
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of the Src SH2 domain and its application in bioassays for mirror-image screening†
Keitou Shuab,
Taro Noguchia,
Kaori Hondac,
Yasumitsu Kondohc,
Hiroyuki Osadac,
Hiroaki Ohnoa,
Nobutaka Fujiia and
Shinya Oishi *a
*a
aGraduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: soishi@pharm.kyoto-u.ac.jp; Fax: +81-75-753-4570; Tel: +81-75-753-4561
bGraduate School of Advanced Integrated Studies in Human Survivability, Kyoto University, Sakyo-ku, Kyoto 606-8306, Japan
cChemical Biology Research Group, RIKEN Center for Sustainable Resource Science, Wako, Saitama 351-0198, Japan
First published on 7th August 2017
Abstract
The Src SH2 domain was synthesized via native chemical ligation of two fragment peptides. The facile protocol was used to prepare the mirror-image SH2 domain (D-Src SH2 domain) and tetramethylrhodamine-labeled SH2 domains. The synthesized SH2 domains were correctly folded and showed activity, and using these proteins we established bioassays to identify novel Src SH2 domain inhibitors from an unexplored mirror-image library of natural products.
Introduction
The Src SH2 domain is a noncatalytic module involved in cellular signaling and is ∼100 amino acids in length.1–4 This domain binds to a phosphorylated tyrosine (pTyr) peptide motif found within target proteins such as FAK, p68, p130 and Hapv middle T antigen (HmT).5–8 To characterize SH2 domain-mediated protein–protein interactions as well as to identify specific pTyr containing peptide(s) towards the SH2 domain, proteomics approaches and screening campaigns using a combinatorial pTyr peptide library have been conducted.9–11 The crystal structure of the Src SH2 domain–pYEEI motif-containing peptide complex revealed valuable information about the interaction interface between the peptide and SH2 domain.12 Since Src is expressed highly in a variety of disease tissues, inhibitors that target Src's function represent promising pharmaceutical agents for cancers and osteoporosis treatment.13 Considerable effort has been devoted to the development of Src SH2 domain inhibitors, which are mainly based on structure-based approaches starting from native peptide pYEEI sequences.14–23 For example, Nam et al. investigated the structure–activity relationships of pYEEI tetrapeptide ligands.21,22 N-terminal modification and incorporation of a conformationally constrained substructure led to the identification of novel potent peptidomimetics with a pYEEI motif. Alternatively, rosmarinic acid (RosA; derived from Prunella vulgaris) inhibits the interaction between the Src SH2 domain and the pYEEI-containing peptide.23 Salvianolic acids A and B, and caftaric acid also exhibit inhibitory activity against the Src SH2 domain in the micromolar range.24 As such, both structure-based design from the pTyr-containing peptides and screening of natural products are promising strategies for the identification of novel Src SH2 domain inhibitors.Recently, we reported a novel screening approach for lead discovery from an unexplored mirror-image library of natural products. In this approach, the bioactivity of existing chiral natural products was evaluated using a synthetic mirror-image MDM2 protein (D-MDM2).25 In the course of this mirror-image screening study, we identified an α-tocopherol derivative as a D-MDM2–D-p53 inhibitor from 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 293 compounds including natural products. The mirror-image structure of the hit compound reproduced the inhibitory activity against the native L-MDM2–L-p53 interaction. Two chemical syntheses of mirror-image molecules of a target protein and hit compound(s) facilitated the lead discovery of novel MDM2–p53 inhibitors without syntheses of a number of unavailable mirror-image natural products. We have also applied this approach to screen systems for novel Grb2 SH2 domain inhibitors.26 For the preparation of the native (L-protein) and mirror-image (D-protein) Grb2 SH2 domain, two synthetic procedures using native chemical ligation(s) (NCLs) of two or three segments were established. The resulting synthetic proteins with an appropriate labeling group were used to develop bioassays to evaluate the inhibitory activity against the interaction between the Grb2 SH2 domain and the counterpart phosphotyrosine (pTyr)-containing peptide. Additionally, Virdee et al. reported the semisynthesis of Src SH2 domain from three peptide fragments including two recombinant proteins and one synthetic peptides.27 With these previous reports in mind, to extend mirror-image screening approach to other SH2 domain proteins, we investigated the chemical synthesis of the Src SH2 domain and the development of several bioassays for mirror-image screening of chiral natural products.
293 compounds including natural products. The mirror-image structure of the hit compound reproduced the inhibitory activity against the native L-MDM2–L-p53 interaction. Two chemical syntheses of mirror-image molecules of a target protein and hit compound(s) facilitated the lead discovery of novel MDM2–p53 inhibitors without syntheses of a number of unavailable mirror-image natural products. We have also applied this approach to screen systems for novel Grb2 SH2 domain inhibitors.26 For the preparation of the native (L-protein) and mirror-image (D-protein) Grb2 SH2 domain, two synthetic procedures using native chemical ligation(s) (NCLs) of two or three segments were established. The resulting synthetic proteins with an appropriate labeling group were used to develop bioassays to evaluate the inhibitory activity against the interaction between the Grb2 SH2 domain and the counterpart phosphotyrosine (pTyr)-containing peptide. Additionally, Virdee et al. reported the semisynthesis of Src SH2 domain from three peptide fragments including two recombinant proteins and one synthetic peptides.27 With these previous reports in mind, to extend mirror-image screening approach to other SH2 domain proteins, we investigated the chemical synthesis of the Src SH2 domain and the development of several bioassays for mirror-image screening of chiral natural products.
