
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMutagenesis and immunological evaluation of group A streptococcal C5a peptidase as an antigen for vaccine development and as a carrier protein for glycoconjugate vaccine design†
Hui Lia,
Subo Wanga,
Yisheng Zhaoa,
Zonggang Chena,
Guofeng Gu *a and
Zhongwu Guo*ab
*a and
Zhongwu Guo*ab
aNational Glycoengineering Research Center, School of Life Science, Shandong University, 27 Shanda Nan Lu, Jinan 250100, China. E-mail: guofenggu@sdu.edu.cn; zguo@chem.ufl.edu; Tel: +86 (531) 88363612 Tel: +1 (352) 392 9133
bDepartment of Chemistry, University of Florida, 214 Leigh Hall, Gainesville, Florida 32611, USA
First published on 30th August 2017
Abstract
A truncated form (AA32-1032) of group A streptococcus (GAS) C5a peptidase (ScpA), an important GAS virulence factor, and its mutants were prepared and examined to find suitable GAS vaccine candidates and conjugate vaccine carriers. Enzymatic evaluation of the recombinant proteins with a MALDI-TOF MS-based method to analyze the reaction indicated that D130 and N295 were not critical for its activity and S512 was significant but not absolutely required either. Therefore, ΔScpAD130A, ΔScpAN295A and ΔScpAS512A were not suitable vaccine and carrier protein candidates due to their remaining enzymatic activity. A single mutation of H193 to Ala abolished the ScpA activity completely, thereby identifying ΔScpAH193A as a promising candidate that was subjected to immunological studies in mouse. It was shown to elicit high titers of antigen-specific IgG1 antibodies and robust T cell-mediated immunities, verifying its potential as a GAS vaccine. Moreover, conjugating the trisaccharide repeating unit of GAS polysaccharide with ΔScpAH193A could convert the nonimmunogenic oligosaccharide into a highly active and T cell-dependent antigen, demonstrating the potential of ΔScpAH193A as a carrier protein to help formulate robust glycoconjugate vaccines.
1. Introduction
The Gram-positive bacterium Streptococcus pyogenes, also known as group A streptococcus (GAS), is one of the most common pathogens, which can cause high morbidity and mortality in humans.1,2 GAS infections are associated with a variety of human diseases from mild pharyngitis and pyoderma to severe necrotizing fasciitis, pneumonia, and toxic shock syndrome, leading in many cases to fatal post-streptococcal sequelae such as rheumatic fever, rheumatic heart disease, and acute glomerulonephritis. It is estimated that more than 700 million people suffer from GAS infections each year, giving rise to over 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 annual deaths.3,4 The estimated cost of GAS pharyngitis among children is up to US$220–540 million every year in the United States alone.5 To date, there is still no commercial vaccine for GAS, thus antibiotic therapy is the only strategy available to treat its infections. However, the insurgence of antibiotic-resistant bacteria,6 together with the high burden of the diseases, makes the development of new therapeutic and preventative strategies, such as vaccines, for the control of GAS infection in urgent demand.
000 annual deaths.3,4 The estimated cost of GAS pharyngitis among children is up to US$220–540 million every year in the United States alone.5 To date, there is still no commercial vaccine for GAS, thus antibiotic therapy is the only strategy available to treat its infections. However, the insurgence of antibiotic-resistant bacteria,6 together with the high burden of the diseases, makes the development of new therapeutic and preventative strategies, such as vaccines, for the control of GAS infection in urgent demand.
For the development of GAS vaccines,7–11 many past endeavors have been focused on the M protein, resulting in several vaccine candidates that entered clinic trials.12–15 However, the antigenic variability of M protein (>100 serotypes) and the safety concerns of its immune response to cause potential cross-reaction with human tissues would affect the applicability of related vaccines.1,16 Therefore, much current attention has been turned towards other GAS virulence factors, such as its toxins,17 polysaccharides,18,19 and C5a peptidase,20,21 etc.
GAS C5a peptidase (ScpA) is a bacterial cell surface-associated peptidase which specifically inhibits the activities of human phagocyte C5a chemotaxin,22,23 a key mediator in the process of phagocyte recruitment to the infection site.24 ScpA cleaves the peptide bond between residues His67 and Lys68 at the leukocyte-binding site of C5a to result in delaying phagocyte permeation and impeding bacterial clearance from the mucosal surface.25 ScpA is thereby recognized as an important virulence factor of streptococci that mediates their colonization in the host. Moreover, ScpA is antigenically conserved among most human GAS isolates and shares a high homology (>95%) with other C5a peptidases from group B, C, and G streptococci.26–28 It was also found that mutagenesis of the catalytic site of ScpA could dramatically decrease its enzymatic activity.29,30 Therefore, ScpA can be an excellent target antigen for vaccine development.
Indeed, it was demonstrated that intranasal immunization of mice with a truncated ScpA49 protein in the ScpA internal deletion form could induce specific IgA and IgG antibodies and prevent streptococcal colonization.31 It was also reported that the sera of healthy human adults contained high titers of anti-ScpA antibodies that could neutralize the activity of ScpA.32 Studies in children further revealed that ScpA was highly immunogenic and the specific immune response could be stimulated irrespective of streptococcal serotypes.21
In the meantime, ScpA is also a promising target for the development of carrier proteins for glycoconjugate vaccines owing to its unique immunological properties. According to reports,33,34 ScpB, a homologous enzyme from GBS, could act as an effective carrier protein to improve the immunogenicity of GBS polysaccharide. It is well documented that most carbohydrates are poorly immunogenic and T cell-independent and cannot be directly used as vaccines.35–37 A conventional strategy used to formulate effective carbohydrate-based vaccines has been to couple carbohydrates with a carrier protein to form glycoconjugates that can have improved immunogenicity and elicit T cell-dependent carbohydrate-specific immunity.38,39 Many antibacterial conjugate vaccines have been successfully developed and commercialized, and the most frequently used carrier proteins in the licensed vaccines were derived from bacterial toxins, such as diphtheria toxoid (DT), tetanus toxoid (TT), a diphtheria toxin mutant CRM197, etc. However, it was reported that long-term use of a carrier protein could cause immunological issues, such as carrier primed enhancement of T lymphocytes, carrier induced carbohydrate epitope suppression and bystander interference.40–43 Consequently, developing new carrier proteins to overcome these problems is currently another important topic.
