
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First-principles calculation of lithium insertion into homogeneous a-SiC2/5O6/5 as high performance anode
Ningbo Liao *,
Beirong Zheng,
Miao Zhang and
Wei Xue
*,
Beirong Zheng,
Miao Zhang and
Wei Xue
College of Mechanical & Electrical Engineering, Wenzhou University, Wenzhou, 325035, P.R.China. E-mail: nliao@wzu.edu.cn; lnb55@163.com; Fax: +86-577-86689138; Tel: +86-577-86689138
First published on 14th June 2017
Abstract
Amorphous silicon oxycarbide is considered as a promising anode material for new generation of lithium-ion batteries, and figuring out the lithiation mechanism is crucial for its application. In this work, first principle calculations are performed to study the atomic structures, formation energy and lithiation voltage of homogeneous SiC2/5O6/5. The interpretation of radial distribution, angular distribution and coordinate number suggests that the Si–O bond tends to break and the Li2O will form at the beginning of lithiation, then the LixO and the LiySi form with increasing Li concentration, which makes a major contribution to the capacity of SiC2/5O6/5. By the Li content dependence of the formation energies curve, the theoretical specific capacity of SiC2/5O6/5 is predicted as 1415 mA h g−1, which is comparable to the reversible capacity of 900 mA h g−1 in experiments. Both the formation energies and the voltage curves suggest lithium is preferable in incorporation with SiC2/5O6/5, and this is attributed to the formation of LixO and LiySi.
1. Introduction
Graphite is widely used as an anode material for commercial lithium-ion batteries (LIBs), however, the low capacity (372 mA h g−1) has limited its further applications. Silicon is a promising material to replace graphite with lower cost and around 10 times the gravimetric capacity.1,2 But silicon exhibits about a 300% volume expansion during cycling, which will lead to mechanical failure, poor cycling performance and irreversible capacity.3,4 Therefore, research efforts are being made to search for new anode materials with high capacities and long life cycle.Amorphous silicon oxycarbide (a-SiCO) presents a high reversible capacity (larger than 800 mA h g−1) and is a promising anode material for LIBs.5–12 Amorphous SiCO ceramics generally include SiCO glass and SiCO glass with free carbon phase, corresponding to homogeneous structure and inhomogeneous structure respectively. Current experiments13 suggested that both the SiCO structures have a very high first insertion capacity, the concentration of lithium residing at the mixed bonds is much greater than the concentration sequestered in the free carbon,7 and the reversible capacity of these mixed bonds is severely diminished when oxygen is substituted by nitrogen.6 Although the experiments provide some information on how the nano-structure influences the electrochemical properties of SiCO, the mechanism for lithium insertion into a-SiCO is still not clearly understood. More insight needs to be gained in terms of atomistic structure and energy, which could be very challenging for current experimental technologies.
First principles calculations can obtain the atomic structure, electronic properties and lithiation characters of anode materials, and were successfully used in analyzing LIB electrodes such as Si,14 SiO,15 Nb2C 16 and graphene sheets.3 In our previous studies,17,18 SiCO models containing mixed-bond tetrahedron were proposed based on crystalline SiO2 configuration,17 the influence of tetrahedron on the performance of SiCO was studied. Moreover, in order to gain deeper insight into amorphous SiCO structure, a more realistic model was established by molecular dynamics simulation of melt-quench, and the effect of carbon segregation on performance of inhomogeneous SiCO was studied.18 In this work, the performance of homogeneous a-SiCO is investigated by first principles calculations, the atomic structures, formation energy and lithiation voltage of SiC2/5O6/5 are calculated and discussed.
2. Methods
Classical molecular dynamics (CMD) based melt quench simulations were used to obtain the initial structures of SiC2/5O6/5. Tersoff potential19 and the potential parameters for SiC19 and SiO2 (ref. 20) were used to describe the atomic interaction in SiC2/5O6/5. The melt-quench procedure proposed in our previous study21 was used to generate the amorphous structures of SiC2/5O6/5 from random atomic configuration, the system temperature was adjusted by velocity scaling and canonical ensemble (NVT) using a Nosé–Hoover thermostat. The CMD simulations were implemented by LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) code.22The first principles simulations were then performed to optimize the SiC2/5O6/5 structures further. The systems were annealed at 1800 K by 8 ps of NVT simulations, and were then quenched to 300 K at a rate of 0.3 K fs−1 by NPT simulations. Finally the amorphous structures are relaxed fully by geometry optimization of lattice parameters and atomic position. A plane-wave cut-off of 300 eV and a 2 × 2 × 2 κ-points mesh were found to give sufficient convergence. The geometry optimization was conducted by: residual force < 0.01 eV Å−1, convergence of energy change per atom < 2 × 10−6 eV, stress < 0.01 GPa. The electronic broadening width was 0.1 eV in a variable occupancy calculation, and the same orbitals for alpha and beta spins were used. The generalized gradient approximation following the Perdew–Bueke–Ernzerhof scheme (GGA-PBE)23 was used. The first principles simulations according to the density functional theory (DFT) ultra-soft pseudo-potential method were conducted in CASTEP code24,25 in Materials Studio 5.5.
