
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acid-mediated sulfonylation of arylethynylene bromides with sodium arylsulfinates: synthesis of (E)-1,2-bis(arylsulfonyl)ethylenes and arylacetylenic sulfones†
Chenshu Dai,
Junqi Wang,
Siqi Deng,
Candong Zhou,
Wenhe Zhang,
Qiuhua Zhu * and
Xiaodong Tang*
* and
Xiaodong Tang*
Guangdong Provincial Key Laboratory of New Drug Screening, School of Pharmaceutical Sciences, Southern Medical University, 1023 South Shatai Road, Baiyun District, Guangzhou 510515, P. R. China. E-mail: zhuqh@smu.edu.cn; tangxdong@smu.edu.cn
First published on 20th July 2017
Abstract
A solvent-dependent sulfonylation of arylethynylene bromides with sodium arylsulfinates has been developed. The (E)-1,2-bis(arylsulfonyl)ethylenes were formed in DMSO, while the arylacetylenic sulfones were obtained in toluene. Utilizing simple and readily available starting materials, the sulfonylation products were generated with good selectivities and yields without the need for a metal catalyst or oxidant.
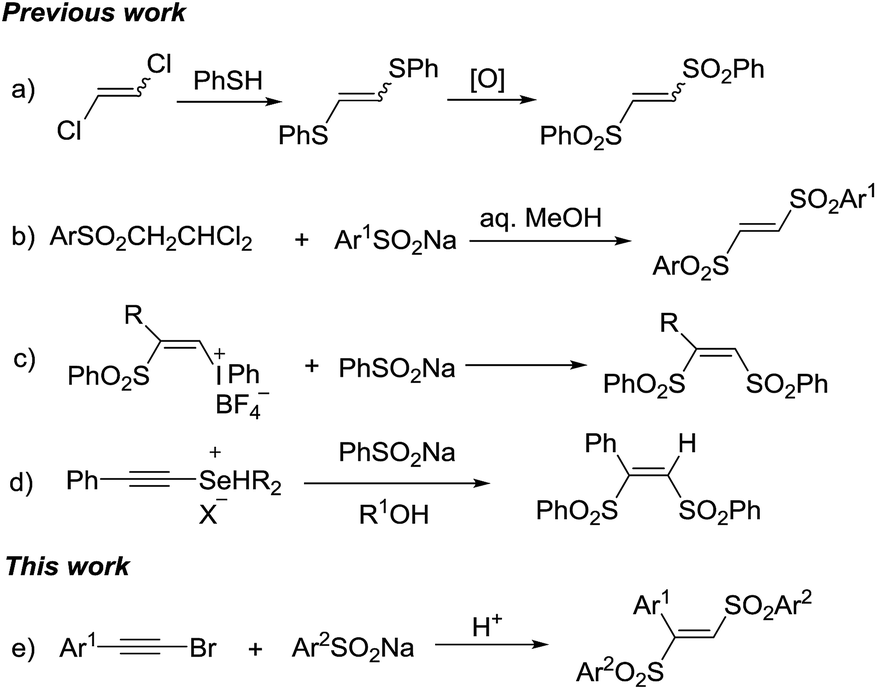
Organosulfone compounds are of great importance in organic chemistry, due to their widely existing in natural products and drug molecules.1 Meanwhile, the sulfonyl group is also extensively used as versatile synthon for other organosulfur compounds synthesis.2 1,2-Bis(arylsulfonyl)ethylenes are important organosulfone compounds and widely studied in synthetic applications. Firstly, they can act as π-deficient alkenes for cycloaddition reaction to synthesize cyclic compounds.3 Secondly, they play the role as leaving group in radical alkenylation reaction, in which various radicals add into the C–C double bond and then eliminate a sulfonyl radical.4 Thirdly, in the presence of organocatalyst, they are able to undergo 1,2-sulfone rearrangement to form various other organosulfone compounds.5 Due to their importance, synthetic chemists have exploring their synthetic methods. However, the methods for synthesis of 1,2-bis(arylsulfonyl)ethylenes are still rarely. The most common method was the reaction of the 1,2-dichloroethylene with phenylthiolate to give 1,2-bis(arylthio)ethylene which was followed by oxidation to furnish 1,2-bis(arylsulfonyl)ethylenes (Scheme 1a).6a While those multi-step synthesis was even impeded by limited symmetric 1,2-bis(arylsulfonyl)ethylenes formation. Reddy et al. reported a one-pot method, through the condensation of 1-arylsulfonyl-2,2-dichloroethanes with sodium sulfinates in aqueous alcohol (Scheme 1b).6b The β-((phenylsulfonyl)alkenyl)iodonium tetrafIuoroborates were used as starting materials to form (Z)-1,2-bis(arylsulfonyl)ethylenes by nucleophilic vinylic substitutions with sodium arylsulfinates. Except the multi-step preparation of vinyl-iodonium salts, stoichiometric amount of iodine benzene as a by-product placed this method in an unfavourable position (Scheme 1c).6c Kataoka et al. also developed a method for 1,2-bis(phenylsulfonyl)ethylene via the reaction of alkynylselenonium salt with sodium benzenesulfinate (Scheme 1d).6d–f In the above methods, all have some obvious disadvantages such as multi-step processes, strong oxidants, unavailable starting materials, toxic by-products and so on. Hence, it is attractive and meaningful to develop a new method for synthesis of 1,2-bis(arylsulfonyl)ethylenes in a direct, simple and green way.