Results and discussion
For the synthesis of the Src SH2 domain, Src(145–251) (Fig. 1), an NCL strategy was designed, in which two fragment peptides would be ligated at the Tyr187–Cys188 peptide bond.28 Initially, the N-terminal fragment Src(145–187) L-1a was prepared on H-Rink amide ChemMatrix resin with a Dawson linker using 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluroniuim hexafluorophosphate (HBTU)/(iPr)2NEt activation by an Fmoc-based solid-phase peptide synthesis (Fmoc-SPPS) protocol, according to our previous report (Scheme 1).29 The more reactive O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU)/(iPr)2NEt was used for coupling of Arg158, Arg159, Arg163, Leu164, and Arg172, because of the less efficient coupling of Fmoc-protected amino acids using HBTU activation. The resin L-1a was subjected to the N-acylurea approach,30 in which the C-terminal Dawson linker was activated by treatment with p-nitrophenyl chloroformate to form the Nbz (N-acyl benzimidazolinone) leaving group of L-2a. Trifluoroacetic acid (TFA)-mediated final deprotection and cleavage from the resin followed by treatment of the crude peptide with 4-mercaptophenylacetic acid (MPAA) afforded the expected L-Src(145–187) thioester L-3a. For the synthesis of the C-terminal fragment, L-Src(188–251) (L-4), the identical Fmoc-SPPS protocol on H-Rink amide ChemMatrix resin followed by TFA-mediated final deprotection gave the expected fragment. The resulting L-Src(145–187) thioester (L-3a) and L-Src(188–251) (L-4) were subjected to native chemical ligation conditions to provide the full-length L-Src(145–251) (L-5a) in 17% yield. To avoid the undesirable intramolecular and intermolecular disulfide bond formation via the three Cys residues, an increase in the concentration of tris(2-carboxyethyl)phosphine (TCEP, 100 mM) was used during ligation. | ||
| Fig. 1 Sequence of Src SH2 domain proteins. The underlined cysteine residue was used for native chemical ligation. | ||
 | ||
| Scheme 1 Synthesis of the Src SH2 domain. Reagents and conditions: (a) Fmoc-Xaa-OH, HBTU, 1-hydroxybenzotriazole (HOBt), (iPr)2NEt, DMF, then 20% piperidine/DMF; (b) Boc-Ser(tBu)-OH, HBTU, HOBt, (iPr)2NEt, DMF (for 1a); 5-carboxytetramethylrhodamine (5-TMR), DIC, HOBt, DMF (for 1b); (c) 0.3 M 4-nitrophenyl chloroformate, DCM; (d) 0.5 M (iPr)2NEt, DMF; (e) TFA/H2O/thioanisole/m-cresol (80:10:5:5); (f) 6 M Gu·HCl, 200 mM MPAA, 20 mM TCEP·HCl in PBS (pH 7.0); (g) 6 M Gu·HCl, 200 mM MPAA, 100 mM TCEP·HCl in PBS (pH 7.0). | ||
The Src SH2 domain with a labeling group required for bioassays was designed and synthesized. The previous report on the crystal structure of the Src SH2 domain in complex with the cognate pTyr-containing peptide revealed that the binding site of the pTyr-containing peptide is located distal from the N-terminus of the Src SH2 domain.13 On the basis of this information, we designed a labeled Src SH2 domain that possessed a single modification with tetramethylrhodamine (TMR) at the N-terminus. The N-terminal TMR group was introduced on the resin after solid-phase synthesis of the N-terminal fragment L-Src(145–187) L-1b. The N-terminally modified fragment L-3b was then used in the identical ligation protocol to afford the TMR-labeled Src SH2 domain L-5b. The mirror-image Src SH2 domains (D-5a and D-5b) were synthesized by the same procedure.
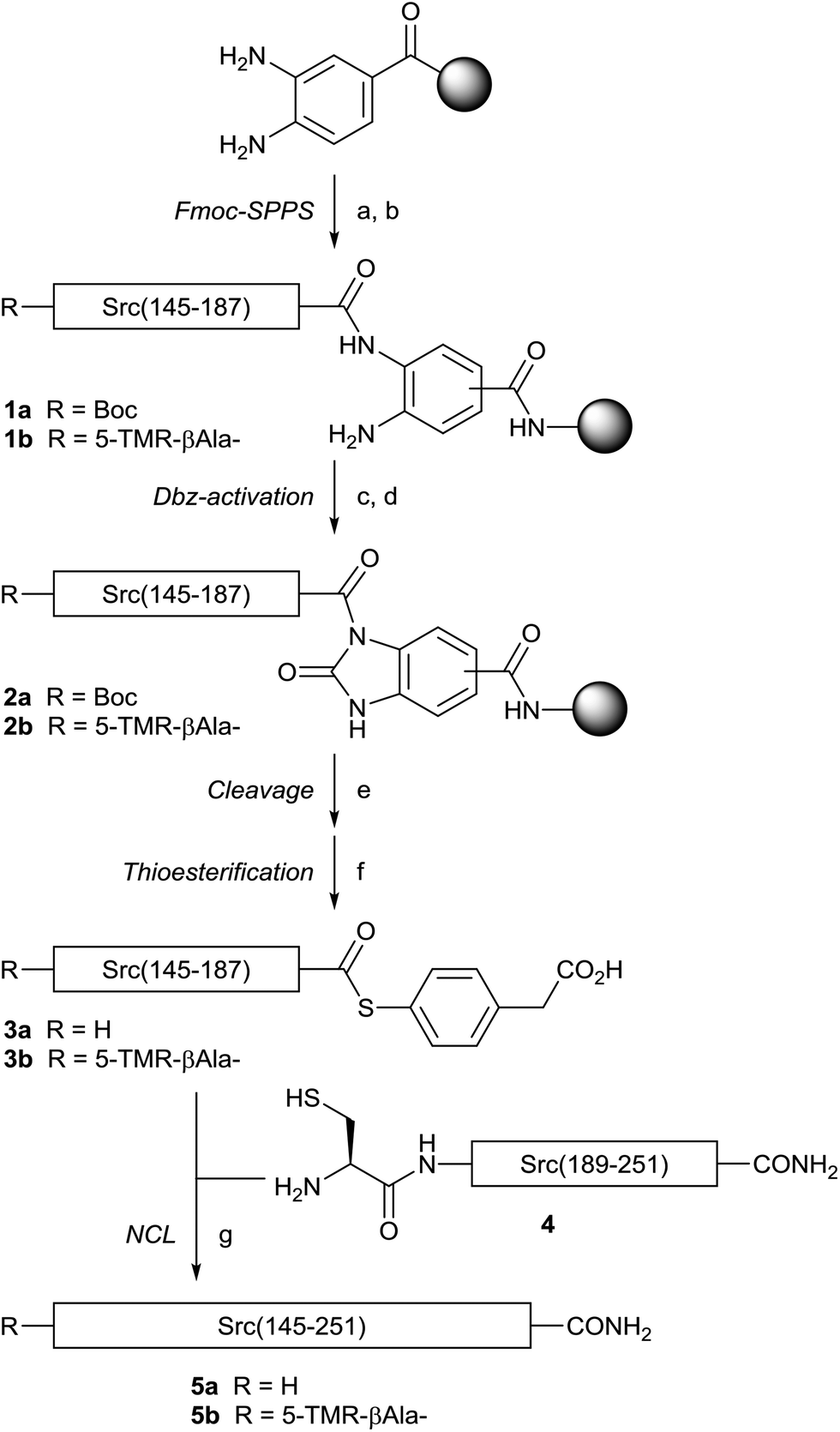
With the different full-length Src SH2 domains in hand, we investigated the refolding conditions based on a previously reported protocol.26,27 Since the three Cys residues (Cys188, Cys241 and Cys248) in Src SH2 are not involved in disulfide bond formation,13 the synthetic proteins were subjected to refolding under reducing conditions in the presence of 0.5 mM TCEP to prevent dimer formation. Dialysis of denatured Src(145–251) (L-5a) against HEPES buffer (20 mM HEPES, 100 mM NaCl, 0.5 mM TCEP, pH 7.4) resulted in a homogenous solution of the Src SH2 domain (L-Src145–251). Circular dichroism (CD) was used to confirm the tertiary structure of the folded Src SH2 domains (L-Src145–251 and D-Src145–251) (Fig. 2). CD spectra of L-Src145–251 was similar to those of other SH2 domains, including the N-terminal SH2 domain of the p85α subunit of the phosphatidylinositol 3-kinase (PI3K) and Btk SH2 domain,31,32 which displayed a broad minimum over the range of 210–220 nm. In addition, the symmetry CD spectra of L-Src145–251 and D-Src145–251 confirmed the mirror-image nature of the structures. The CD results indicated that the Src SH2 domains are correctly folded and should have biological activity.
