FTO/WO3 photoanode was measured, where the same setup as the voltammetric measurement was employed (cf. Scheme S1†). The resulting action spectrum acquired at the photoanode of WO3 was consistent with its absorption spectrum (Fig. S1†), indicating that a photocurrent can be generated due to the band-gap excitation of WO3 [cf. the magnitude of its band-gap was in accordance with the reported value (ca. 2.7 eV) corresponding to the absorption edge of ca. 460 nm18,21,23,42,43]. As for the ITO/ZnPc/C60–Pt photocathode capable of H2 evolution, its action spectrum has previously been clarified to originate in the entire visible-light absorption of both ZnPc and C60.40
According to Scheme 1, a photoelectrochemical water splitting was studied by applying bias voltages to the system. As a result, H2 evolution was found to occur along with O2 evolution, particularly at bias voltages less than 1.23 V (the theoretical voltage for water splitting). In other words, the stoichiometric decomposition of water took place in the present system. Fig. 1 shows the relationships between the amounts of H2 and O2 evolved and the applied voltages. Note that the application of only a low bias voltage of 0.1 V to the system led to water splitting, although voltages more than 0.3 V are usually needed to be applied to the reference system of WO3 photoanode and Pt counter (vide infra).11,15–17,21,44,45 The H2 and O2 amounts essentially increased with elevating applied voltages; however, the evolved amounts became gentle at voltages larger than 0.6 V. Based on the data of water splitting, the light-to-hydrogen conversion efficiency (η, see the ESI† concerning the calculation method) was estimated with respect to the applied voltages (Fig. 1). The most efficient water splitting occurred at 0.6 V with ca. 0.07% while the η value decreased with higher voltages. Applied bias voltages can lead to efficient charge separation along with an efficient charge transfer between both photoelectrodes, where the amounts of H2 and O2 may increase to involve the increasing concentration of carriers available for water decomposition. A decreasing value of η is attributed to a non-linear increase of carrier concentration with applied voltages, thus resulting in a moderate enhancement of kinetics for H2 and O2 formation. Such a phenomenon was also represented in another system of photoelectrochemical water splitting.4,44–49
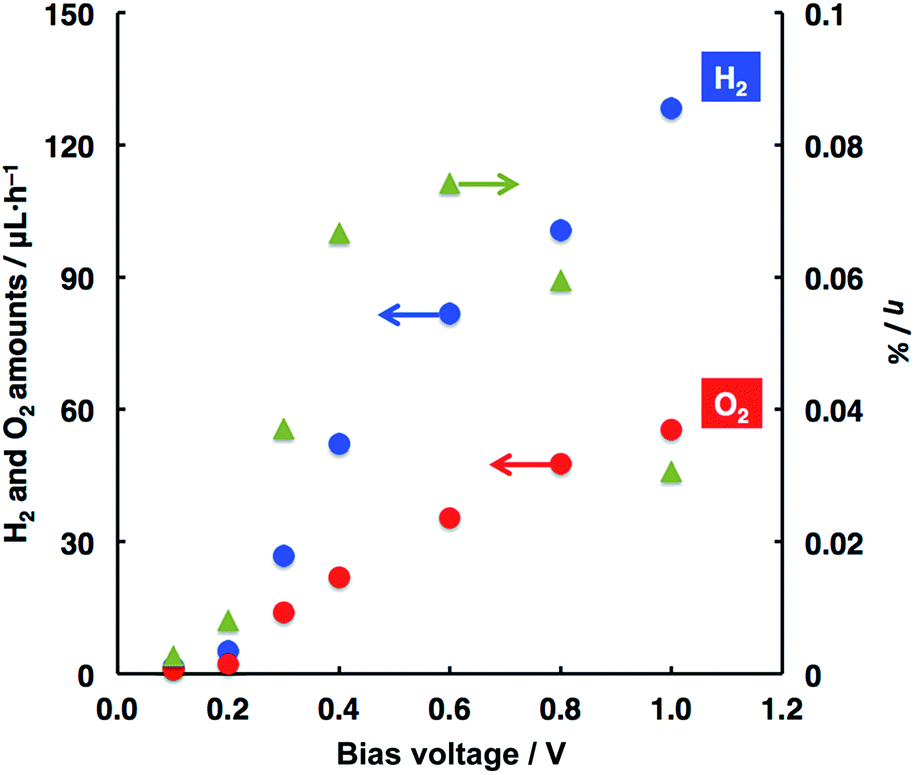 | ||
| Fig. 1 Relationships of the evolved H2 and O2 amounts and η values with applied voltages. This study was conducted in the two-electrode system depicted in Scheme 1. Faradaic efficiencies for H2 and O2 evolution were typically >90% and >80%, respectively (see ESI†). Photoanode, WO3 (geometrical area, 1 cm2); photocathode, ITO/ZnPc (75 nm)/C60 (125 nm)–Pt (geometrical area, 1 cm2); electrolyte, H3PO4 solution (pH = 2); light intensity (for photoanode), ca. 50 mW cm−2; light intensity (for photocathode), ca. 90 mW cm−2. | ||
A prolonged study of water splitting was conducted to examine the durability of the present system, where both photoanode and photocathode were repeatedly used with the cycle number. As shown in Fig. 2, the amounts of H2 and O2 evolved linearly increased with the cycle number, demonstrating stable and durable performance for water splitting.