Inspired by the aforementioned discoveries, we became interested in developing ScpA-based GAS vaccines and using ScpA as a carrier protein of glycoconjugate vaccines. In this regard, it is necessary to find a proper mutant of ScpA that does not have the original enzymatic activity but possesses robust immunostimulant function. Accordingly, a truncated form of ScpA and a series of its mutants were expressed and studied in this research. A mutant protein that was proved to be non-active to the human C5a peptide substrate was identified and evaluated in vivo as a potential GAS vaccine candidate and as a glycoconjugate vaccine carrier.
2. Results and discussion
It has been disclosed that ScpA is a protein composed of 1167 amino acids having a catalytic domain at the N-terminus (residues 97-583) and three tandemly arranged fibronectin domains (Fn1, Fn2 and Fn3) at the C-terminus (residues 584-1032).44 Its amino acid residues Asp130, His193, and Ser512 in the N-terminal sequence forms a catalytic triad at the active site. Furthermore, the Asn295 residue is involved in the formation of an oxyanion-hole and thus is also considered being critical for its catalytic activity.45 Based on these findings, we planned a systematic study on the mutagenesis of ScpA by replacing Asp130, His193, Asn295, and Ser512 at the active site with alanine residues in the truncated form of ScpA containing amino acid residues 32-1032. Therefore, four single-site (Asp130Ala, His193Ala, Asn295Ala, Ser512Ala) and one two-site (Asp130Ala and Ser512Ala) mutants of this protein were prepared and investigated.2.1 Expression and purification of recombinant proteins
The truncated gene of wild-type ScpA encoding amino acid residues 32-1032 and the genes of its mutants with Asp130Ala, His193Ala, Asn295Ala, Ser512Ala, and Asp130Ala/Ser512Ala mutations were synthesized and then inserted into expression vector pGEX-6p-3 between the restriction enzyme cutting sites of BamH I and Xho I by standard protocols. The recombinant plasmids were transferred into Escherichia coli BL21(DE3), and the transformants were cultured in the Luria–Bertani (LB) medium containing ampicillin at 37 °C overnight. The target proteins should contain the GST-tag at their N-termini. Protein expression was induced by the addition of IPTG (0.3 mM). The cells were continuously cultivated at 16 °C for 16 h, harvested when the cell culture reached an optical density (OD) value of ca. 1.0 at 600 nm, and finally disrupted by ultrasonication. The crude GST-tagged protein was isolated in the supernatant after centrifugation of the cell lysate at 12000 rpm and 4 °C for 30 min and was purified to homogeneity by GSTrap affinity chromatography.
The SDS-PAGE results of ΔScpAH193A depicted in Fig. 1 showcased the purification process of recombinant proteins. The band of the target protein in accordance with the theoretical mass prediction (140 kDa) of GST-tagged ΔScpAH193A was enriched after the initial purification on a GSTrap column (lane 4 and 6, Fig. 1). The GST tag of the purified fusion protein was then cleaved by PreScission protease, a highly restricted enzyme that hydrolyzes amino acid sequence LEVLFQGP between Q and G, at 4 °C overnight, affording the recombinant ΔScpAH193A protein after GSTrap column separation to remove GST tag (lane 2, Fig. 1). The protein was finally purified with a Q Sepharose anion exchanger column to give a pure protein (∼110 kDa) as shown by SDS-PAGE (lane 1, Fig. 1). All other recombinant proteins were purified to homogeneity by the same protocols and procedure.
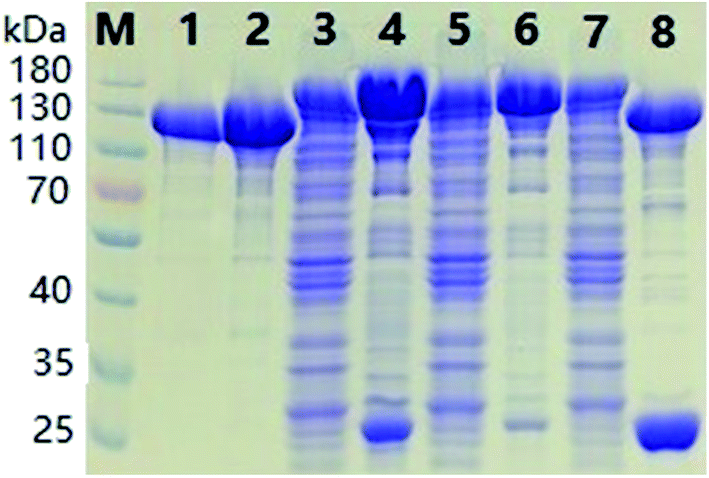 | ||
| Fig. 1 SDS-PAGE results for ΔScpAH193A protein purification. Lane 1: purified protein after the Q Sepharose column; lane 2: purified protein after the second GSTrap column; lanes 3, 5 and 7: flow through of the first GSTrap column; lanes 4 and 6: GST-tagged ΔScpAH193A after the first GSTrap column; lane 8: mixture of the GST tag and ΔScpAH193A after treatment with PreScission protease; lane M: protein standards with the molecular weights in kDa labeled on the left side of the figure. | ||
2.2 Enzymatic activities of ΔScpA proteins
Literature reported methods for assaying the enzymatic activities of ScpA were based upon SDS-PAGE or reverse-phase HPLC analysis.29,30 These methods suffer from drawbacks such as relatively low sensitivity and time-consuming operations. Furthermore, the former method also requires a C5a-green fluorescent fusion protein as the substrate. To facilitate the enzymological study of the recombinant ΔScpA proteins, we developed herein a simple and very sensitive mass spectrometry-based method to monitor and analyze the enzymatic activity.The human C5a peptide is a 74-amino acid fragment of a complement component, which can be hydrolyzed by the highly restricted ScpA between His67 and Ly68 at the leukocyte-binding site, resulting in the cleavage of seven amino acid residues at the C-terminus of C5a peptide and decrease of its molecular weight by about 830 Da.44 Commercially available recombinant human C5a peptide was employed in this study to assess the enzymatic activities of ΔScpA proteins by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS).