 | ||
| Fig. 2 Circular dichroism spectra of the folded L-Src SH2 domain and D-Src SH2 domain. | ||
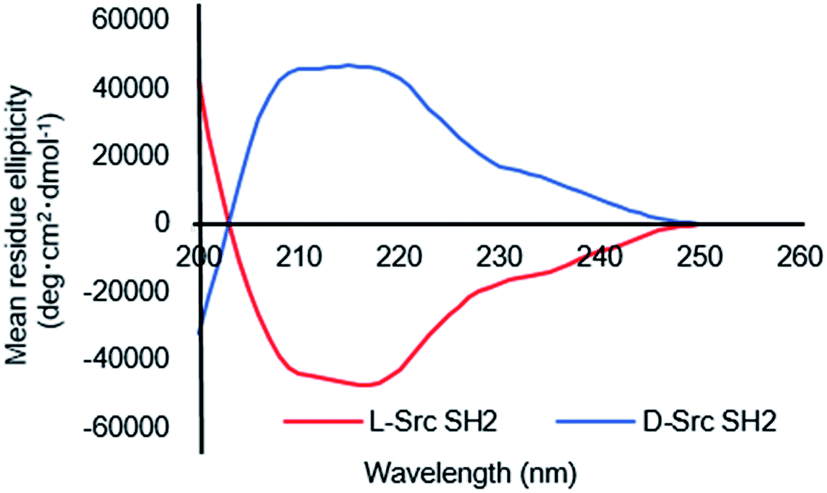
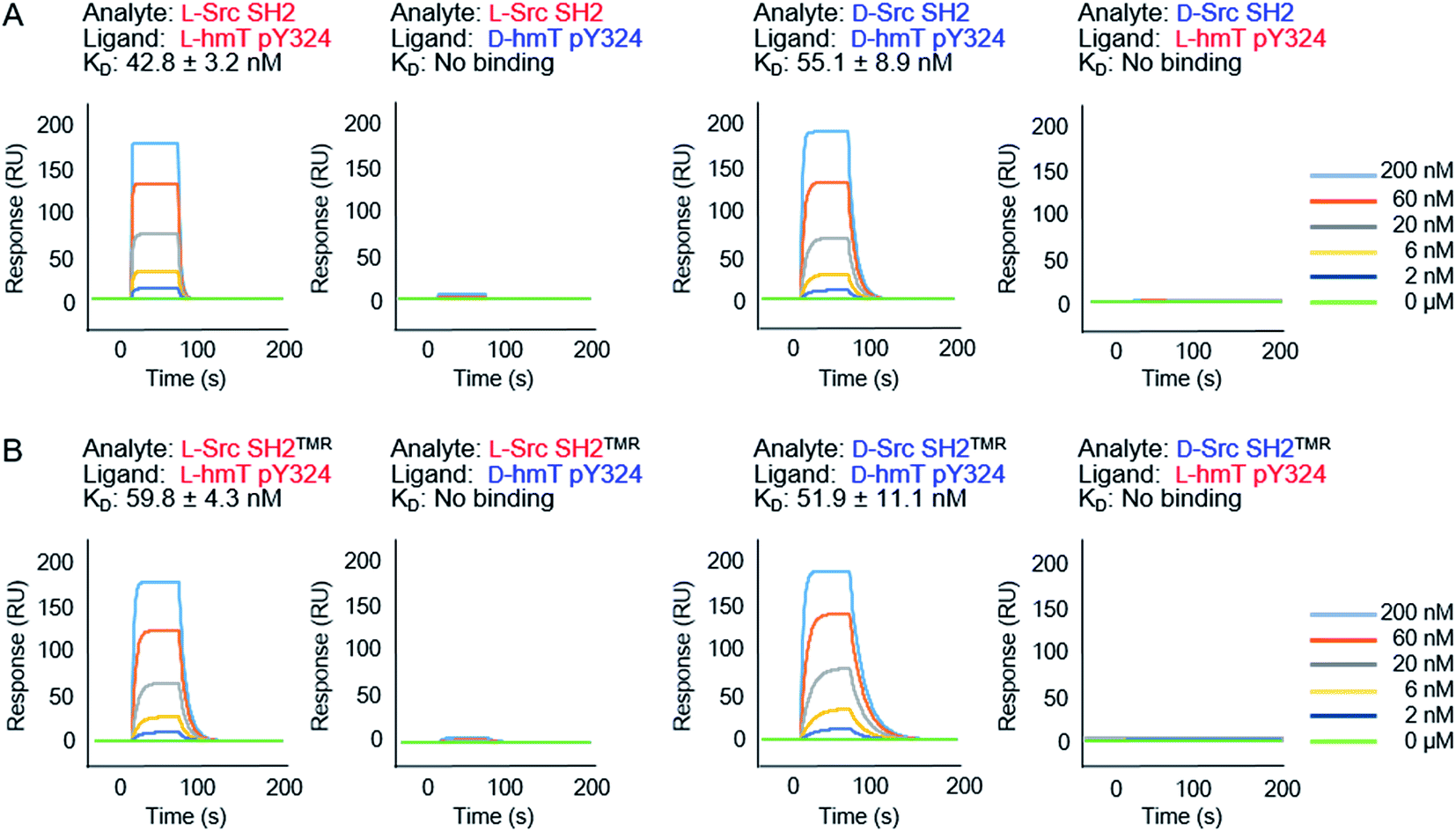
We next evaluated the binding affinity of the synthetic Src SH2 domains toward a pTyr-containing peptide by surface plasmon resonance (SPR) analysis. We chose hmT pY324 (H-EPQpYEEIPIYL-NH2) as a target sequence, which is derived from the hamster polyoma middle-sized tumor antigen (hmT antigen).1 After the biotinylated hmT pY324 was immobilized on the streptavidin coated sensor chip, interaction between the immobilized hmT pY324 ligand and a Src SH2 domain analyte was evaluated. L-Src145–251 bound the L-peptide form of hmT pY324 (L-7) with high affinity [KD (L-Src145–251–L-hmT pY324 peptide): 42.8 nM], whereas no interaction with the D-peptide (D-7) was observed. In a similar manner, D-Src145–251 showed potent affinity toward D-hmT pY324 selectively [KD (D-Src145–251–D-hmT pY324 peptide): 55.1 nM] (Fig. 3A). The bioactivity of the synthetic Src SH2 domains was also evaluated by a fluorescence polarization assay using a fluorescent peptide FMT1 (FAM-GpYEEIA-NH2).33 L-Src145–251 and D-Src145–251 interacted with L-FMT1 (L-8) and D-FMT1 (D-8) peptides, respectively, with submicromolar affinities [KD (L-Src145–251–L-FMT1): 162 nM; KD (D-Src145–251–D-FMT1): 182 nM] (Fig. 4). These results suggested the synthetic Src SH2 domains after an appropriate refolding process reproduced biological functions, and exhibited stereoselective interaction with the counterpart pTyr-containing sequence.
 | ||
| Fig. 3 Surface plasmon resonance analysis of synthetic folded Src SH2 derivatives binding to the hmT pY324 peptide. Binding kinetics were evaluated by immobilizing a biotinylated hmT pY324 peptide on an NLC sensor chip. | ||
 | ||
| Fig. 4 Binding of Src SH2 to pTyr-containing peptides. (A) Binding curve of the L-FMT–L-Src SH2 complex. KD (L-Src–L-FMT1): 162 ± 5 nM. (B) Binding curve of the D-FMT1–D-Src SH2 complex. KD (D-Src–D-FMT1): 182 ± 3 nM. KD values were determined from the saturation curves generated from triplicate experiments of the fluorescence polarization assay using the FMT1 peptide (20 nM). Representative saturation curves of the binding experiments are shown. | ||
The SPR analysis was carried out using the TMR-labeled Src SH2 domains. L-Src SH2TMR had similar binding affinity for L-hmT pY324 peptide (L-7) when compared with that of unlabeled L-Src145–251 [KD (L-Src SH2TMR–L-hmT pY324 peptide): 59.8 nM], indicating that the N-terminal TMR modification had no effect on either the folding of the Src SH2 domain or the interaction with the target pTyr peptide. The mirror-image interaction between D-Src SH2TMR and D-hmT pY324 peptide (D-7) was also similarly observed with high affinity [KD (D-Src SH2TMR–D-hmT pY324 peptide): 51.9 nM] (Fig. 3B). These fluorescent-labeled proteins should be suitable in a number of in vitro bioassays for screening campaigns.
Chemical array screening is an ultrahigh-throughput screening technology for drug discovery (Fig. 5A).34 The affinity-based selection by microarray technology facilitates the identification of potential inhibitors against a protein–protein interaction(s) from a library of small molecules. In our previous mirror-image screening studies for MDM2 inhibitors and Grb2 SH2 domain inhibitors,25,26 this chemical array analysis was employed as the initial screening of natural products, which were immobilized via carbene-mediated covalent bond formation.35–37 Using this unique chemical array technique, the binding compound(s) for the alternative or unprecedented pockets as well as previously recognized binding pockets of the target protein were identified. To demonstrate the applicability of the TMR-labeled Src SH2 domains to chemical array screening, binding with the target hmT pY324 peptide on a chemical array was assessed. The L-peptide and D-peptide of hmT pY324 (L-6 and D-6) were immobilized on a chemical array by carbene-mediated covalent bond formation at different concentrations. When the array was treated with TMR-labeled Src SH2 domains, L-Src SH2TMR and D-Src SH2TMR bound to L-hmT pY324 and D-hmT pY324, respectively (Fig. 6). In contrast, no significant binding to the mismatch target peptides was observed, indicating the enantioselective recognition by the synthetic TMR-labeled proteins. Thus, the synthetic TMR-labeled Src SH2 domains should be applicable to mirror-image screening using the chemical array technology.
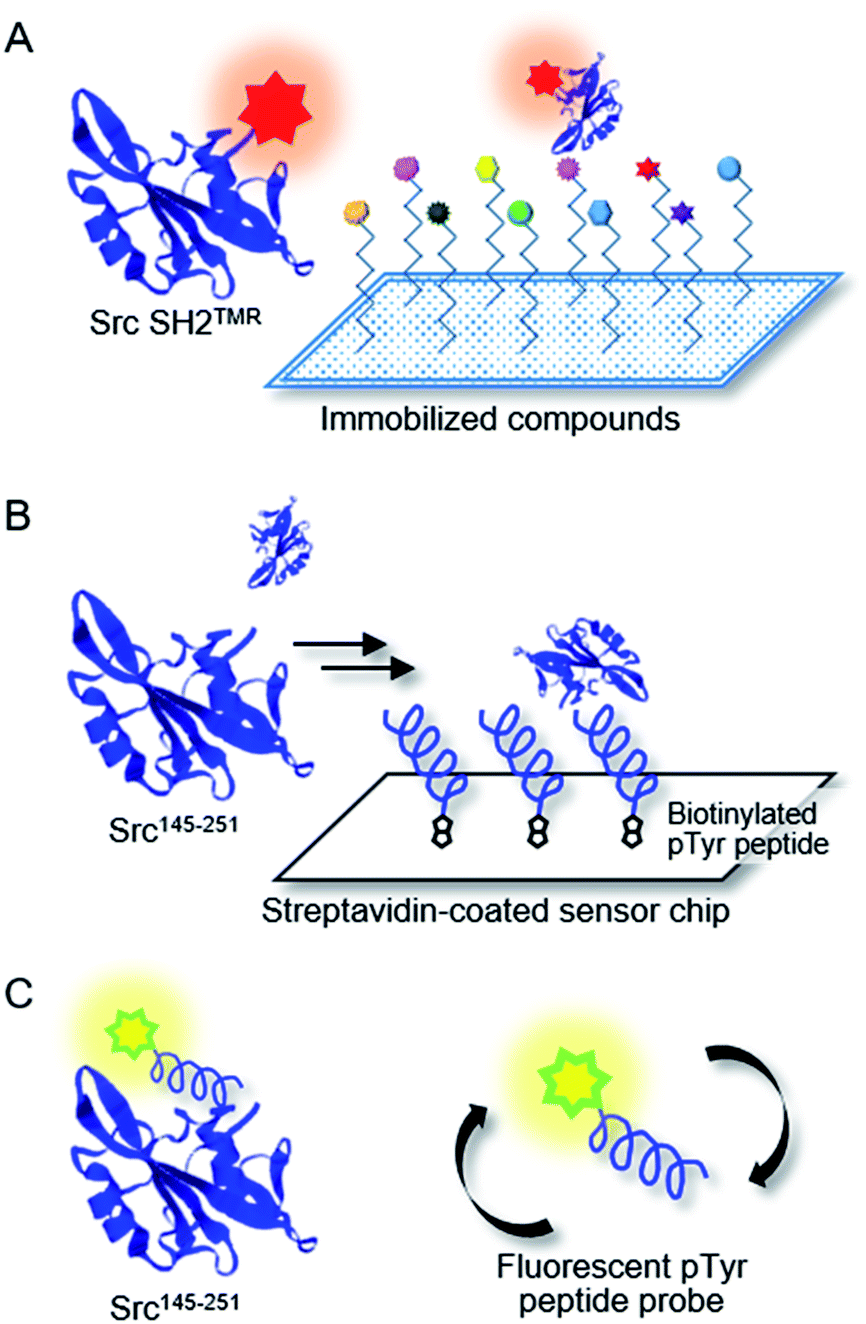 | ||
| Fig. 5 Design of the bioassay systems using synthetic proteins. (A) Chemical array analysis for the first screening. Binding with immobilized compound(s) on glass slides is detected by fluorescence signal of Src SH2TMR. (B) Surface plasmon resonance (SPR) analysis of Src SH2 domain binding with hmT pY324 (a pTyr-containing peptide) immobilized on the streptavidin-coated sensor chip. The Src SH2 binding to hmT pY342 is detected by the increased SPR signal. The inhibitory activity is evaluated in the presence of the potential inhibitor as an analyte. (C) Fluorescence polarization (FP) assay for Src SH2 domain binding with fluorescent FMT1 peptide probe. The FMT1 binding to Src SH2 binding is detected by the increased FP signal, which is derived from the impaired mobility of the fluorescent probe. The inhibitory activity is evaluated in the presence of the potential inhibitor. | ||
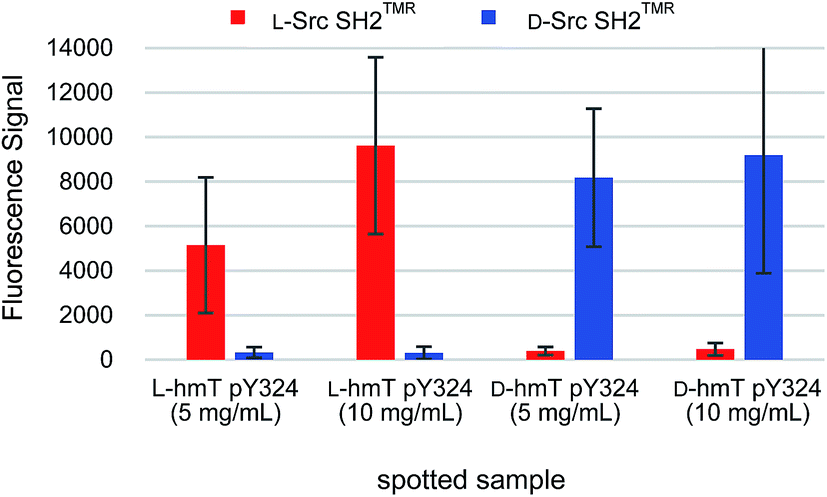 | ||
| Fig. 6 Binding activities of the Src SH2 domain towards hmT pY324 peptides immobilized on a chemical array. The binding of L-Src SH2TMR and D-Src SH2TMR were assessed using a chemical array, where L-hmT pY324 or D-hmT pY324 peptides were spotted at various concentrations. | ||
For secondary analysis to identify compounds that bind to the pTyr peptide binding pocket in the Src SH2 domain, a competitive binding assay using SPR was established (Fig. 5B). In the SPR analysis, specific and stereoselective binding of the Src SH2 domain (Src145–251) to the pTyr-containing peptide (biotinylated hmT pY324) was observed, which was immobilized on the streptavidin-coated sensor chip (Fig. 3).38 Using this system in the presence of a potential inhibitor(s), the inhibitory effect against the Src SH2 domain–pTyr peptide interaction could be measured. Since the detection of the Src SH2 domain–pTyr peptide interaction by SPR does not involve a colored material(s), the possibility of observing false-positives and false-negatives was minimized. The SPR response of L-Src145–251 onto the immobilized L-pTyr peptide (L-7) decreased in the presence of the unlabeled L-hmT pY234 peptide (L-6) in a dose-dependent manner (IC50: 323 nM) (Fig. 7A). The inhibitory activity of D-hmT pY324 peptide (D-6) against the D-Src145–251–D-pTyr peptide interaction was determined as well (IC50 387 nM). We also investigated the development of a competitive inhibition assay for Src SH2 domain inhibitors (Fig. 7B) using fluorescence polarization (FP) based on a reported protocol (Fig. 5C).33 The homogeneous assay by FP experiments provides an alternative high-throughput screening approach using non-labeled protein.39 The fluorescent pTyr-peptide probe (FMT1, 20 nM) and unlabeled Src protein (Src145–251, 300 nM) were used for the inhibition experiment. Dose-dependent inhibition against the L-Src145–251–L-pTyr peptide interaction by the unlabeled L-hmT pY324 peptide (L-6) was observed (IC50: 860 nM). The mirror-image interaction between D-Src145–251 and D-pTyr peptide was also inhibited by the D-hmT pY324 peptide (D-6) (IC50: 946 nM). These two complementary bioassay systems to determine the inhibitory activity should be suitable for identifying potential inhibitors that bind to the pTyr binding pocket in the Src SH2 domain following the primary comprehensive chemical array-based screening to explore and identify compounds that bind to the Src SH2 domain.
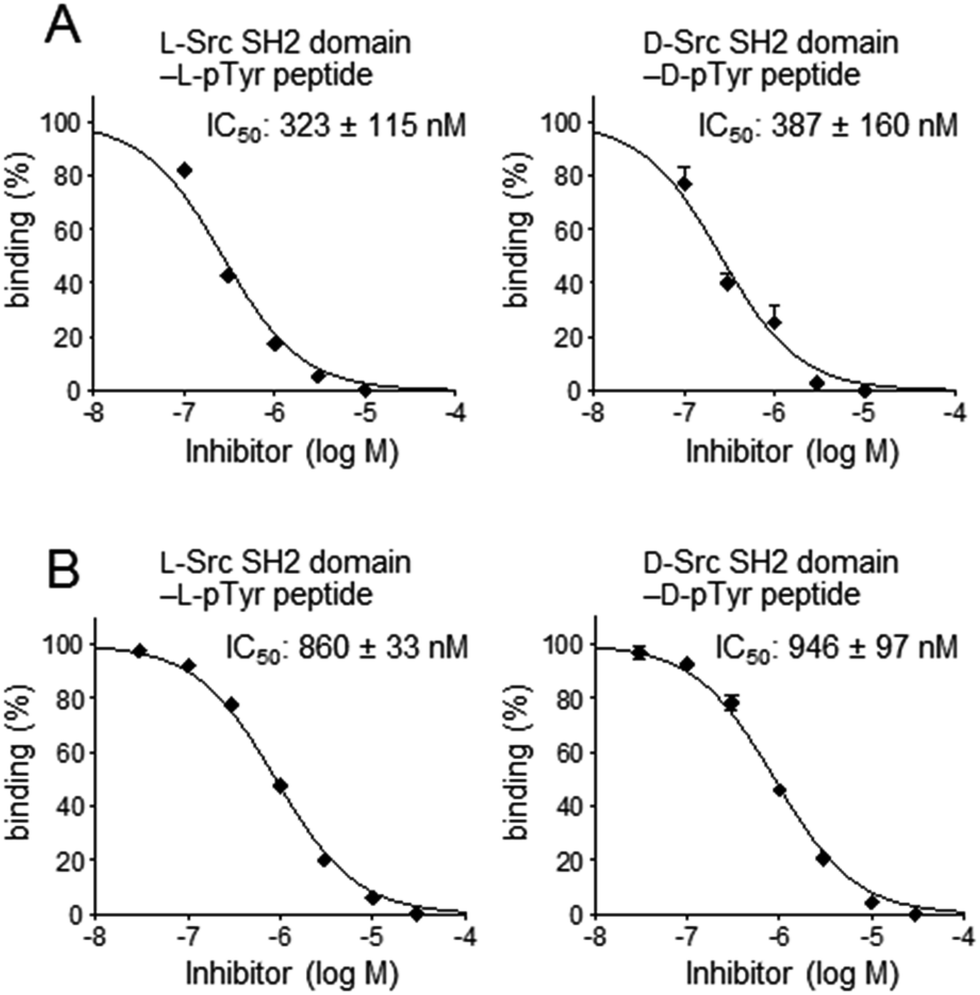 | ||
| Fig. 7 Inhibitory activity of the hmT pY324 peptide against the Src SH2 domain–pTyr-containing peptide interaction. (A) SPR analysis was carried out by Src145–251 (200 nM) in the presence of the inhibitor as an analyte on the pTyr peptide-immobilized NLC sensor chip. (B) FP assay was carried out using the fluorescent FMT1 peptide probe (20 nM) and Src145–251 (300 nM). IC50 values were derived from the dose–response curves generated from triplicate experiments. Representative dose–response curves of the inhibition experiments are shown. | ||
Conclusions
We accomplished total synthesis of the Src SH2 domain by conjugating two fragment peptides by native chemical ligation at Cys188. The TMR-labeled Src SH2 domain was also synthesized using the same procedure with the N-terminal peptide fragment ligated to a fluorescent group at the N-terminus. The synthetic L-Src SH2 and D-Src SH2 domains were converted into functional proteins under appropriate conditions and were found to recognize the target pTyr sequence in a stereoselective manner. Taking advantage of the resulting domain prepared, we established three bioassays, namely SPR analysis, chemical array analysis, and fluorescence polarization (FP), to detect the interaction between the Src SH2 domain and the counterpart pTyr peptide. Additionally, two competitive inhibition assays were established using SPR analysis and FP to identify Src SH2 domain inhibitors, which bound into the pTyr-binding pocket. These mirror-image bioassays should facilitate efficient screening of potential Src SH2 inhibitors from unexplored mirror-image compounds of natural product resources.Experimental
Peptide synthesis
Protected peptide-resins were constructed by Fmoc-based solid-phase peptide synthesis using automatic peptide synthesizer (PSSM-8, Shimadzu Corporation) unless otherwise stated. tBu ester for Asp and Glu; 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Pbf) for Arg; tBu for Thr, Tyr and Ser; Boc for Lys and Trp; Trt for Gln, Asn, His, and Cys; mono Bzl for pTyr were employed for side-chain protection. Fmoc–amino acids were coupled using HBTU (5 eq.)/HOBt·H2O (5 eq.)/(iPr)2NEt (10 eq.) activation to free amino group in DMF for 45 min. The coupling reaction was repeated twice. Fmoc deprotection was performed by 20% piperidine in DMF (2 × 6 min). The resulting protected resin was treated with TFA/H2O/thioanisole/m-cresol/1,2-ethanedithiol (80:5:5:5:5) at room temperature for 2 h. After removal of the resin by filtration, the filtrate was poured into ice-cold dry Et2O. The resulting powder was collected by centrifugation and washed with ice-cold dry Et2O. The crude product was purified by preparative HPLC on a Cosmosil 5C18-AR300 preparative column (Nacalai Tesque, 20 × 250 mm, flow rate 8 mL min−1). For analytical HPLC of the FMT1 peptide, Cosmosil 5C18-ARII column (4.6 × 250 mm, Nacalai Tesque) was employed with a linear gradient of CH3CN containing 0.1% (v/v) TFA at a flow rate of 1 mL min−1. For analytical HPLC for other peptides, Cosmosil 5C18-AR300 (4.6 × 250 mm, Nacalai Tesque) was employed with a linear gradient of CH3CN containing 0.1% (v/v) TFA at a flow rate of 1 mL min−1. All peptides were characterized by an ESI-MS (micromass ZQ, Waters) or MALDI-TOF-MS (AXIMA-CFR plus, Shimadzu).
L-Src(145–187) (L-3a)
3-[(9-Fluorenylmethyloxycarbonyl)amino]-4-aminobenzoic acid (Fmoc-Dbz-OH, 37.4 mg, 0.1 mmol) was manually coupled on H-Rink amide-ChemMatrix resin (0.4–0.6 mmol g−1, 40 mg, 0.016–0.024 mmol) using HBTU (37.8 mg, 0.1 mmol), HOBt·H2O (15.2 mg, 0.1 mmol), and (iPr)2NEt (0.0348 mL, 0.2 mmol) in DMF for 2.5 h. This treatment was repeated four times. The peptide sequence was constructed by the standard protocol. Because of the less efficient coupling of Arg158, Arg159, Arg163, Leu164, and Arg172, HATU (38.0 mg, 0.1 mmol) and (iPr)2NEt (0.0348 mL, 0.2 mmol) were employed for activation and the coupling reactions for these amino acids were repeated three times. A solution of p-nitrophenyl chloroformate (22.5 mg, 0.11 mmol) in CH2Cl2 (0.38 mL) was added to the protected peptide resin L-1a, and the mixture was agitated for 40 min at room temperature. Then, the resin was treated with a solution of 0.5 M (iPr)2NEt in DMF (0.38 mL) for 15 min. Three portions of the resulting peptide resin were combined and the total peptide resin L-2a (0.048–0.072 mmol) was treated with TFA/H2O/thioanisole/m-cresol (80:10:5:5) at room temperature for 2 h. After removal of the resin by filtration, the filtrate was poured into ice-cold dry Et2O. The precipitate was washed with ice-cold dry Et2O three times. The crude peptide was dissolved in 1 M phosphate buffer (pH 7.0) containing 6 M GuHCl, 200 mM, 4-mercaptophenyl acetic acid (MPAA), and 20 mM TCEP. The solution was kept at 37 °C for 30 min and purified by preparative HPLC (a linear gradient of 25–55% CH3CN in H2O containing 0.1% (v/v) TFA over 90 min) to afford the thioester L-3a (10.5 mg, 2.9% yield). MS(ESI): calcd for C231H357N64O71S (MH+): 5198.83; observed: [M + 6H]6+ m/z = 867.2, [M + 5H]5+ m/z = 1040.7, [M + 4H]4+ m/z = 1300.4, [M + 3H]3+ m/z = 1733.3.
L-Src(188–251) (L-4)
By the standard protocol for peptide synthesis, peptide L-4 was synthesized (8.91 mg, 1.8% yield) from H-Rink amide-ChemMatrix resin (40 mg × 3 portions, 0.048–0.072 mmol). MS(ESI): calcd for C318H500N91O94S3 (MH+): 7198.22; observed: [M + 8H]8+ m/z = 900.7, [M + 7H]7+ m/z = 1029.1, [M + 6H]6+ m/z = 1200.4, [M + 5H]5+ m/z = 1440.4, [M + 4H]4+ m/z = 1800.0.Native chemical ligation: synthesis of L-Src(145–251) (L-5a)
The L-Src(145–187) thioester (L-3a, 4.5 mg) and L-Src(188–251) (L-4, 4.8 mg) were dissolved in 0.200 mL of ligation buffer [1 M phosphate buffer (pH 7.0) containing 6 M Gu·HCl, 200 mM MPAA, 100 mM TCEP], and the ligation reaction was continued for 1 h at 37 °C. Then, 1.80 mL of TCEP solution (6 M GuHCl and 100 mM TCEP) was added to the mixture and the reaction was continued for 15 min at room temperature. The crude product was purified by preparative HPLC (a linear gradient of 25–55% CH3CN in H2O containing 0.1% (v/v) TFA over 90 min) to provide peptide L-5a (1.4 mg, 17% yield). MS(ESI): calcd for C541H848N155O163S3 (MH+): 12227.84; observed (ESI): [M + 14H]14+ m/z = 874.4, [M + 13H]13+ m/z = 941.7, [M + 12H]12+ m/z = 1020.1, [M + 11H]11+ m/z = 1112.7, [M + 10H]10+ m/z = 1223.9, [M + 9H]9+ m/z = 1359.7, [M + 8H]8+ m/z = 1529.8, [M + 7H]7+ m/z = 1747.8.
L-hmT pY324 (L-6, H-EPQpYEEIPIYL-NH2)
The peptide resin was manually constructed by Fmoc-SPPS on Rink-amide resin (0.6 mmol g−1, 75 mg, 0.03 mmol). Fmoc-protected amino acids (3 eq.) were coupled by using DIC (0.209 mL, 0.135 mmol) and HOBt·H2O (21.2 mg, 0.135 mmol) in DMF. For coupling of the N-terminal Glu, Pro, Gln and pTyr, Fmoc-protected amino acid (5 eq.), HATU (85.5 mg, 0.225 mmol) and (iPr)2NEt (0.783 mL, 0.45 mmol) were employed. Fmoc-protecting group was removed by treatment of the resin with 20% piperidine in DMF. The resulting protected peptide resin was treated with TFA/thioanisole/m-cresol/1,2-ethanedithiol/H2O (80:5:5:5:5) at room temperature for 2 h. After removal of the resin by filtration, the filtrate was poured into ice-cold dry Et2O. Purification by preparative HPLC on a Cosmosil 5C18-ARII column (Nacalai Tesque, 20 × 250 mm, a linear gradient of 20–50% CH3CN containing 0.1% (v/v) TFA aq. over 90 min) provided the peptide L-6 (22.4 mg, 31% yield). MS(ESI): calcd for C66H99N13O23P: 1473.56; observed: [M + H]+ m/z = 1473.0.
Folding of synthetic Src SH2 domains
Folding of synthetic Src SH2 derivatives were carried out by dialysis using Slide-A-Lyzer G2 dialysis cassette (cutoff 3.5 kDa, Thermo).26,40 Lyophilized polypeptide (1 mg mL−1) was dissolved in guanidine solution (6 M Gu·HCl, 20 mM HEPES, 100 mM NaCl, pH 8.5) and the solution was dialyzed against a 200-fold volume of dialysis buffer (20 mM HEPES, 100 mM NaCl, and 0.5 mM TCEP, pH 7.4) for 1.5 h at 4 °C. The additional dialysis was repeated against the dialysis buffer for an additional 1.5 h at 4 °C and overnight at 4 °C.CD spectra of L-Src SH2 and D-Src SH2 domains
Using the identical procedure for the folding of the protein, we dialyzed the protein solution in HEPES buffer against PBS buffer [1 M phosphate buffer (pH 7.4) containing 1 mM DTT]. CD spectra of L-Src145–251 and D-Src145–251 were recorded on a JASCO J-720 circular dichroism spectrometer at 20 °C.Surface plasmon resonance analysis
SPR analysis was carried out using a ProteOn XPR36 surface plasmon resonance (SPR) system (Bio-Rad) in the running buffer [10 mM HEPES (pH 7.4), 150 mM NaCl containing 0.005% Tween-20] at 25 °C. For binding analysis of synthetic Src145–251 (0–200 nM) and Src SH2TMR (0–200 nM) proteins, hmT pY324biotin (5 nM, 5 min) was immobilized on a ProteOn NLC sensor chip. All analytes were evaluated for 60 s as contact time, followed by 600 s dissociation at a flow rate of 0.050 mL min−1. For competitive experiments, Src SH2 (200 nM) in the presence of varying concentrations of unlabeled hmT pY324 (0–10 μM) in running buffer were injected onto the ProteOn NLC sensor chip, where hmT pY324biotin was immobilized. The data were analyzed using GraphPad Prism software.Fluorescence polarization assay
Fluorescence polarization (FP) assays were carried out in the assay buffer (PBS containing 2 mM DTT, 0.1% bovine gamma globulin, and 2% DMSO) using FMT1FAM (20 nM) in 96-well non-binding surface black assay plates (Corning).2 For binding titration analysis, probe FMT1FAM (20 nM) was incubated with the synthetic Src SH2 domain at increasing concentrations (0–30 μM) in 0.10 mL of assay buffer. The KD was obtained by nonlinear least-squares fitting to a single site binding model and Scatchard plot. For the competitive inhibition experiment, the hmT pY324 (inhibitor) and FMT1FAM were diluted five-fold with PBS in advance. The Src SH2 domain (300 nM, 0.090 mL per well) was incubated with hmT pY324 (0.005 mL) for 30 min. Then, FMT1FAM (0.005 mL) was added and the mixture was incubated for 30 min. FP signals were detected using an EnVision Xcite plate reader (PerkinElmer) with a 480 nm excitation filter and a 535 nm emission filter. The data were analysed using GraphPad Prism software.Chemical array analysis
Photoaffinity linker-coated (PALC) slides were prepared according to previous reports25,35,37 using amine-coated slides and the photoaffinity PEG linker. A solution of compounds in DMSO was spotted onto the PALC glass slides with a chemical arrayer equipped with 24 stamping pins or a MultiSPRinter spotter (Toyobo) equipped with a single stamping pin. The slides were exposed to UV irradiation of 4 J cm−2 at 365 nm using a CL-1000L UV crosslinker (UVP, CA) for immobilization. The slides were washed successively with DMSO, DMF, acetonitrile, THF, dichloromethane, EtOH, and ultra-pure water (5 min, three times each), and dried. L-Src SH2TMR or D-Src SH2TMR (3 μM in 1% skimmed-milk-TBS-T) was incubated with the glass slide for 1 h and then washed with TBS-T [10 mM Tris–HCl (pH 8.0), 150 mM NaCl, 0.05% Tween-20] (5 min, three times). The slides were dried and scanned at 532 nm on a GenePix scanner. The fluorescence signals were quantified with GenePixPro.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research from JSPS, Japan (15H04653, 26-7738); Platform for Drug Discovery, Informatics, and Structural Life Science from MEXT, Japan; the Tokyo Biochemical Research Foundation; Mochida Memorial Foundation for Medical and Pharmaceutical Research; and the Kato Memorial Bioscience Foundation. T. N. is grateful for JSPS Research Fellowships for Young Scientists.References
- Z. Songyang, S. E. Shoelson, M. Chaudhuri, G. Gish, T. Pawson, W. G. Haser, F. King, T. Roberts, S. Ratnofsky, R. J. Lechleider, B. G. Neel, R. B. Birge, J. E. Fajard, M. M. Chou, H. Hanafusa, B. Schaffhausen and L. C. Cantley, Cell, 1993, 72, 767–778 CrossRef CAS PubMed.
- P. L. Schwartzberg, L. Xing, O. Hoffmann, C. A. Lowell, L. Garrett, B. F. Boyce and H. E. Varmus, Genes Dev., 1997, 11, 2835–2844 CrossRef CAS PubMed.
- K. B. Kaplan, J. R. Swedlow, D. O. Morgan and H. E. Varmus, Genes Dev., 1995, 9, 1505–1517 CrossRef CAS PubMed.
- Y. Fukui, M. C. O'Brien and H. Hanafusa, Mol. Cell. Biol., 1991, 11, 1207–1213 CrossRef CAS PubMed.
- M. D. Schaller, J. D. Hildebrand, J. D. Shannon, J. W. Fox, R. R. Vines and J. T. Parsons, Mol. Cell. Biol., 1994, 14, 1680–1688 CrossRef CAS PubMed.
- S. J. Taylor and D. Shalloway, Nature, 1994, 368, 867–871 CrossRef CAS PubMed.
- L. A. Petch, S. M. Bockholt, A. Bouton, J. T. Parsons and K. Burridge, J. Cell Sci., 1995, 108, 1371–1379 CAS.
- S. A. Courtneidge, L. Goutebroze, A. Cartwright, A. Heber, S. Scherneck and J. Feunteun, J. Virol., 1991, 65, 3301–3308 CAS.
- P. Majkut, I. Claußnitzer, H. Merk, C. Freund, C. P. R. Hackenberger and M. Gerrits, PLoS One, 2013, 8, e82352 Search PubMed.
- K. D. Beebe, P. Wang, G. Arabaci and D. Pei, Biochemistry, 2000, 39, 13251–13260 CrossRef CAS PubMed.
- K. Machida, C. M. Thompson, K. Dierck, K. Jablonowski, S. Kärkkäinen, B. Liu, H. Zhang, P. D. Nash, D. K. Newman, P. Nollau, T. Pawson, G. H. Renkema, K. Saksela, M. R. Schiller, D. Shin and B. J. Mayer, Mol. Cell, 2007, 26, 899–915 CrossRef CAS PubMed.
- G. Waksman, S. E. Shoelson, N. Pant, D. Cowburn and J. Kuriyan, Cell, 1993, 72, 779–790 CrossRef CAS PubMed.
- M. T. Brown and J. A. Cooper, Biochim. Biophys. Acta, 1996, 1287, 121–149 Search PubMed.
- M. S. Plummer, D. R. Holland, A. Shahripour, E. A. Lunney, J. H. Fergus, J. S. Marks, P. McConnell, W. T. Mueller and T. K. Sawyer, J. Med. Chem., 1997, 40, 3719–3725 CrossRef CAS PubMed.
- J. L. Buchanan, R. S. Bohacek, G. P. Luke, M. Hatada, X. Lu, D. C. Dalgarno, S. S. Narula, R. Yuan and D. A. Holt, Bioorg. Med. Chem. Lett., 1999, 9, 2353–2358 CrossRef CAS PubMed.
- W. Shakespeare, M. Yang, R. Bohacek, F. Cerasoli, K. Stebbins, R. Sundaramoorthi, M. Azimioara, C. Vu, S. Pradeepan, C. Metcalf 3rd, C. Haraldson, T. Merry, D. Dalgarno, S. Narula, M. Hatada, X. Lu, M. R. van Schravendijk, S. Adams, S. Violette, J. Smith, W. Guan, C. Bartlett, J. Herson, J. Iuliucci, M. Weigele and T. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9373–9378 CrossRef CAS.
- R. Sundaramoorthi, C. Siedem, C. B. Vu, D. C. Dalgarno, E. C. Laird, M. C. Botfield, A. B. Combs, S. E. Adams, R. W. Yuan, M. Weigele and S. S. Narula, Bioorg. Med. Chem. Lett., 2001, 11, 1665–1669 CrossRef CAS PubMed.
- J. P. Davidson, O. Lubman, T. Rose, G. Waksman and S. F. Martin, J. Am. Chem. Soc., 2002, 124, 205–215 CrossRef CAS PubMed.
- S. Park, J. Won and K. Lee, Bioorg. Med. Chem. Lett., 2002, 12, 2711–2714 CrossRef CAS PubMed.
- G. Lange, D. Lesuisse, P. Deprez, B. Schoot, P. Loenze, D. Bénard, J. Marquette, P. Broto, E. Sarubbi and E. Mandine, J. Med. Chem., 2002, 45, 2915–2922 CrossRef CAS PubMed.
- N. Nam, R. L. Pitts, G. Sun, S. Sardari, A. Tiemo, M. Xie, B. Yan and K. Parang, Bioorg. Med. Chem., 2004, 12, 779–787 CrossRef CAS PubMed.
- N. Nam, G. Ye, G. Sun and K. Parang, J. Med. Chem., 2004, 47, 3131–3141 CrossRef CAS PubMed.
- J. Won, Y. G. Hur, E. M. Hur, S. H. Park, M. A. Kang, Y. Choi, C. Park, K. H. Lee and Y. Yun, Eur. J. Immunol., 2003, 33, 870–879 CrossRef CAS PubMed.
- B. Sperl, M. H. J. Seifert and T. Berg, Bioorg. Med. Chem. Lett., 2009, 19, 3305–3309 CrossRef CAS PubMed.
- T. Noguchi, S. Oishi, K. Honda, Y. Kondoh, T. Saito, H. Ohno, H. Osada and N. Fujii, Chem. Commun., 2016, 52, 7653–7656 RSC.
- T. Noguchi, H. Ishiba, K. Honda, Y. Kondoh, H. Osada, H. Ohno, N. Fujii and S. Oishi, Bioconjugate Chem., 2016, 28, 609–619 CrossRef PubMed.
- S. Virdee, D. Macmillan and G. Waksman, Chem. Biol., 2010, 17, 274–284 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CAS.
- T. Noguchi, S. Oishi, K. Honda, Y. Kondoh, T. Saito, T. Kubo, M. Kaneda, H. Ohno, H. Osada and N. Fujii, Bioorg. Med. Chem. Lett., 2013, 23, 3802–3805 CrossRef CAS PubMed.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851–6855 CrossRef CAS PubMed.
- G. Panayotou, B. Bax, I. Gout, M. Federwisch, B. Wroblowski, R. Dhand, M. J. Fry, T. L. Blundell, A. Wollmer and M. D. Waterfield, EMBO J., 1992, 11, 4261–4272 CAS.
- P. T. Mattsson, I. Lappalainen, C. M. Bäckesjö, E. Brockmann, S. Laurén, M. Vihinen and C. I. E. Smith, J. Immunol., 2000, 164, 4170–4177 CrossRef CAS.
- B. A. Lynch, K. A. Loiacono, C. L. Tiong, S. E. Adams and I. A. MacNeil, Anal. Biochem., 1997, 247, 77–82 CrossRef CAS PubMed.
- For a review, see: Y. M. Foong, J. Fu, S. Q. Yao and M. Uttamchandani, Curr. Opin. Chem. Biol., 2012, 16, 234–242 CrossRef CAS PubMed.
- N. Kanoh, S. Kumashiro, S. Simizu, Y. Kondoh, S. Hatakeyama, H. Tashiro and H. Osada, Angew. Chem., Int. Ed., 2003, 42, 5584–5587 CrossRef CAS PubMed.
- N. Kanoh, A. Asami, M. Kawatani, K. Honda, S. Kumashiro, H. Takayama, S. Simizu, T. Amemiya, Y. Kondoh, S. Hatakeyama, K. Tsuganezawa, R. Utata, A. Tanaka, S. Yokoyama, H. Tashiro and H. Osada, Chem.–Asian J., 2006, 1, 789–797 CrossRef CAS PubMed.
- Y. Kondoh, K. Honda and H. Osada, Methods Mol. Biol., 2015, 1263, 29–41 CAS.
- T. Bravman, V. Bronner, K. Lavie, A. Notcovich, G. A. Papalia and D. G. Myszka, Anal. Biochem., 2006, 358, 281–288 CrossRef CAS PubMed.
- C. M. Labbé, G. Laconde, M. A. Kuenemann, B. O. Villoutreix and O. Sperandio, Drug Discovery Today, 2013, 18, 958–968 CrossRef PubMed.
- P. Ettmayer, D. France, J. Gounarides, M. Jarosinski, M. Martin, J. Rondeau, M. Sabio, S. Topiol, B. Weidmann, M. Zurini and K. W. Bair, J. Med. Chem., 1999, 42, 971–980 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07445j |
| This journal is © The Royal Society of Chemistry 2017 |