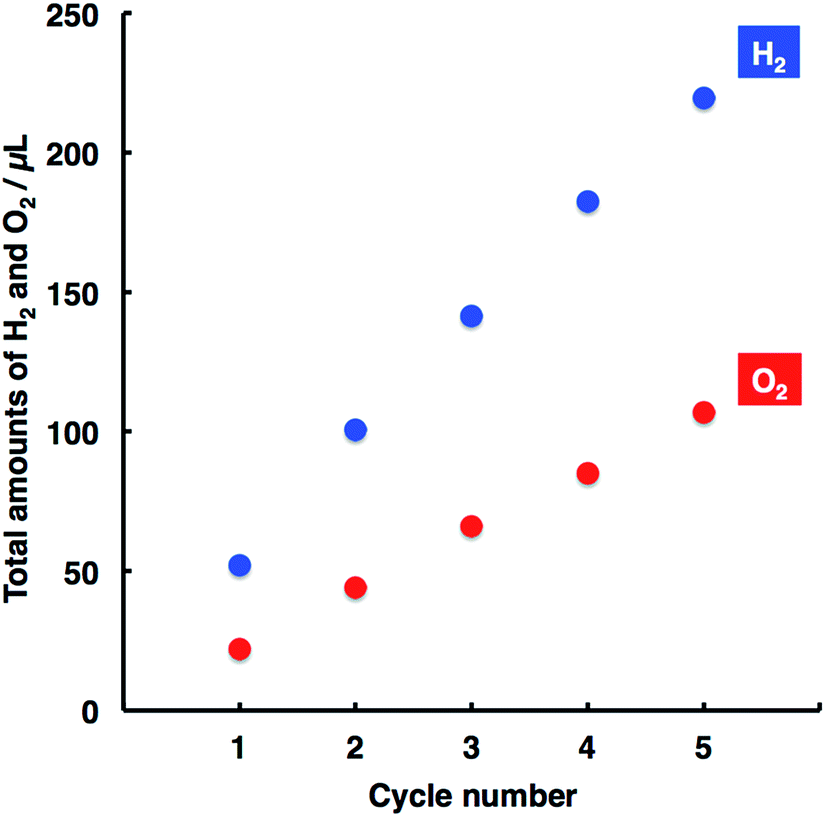 | ||
| Fig. 2 Relationships between the H2 and O2 amounts with cycle number. Experimental conditions were the same as those used in Fig. 1. The bias voltage of 0.4 V was applied to the system, and the irradiation time was 1 h per one cycle. | ||
Control experiments were conducted in the presence of methanol (electron donor) or Fe3+ ion (electron acceptor), which are compared with a typical result of photoelectrochemical water splitting (Table 1). Irrespective of the presence of Fe3+ ion, the amount of O2 evolved was almost constant (Entries 1 and 2). The evolved amount of H2 noticeably increased in the presence of methanol (Entry 3). Therefore, these results suggest that the present system (i.e. Entry 1) is kinetically dominated by the evolution of O2 from water.