Assays of the enzymatic activities of ΔScpA proteins were performed in Tris–HCl buffer (50 mM, 20 μL, pH 7.5) containing 100 mM of NaCl, 5 mM of CaCl2 and 30 μg mL−1 of C5a peptide, using different concentrations of each ΔScpA protein (1, 3, 30, and 300 μg mL−1). The reaction mixture was kept at 20 °C for 30 min before it was subjected to MALDI-TOF MS analysis. The MS data were collected in a linear-positive mode using sinapinic acid as the matrix.
As shown in Fig. 2 (trace A and ESI Fig. S1†), under the above mentioned condition, the wild-type ΔScpA could completely transform the substrate C5a peptide (m/z ∼ 8600 Da) into the product (m/z ∼ 7760 Da) at the lowest tested concentration (1 μg mL−1), verifying its activity. The ΔScpAD130A and ΔScpAN295A mutants (traces B and C, Fig. 2, ESI S2 and S3†) could also hydrolyze C5a peptide completely at low concentrations, thus exhibiting similar activities as the wild-type ΔScpA. The results suggested that the Asp130 and Asn295 residues might not play a key role in the enzymatic activity of ScpA. No obvious reaction was found with the ΔScpAS512A mutant at low concentrations (1 and 3 μg mL−1), but its enzymatic activity was evident at high concentrations (30 and 300 μg mL−1) (trace D, Fig. 2 and ESI S4†). Similarly, the two-site mutant ΔScpAD130A,S512A also showed some enzymatic activity only at high concentrations (trace E, Fig. 2 and ESI S5†). These results demonstrated that the Ser512 residue was important but not absolutely required for the catalytic activity of ScpA. In contrast, the ΔScpAH193A mutant exhibited no enzymatic activity at all even at 300 μg mL−1 concentration (trace F, Fig. 2 and ESI S6†), indicating that the His193 residue was crucial for the catalytic activity of ScpA.29
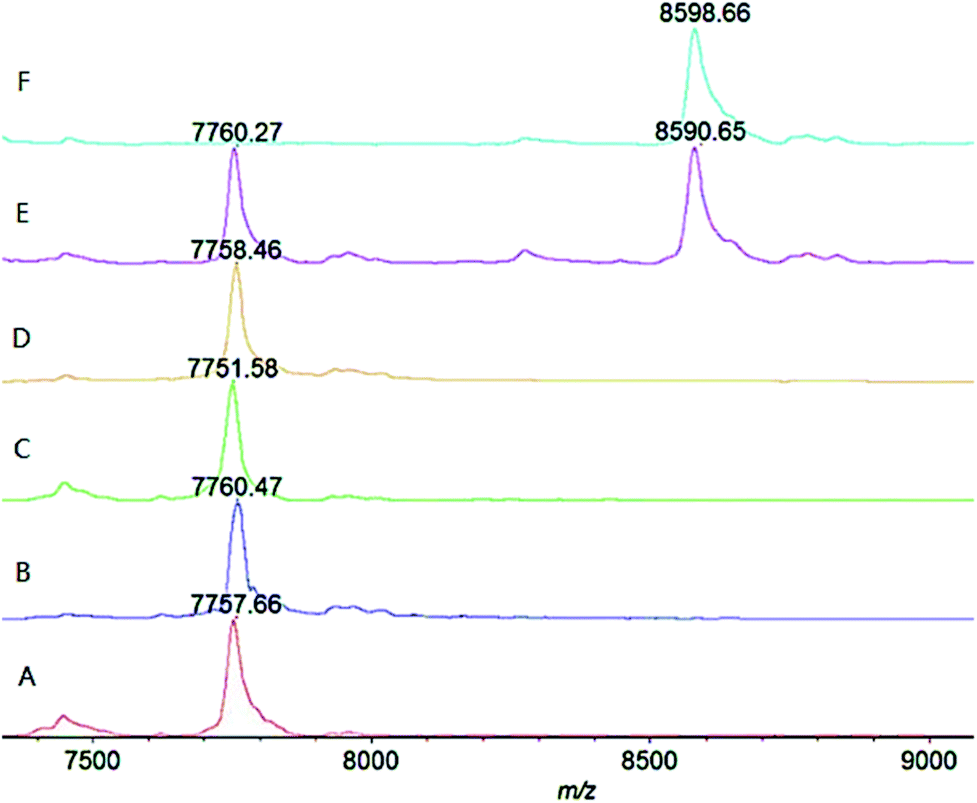 | ||
| Fig. 2 MALDI-TOF MS results for the hydrolysis of human C5a peptide (30 μg mL−1) catalyzed by (A) truncated wild-type ΔScpA (1 μg mL−1) and its various mutants, including (B) ΔScpAD130A (1 μg mL−1), (C) ΔScpAN295A (1 μg mL−1), (D) ΔScpAS512A (30 μg mL−1), (E) ΔScpAD130A,S512A (30 μg mL−1) and (F) ΔScpAH193A (30 μg mL−1), after reaction at 20 °C for 30 min. | ||
2.3 Immunization of mice with the ΔScpAH193A mutant
The above results showed that His193Ala mutation could completely deactivate ΔScpA and suppress its toxicity. Thus, the ΔScpAH193A mutant was selected for immunological evaluations as a potential vaccine candidate using female C57BL/6 mouse. An emulsion of ΔScpAH193A with the complete Freund adjuvant (CFA) for the initial immunization or with the incomplete Freund adjuvant (IFA) for subsequent immunizations was subcutaneously (s.c.) injected to a group of six mice on days 1, 14, 21, 28, and 42, respectively. Blood samples were collected from these mice prior to and after the immunizations on days 0 and 56 to prepare antisera, which were analyzed by enzyme-linked immunosorbent assays (ELISA) to determine ScpA-specific total (anti-kappa), IgM, IgG1, IgG2b, IgG2c, and IgG3 antibodies titers using ΔScpAH193A as the capture antigen. Antibody titers were calculated from a linear regression analysis of the curves of the observed optical density (OD) values against serum dilution numbers, and were defined as the dilution number yielding an OD value of 0.2.As shown in Fig. 3, the ΔScpAH193A protein elicited high titers of mainly antigen-specific IgG, including IgG1, IgG2b and IgG2c, antibodies in all of the six tested mice, suggesting that ΔScpAH193A could provoke a robust immune response. The production of higher titers of IgG1, IgG2b and IgG2c antibodies, especially the IgG1 antibody, suggested a T cell-mediated immune response.46,47 These results suggested that ΔScpAH193A can be a promising vaccine candidate and a promising carrier protein for glycoconjugate vaccines.