 | ||
| Scheme 1 Synthetic methods for 1,2-bis(arylsulfonyl)ethylenes. | ||
Haloalkynes are easily available building blocks which can be prepared in quantitative yield in mol-scale on the bench top. Due to the electron-deficiency of haloalkyne, it usually used as an activated alkyne for addition reaction to give the sole regio-selectivity.7 In recent years, some practical synthetic methods involving haloalkynes have been developed, including nucleophilic addition,8 cross-coupling reactions9 and cycloaddition reactions.10 In comparison with sulfonyl chlorides, sodium sulfinates as mild sulfone moieties have many advantages, such as low toxicity, ready accessibility and stability. The sodium sulfinates are not only used as the simple nucleophilic reagents but also sulfonyl radicals to participate in the sulfonylation reaction. Recently, many endeavors have been made towards utilizing sodium sulfinates to synthesize organosulfone compounds.11 Based on the reaction development of haloalkynes and sodium sulfinates, we developed a direct synthesis of 1,2-bis(arylsulfonyl)ethylenes through tandem reaction between haloalkynes and sodium sulfinates under acidic conditions. Herein, we presented a acid-mediated synthesis of 1,2-bis(arylsulfonyl)ethylenes from bromoalkynes and sodium sulfinates (Scheme 1e).
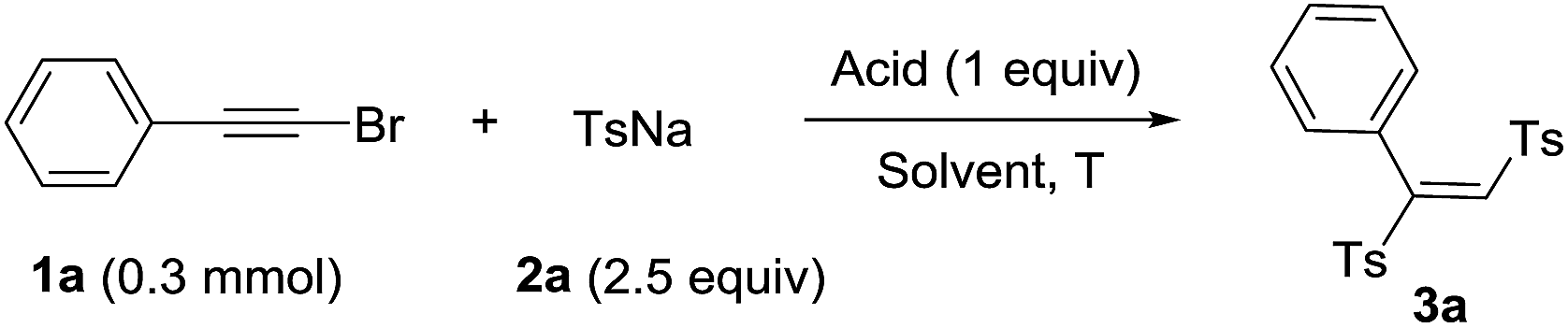
We used the reaction between phenylethynyl bromide (1a) and sodium p-tolylsulfinate (2a) as a model to examine various reaction parameters and the results were summarized in Table 1. On the first trial, 1a and 2a (2.5 equiv.) were treated with 1 M HCl (1 equiv.) in DMSO as solvent at 60 °C for 12 h. To our delight, the target product 1,2-bis(tolylsulfonyl)phenylethene (3a) was obtained in 60% yield (entry 1). And the structure was confirmed by single-crystal X-ray analysis, in which the double bond is E-type configuration.12 Afterwards, we examined the concentration of hydrochloric acid and the results indicated that the yields increased with higher concentration HCl (entries 2–5). The yield was raised up to 91% when 12 M HCl was used (entry 5). Next, the screening of reaction temperature showed that neither increasing nor decreasing the temperature led to a lower yield of product (entries 6–9). Other Brønsted acids such as H2SO4, TsOH, TfOH, TFA and HOAc were tested to give the product in decent yields (entries 10–14). Control experiment indicated that acid was essential for this transformation (entry 15). The screening of different solvents showed that the solvents played a critical role, the polar solvents were beneficial for the transformation (entries 16–24).