| System | H2 amount (μL h−1) | O2 amount (μL h−1) | Note |
|---|---|---|---|
a Bias voltage of 0.6 V was applied for the system with experimental conditions similar to those in Fig. 1.b Data from Fig. 1.c A methanol solution (methanol/water(v/v) = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, pH = 2) was used.d An aqueous solution of Fe(NO3)3 (5 mM, pH = 2) was employed. :1, pH = 2) was used.d An aqueous solution of Fe(NO3)3 (5 mM, pH = 2) was employed. |
|||
| Entry 1b | 81.8 | 35.4 | No control system |
| Entry 2c | 150 | — | In the presence of methanol |
| Entry 3d | — | 34.0 | In the presence of Fe3+ |
A reference system of WO3 photoanode and Pt counter (denoted as WO3–Pt system) was examined for photoelectrochemical water splitting. The results are summarized in Fig. S3.† The WO3–Pt system resulted in the evolution of both H2 and O2 only when high voltages (i.e. ≥0.4 V) were applied to the system. This is reasonable since the bottom edge of the conduction band of WO3, corresponding to the position of reducing power, is 0.3 V positive for the formal potential of H+/H2 (vide supra).18–21 Fig. S3† showed similar dependencies of both the evolved amounts and η values on bias voltages to Fig. 1. In a separate experiment, the rate-limiting reaction in the WO3–Pt system was investigated in the presence of methanol or Fe3+ ion. As shown in Table S1 (in the ESI†), it was confirmed that the rate-limiting O2 evolution occurs in the reference system.
As for WO3 photoanode10–17 and ITO/ZnPc/C60–Pt photocathode,40 those photoelectrochemical reactivities for water reaction have previously been elucidated. The most typical characteristics of the present system are that oxidizing and reducing powers are separately generated at WO3 and ITO/ZnPc/C60–Pt, respectively. Furthermore, as indicated in Scheme 1, the excited electron generated at WO3 can be transferred to the hole-remained valence band of ZnPc for accomplishing photoelectrochemical water splitting, through which the pristine species of WO3 and ZnPc can be regenerated. This is distinct from the reference WO3–Pt system where the reducing power photogenerated at WO3 can directly participate in the evolution of H2 from H+. In Table 2, the typical comparison of the WO3–Pt system with the WO3 and ITO/ZnPc/C60–Pt system is shown. As aforementioned, those systems involve the rate-limiting O2 evolution. The amounts of H2 and O2 originating from water splitting were greater in the latter system. The difference between those systems is attributable to the magnitude of charge separation. In other words, the steady concentration of holes available for O2 evolution can be considered to be higher in the WO3 and ITO/ZnPc/C60–Pt system. This may indicate that the electron transport between the photoelectrodes employed (vide supra) efficiently occurs for producing the higher concentration of carriers.
| System | Photoanode | Cathode | H2 amount (μL h−1) | O2 amount (μL h−1) |
|---|---|---|---|---|
| a Bias voltage of 0.6 V was applied to the system, and other experimental conditions were similar to those of Fig. 1.b Data from Fig. 1.c Instead of ITO/ZnPc/C60–Pt, a Pt wire was employed as a cathode; data listed in this table can also be seen in Fig. S3.† | ||||
| Entry 1b | WO3 | ITO/ZnPc/C60–Pt | 81.8 | 35.4 |
| Entry 2c | WO3 | Pt wire | 13.5 | 7.82 |
Conclusions
This work studied the photoelectrochemical water-splitting system featuring an organo-photocathode along with a WO3 photoanode, where the stoichiometric decomposition of water into H2 and O2 was found to occur. Based on the action spectral characteristics of both ITO/ZnPc/C60–Pt photocathode and WO3 photoanode, a visible-light energy was available for water splitting. In the reference WO3–Pt system, the bias voltages more than 0.4 V needed to be applied for evolving H2 and O2; however, the present system demonstrated that extremely low-biased water splitting can occur. Distinct from the reference system, oxidizing and reducing powers for water splitting were separately generated at WO3 photoanode and ITO/ZnPc/C60–Pt photocathode, respectively. Irrespective of the systems studied, the evolution of O2 from water was the rate-limiting step. However, in the present system, the electron transfer from WO3 (CB) to hole-remained ZnPc (VB) can efficiently occur for producing a high concentration of carriers available for O2 evolution. The application of two types of materials for water splitting is an effective method, which can be seen in the Z-scheme type photocatalytic water-splitting system50–52 as well as the present system. In these systems, the materials, which cannot solely participate in the overall decomposition of water, can play active parts. The utilization of distinct types of materials may also have merits in terms of harvesting of solar energy. Photoelectrochemical water decomposition is one of the most promising approaches for acquiring solar hydrogen. In this context, developing efficient photoelectrodes is a vital issue for establishing a practical water-splitting system.Acknowledgements
This work was partly supported by a grant from The Murata Science Foundation (T. A.), Yashima Environment Technology Foundation (T. A.), and the Cooperative Research Program of “Network Joint Research Center for Materials and Devices” (T. A.).References
- J. Su, L. Zhu and G. Chen, Appl. Catal., B, 2016, 186, 127–135 CrossRef CAS.