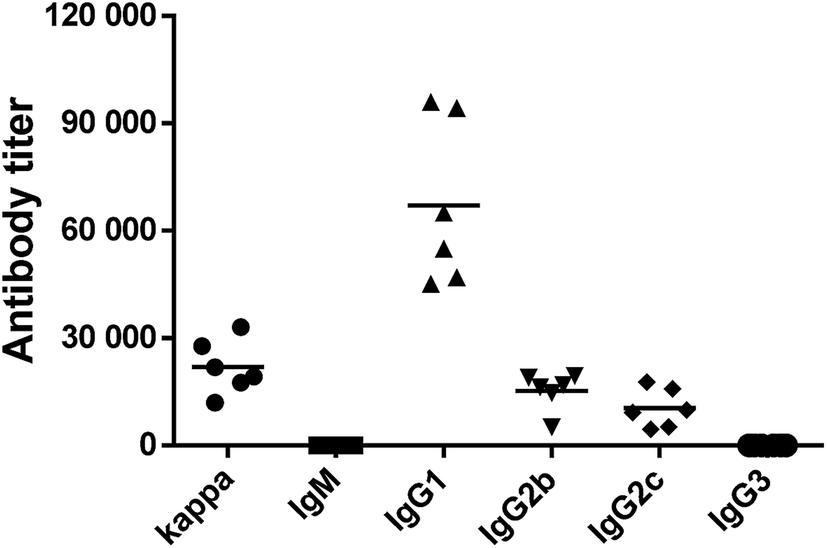 | ||
| Fig. 3 ELISA results showing the total (anti-kappa) and IgM, IgG1, IgG2b, IgG2c, and IgG3 antibody titers in the day 56 antisera of mice immunized with ΔScpAH193A. Each dot represents the result of an individual mouse, and the horizontal bar represents the average of antibody titer for the group of six mice. | ||
2.4 Immunization of mice with GAS trisaccharide–ΔScpAH193A conjugate
For the preparation of neoglycoprotein conjugate vaccines, carbohydrate antigens are usually covalently linked to the ε-amino group of lysine residues in the carrier protein through reductive amination, acylation, and various other reactions. Therefore, the number of Lys residues on the molecule surface is an important factor for the carrier protein as this can significantly affect the carbohydrate antigen loading efficiency. To preliminarily evaluate the feasibility of ΔScpAH193A as a conjugate vaccine carrier, we calculated and predicted the number of lysine residues present on its surface using the PyMOL software. It was shown that among a total of 87 Lys residues in the structure of ΔScpAH193A, approximately 30 are on the protein surface (Fig. 4), which can provide a sufficient number of conjugating sites for carbohydrate antigens.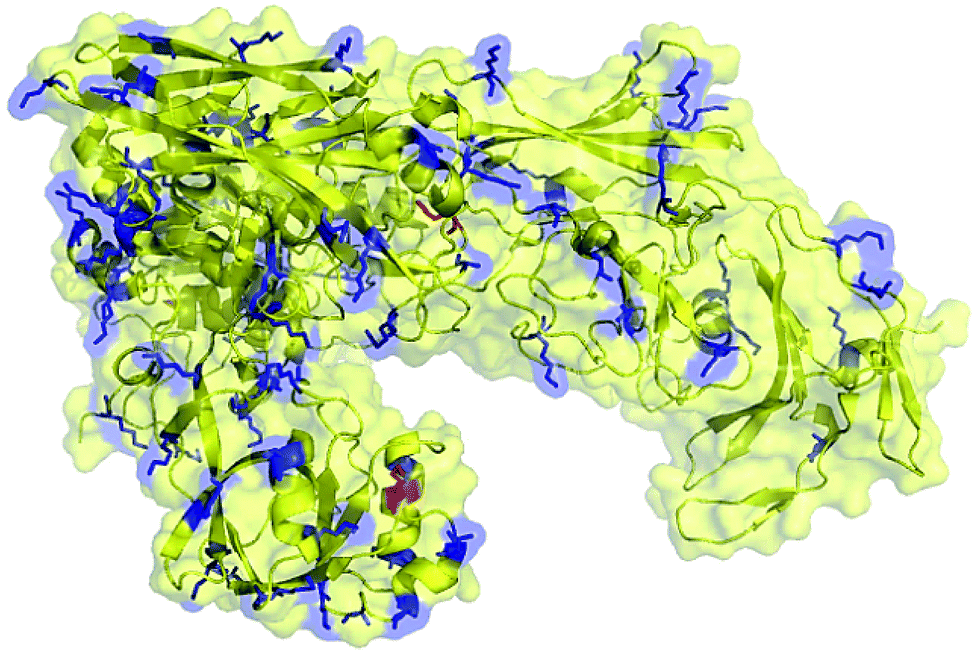 | ||
| Fig. 4 Predicted Lys residues present on the surface (marked in blue color) of the ΔScpAH193A protein. | ||
The strong immunological activity of ΔScpAH193A to elicit a robust T cell-mediated immune response and the large number of Lys residues present on its surface to facilitate its conjugation with carbohydrate antigens support the potential of ΔScpAH193A as a conjugate vaccine carrier. To investigate and verify this possibility, a nonimmunogenic trisaccharide derivative of the GAS polysaccharide,48,49 α-L-Rha-(1→2)-[2-deoxy-2-acetamido-D-Glc-(1→3)]-α-L-Rha-OCH2CH2NH2 (1), which was fully characterized by the NMR and MS data (ESI Fig. S7†), was selected as a carbohydrate hapten. It was conjugated with ΔScpAH193A via a di(N-succinimidyl)glutarate (DSG) linker (ESI Fig. S8†), and the immunological properties of resulting trisaccharide–ΔScpAH193A conjugate 2 (Fig. 5A) were evaluated in mice. Therefore, each group of six female C57BL/6 mice were respectively immunized through s.c. injection of a CFA/IFA emulsion of conjugate 2 or free GAS trisaccharide 1 on days 1, 14, 21, and 28, as described above. Blood samples were collected from the mice on day 33 and utilized to prepare antisera by the standard protocol. The antisera were then analyzed by ELISA to determine GAS trisaccharide-specific antibodies using the bovine serum albumin (BSA) conjugate 3 of the same trisaccharide (ESI Fig. S8†) as capture antigen and the linker as a negative control to eliminate its potential influence.