|
||||
|---|---|---|---|---|
| Entry | Acid | Solvent | T (°C) | Yield (%) |
| a Reaction were performed with 1a (0.3 mmol), 2a (0.75 mmol), acid (0.3 mmol) in solvent (3.0 mL) for 12 h. Isolated yield. n.d. = not determined. | ||||
| 1 | 1 M HCl | DMSO | 60 | 60 |
| 2 | 3 M HCl | DMSO | 60 | 68 |
| 3 | 6 M HCl | DMSO | 60 | 74 |
| 4 | 9 M HCl | DMSO | 60 | 83 |
| 5 | 12 M HCl | DMSO | 60 | 91 |
| 6 | 12 M HCl | DMSO | 40 | 42 |
| 7 | 12 M HCl | DMSO | rt | 13 |
| 8 | 12 M HCl | DMSO | 80 | 80 |
| 9 | 12 M HCl | DMSO | 100 | 78 |
| 10 | H2SO4 | DMSO | 60 | 71 |
| 11 | TsOH | DMSO | 60 | 58 |
| 12 | TfOH | DMSO | 60 | 68 |
| 13 | TFA | DMSO | 60 | 65 |
| 14 | HOAc | DMSO | 60 | 10 |
| 15 | — | DMSO | 60 | n.d. |
| 16 | 12 M HCl | CH3CN | 60 | 61 |
| 17 | 12 M HCl | DMF | 60 | 50 |
| 18 | 12 M HCl | Acetone | 60 | 38 |
| 19 | 12 M HCl | CH3NO2 | 60 | 27 |
| 20 | 12 M HCl | 1,4-Dioxane | 60 | 15 |
| 21 | 12 M HCl | CHCl3 | 60 | n.d. |
| 22 | 12 M HCl | EtOAc | 60 | n.d. |
| 23 | 12 M HCl | Toluene | 60 | n.d. |
| 24 | 12 M HCl | DCE | 60 | n.d. |
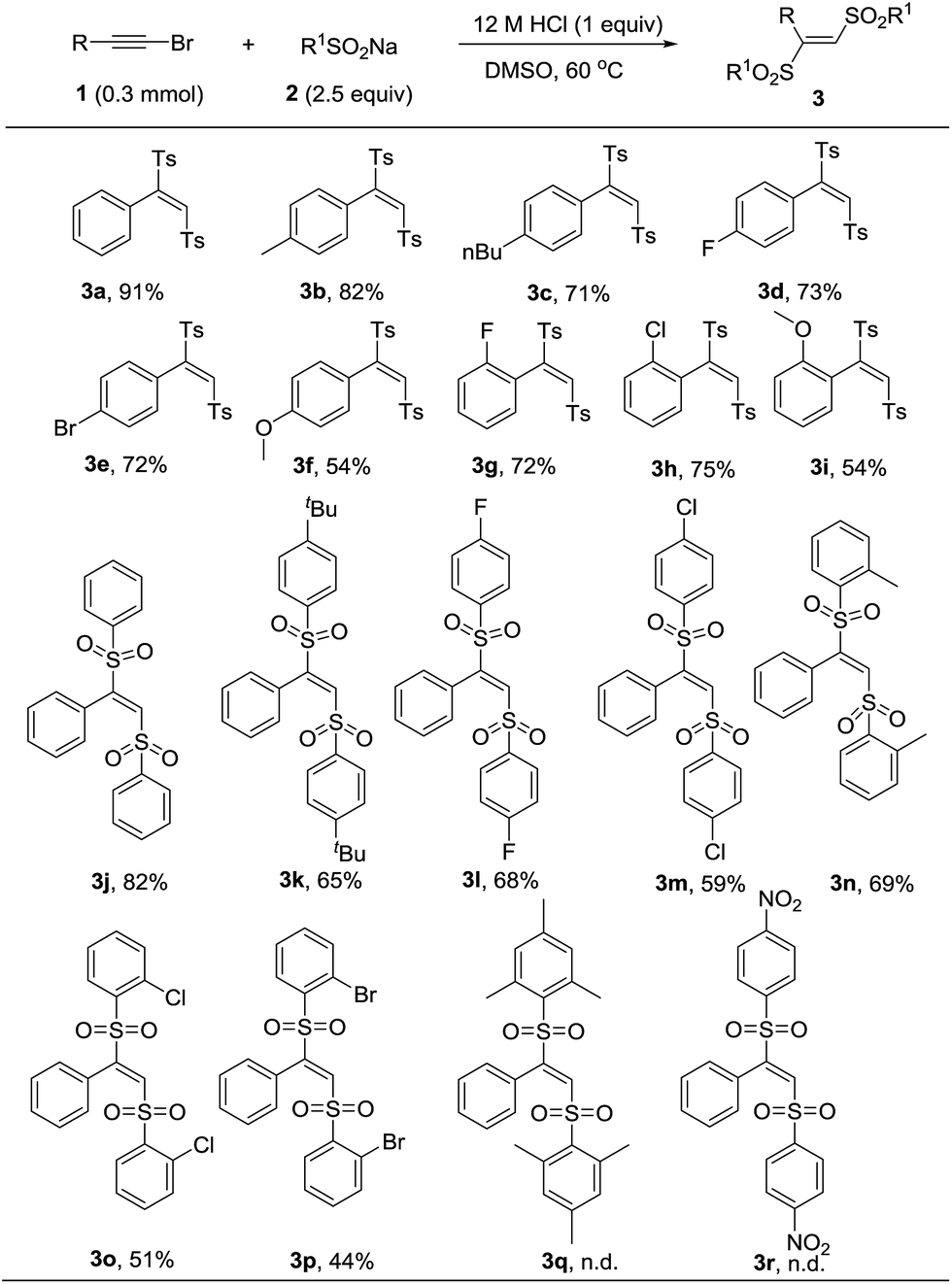
With the optimal reaction condition in hand, we next conducted a survey of various substrates to explore the scope of this transformation. As listed in Table 2, the reaction had a good substrate suitability and various substituted (E)-1,2-bis(arylsulfonyl)ethylenes were obtained in moderate to excellent yields. We firstly evaluated the scope of bromoalkynes. Various alkyl and halogen substitutions on benzene ring of phenylacetylene bromides were tolerated for this reaction. Strong electron-donating substitute such as methoxyl slightly decreased the yield of desired product (Table 2, 3f and 3i). For the substrate scope of sodium sulfinates, different para-substituted sodium phenylsulfinates could be converted into the corresponding products in moderate to good yields (Table 2, 3j–3m). Even the steric hindered ortho-substituted sodium phenylsulfinates also worked efficiently to give the products in 44% to 69% yields (Table 2, 3n–3p). Unfortunately, more steric hindered mesitylenesulfinate and electron-deficient nitrobenzenesulfinate were failed to transform into the corresponding products (Table 2, 3q and 3r).