The SiC2/5O6/5 studied here refers to the stoichiometric glass composed of 1/3(SiC) and 2/3(SiO2). The formation energy of LixSiC2/5O6/5 is obtained by subtracting the reference energies of SiC2/5O6/5 and Li from the energy of LixSiC2/5O6/5 model. The formation energy per Si atom is given by:15
| Ef = E(LixSiC2/5O6/5) − (xE(Li) + E(SiC2/5O6/5)) | (1) |
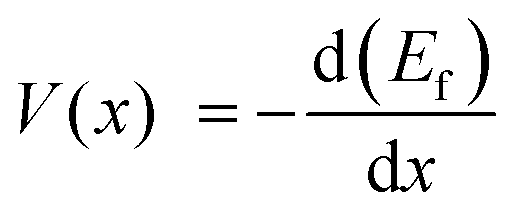 | (2) |
3. Results and discussion
The calculated properties of SiC2/5O6/5 are listed in Table 1 and compared to the experimental data. The peaks values in radial distribution functions (RDFs) and angular distribution are obtained by Gaussian multi-peak fitting.26 The bond length of Si–O, Si–C and C–C are obtained by the sharp peaks of RDFs, observed at rSiO = 1.64 Å, rSiC = 1.89 Å and rCC = 1.51 Å respectively, correspond to the experimental data of rSiO = 1.62 Å, rSiC = 1.88 Å and rCC = 1.49 Å.27,28 The tetrahedrons are mainly linked to each other by vertex and form a broad angular distribution Si–O–Si, centered approximately at 146°, it corresponds to the peak of 140–144° in experiments.27 The Young's modulus (E) of SiC2/5O6/5 is 99 GPa and within the range of 90–110 GPa in experiments.29,30 The glass transition temperature (Tg) is determined by the temperature corresponds to the change in slope of temperature-dependent Young's modulus curve. The obtained Tg of SiC2/5O6/5 is 1460 K and is closed to the range of 1573–1673 K in experiments.31–33| This work | Experimental results27,28,30–36 | |
|---|---|---|
| Si–O [Å] | 1.64 | 1.62 |
| Si–C [Å] | 1.89 | 1.88 |
| C–C [Å] | 1.51 | 1.49 |
| Si–O–Si [deg] | 146 | 140–144 |
| E [GPa] | 99 | 90–110 |
| Tg [K] | 1460 | 1573–1673 |
| Density [g m−3] | 2.58 | 2.4–2.64 |
In order to describe the structural evolution of SiC2/5O6/5 under Li insertion, a series of configuration snapshots of LixSiC2/5O6/5 are presented in Fig. 1. At low Li concentration, the Si atoms are slightly displaced from their tetrahedral positions, and formation of LixSiOy-like structures can be observed. With increasing Li concentration, some of the silicon-centered tetrahedrons are destroyed, which leads to the breaking of Si–O bonds and the formation of LixO.
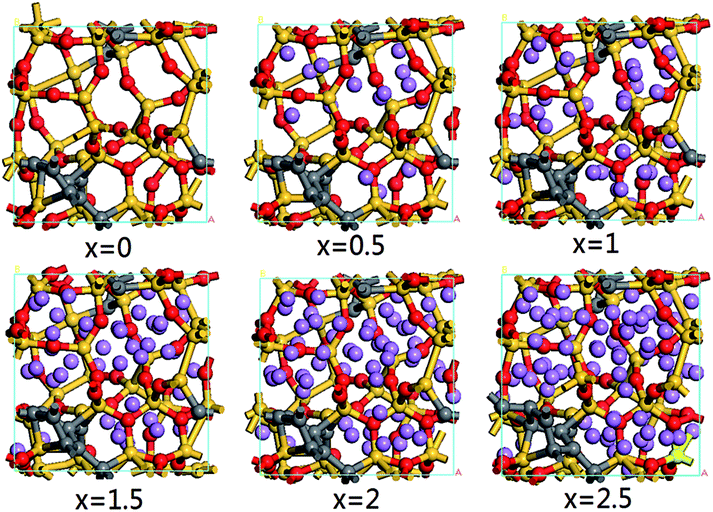 | ||
| Fig. 1 Atomic configurations of LixSiC2/5O6/5 with increasing Li concentration. The yellow, grey, red and purple spheres represent the silicon, carbon, oxygen and lithium atoms respectively. | ||
The atomic correlations of the lithiated SiCxO6/5 are inferred by means of RDFs, as shown in Fig. 2. The bond length of Si–O and C–C for lithiated SiC2/5O6/5 are determined by the first peaks of RDFs around 1.64 Å and 1.51 Å respectively. With increasing Li concentration, the first peak of Si–O slightly shift to a larger value and its long distance correlation becomes more predominate, this is an indicative of stretched Si–O bonds and rupture of Si–O–Si units, which is accorded with the above observations. The small peak of Si–O around 1.85 Å corresponds to the Si–O bond near the edge of free carbon. The C–C network tends to be more close together after the rupture of Si–O–Si units, it results in a very slight decrease on C–C bond length, which proves the structural stability of free carbon during lithiation.