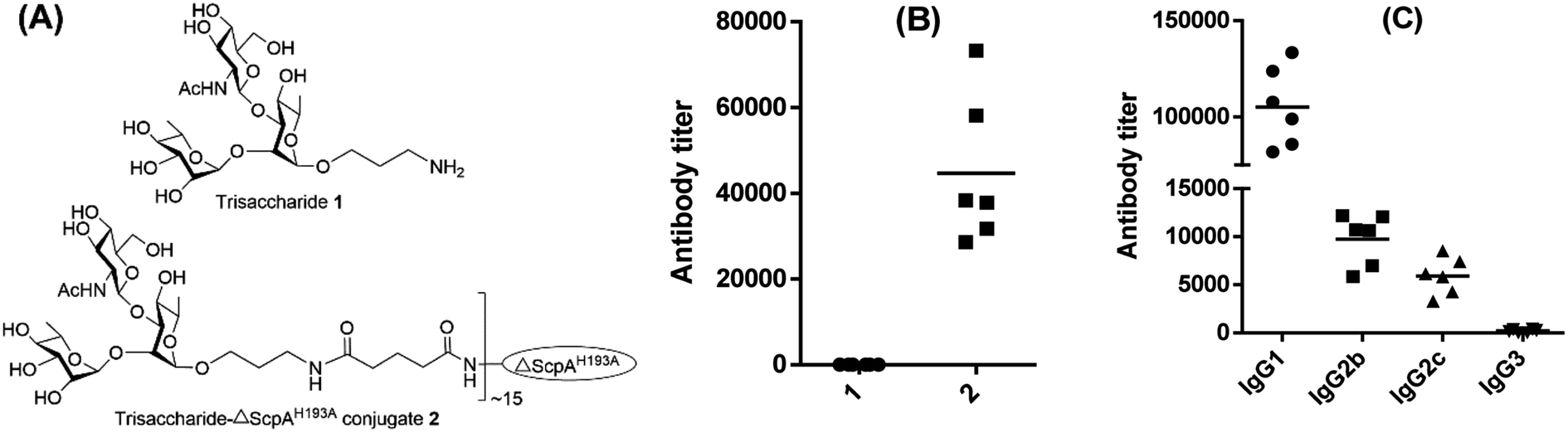 | ||
| Fig. 5 The chemical structures of (A) the GAS trisaccharide 1 and trisaccharide-ΔScpAH193A conjugate 2, and ELISA results of mouse antisera showing (B) the total (anti-kappa) antibodies elicited by GAS trisaccharide 1 and trisaccharide–ΔScpAH193A conjugate 2, respectively, and (C) IgG1, IgG2b, IgG2c and IgG3 antibodies elicited by trisaccharide–ΔScpAH193A conjugate 2. Each dot represents the result of an individual mouse, and the horizontal bar represents the average antibody titer for each group of six mice. | ||
The ELISA results depicted in Fig. 5B showed clearly that while free GAS trisaccharide 1 did not have any immunological activity, its ΔScpAH193A conjugate 2 elicited very high titers of carbohydrate antigen-specific total antibodies (Fig. 5B). Our detailed assessment of the antibody isotypes further revealed that conjugate 2 elicited predominantly IgG antibodies, including IgG1, IgG2b, IgG2c and IgG3, especially IgG1 (Fig. 5C). These results indicated that an immunologically inactive oligosaccharide could be converted into a highly immunogenic antigen after conjugating with ΔScpAH193A and the resulting conjugate elicited extremely robust T cell-dependent immune responses. Consequently, ΔScpAH193A was verified in vivo to be a promising carrier protein for the formulation of glycoconjugate vaccines.
3. Conclusion
ScpA is one of the promising protein antigens in the development of anti-GAS vaccines and has attracted significant attention in recent years. It was reported that in general the amino acids Asp130, His193, Ser512 and Asn295 of ScpA were critical for its catalytic activities44,45 and that its mutants had significantly decreased activity as compared to the wild-type enzyme.29,30 It was further discovered that some mutants of truncated ScpA, such as ΔScpAD130A,S512A, could induce robust immunities to provide protection against GAS infections.20 However, currently systematic studies on the mutagenesis and immunology of this enzyme and its mutants are missing.In this work, several ΔScpA mutants were designed, expressed, and investigated to probe the structure–enzymatic activity relationship of ScpA and its application potential in the development of GAS vaccines and as a carrier protein of glycoconjugate vaccines. To facilitate the evaluation of the enzymatic activities of these recombinant proteins, a MALDI-TOF MS-based simple and sensitive detection method was developed for analyzing the reaction. This assisted to reveal that for the enzymatic activity of ScpA, amino residues D130 and N295 may not be critical and S512, although important, is not absolutely required either because the ΔScpAS512A mutant still retained some enzymatic activity. These results were different from the literature predictions made on the basis of the ScpA structure as mentioned above. Accordingly, the ΔScpAD130A and ΔScpAS512A mutants may not be the ideal candidates for vaccine development or other applications. More importantly, we discovered that a single mutation of the amino acid residue H193 to Ala could completely abolish the enzymatic activity of ΔScpA, making its applications safe. Consequently, the ΔScpAH193A mutant was selected to conduct further investigations.
The preliminary results of our immunological studies revealed that both ΔScpAH193A and its conjugate 2 with the trisaccharide repeating unit of a GAS polysaccharide elicited very high titers of antigen-specific IgG1 antibodies in mice, suggesting the induction of robust T cell-mediated immune response. Although the strategy to improve the immunogenicity of carbohydrate antigens by forming covalently linked conjugates with various carrier proteins has been widely used in the development of vaccines, the exact mechanism of action is largely unclear. It is believed that the carrier protein may help the processing of carbohydrates by antigen presenting cells, but there is limited data available to verify the hypothesis.50,51 Nevertheless, the results of our immunological studies on ΔScpAH193A and its carbohydrate conjugate 2 have demonstrated its great promise in the design and development of novel anti-GAS vaccines and as a carrier protein to help convert immunologically inactive carbohydrates into immunogenic and T cell-dependent antigens for the formulation of functional conjugate vaccine. To fully exploit this opportunity, more in-depth and extensive immunological, antibody-antigen/pathogen binding, in vitro and in vivo antibacterial studies are necessary, which are currently pursued in our laboratory.