| a Reactions were performed with 1 (0.3 mmol), 2 (0.75 mmol), 12 M HCl (0.3 mmol), and DMSO (3 mL) at 60C for 12 h. Yields was referred to isolated yields. |
|---|
 |
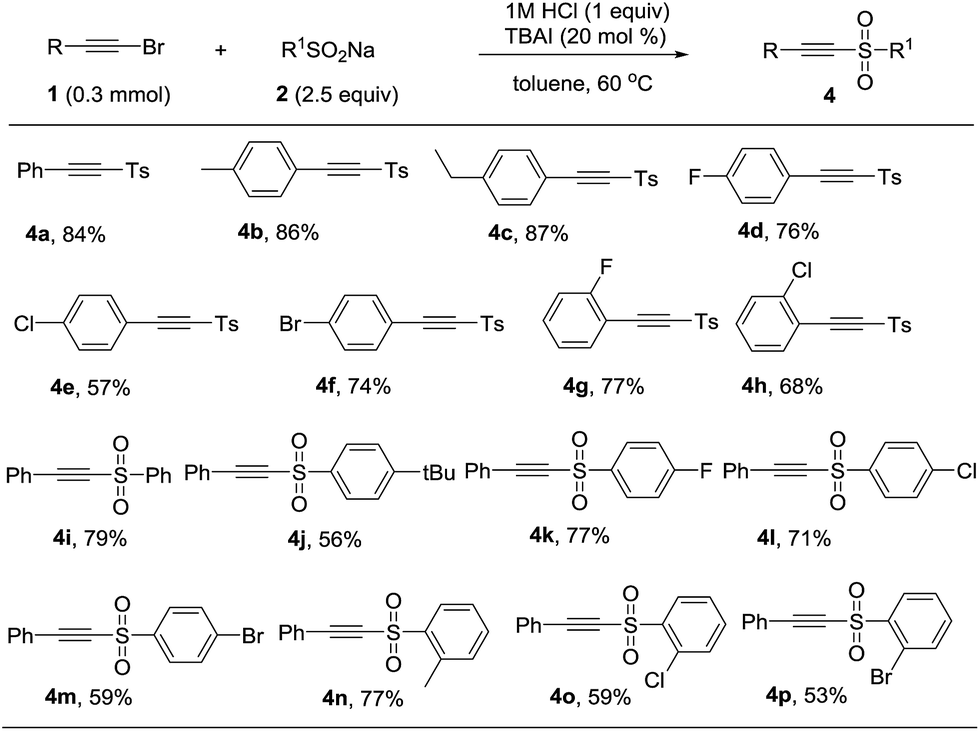
It was surprising that when 1a and 2a was treated with 12 M HCl (1 equiv.) in toluene as solvent at 60 °C for 12 h, the product acetylenic sulfone (4a) was formed in 48% yield.13 Though some methods have been reported for the synthesis of acetylenic sulfones, they had some disadvantages such as unavailable starting materials and strong oxidant.14 Thus, we optimized the reaction conditions and the best reaction conditions were as followed: 1a (0.3 mmol), 2a (0.75 mmol), 1 M HCl (0.3 mmol), TBAI (20 mol%), in toluene (3 mL) at 60 °C for 12 h (see the ESI† for details). After establishing the optimized reaction conditions, the generality and limitations of various substrates were investigated. Firstly, sodium p-tolylsulfinate (2a) was treated with different substituted phenylacetylene bromides. Different para-substituted phenylacetylene bromides including alkyl group (Me, Et) and halides (F, Cl, Br) were well tolerated under the optimized condition to afford the products in 57% to 87% (Table 3, 4a–4f). Subsequently, the effect of ortho-substituents on the phenyl ring of phenylacetylene bromides were investigated. The product 4g and 4h were formed in 77% and 68% yields, respectively. Further investigating the scope of this transformation included various substitutions effect on sodium phenylsulfinates. Various sodium sulfinates could be proceeded smoothly and afforded the corresponding products in moderate to good yields (Table 3, 4i–4p). It was a pity that sodium alkylsulfinates was not appropriate for the transformation at this stage.
| a Reaction conditions: 1 (0.3 mmol), 2 (0.75 mmol), 1 M HCl (0.3 mmol), TBAI (20 mol%), and toluene (3 mL) at 60 °C for 12 h. Yields was referred to isolated yields. |
|---|
 |