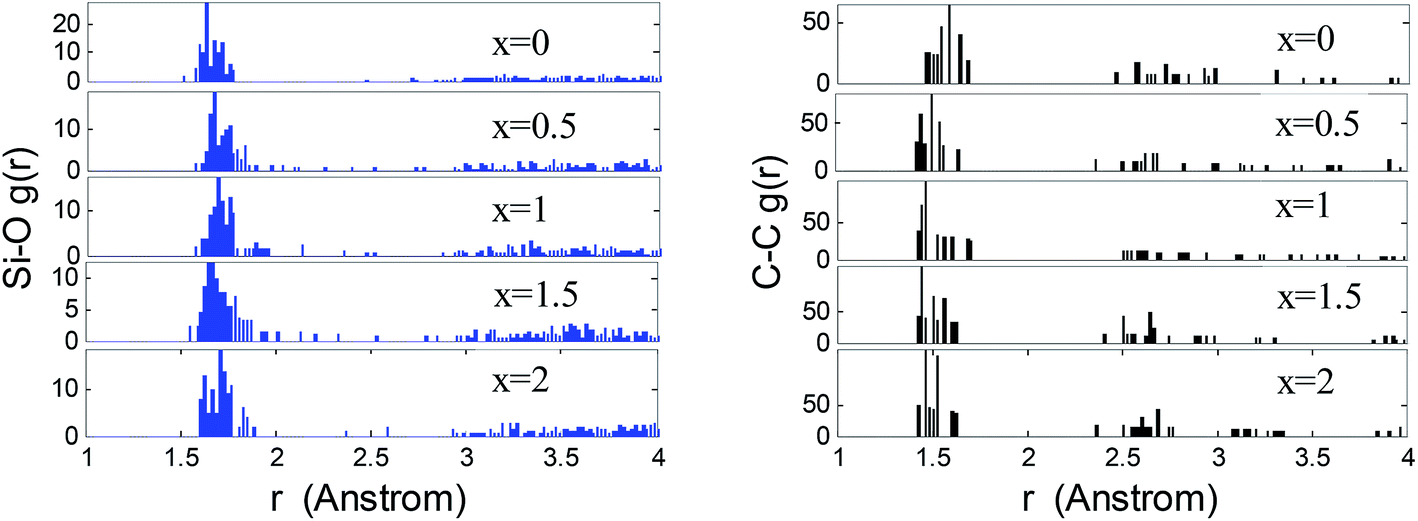 | ||
| Fig. 2 Calculated Si–O and C–C RDFs of a-SiC2/5O6/5 at several degrees of lithiation. | ||
The coordination numbers are obtained by integrating the first peak of radial distribution function, as shown in Fig. 3. Based on the average bond length of O–Si, Si–Li and O–Li,15,37–40 1.85 Å, 3.08 Å and 2.16 Å are chosen as maximum cutoff radii for the calculations of CNO–Si, CNSi–Li and CNO–Li respectively. Consisting with the above results, with increasing concentration of lithium atoms in the mixture, a quasi-linear drop of CNO–Si from 1.9 to 1.4 confirm the disintegration of Si–O bonds. The increasing of CNSi–Li and CNO–Li is attributed to increasing Li atoms surround the Si and O atoms, it indicates the formation of LixO and LiySi structures. The Si and O atoms prefer to interact with the Li atoms rather than with each other, specially at high Li concentration. When x = 2, the insertion of Li gradually disintegrates the a-SiC2/5O6/5 host mixture with O and Si atoms coordinating to 2.8 and 3.5 Li atoms.