4. Materials and methods
4.1 Bacterial strains
The E. coli strains DH5α and BL21(DE3) were used for the plasmid maintenance and protein expression, respectively. Bacteria were cultured in the LB medium in the presence of antibiotic ampicillin (finial concentration of 100 μg mL−1) under conditions described below in details.4.2 Ethical statement
Female C57BL/6 mice (6–8 week old) were purchased from Shandong University Laboratory Animal Center. All care and handling of animals in this study were performed in strict accordance with the National Institute for Health Guide for the Care and Use of Laboratory Animals (National Research Council, 8th Ed. National Academies Press (US); Washington DC: 2011), and approved by the Institutional Animal Care and Use Committee (IACUC) of Shandong University.4.3 Mutagenesis of ΔScpA
Expression vector pGEX-6p-3 carrying the ΔScpA gene (bases 96-3096) of S. pyogenes B220 encoding amino acid residues 32-1032 was provided as a gift by Professor Jakki C. Cooney's lab in University of Limerick, Ireland. Site-specific mutagenesis of this truncated ΔScpA with amino acid residues D130, H193, N295, and S512 substituted with an alanine residue, respectively, was performed with a Fast Mutagenesis System (TransGen Biotech) according to standard protocols. Briefly, using the plasmid pGEX-6p-3 carrying the ΔScpA gene as a temple, forward and reverse primers (ESI Table S1†) were designed, synthesized, and utilized to replace the designated residue. PCR reactions were performed to obtain the amplified fragments, which were then treated with the Dpn I enzyme to remove the template and transformed into E. coli DH5α. The mutant plasmids were extracted from E. coli, and the mutations were confirmed by gene sequencing. The mutant proteins were expressed in E. coli BL21(DE3) host cells.4.4 Expression and purification of recombinant proteins
After recombinant pGEX-6p-3-ΔScpA and mutant gene plasmids were transformed into the E. coli BL21(DE3) host, the recombinant strains were cultured in the LB medium at 37 °C overnight and were allowed to grow to reach an OD600 value of ∼1.0. The cultures were cooled to 16 °C, and then induced with 0.3 mM of isopropyl 1-thio-β-D-galactoside (IPTG) for another 16 h. The BL21 cells were harvested through centrifugation, and then re-suspended in the binding buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl), which was followed by disruption with sonication and centrifugation at 12000 rpm and 4 °C for 30 min to remove cell debris.
Initial protein purification was achieved via affinity chromatography using a GSTrap column (GE Healthcare Life Sciences Co.). Accordingly, after the cell supernatants obtained above were loaded onto GST trap columns, the columns were washed first with Tris–HCl binding buffer and then with 10 mM glutathione elution buffer (50 mM Tris–HCl containing 150 mM NaCl) to elute GST-tagged target proteins. The protein solution was passed through a Sephadex G-25 column to desalt and remove glutathione with water as the eluent and then treated with PreScission Protease at 4 °C overnight. The resulting protein solution was loaded again onto a GST trap column, which were washed with Tris–HCl binding buffer to give the GST tag-deleted target protein. The proteins were eventually purified with a strong anion exchanger Q Sepharose column using gradient NaCl solution (0–1.0 M) in 50 mM Tris–HCl buffer as an eluent. Protein samples were analyzed with 10% SDS-PAGE and visualized by Coomassie Brilliant Blue staining. Protein concentrations were determined with a Nanodrop apparatus (Thermo Fisher Inc.) at 280 nm and calculated according to the Beer–Lambert equation. The proteins were stored in 50 mM Tris–HCl buffer at −80 °C.
4.5 Evaluation of the enzymatic activities of ΔScpA and its mutants
To a solution of the human C5a peptide (0.6 μg) dissolved in 50 mM Tris–HCl buffer (pH 7.5, 20 μL) containing 100 mM of NaCl and 5 mM of CaCl2 were added varying amounts (0.02, 0.06, 0.6, 6 μg) of ΔScpA and its mutants. After the solution was incubated at 20 °C for 30 min, 1 μL of the reaction mixture was spotted onto a stainless-steel MALDI-TOF MS plate and then covered with sinapinic acid. After solvent was evaporated, the sample plate was subjected to MS analysis with a MALDI-TOF mass spectrometer (AXIMA Confidence, Shimadzu Co.) using the linear-positive mode.4.6 Preparation of the GAS trisaccharide–protein conjugates
A mixture of trisaccahride 1 (2 mg) and di(N-succinimidyl)glutarate (DSG, 15 equiv.) in a mixture of DMF and PBS buffer (0.1 M, pH 8.0) (v/v 4:1, 0.5 mL) was gently stirred at rt for 4 h. Thereafter, the solvents were removed under reduced pressure, and ethyl acetate (4.5 mL) was added to wash the activated ester 10 times and remove excessive DSG. The residue was dried under high vacuum. The activated trisaccharide and ΔScpAH193A or BSA (oligosaccharide/protein mass ratio of 2:3) were dissolved in PBS buffer (0.1 M, 0.5 mL, pH 8.0), and the solution was gently stirred at rt for 4 days. When analysis by MALDI-TOF mass spectrometry showed no further increase in molecular mass of the conjugate, the reaction mixture was dialyzed against distilled water (3 × 8 mL) using an Amicon ulfrafiltration cell equipped with a Diaflo membrane. The residues were subjected to Biogel A0.5 column chromatography and then dialysis against distilled water. The dialysates were lyophilized to give the glycoconjugates 2 (2.8 mg) and 3 (2.5 mg), respectively, as white fluffy powders. MALDI-TOF MS data (sinapinic acid matrix, 0.1% TFA in 1:1 CH3CN/H2O): ΔScpAH193A 110376 Da (ESI Fig. S9A†); trisaccharide–ΔScpAH193A conjugate 2120868 Da (ESI Fig. S9B†); BSA 66924 Da (ESI Fig. S10A†); trisaccharide–BSA conjugate 3 71856 Da (ESI Fig. S10B†).