To gain more insight into the mechanism, the control experiments were carried out and shown in Scheme 2. When we utilized phenylacetylene, phenylethynyl chloride or phenylethynyl iodine to react with 2a under the standard conditions. Phenylethynyl iodide gave the product 4a in 82% yield, while phenylacetylene and phenylethynyl chloride were failed to convert into the product (Scheme 2, eqn (1)). The presence of TEMPO or BHT strongly suppressed the product formation, respectively gave the product 4a in 0% and 15% yields (Scheme 2, eqn (2)). These results indicated that a radical pathway may be involved. However, we did not detect the additive products of radicals coupling with TEMPO or BHT. When the 4-methylbenzenesulfinic acid 5 reacted with 2a in absence of 12 M HCl, the product 3a was obtained in 45% yield (Scheme 2, eqn (3)). The vinyl sulfone 6 could not transformed into the product with 2a under standard reaction condition (Scheme 2, eqn (4)). We next investigated whether 4a was the reaction intermediate for the formation product 3a. Sodium sulfinates could add into 4a to form 3a in excellent yield in the presence of 12 M HCl (Scheme 2, eqn (5)).
 | ||
| Scheme 2 Control experiments. | ||
According to previous studies15,16 and our control experiments, the proposed mechanisms were shown in Scheme 3. One proposed mechanism was a addition–elimination process (path a).15 Firstly, the nucleophilic attack of sulfinate ion to 1a formed the intermediate A, which was followed by protolysis to give the intermediate B. Finally, the intermediate B eliminated hydrogen bromide to produce the product 4a. The other mechanism was a radical process (path b). The p-tolylsulfonyl radical C could be generated from sodium p-tolylsulfinate in acid under heated condition.16a,b Subsequently, a radical addition of p-tolylsulfonyl radical to 1a formed a bromovinyl radical D.16c Then, the product 4a was obtained via bromine radical elimination from D.16c Finally, the bromine radical oxidized sodium p-tolylsulfinate to afford p-tolylsulfonyl radical C with releasing NaBr.16c The 4a could be transformed into the product 3a through nucleophilic addition. The polarity of the solvent determined the final product was 4a or 3a. The final product was 4a in the low polar solvents such as toluene, CHCl3, DCE and so on. The high polar solvent was good for nucleophilic addition process. So when the high polar solvents such as DMSO, DMF, CH3CN were used, the final product was 3a.