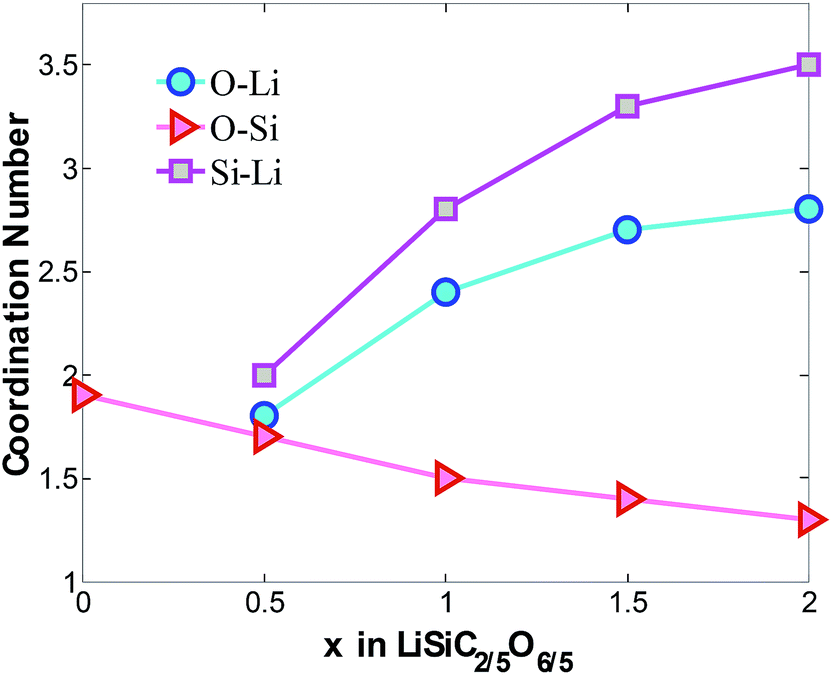 | ||
| Fig. 3 Average coordinate number of O–Li, O–Si and Si–Li atom pairs in LixSiC2/5O6/5. The cutoff radii for CNO–Si, CNSi–Li and CNO–Li are 1.85 Å, 3.08 Å and 2.16 Å respectively. | ||
Detailed views on formation of LixO and LiySi complexes in lithiated SiC2/5O6/5 (x = 2) are shown in Fig. 4. As the Li concentration increases, more O atoms are involved in formation of LixO (x = 2, 3, 4) and LiySi (y = 3, 4, 5) complexes. Stable formation of both LixO and LiySi complexes in high Li concentration structure is attributed to the Si–C/O mixed-bond tetrahedrons in the original structure. According to the first-principle calculation of silicon dioxide,41 after lithiation and some of the Si–O bonds broke, only LixO was found in the lithiated structure, indicating the broken Si atoms still connected with other O atoms and could not form LiySi compound in silicon dioxide. While the mixed bond tetrahedrons in SiCO provide the possibility to generate broken Si and O for the formation of LixO and LiySi complexes.
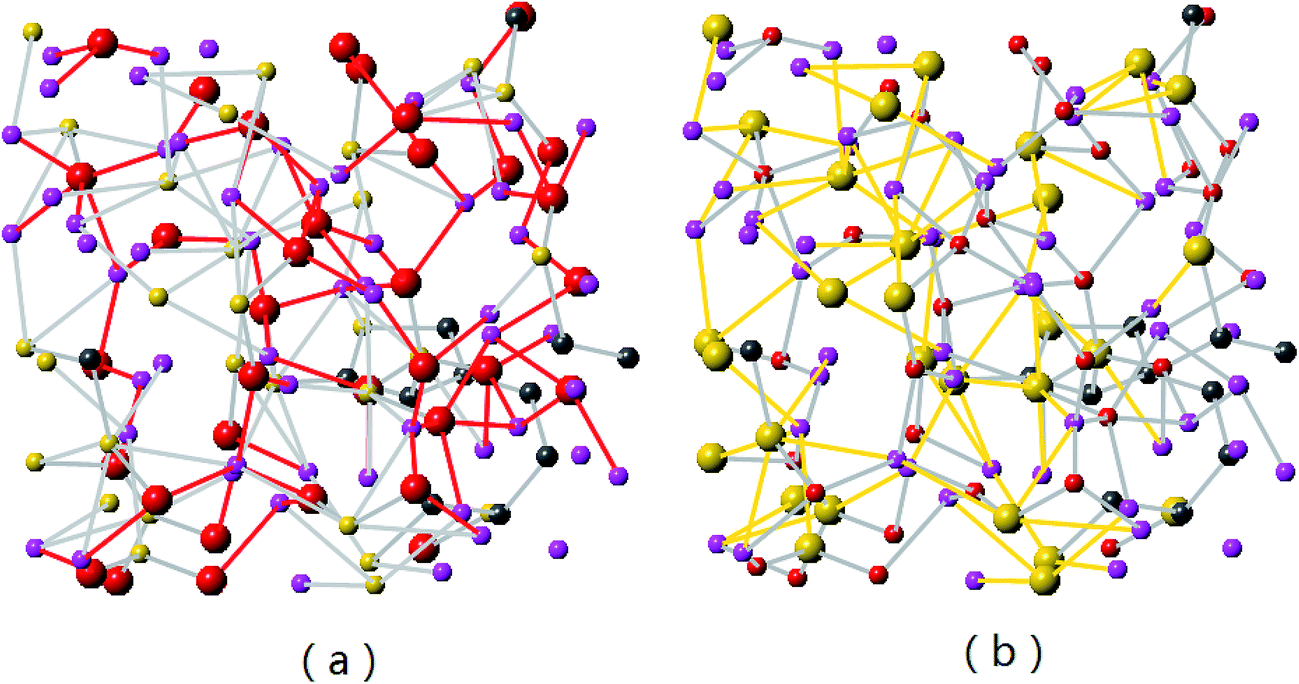 | ||