- M. Kodera, H. Urabe, M. Katayama, T. Hisatomi, T. Minegishi and K. Domen, J. Mater. Chem. A, 2016, 4, 7658–7664 CAS.
- S. Ida, K. Yamada, M. Matsuka, H. Hagiwara and T. Ishihara, Electrochim. Acta, 2012, 82, 397–401 CrossRef CAS.
- T. Abe, K. Fukui, Y. Kawai, K. Nagai and H. Kato, Chem. Commun., 2016, 52, 7735–7737 RSC.
- K. Tsuji, O. Tomita, M. Higashi and R. Abe, ChemSusChem, 2016, 9, 2201–2208 CrossRef CAS PubMed.
- T. Grewe and H. Tüysüz, J. Mater. Chem. A, 2016, 4, 3007–3017 CAS.
- J. Yan, H. Wu, H. Chen, Y. Zhang, F. Zhang and S. Liu, Appl. Catal., B, 2016, 190, 130–137 CrossRef.
- R. Kobayashi, T. Takashima, S. Tanigawa, S. Takeuchi, B. Ohtani and H. Irie, Phys. Chem. Chem. Phys., 2016, 18, 27754–27760 RSC.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- J. Zhang, H. Ma and Z. Liu, Appl. Catal., B, 2017, 201, 84–91 CrossRef CAS.
- C. Fàbrega, S. Murcia-López, D. Monllor-Satoca, J. D. Prades, M. D. Hernández-Alonso, G. Penelas, J. R. Morante and T. Andreu, Appl. Catal., B, 2016, 189, 133–140 CrossRef.
- X. Shi, I. Herraiz-Cardona, L. Bertoluzzi and P. Lopez-Varo, Phys. Chem. Chem. Phys., 2016, 18, 9255–9261 RSC.
- J. Zhao, E. Olide and F. E. Osterloh, J. Electrochem. Soc., 2015, 162, H65–H71 CrossRef CAS.
- D. Chandra, K. Saito, T. Yui and M. Yagi, Angew. Chem., Int. Ed., 2013, 52, 12606–12609 CrossRef CAS PubMed.
- A. Kleiman-Shwarsctein, A. B. Laursen, F. Cavalca, W. Tang, S. Dahla and I. Chorkendorff, Chem. Commun., 2012, 48, 967–969 RSC.
- R. Liu, Y. Lin, L.-Y. Chou, S. W. Sheehan, W. He, F. Zhang, H. J. M. Hou and D. Wang, Angew. Chem., Int. Ed., 2011, 50, 499–502 CrossRef CAS PubMed.
- C. Santato, M. Ulmann and J. Augustynski, J. Phys. Chem. B, 2001, 105, 936–940 CrossRef CAS.
- B. D. Alexander, P. J. Kulesza, I. Rutkowska, R. Solarskac and J. Augustynski, J. Mater. Chem., 2008, 18, 2298–2303 RSC.
- S. Sfaelou, L.-C. Pop, O. Monfort, V. Dracopoulos and P. Lianos, Int. J. Hydrogen Energy, 2016, 41, 5902–5907 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CAS.
- T. Jin, P. Diao, Q. Wu, D. Xu, D. Hu, Y. Xie and M. Zhang, Appl. Catal., B, 2014, 148–149, 304–310 CrossRef CAS.
- Q. Mi, A. Zhanaidarova, B. S. Brunschwig, H. B. Gray and N. S. Lewis, Energy Environ. Sci., 2012, 5, 5694–5700 CAS.
- D. Monllor-Satoca, L. Borja, A. Rodes, R. Gómez and P. Salvador, ChemPhysChem, 2006, 12, 2540–2551 CrossRef PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- L. N. Quan, Y. H. Jang, K. A. Stoerzinger, K. J. May, Y. J. Jang, S. T. Kochuveedu, Y. Shao-Horn and D. H. Kim, Phys. Chem. Chem. Phys., 2014, 16, 9023–9030 RSC.
- H. Li, J. Chen, Z. Xia and J. Xing, J. Mater. Chem. A, 2015, 3, 699–705 CAS.
- S. Hernández, D. Hidalgo, A. Sacco, A. Chiodoni, A. Lamberti, V. Cauda, E. Tresso and G. Saracco, Phys. Chem. Chem. Phys., 2015, 17, 7775–7786 RSC.
- M. S. Wrighton, D. S. Ginley, P. T. Wolczanski, A. B. Ellis, D. L. Morse and A. Linz, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 1518–1522 CrossRef CAS.