The carbohydrate loading of each glycoconjugate was determined by means of MALDI-TOF MS and calculated according to the equation below:
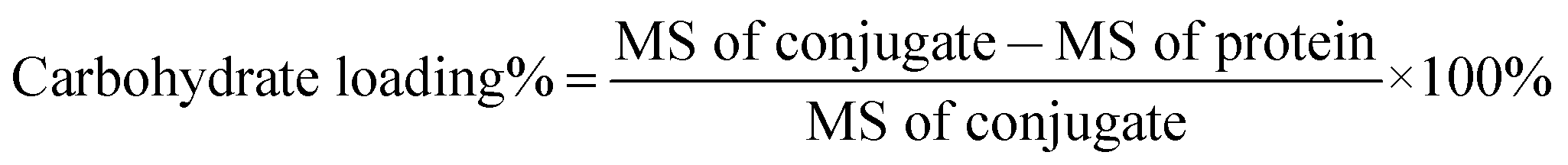
The results are listed in Table S2.† The carbohydrate loading levels for trisaccharide–ΔScpAH193A conjugate 2 and trisaccharide–BSA conjugate 3 were 8.7% and 6.8% (w/w), respectively.
4.7 Protocols for immunological studies
:300 to 1:4687500 in PBS (100 μL per well) was added to the coated plates, and the plates were incubated at 37 °C for 2 h. After being washed with PBST, the plates were incubated with a 1:1000 diluted solution of an alkaline phosphatase-linked goat anti-mouse kappa, IgM, IgG1, IgG2b, IgG2c or IgG3 antibody (Abcam), respectively, at rt for 1 h. The plates were again washed with PBST three times and developed with a p-nitrophenylphosphate (PNPP) solution (1.67 mg mL−1 in buffer, 100 μL) at rt for 30 min. The reaction was quenched by adding 25 μL of the quenching solution (3 M NaOH) to each well. Finally, the plates were examined with a microplate reader at 405 nm wavelength. The OD values after deducting the background OD values obtained with the day 0 sera were plotted against dilution numbers, and the best-fit equation was obtained for each set of data and used to calculate the antibody titer, defined as the dilution number giving an adjusted OD value of 0.20.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by grants from the Shandong Provincial Natural Science Foundation (No. ZR2015BM020) and Science and Technology Development Projects of Shandong Province (No. 2015GSF118019 and 2016GGH4502). We thank Professor Jakki C. Cooney at University of Limerick for kindly supplying expression vector pGEX-6p-3 carrying the ΔScpA gene (bases 96-3096) of S. pyogenes B220.References
- M. W. Cunningham, Clin. Microbiol. Rev., 2000, 13, 470–511 CrossRef CAS PubMed.
- N. N. Lynskey, R. A. Lawrenson and S. Sriskandan, Curr. Opin. Infect. Dis., 2011, 24, 196–202 CrossRef PubMed.
- J. R. Carapetis, A. C. Steer, E. K. Mulholland and M. Weber, Lancet Infect. Dis., 2005, 5, 685–694 CrossRef PubMed.
- A. P. Ralph and J. R. Carapetis, Curr. Top. Microbiol. Immunol., 2013, 368, 1–27 CAS.
- E. Pfoh, M. R. Wessels, D. Goldmann and G. M. Lee, Pediatrics, 2008, 121, 229–234 CrossRef PubMed.
- K. Wright, Science, 1990, 249, 23–24 Search PubMed.
- WHO, 2005, WHO/FCH/CAH/05.09.
- A. C. Steer, M. R. Batzloff, K. Mulholland and J. R. Carapetis, Curr. Opin. Infect. Dis., 2009, 22, 544–552 CrossRef CAS PubMed.
- J. B. Dale, V. A. Fischetti, J. R. Carapetis, A. C. Steer, S. Sow, R. Kumar, B. M. Mayosi, F. A. Rubin, K. Mulholland, J. M. Hombach, F. Schodel and A. M. Henao-Restrepo, Vaccine, 2013, S31, B216–B222 CrossRef PubMed.
- M. M. Georgousakis, D. J. McMillan, M. R. Batzloff and K. S. Sriprakash, Expert Rev. Vaccines, 2009, 8, 747–760 CrossRef CAS PubMed.
- A. C. Steer, J. B. Dale and J. R. Carapetis, Pediatr. Infect. Dis. J., 2013, 32, 180–182 CrossRef PubMed.
- S. A. McNeil, S. A. Halperin, J. M. Langley, B. Smith, A. Warren, G. P. Sharratt, D. M. Baxendale, M. A. Reddish, M. C. Hu, S. D. Stroop, J. Linden, L. F. Fries, P. E. Vink and J. B. Dale, Clin. Infect. Dis., 2005, 41, 1114–1122 CrossRef CAS PubMed.
- J. B. Dale, T. A. Penfound, E. Y. Chiang and W. J. Walton, Vaccine, 2011, 29, 8175–8178 CrossRef CAS PubMed.
- M. R. Batzloff, W. A. Hayman, M. R. Davies, M. Zeng, S. Pruksakorn, E. R. Brandt and M. F. Good, J. Infect. Dis., 2003, 187, 1598–1608 CrossRef CAS PubMed.
- L. Guilherme, E. Postol, S. Freschi de Barros, F. Higa, R. Alencar, M. Lastre, C. Zayas, C. R. Puschel, W. R. Silva, L. C. Sa-Rocha, V. M. Sa-Rocha, O. Perez and J. Kalil, Methods, 2009, 49, 316–321 CrossRef CAS PubMed.
- M. W. Cunningham, N. K. Hall, K. K. Krisher and A. M. Spanier, J. Immunol., 1986, 136, 293–298 CAS.
- R. G. Ulrich, J. Immune Based Ther. Vaccines, 2008, 6, 8 CrossRef PubMed.
- H. Sabharwal, F. Michon, D. Nelson, W. L. Dong, K. Fuchs, R. C. Manjarrez, A. Sarkar, C. Uitz, A. Viteri-Jackson, R. S. R. Suarez, M. Blake and J. B. Zabriskie, J. Infect. Dis., 2006, 193, 129–135 CrossRef CAS PubMed.
- A. Kabanova, I. Margarit, F. Berti, M. R. Romano, G. Grandi, G. Bensi, E. Chiarot, D. Proietti, E. Swennen, E. Cappelletti, P. Fontani, D. Casini, R. Adamo, V. Pinto, D. Skibinski, S. Capo, G. Buffi, M. Gallotta, W. J. Christ, A. S. Campbell, J. Pena, P. H. Seeberger, R. Rappuoli and P. Costantino, Vaccine, 2010, 29, 104–114 CrossRef CAS PubMed.