| Fig. 4 Formation of LixO and LiySi in Li2SiC2/5O6/5 (the Li–O and Li–Si bonds are presented by red and yellow colors respectively). | ||
The total density of states (TDOS) and partial density of states (PDOS) of SiC2/5O6/5 and lithiated SiC2/5O6/5 around the Fermi level are shown in Fig. 5. The insertion of Li makes two main contributions to the TDOS: one is located at around −4 eV while the other is near 1.2 eV, which can be viewed as bonding and anti-bonding states introduced by Li insertion. The TDOS around the Fermi level is mainly composed of Si-s, Si-p, C-p, O-p and a little Li-p components. In particular, the non-zero s component of Li PDOS is almost the same as the p orbit of Si atoms. Above Fermi level, Si-p makes a major contribution to TDOS with some Li-s, Si-s and C-p components, it means that the bond of Li–Si is mainly covalency with little ionicity. The relative volumes of SiC2/5O6/5 with different degree of lithiation were calculated and compared with those of Si,14 as shown in Fig. 6. The results show that the profile of relative volume for LixSiC2/5O6/5 is considerably lower compared to the value of LixSi, SiC2/5O6/5 presented a better expansion performance than Si.
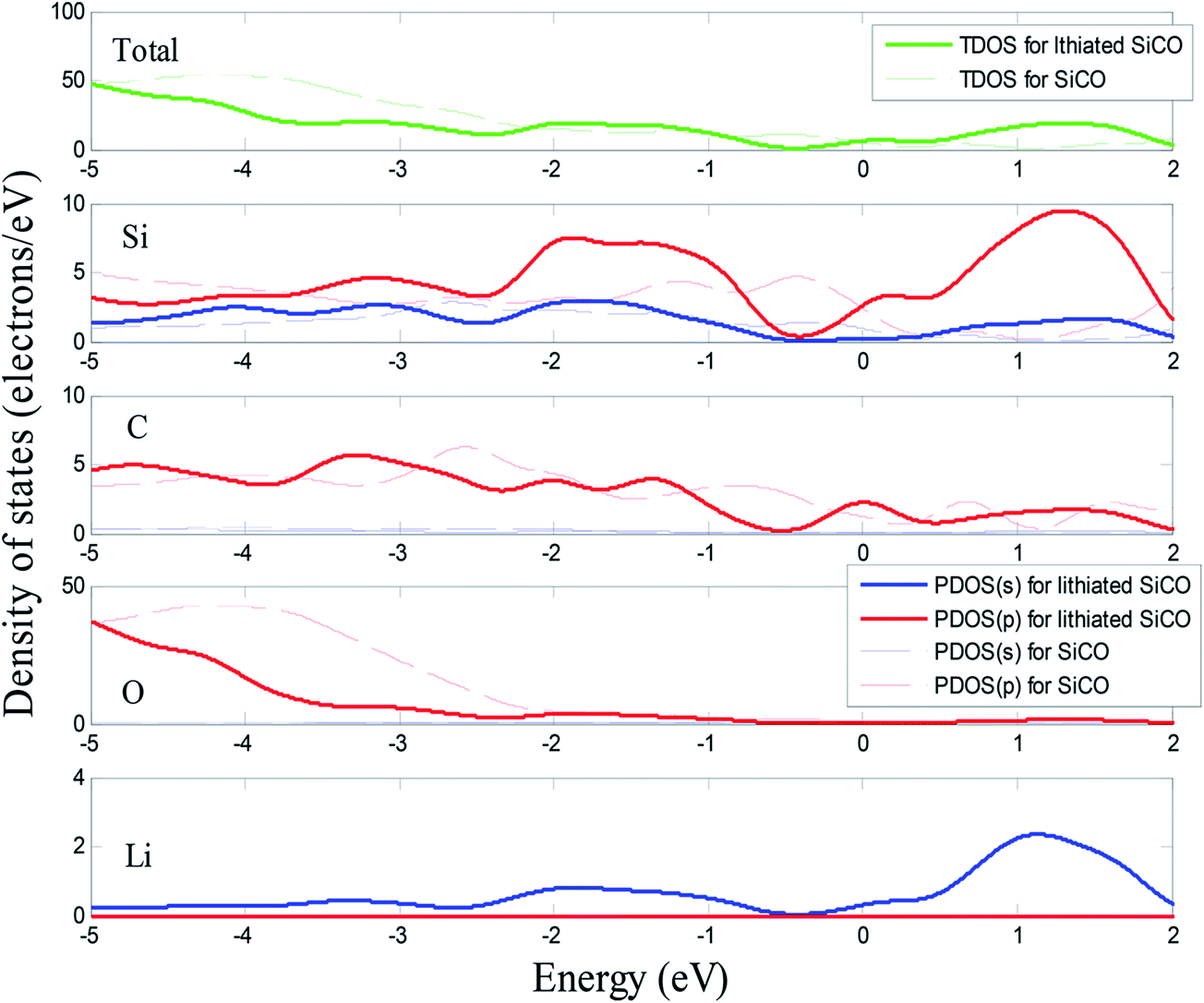 | ||
| Fig. 5 Calculated total and partial density of states of SiC2/5O6/5 and lithiated SiC2/5O6/5. | ||
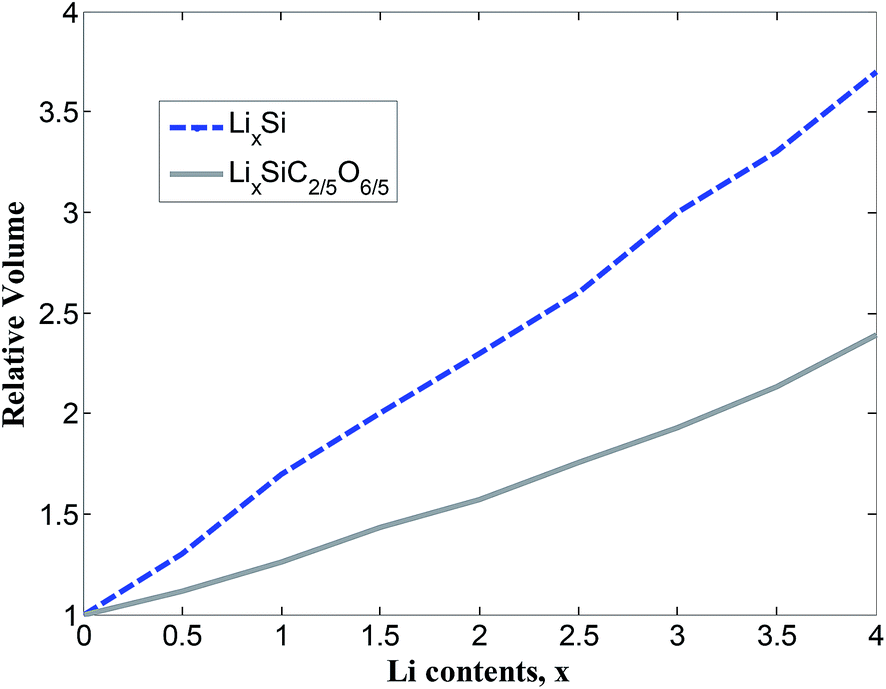 | ||
| Fig. 6 Evolution of relative volume of LixSiC2/5O6/5 and LixSi 14. | ||
Fig. 7 showed the formation energies of LixSiC2/5O6/5 as a function of x. The formation energies decrease with increasing Li concentration and reach the minimum when the structure is fully lithiated. The lithiation is favorable until a Li concentration of x = 2.75, an additional Li insertion is thermodynamically unfavorable. It corresponds to a specific capacity of 1415 mA h g−1, considering that the ratio of irreversible and reversible capacity is about 1/3 for the SiCO with similar composition,42 a reversible capacity of 1061 mA h g−1 is expected and this is comparable to reversible capacity of 900 mA h g−1 in experiments.42 The composition–voltage curve if LixSiC2/5O6/5 is also calculated and compared to the experimental data.5 The lithiation voltage relative is given by the negative of the reaction free energy per Li.43 The lithiation voltage for SiC2/5O6/5 is predicted to be around 0.42 to 1.12 V.