- A. Fujishima, K. Kohayakawa and K. Honda, J. Electrochem. Soc., 1975, 122, 1487–1489 CrossRef CAS.
- T. Abe, K. Nagai, S. Kabutomori, M. Kaneko, A. Tajiri and T. Norimatsu, Angew. Chem., Int. Ed., 2006, 45, 2778–2781 CrossRef CAS PubMed.
- K. Nagai, T. Abe, Y. Kaneyasu, Y. Yasuda, I. Kimishima, T. Iyoda and H. Imaya, ChemSusChem, 2011, 4, 727–730 CrossRef CAS PubMed.
- K. Nagai, Y. Yasuda, T. Iyoda and T. Abe, ACS Sustainable Chem. Eng., 2013, 1, 1033–1039 CrossRef CAS.
- T. Abe, M. Okumura, Y. Kikuchi, T. Itoh and K. Nagai, J. Mater. Chem. A, 2017, 5, 7445–7450 CAS.
- P. Arunachalam, S. Zhang, T. Abe, M. Komura, T. Iyoda and K. Nagai, Appl. Catal., B, 2016, 193, 240–247 CrossRef CAS.
- D. Mendori, T. Hiroya, M. Ueda, M. Sanyoushi, K. Nagai and T. Abe, Appl. Catal., B, 2017, 205, 514–518 CrossRef CAS.
- T. Abe, N. Taira, Y. Tanno, Y. Kikuchi and K. Nagai, Chem. Commun., 2014, 50, 1950–1952 RSC.
- T. Abe, Y. Tanno, N. Taira and K. Nagai, RSC Adv., 2015, 5, 46325–46329 RSC.
- T. Abe, K. Nagai, K. Sekimoto, A. Tajiri and T. Norimatsu, Electrochem. Commun., 2005, 7, 1129–1132 CrossRef CAS.
- T. Abe, S. Tobinai, N. Taira, J. Chiba, T. Itoh and K. Nagai, J. Phys. Chem. C, 2011, 115, 7701–7705 CAS.
- T. Abe, Y. Hiyama, K. Fukui, K. Sahashi and K. Nagai, Int. J. Hydrogen Energy, 2015, 40, 9165–9170 CrossRef CAS.
- R. Saito, H. Ueno, J. Nemoto, Y. Fujii, A. Izuoka and M. Kaneko, Chem. Commun., 2009, 2009, 3231–3233 RSC.
- F. Amano, E. Ishinaga and A. Yamakata, J. Phys. Chem. C, 2013, 117, 22584–22590 CAS.
- T. Jin, P. Diao, D. Xu and Q. Wu, Electrochim. Acta, 2013, 114, 271–277 CrossRef CAS.
- W. Zhao, Z. Wang, X. Shen, J. Li, C. Xu and Z. Gan, Int. J. Hydrogen Energy, 2012, 37, 908–915 CrossRef CAS.
- Y. Hou, F. Zuo, A. P. Dagg, J. Liu and P. Feng, Adv. Mater., 2014, 26, 5043–5049 CrossRef CAS PubMed.
- I. Fujimoto, N. Wang, R. Saito, Y. Miseki, T. Gunji and K. Sayama, Int. J. Hydrogen Energy, 2014, 39, 2454–2461 CrossRef CAS.
- J. Akikusa and S. U. M. Khan, Int. J. Hydrogen Energy, 2002, 27, 863–870 CrossRef CAS.
- M. Radecka, M. Rekas, A. Trenczek-Zajac and K. Zakrzewska, J. Power Sources, 2008, 181, 46–55 CrossRef CAS.
- M. Moriya, T. Minegishi, H. Kumagai, M. Katayama, J. Kubota and K. Domen, J. Am. Chem. Soc., 2013, 135, 3733–3735 CrossRef CAS PubMed.
- R. Abe, M. Higashi and K. Domen, ChemSusChem, 2011, 4, 228–237 CrossRef CAS PubMed.
- K. Maeda, M. Higashi, D. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858–5868 CrossRef CAS PubMed.
- H. Kato, Y. Sasaki, N. Shirakura and A. Kudo, J. Mater. Chem. A, 2013, 1, 12327–12333 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, cyclic voltammograms at photoelectrodes, action spectrum for photocurrents generated at WO3, and its absorption spectrum and SEM images, and data of photoelectrochemical water splitting in the reference WO3–Pt system. See DOI: 10.1039/c7ra05272c |
| This journal is © The Royal Society of Chemistry 2017 |