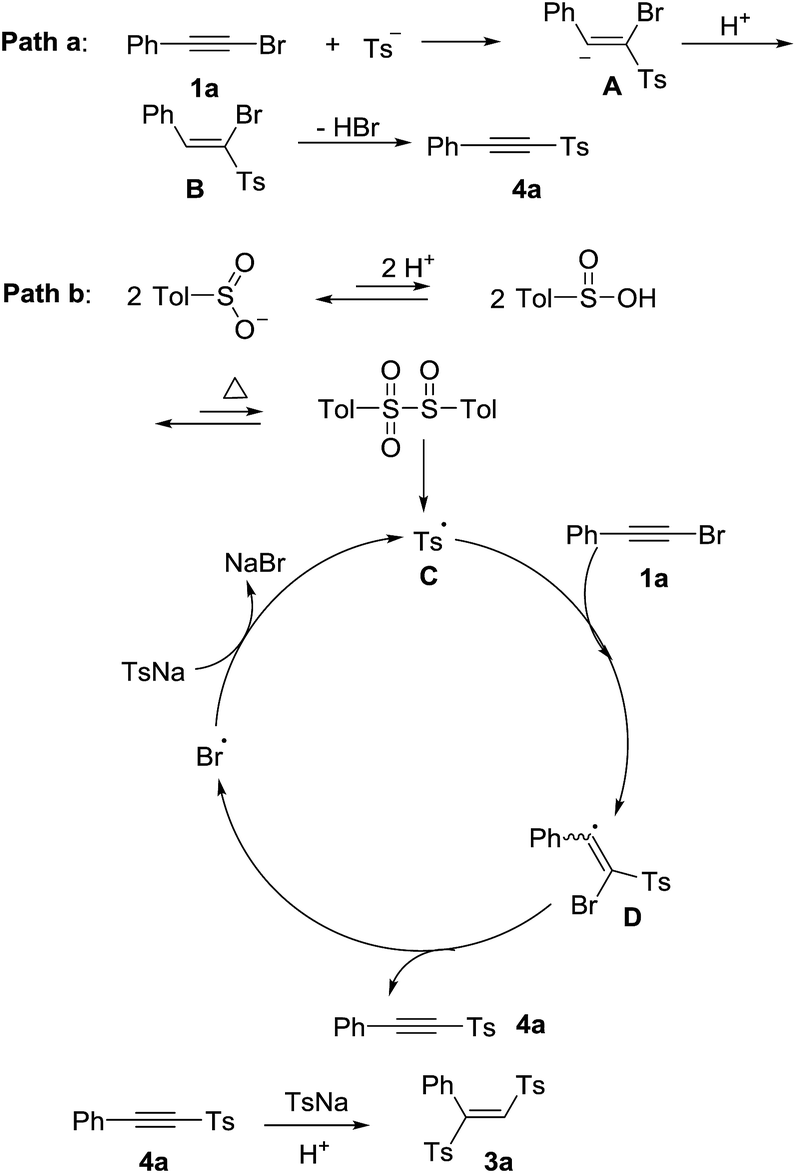 | ||
| Scheme 3 Possible reaction mechanism. | ||
In conclusion, we have developed a practical and novel procedure for the synthesis of (E)-1,2-bis(arylsulfonyl)ethylenes and arylacetylenic sulfones by sulfonylation of arylethynylene bromides with sodium arylsulfinates in different solvents. The method obviated the need for unavailable starting materials or strong oxidants with simple operation. Further research for the mechanism and the synthetic applications are ongoing in our laboratory.
Acknowledgements
The authors thank the High-level Talent Introduction Foundation of Southern Medical University (C1033520), and the Science and Technology Program of Guangdong Province (2015A010105015) for financial support.Notes and references
- (a) E. Dunny, W. Doherty, P. Evans, J. P. G. Malthouse, D. Nolan and A. J. S. Knox, J. Med. Chem., 2013, 56, 6638 CrossRef CAS PubMed; (b) H. Rueeger, R. Lueoend, O. Rogel, J.-M. Rondeau, H. Möbitz, R. Machauer, L. Jacobson, M. Staufenbiel, S. Desrayaud and U. Neumann, J. Med. Chem., 2012, 55, 3364 CrossRef CAS PubMed; (c) M. Uttamchandani, K. Liu, R. C. Panicker and S. Q. Yao, Chem. Commun., 2007, 1518 RSC; (d) Y. Shen, C. A. Zificsak, J. E. Shea, X. Lao, O. Bollt, X. Li, J. G. Lisko, J. P. Theroff, C. L. Scaife, M. A. Ator, B. A. Ruggeri, B. D. Dorsey and S. K. Kuwada, J. Med. Chem., 2015, 58, 1140 CrossRef CAS PubMed.
- (a) M.-Y. Chang and Y.-C. Cheng, Org. Lett., 2016, 18, 1682 CrossRef CAS PubMed; (b) M.-Y. Chang, Y.-C. Cheng and Y.-J. Lu, Org. Lett., 2014, 16, 6252 CrossRef CAS PubMed; (c) J.-C. Wu, L.-B. Gong, Y. Xia, R.-J. Song, Y.-X. Xie and J.-H. Li, Angew. Chem., Int. Ed., 2012, 51, 9909 CrossRef CAS PubMed.
- (a) R. Robles-Machín, A. López-Pérez, M. González-Esguevillas, J. Adrio and J. C. Carretero, Chem.–Eur. J., 2010, 16, 9864 CrossRef PubMed; (b) Z. Li, H. Yu, H. Liu, L. Zhang, H. Jiang, B. Wang and H. Guo, Chem.–Eur. J., 2014, 20, 1731 CrossRef CAS PubMed; (c) E. Conde, I. Rivilla, A. Larumbe and F. P. Cossío, J. Org. Chem., 2015, 80, 11755 CrossRef CAS PubMed; (d) J. Mancebo-Aracil, C. Nájera and J. M. Sansano, Org. Biomol. Chem., 2013, 11, 662 RSC.
- (a) V. Corcé, L.-M. Chamoreau, E. Derat, J.-P. Goddard, C. Ollivier and L. Fensterba, Angew. Chem., Int. Ed., 2015, 54, 11414 CrossRef PubMed; (b) A.-P. Schaffner, V. Darmency and P. Renaud, Angew. Chem., Int. Ed., 2006, 45, 5847 CrossRef CAS PubMed; (c) Y. Amaoka, M. Nagatomo, M. Watanabe, K. Tao, S. Kamijo and M. Inoue, Chem. Sci., 2014, 5, 4339 RSC; (d) R. Beniazza, V. Liautard, C. Poittevin, B. Ovadia, S. Mohammed, F. Robert and Y. Landais, Chem.–Eur. J., 2017, 23, 2439 CrossRef CAS PubMed; (e) C. Poittevin, V. Liautard, R. Beniazza, F. Robert and Y. Landais, Org. Lett., 2013, 15, 2814 CrossRef CAS PubMed.
- (a) A. Quintard and A. Alexakis, Org. Biomol. Chem., 2011, 9, 1407 RSC; (b) A. Quintard and A. Alexakis, Chem.–Eur. J., 2009, 15, 11109 CrossRef CAS PubMed; (c) A. Quintard, S. Belot, E. Marchal and A. Alexakis, Eur. J. Org. Chem., 2010, 927 CrossRef CAS.
- (a) O. D. Lucchi, V. Lucchini, L. Pasquato and G. Modena, J. Org. Chem., 1984, 49, 596 CrossRef; (b) D. B. Reddy, N. C. Babu, V. Padmavathi and R. P. Sumathi, Synthesis, 1999, 491 CrossRef CAS; (c) M. Ochiai, K. Oshima, Y. Masaki, M. Kunishima and S. Tani, Tetrahedron Lett., 1993, 34, 4829 CrossRef CAS; (d) T. Kataoka, Y. Banno, S.-I. Watanabe, T. Iwamura and H. Shimizu, Tetrahedron Lett., 1997, 38, 1809 CrossRef CAS; (e) S.-I. Watanabe, K. Yamamoto, Y. Itagaki and T. Kataoka, J. Chem. Soc., Perkin Trans. 1, 1999, 2053 RSC; (f) S.-I. Watanabe, K. Yamamoto, Y. Itagaki, T. Iwamura, T. Iwama, T. Kataoka, G. Tanabe and O. Muraoka, J. Chem. Soc., Perkin Trans. 1, 2001, 239 RSC.
- W. Wu and H. Jiang, Acc. Chem. Res., 2014, 47, 2483 CrossRef CAS PubMed.
- For selected examples: (a) G. Jiang, C. Zhu, J. Li, W. Wu and H. Jiang, Adv. Synth. Catal., 2017, 359, 1208 CrossRef CAS; (b) N. Rajesh and D. Prajapati, RSC Adv., 2014, 4, 32108 RSC.
- (a) N. Matsuyama, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 4156 CrossRef CAS PubMed; (b) Y.-J. Liu, Y.-H. Liu, S.-Y. Yan and B.-F. Shi, Chem. Commun., 2015, 51, 6388 RSC; (c) J. Li, W. Hu, C. Li, S. Yang, W. Wu and H. Jiang, Org. Chem. Front., 2017, 4, 373 RSC; (d) Y. Dong, S. Sun, F. Yang, Y. Zhu, W. Zhu, H. Qiao, Y. Wu and Y. Wu, Org. Chem. Front., 2016, 3, 720 RSC.
- (a) A. Allen, K. Villeneuve, N. Cockburn, E. Fatila, N. Riddell and W. Tam, Eur. J. Org. Chem., 2008, 4178 CrossRef CAS; (b) Y. Gao, M. Yin, W. Wu, H. Huang and H. Jiang, Adv. Synth. Catal., 2013, 355, 2263 CrossRef CAS.
- For selected examples: (a) Y. Gao, W. Wu, Y. Huang, K. Huang and H. Jiang, Org. Chem. Front., 2014, 1, 361 RSC; (b) W. Wei, H. Cui, D. Yang, X. Liu, C. He, S. Dai and H. Wang, Org. Chem. Front., 2017, 4, 26 RSC; (c) F. Xiao, H. Chen, H. Xie, S. Chen, L. Yang and G.-J. Deng, Org. Lett., 2014, 16, 50 CrossRef CAS PubMed; (d) X. Pan, J. Gao, J. Liu, J. Lai, H. Jiang and G. Yuan, Green Chem., 2015, 17, 1400 RSC; (e) V. G. Pandya and S. B. Mhaske, Org. Lett., 2014, 16, 3836 CrossRef CAS PubMed; (f) S. Liang and G. Manolikakes, Adv. Synth. Catal., 2016, 358, 2371 CrossRef CAS; (g) X. Tang, L. Huang, Y. Xu, J. Yang, W. Wu and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4205 CrossRef CAS PubMed; (h) J. Shi, X.-D. Tang, Y.-C. Wu, J.-F. Fang, L. Cao, X.-Y. Chen and Z.-Y. Wang, RSC Adv., 2016, 6, 25651 RSC.
- CCDC 1502249 (3a) contains the supplementary crystallographic data for this paper.†.
- For selected examples: (a) H. Chen and L. Zhang, Angew. Chem., Int. Ed., 2015, 54, 11775 CrossRef CAS PubMed; (b) J. L. G. Ruano, J. Alemán, L. Marzo, C. Alvarado, M. Tortosa, S. Díaz-Tendero and A. Fraile, Chem.–Eur. J., 2012, 18, 8414 CrossRef PubMed; (c) L. Marzo, A. Parra, M. Frías, J. Alemán and J. L. G. Ruano, Eur. J. Org. Chem., 2013, 4405 CrossRef CAS; (d) R. Ren, Z. Wu, Y. Xu and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 2866 CrossRef CAS PubMed; (e) N. Riddell and W. Tam, J. Org. Chem., 2006, 71, 1934 CrossRef CAS PubMed.
- For selected examples: (a) W. E. Truce, H. E. Hill and M. M. Boudakian, J. Am. Chem. Soc., 1956, 78, 2760 CrossRef CAS; (b) J. W. Lee and D. Y. Oh, Synlett, 1990, 290 CrossRef CAS; (c) J. W. Lee, T. H. Kim and D. Y. Oh, Synth. Commun., 1989, 19, 2633 CrossRef CAS; (d) D. J. Hamnett and W. J. Moran, Org. Biomol. Chem., 2014, 12, 4156 RSC; (e) J. Meesin, P. Katrun, C. Pareseecharoen, M. Pohmakotr, V. Reutrakul, D. Soorukram and C. Kuhakarn, J. Org. Chem., 2016, 81, 2744 CrossRef CAS PubMed; (f) R. Singh, B. K. Allam, N. Singh, K. Kumari, S. K. Singh and K. N. Singh, Org. Lett., 2015, 17, 2656 CrossRef CAS PubMed.
- (a) G. R. Ziegler, C. A. Welch, C. E. Orzech, S. Kikkawa and S. I. Miller, J. Am. Chem. Soc., 1963, 85, 1648 CrossRef CAS; (b) S. I. Miller, C. E. Orzech, C. A. Welch, G. R. Ziegler and J. I. Dickstein, J. Am. Chem. Soc., 1962, 84, 2020 CrossRef CAS.
- (a) J. L. Kice and N. E. Pawlowski, J. Am. Chem. Soc., 1964, 86, 4898 CrossRef CAS; (b) C.-P. Chuang, Tetrahedron Lett., 1992, 33, 6311 CrossRef CAS; (c) J. Zhang, P. Li and L. Wang, Org. Biomol. Chem., 2014, 12, 2969 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization of all compounds, Fig. S1, copies of 1H and 13C NMR spectra for all target compounds. CCDC 1502249. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra07105a |
| This journal is © The Royal Society of Chemistry 2017 |