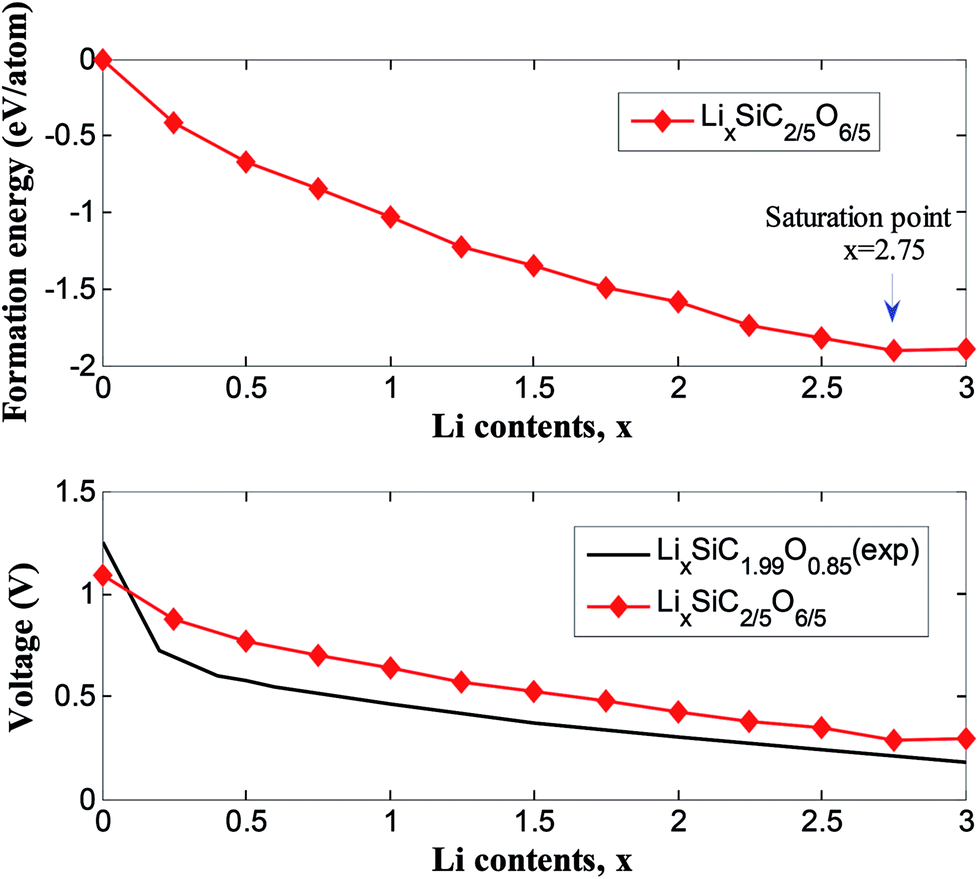 | ||
| Fig. 7 Formation energy and voltage profiles for LixSiC2/5O6/5 in comparison to experimental results of LixSiC1.99O0.85 (ref. 5). | ||
4. Conclusions
The present first-principle simulations offer an insights into the lithiated structures and lithiation mechanism of a-SiC2/5O6/5. Our interpretation of radial distribution, angular distribution and coordinate number suggests that the Si–O bond tends to break and the Li2O will forms at the beginning of lithiation, then LixO and LiySi form with increasing of Li concentration, which make major contribution to the capacity of the a-SiC2/5O6/5. The Li concentration dependent of formation energies curve indicates that the lithiation of SiC2/5O6/5 is favorable until a Li concentration of x = 2.75, which corresponds to a specific capacity of 1415 mA h g−1. Both the formation energies and the voltage curves suggest lithium is preferable in incorporation with SiC2/5O6/5, and this is attributed to the formation of LixO and LiySi.Acknowledgements
The authors would like to acknowledge the support of the National Natural Science Foundation of China (51675384), Public Welfare Science and Technology Project of Wenzhou City (G20160011) and High Performance Computing System of Wenzhou University.References
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- Y. Bie, J. Yang, Y. Nuli and J. Wang, RSC Adv., 2016, 6, 97084–97088 RSC.
- Y. J. Cho, H. S. Kim, H. Im, Y. Myung, G. B. Jung, C. W. Lee, J. Park, M.-H. Park, J. Cho and H. S. Kang, J. Phys. Chem. C, 2011, 115, 9451–9457 CAS.
- A. G. Kannan, S. H. Kim, H. S. Yang and D.-W. Kim, RSC Adv., 2016, 6, 25159–25166 RSC.
- D. Ahn and R. Raj, J. Power Sources, 2010, 195, 3900–3906 CrossRef CAS.
- D. Ahn and R. Raj, J. Power Sources, 2011, 196, 2179–2186 CrossRef CAS.
- P. E. Sanchez-Jimenez and R. Raj, J. Am. Ceram. Soc., 2010, 93, 1127–1135 CrossRef CAS.
- R. Bhandavat and G. Singh, J. Phys. Chem. C, 2013, 117, 11899–11905 CAS.
- H. Fukui, Y. Harimoto, M. Akasaka and K. Eguchi, ACS Appl. Mater. Interfaces, 2014, 6, 12827–12836 CAS.
- S. Choi, D. S. Jung and J. W. Choi, Nano Lett., 2014, 14(12), 7120–7125 CrossRef CAS PubMed.
- M. Wilamowska-Zawlocka, P. Puczkarski, Z. Grabowska, J. Kaspar, M. Graczyk-Zajac, R. Riedel and G. D. Sorarù, RSC Adv., 2016, 6, 104597–104607 RSC.