- P. P. Cleary, Y. V. Matsuka, T. Huynh, H. Lam and S. B. Olmsted, Vaccine, 2004, 22, 4332–4341 CrossRef CAS PubMed.
- A. Shet, E. L. Kaplan, D. R. Johnson and P. P. Cleary, J. Infect. Dis., 2003, 188, 809–817 CrossRef CAS PubMed.
- P. P. Cleary, U. Prahbu, J. B. Dale, D. E. Wexler and J. Handley, Infect. Immun., 1992, 60, 5219–5223 CAS.
- H. D. Manthey, T. M. Woodruff, S. M. Taylor and P. N. Monk, Int. J. Biochem. Cell Biol., 2009, 41, 2114–2117 CrossRef CAS PubMed.
- D. E. Wexler, D. E. Chenoweth and P. P. Cleary, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 8144–8148 CrossRef CAS.
- Y. Ji, L. McLandsborough, A. Kondagunta and P. P. Cleary, Infect. Immun., 1996, 64, 503–510 CAS.
- P. P. Cleary, J. Handley, A. N. Suvorov, A. Podbielski and P. Ferrieri, Infect. Immun., 1992, 60, 4239–4244 CAS.
- P. P. Cleary, J. Peterson, C. Chen and C. Nelson, Infect. Immun., 1991, 59, 2305–2310 CAS.
- I. Chmouryguina, A. Suvorov, P. Ferrieri and P. P. Cleary, Infect. Immun., 1996, 64, 2387–2390 CAS.
- D. K. Stafslien and P. P. Cleary, J. Bacteriol., 2000, 182, 3254–3258 CrossRef CAS PubMed.
- E. T. Anderson, M. G. Wetherell, L. A. Winter, S. B. Olmsted, P. P. Cleary and Y. V. Matsuka, Eur. J. Biochem., 2002, 269, 4839–4851 CrossRef CAS PubMed.
- Y. D. Ji, B. Carlson, A. Kondagunta and P. P. Cleary, Infect. Immun., 1997, 65, 2080–2087 CAS.
- S. P. Oconnor, D. Darip, K. Fraley, C. M. Nelson, E. L. Kaplan and P. P. Cleary, J. Infect. Dis., 1991, 163, 109–116 CrossRef CAS.
- Q. Cheng, B. Carlson, S. Pillai, R. Eby, L. Edwards, S. B. Olmsted and P. Cleary, Infect. Immun., 2001, 69, 2302–2308 CrossRef CAS PubMed.
- Q. Cheng, S. Debol, H. Lam, R. Eby, L. Edwards, Y. Matsuka, S. B. Olmsted and P. P. Cleary, Infect. Immun., 2002, 70, 6409–6415 CrossRef CAS PubMed.
- J. J. Mond, A. Lees and C. M. Snapper, Annu. Rev. Immunol., 1995, 13, 655–692 CrossRef CAS PubMed.
- H. J. Jennings and R. K. Sood, Synthetic glycoconjugates as human vaccines, in Neoglycoconjugates: Preparation and Applications, ed. Y. C. Lee and R. T. Lee, Academic Press, San Diego, 1994, pp. 325–371 Search PubMed.
- Z. Guo and G.-J. Boons, Carbohydrate-Based Vaccines and Immunotherapies, John Wiley & Sons, Inc., Hoboken, 2009 Search PubMed.
- C. Anish, B. Schumann, C. L. Pereira and P. H. Seeberger, Chem. Biol., 2014, 21, 38–50 CrossRef CAS PubMed.
- R. Rappuoli and E. De Gregorio, Nat. Med., 2011, 17, 1551–1552 CrossRef CAS PubMed.
- K. Pobre, M. Tashani, I. Ridda, H. Rashid, M. Wong and R. Booy, Vaccine, 2014, 32, 1423–1430 CrossRef CAS PubMed.
- R. Dagan, J. Poolman and C. A. Siegrist, Vaccine, 2010, 28, 5513–5523 CrossRef CAS PubMed.
- S. Pecetta, P. Lo Surdo, M. Toritini, D. Proietti, C. Zambonelli, M. J. Bottomley, M. Biagini, F. Berti, P. Costantino, M. R. Romano and S. Grp, Vaccine, 2015, 33, 314–320 CrossRef CAS PubMed.
- M. Tontini, F. Berti, M. R. Romano, D. Proietti, C. Zambonelli, M. J. Bottomley, E. De Gregorio, G. Del Giudice, R. Rappuoli, P. Costantino, G. Brogioni, C. Balocchi, M. Biancucci, E. Malito and S. Grp, Vaccine, 2013, 31, 4827–4833 CrossRef CAS PubMed.
- T. F. Kagawa, M. R. O'Connell, P. Mouat, M. Paoli, P. W. O'Toole and J. C. Cooney, J. Mol. Biol., 2009, 386, 754–772 CrossRef CAS PubMed.
- R. J. Siezen, W. M. de Vos, J. A. Leunissen and B. W. Dijkstra, Protein Eng., 1991, 4, 719–737 CrossRef CAS PubMed.
- T. Mazumdar, K. Anam and N. Ali, Vaccine, 2004, 22, 1162–1171 CrossRef CAS PubMed.
- T. Mygind, B. Vandahl, A. S. Pedersen, G. Christiansen, P. Hollsberg and S. Birkelund, FEMS Immunol. Med. Microbiol., 2004, 40, 129–137 CrossRef CAS PubMed.
- J. E. Coligan, T. J. Kindt and R. M. Krause, Immunochemistry, 1978, 15, 755–760 CrossRef CAS PubMed.
- D. H. Huang, N. Rama Krishna and D. G. Pritchard, Carbohydr. Res., 1986, 155, 193–199 CrossRef CAS PubMed.
- A. M. Vlad, S. Muller, M. Cudic, H. Paulsen, L. Otvos Jr, F. G. Hanisch and O. J. Finn, J. Exp. Med., 2002, 196, 1435–1446 CrossRef CAS PubMed.
- B. A. Cobb, Q. Wang, A. O. Tzianabos and D. L. Kasper, Cell, 2004, 117, 677–687 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07923k |
| This journal is © The Royal Society of Chemistry 2017 |