- M. Ma, H. Wang, M. Niu, L. Su, X. Fan, J. Deng, Y. Zhang and X. Du, RSC Adv., 2016, 6, 43316–43321 RSC.
- V. Pradeep, M. Graczyk-Zajac, R. Riedel and G. Soraru, Electrochim. Acta, 2014, 119, 78–85 CrossRef CAS.
- M. K. Chan, C. Wolverton and J. P. Greeley, J. Am. Chem. Soc., 2012, 134, 14362–14374 CrossRef CAS PubMed.
- C.-Y. Chou and G. S. Hwang, Chem. Mater., 2013, 25, 3435–3440 CrossRef CAS.
- J. Hu, B. Xu, C. Ouyang, Y. Zhang and S. A. Yang, RSC Adv., 2016, 6, 27467–27474 RSC.
- N. Liao, B. Zheng, H. Zhou and W. Xue, J. Mater. Chem. A, 2015, 3, 5067–5071 CAS.
- N. Liao, B. Zheng, H. Zhou and W. Xue, J. Power Sources, 2016, 334, 39–43 CrossRef CAS.
- J. Tersoff, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 5566 CrossRef.
- S. Munetoh, T. Motooka, K. Moriguchi and A. Shintani, Comput. Mater. Sci., 2007, 39, 334–339 CrossRef CAS.
- N. Liao, W. Xue and M. Zhang, J. Eur. Ceram. Soc., 2012, 32, 1275–1281 CrossRef CAS.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892 CrossRef.
- S. J. Clark, M. D. Segall and C. J. Pickard, Z. Kristallogr., 2005, 220, 567–570 CAS.
- W.-S. Feng, Y. Fang, J.-X. XU, C.-H. Fang, Q.-J. Jia, H.-H. Wang and X.-M. Jiang, Acta Phys.-Chim. Sin., 2008, 24, 497–501 CAS.
- H. Bréquel, J. Parmentier, S. Walter, R. Badheka, G. Trimmel, S. Masse, J. Latournerie, P. Dempsey, C. Turquat and A. Desmartin-Chomel, Chem. Mater., 2004, 16, 2585–2598 CrossRef.
- H. Brequel, G. Soraru, L. Schiffini and S. Enzo, Mater. Sci. Forum, 2000, 343, 677–682 CrossRef.
- G. D. Sorarù, G. D'Andrea and A. Glisenti, Mater. Lett., 1996, 27, 1–5 CrossRef.
- C. Moysan, R. Riedel, R. Harshe, T. Rouxel and F. Augereau, J. Eur. Ceram. Soc., 2007, 27, 397–403 CrossRef CAS.
- M. G. Segatelli, A. T. N. Pires and I. V. P. Yoshida, J. Eur. Ceram. Soc., 2008, 28, 2247–2257 CrossRef CAS.
- B. Papendorf, E. Ionescu, H. J. Kleebe, C. Linck, O. Guillon, K. Nonnenmacher and R. Riedel, J. Am. Ceram. Soc., 2013, 96, 272–280 CrossRef CAS.
- T. Rouxel, G. D. Soraru and J. Vicens, J. Am. Ceram. Soc., 2001, 84, 1052–1058 CrossRef CAS.
- G. D. Sorarù, E. Dallapiccola and G. D'Andrea, J. Am. Ceram. Soc., 1996, 79, 2074–2080 CrossRef.
- G. D. Soraru, G. D'andrea, R. Campostrini, F. Babonneau and G. Mariotto, J. Am. Ceram. Soc., 1995, 78, 379–387 CrossRef CAS.
- R. Angel, D. Allan, R. Miletich and L. Finger, J. Appl. Crystallogr., 1997, 30, 461–466 CrossRef CAS.
- B. Schiemenz and P. P. Power, Angew. Chem., Int. Ed. Engl., 1996, 35, 2150–2152 CrossRef CAS.
- M. M. Islam, T. Bredow and P. Heitjans, J. Phys. Chem. C, 2011, 115, 12343–12349 CAS.
- E. Martinez, J. Plans and F. Yndurain, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 8043 CrossRef CAS.
- H. Chen, P. Jutzi, W. Leffers, M. Olmstead and P. Power, Organometallics, 1991, 10, 1282–1286 CrossRef CAS.
- S. Perez-Beltran, G. E. Ramírez-Caballero and P. B. Balbuena, J. Phys. Chem. C, 2015, 119, 16424–16431 CAS.
- X. Liu, K. Xie, C.-m. Zheng, J. Wang and Z. Jing, J. Power Sources, 2012, 214, 119–123 CrossRef CAS.
- M. Aydinol, A. Kohan, G. Ceder, K. Cho and J. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1354 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |