
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCore–shell and alloy integrating PdAu bimetallic nanoplates on reduced graphene oxide for efficient and stable hydrogen evolution catalysts†
Yi Jiang ,
Yucong Yan,
Yu Han,
Hui Zhang* and
Deren Yang
,
Yucong Yan,
Yu Han,
Hui Zhang* and
Deren Yang
State Key Laboratory of Silicon Materials, School of Materials Science & Engineering, Zhejiang University, Hangzhou, Zhejiang 310027, People's Republic of China. E-mail: msezhanghui@zju.edu.cn
First published on 7th September 2017
Abstract
PdAu bimetallic nanoplates are synthesized by titrating HAuCl4 into an aqueous solution containing in situ generated Pd square nanoplates (PdSPs) on reduced graphene oxide (rGO) in the presence of ascorbic acid (AA), serving as a reducing agent, at different injection rates. At a high injection rate (e.g., 45 mL min−1), PdAu core–shell nanoplates on rGO are generated by strong galvanic replacement between PdSPs and the Au precursor, followed by reduction of the Pd precursor. In contrast, PdAu nanoplates with a core–shell and alloy integrating structure are obtained on rGO by co-reduction of the Au and Pd precursors by AA due to the inhibition of the abovementioned galvanic replacement at a slow injection rate (e.g., 0.5 mL min−1). The PdAu nanoplates with a core–shell and alloy integrating structure on rGO exhibit a substantially enhanced catalytic activity towards the hydrogen evolution reaction (HER) relative to the PdAu core–shell nanoplates and PdSPs on rGO, and is comparable to the commercial Pt/C. Significantly, these core–shell and alloy integrating nanoplates on rGO have much superior durability over Pt/C for catalyzing the HER under acidic conditions. The remarkable enhancement in activity and durability can be attributed to the cooperative function of the PdAu alloy and electron coupling between nanoplates and graphene.
Introduction
Searching for clean and sustainable energy sources has recently been driven by the serious and various environment problems arising from the extensive use of limited fossil fuels. Amongst several attractive candidates, molecular hydrogen (H2) is considered to be the most promising solution to replace fossil fuels owing to its high-energy density, high utilization efficiency and the fact that it does not release pollutants.1,2 Nowadays, steam reforming of fossil fuels is the primary method for the large-scale production of H2. However, this process not only consumes a large amount of fossil fuel resources, but it also contributes to the serious problem of global greenhouse gas (e.g., CO2) emission.3 Therefore, clean production of H2 has received abundant research interest over the last few decades.4 In this respect, water splitting (H2O → H2 + 1/2O2) provides an efficient and clean way to meet the future energy demand because water is easily available all over the world and no greenhouse gas is emitted during the production.5 The hydrogen evolution reaction (HER) is the reductive half reaction (H+ + e− → 1/2H2) of water splitting. In order to minimize the overpotential and promote the faradaic efficiency, an electrocatalyst is required for HER.6,7 Of them, Pt-based nanocrystals have proven to be the most effective catalysts for HER owing to their low reduction overpotential and fast reaction kinetics.8–10 Unfortunately, the scarce abundance and high cost associated with Pt limit its extensive use as catalysts in HER. To overcome these limitations, a rich variety of effective and low-cost non-Pt catalysts for HER are widely studied.11–13 Specifically, Pd is considered as the most promising substitute for Pt because of its excellent potentials for improvement in catalytic performance as well as relatively ample reserves.14–16 However, palladium hydride is usually formed during the catalytic reaction owing to the appreciable solubility of hydrogen atoms in Pd lattice, leading to the severe decrease in the catalytic performance in terms of activity and stability.17 As such, it is highly desirable to suppress the formation of hydride phase by modification of surface and electronic structures of Pd catalysts with an aim to eventually improve the catalytic properties.It should be pointed out that the absorption of hydrogen on the catalyst surface is an important intermediate process of HER.18 According to the well-known volcano plot,19 the adsorption energy of hydrogen on the surface of catalysts should be well tuned to achieve the best catalytic activity. For this purpose, massive efforts have been focused on the enhancement in HER performance of Pd-based catalysts by alloying with secondary metal, designing special nanostructures, and compositing with some particular support materials in the last few years.20–24 For example, Li and co-workers demonstrated the synthesis of the core–shell structured PdCu@Pd nanocubes by selectively etching Cu on the surface of highly monodispersed PdCu nanocubes, which exhibited a fascinating HER performance close to commercial Pt/C.25 In another study, Liu and co-workers reported the synthesis of concave Pd@PdPt octahedra serving as a bifunctional catalyst for both HER and oxygen reduction reaction (ORR) with excellent catalytic activity and stability.26 It is clear that the incorporation of Au can tune the adsorption strength of hydrogen on Pd through ligand and geometry effects,27,28 making PdAu alloy nanocrystals the attractive HER catalysts.29–31 To this end, Darabdhara synthesized PdAu bimetallic nanoparticles on reduced graphene oxide (rGO), showing the enhanced catalytic properties towards HER compared to Au nanoparticles/rGO and Pd nanoparticles/rGO.32 In spite of these great progresses in PdAu-based HER catalysts, precise control over their structure and shape achieves only limited success.
Two-dimensional (2D) metal nanocrystals have received great attention in catalysis in recent years due to their unique physicochemical properties.33–37 However, the close-packed structure in three-dimension (3D) associated with noble-metals such as Pd makes it very difficult to spontaneously generate 2D nanostructures. Most recently, we have reported the synthesis of single-crystalline Pd square nanoplates (PdSPs) enclosed by {100} facets with GO containing carbonyl groups as the template.38 Interestingly, the previous report experimentally and theoretically indicated that Pd {100} facets are more active than the {111} facets towards HER since the {100} facets possess higher electron density.39 In addition, electrons resulting from rGO incline to accumulate on the surface of Pd in Pd/rGO nanocomposites due to the smaller redox potential of rGO relative to Pd (rGO: 0.38 V, PdCl42−/Pd: 0.62 V, Pd2+/Pd: 0.95 V vs. RHE), leading to an enhanced HER properties.16 As such, the nanocomposites made of rGO and PdSPs enclosed by {100} facets provide a promising platform to exploit excellent HER catalysts.
Here we report the synthesis of {100} faceted PdAu nanoplates with different structures (i.e., core–shell and integrating structure consisting of core–shell and alloy) by using in situ generated PdSP/rGO as the template. The structures of the PdAu nanoplates are easily controlled by varying the injection rate of the Au precursor. The PdAu nanoplates with a core–shell and alloy integrating structure exhibit a Pt-like catalytic activity and substantially enhanced durability towards HER relative to commercial Pt/C.
Experimental
Chemicals and materials
Graphite oxide powder was purchased from XFNANO at Nanjing. Na2PdCl4 (≥99.99%), HAuCl4 (≥99.9%), ascorbic acid (AA, 99%), poly(vinyl pyrrolidone) (PVP, MW ≈ 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), KBr (≥99%) and Nafion 117 were purchased from Sigma-Aldrich. All the chemicals and materials were used as received.
000), KBr (≥99%) and Nafion 117 were purchased from Sigma-Aldrich. All the chemicals and materials were used as received.
Synthesis of graphene oxide aqueous suspension
Graphene oxide (GO) aqueous suspension was fabricated by sonicating graphite oxide powder in de-ionized (DI) water (Millipore, 18.2 MΩ) for 2 h. After centrifugation, the supernatant solution was freezing dried and the GO powder was collected. Subsequently, the obtained GO was annealed at 60 °C for 1 h in Ar atmosphere. The annealed GO was re-dispersed in DI water for further use.Synthesis of PdAu@rGO-1
In a standard procedure, 120 mg of KBr, 12 mg of AA and 50 mg of PVP were dissolved into 8 mL of DI water containing 12.96 mg of GO in a 25 mL flask. The mixed solution was preheated at 80 °C in the air with magnetic stirring for 10 min. After that, 1.5 mL Na2PdCl4 solution (8 mg mL−1) was quickly injected into the mixed solution, followed by the injection of 1.5 mL HAuCl4 solution (2.78 mg mL−1) with a pipette after 1 min (the inject rate is ca. 45 mL min−1). The flask was tightly capped and kept at 80 °C for 3 h before cooled down in an ice water. The product was collected and washed by centrifugation with DI water and ethanol for over 10 times. The catalysts (denoted as PdAu@rGO-1) were re-dispersed in 5 mL of 10% ethanol solution and dried by freeze-drying.Synthesis of PdAu@rGO-2
The PdAu@rGO-2 nanoplates were generated by using the standard procedure, except for the injection of HAuCl4 solution at a slow rate (0.5 mL min−1) with a syringe pump.Synthesis of PdSP@rGO
The synthesis of PdSP@rGO followed up the standard procedure, except that no HAuCl4 was added into the solution.Morphological, structural, and compositional characterizations
Transmission electron microscopy (TEM) images were obtained using a Hitachi HT-7700 microscope operated at 100 kV. High-resolution transmission electron microscopy (HRTEM), high-angle annular dark-field scanning TEM (HAADF-STEM) and X-ray energy dispersive spectroscopy (EDS) mapping were performed using a FEI Tecnai G2 F20 microscope operated at 200 kV. The XRD patterns were recorded on a Bruker D8 focus diffractometer in a scan range of 10–80° at a scan rate of 1.0° min−1. The X-ray photoelectron spectroscopy (XPS) data were obtained using a scanning X-ray microprobe (Axis Supra, Kratos Inc.) with Al Kα radiation. The corresponding binding energies were calibrated with a C 1s peak of 284.6 eV.Electrochemical measurement
To prepare catalyst ink, 5 mg of catalysts were ultrasonically dispersed into 5 mL mixed solution containing 4 mL of DI water, 1 mL of isopropanol, and 25 μL of Nafion 117. For preparation of working electrodes, 10 μL of this ink was pipetted on the surface of glassy carbon (GC) electrode, and then blow-dried under atmospheric conditions. The electrochemical measurements were carried out in the three-electrode system at room temperature using a rotating disk electrode (RDE, Pine Research Instrumentation, United States) connected to an electrochemical workstation (CHI 760E). A carbon electrode and a Ag/AgCl electrode were used as the counter electrode and reference electrode, respectively. The as-received data were finally converted to reversible hydrogen electrode (RHE) as the reference. Before electrochemical measurement, the working electrode was cleaned in the Ar-saturated 0.5 M H2SO4 solution with a steady-state cyclic voltammograms (CV) scan for 100 cycles in the range of 0–1 V vs. RHE at a rate of 50 mV s−1. The electrochemical active surface area (ECSA) was determined by integrating the carbon monoxide oxidation charge via CO stripping measurements. The CO stripping measurements were conducted in CO-saturated 0.5 M H2SO4 solution at a scan rate of 50 mV s−1 in the range of 0–1.2 V vs. RHE. Linear sweep voltammetry (LSV) was conducted at a scan rate of 10 mV s−1 with a flow of Ar gas maintained during the HER measurement to eliminate dissolved oxygen. The working electrode was rotated at 1600 rpm to remove hydrogen gas bubbles formed on the catalyst surface. To investigate the cycling stability of the catalysts, 10000 cycles of CV were conducted in the range of −0.3 to 0.1 V vs. RHE at a scan rate of 100 mV s−1. The chronoamperometry (I–t) curves were measured at the potential with the current density being nearly 10 mA cm−2 for 10000 s. Electrochemical impedance spectroscopy (EIS) was performed in a frequency range of 1–105 Hz with a 5 mV amplitude at a bias potential of 0 V vs. RHE.
Results and discussion
Fig. 1 shows morphological, structural, and compositional characterizations of the PdAu@rGO-1 prepared using the standard procedure at a high injection rate of HAuCl4 solution. From TEM images in Fig. 1A and S1A,† the square nanoplates with edge length of about 100 nm are obtained on rGO. This demonstration is supported by the HAADF-STEM-EDX mapping image in Fig. 1B. Careful observation indicates a clear contrast between the interior with a cross shape and the surface of the nanoplate. EDX mapping analysis further shows that the interior is dominated by Au element and the surface is occupied by Pd element. As such, this result confirms the formation of the PdAu core–shell nanoplates on rGO. The HRTEM image (Fig. 1C) of an individual nanoplate shows well-resolved, ordered fringes in the same orientation, indicating that the nanoplate is a single crystal. The fringes with a lattice spacing of 0.196 and 0.202 nm can be indexed to {200} facets of Pd and Au with a face-centered cubic (fcc) structure, respectively. The dark part in HRTEM image is considered as the interjunction between Pd and Au. Moreover, the fcc structure of PdAu@rGO-1 is revealed by XRD pattern, as shown in Fig. 1D. The peaks at 38.25° and 44.46° are attributed to {111} and {200} facets of Au while the peaks at 40.14° and 46.69° belong to {111} and {200} facets of Pd. There is no shift of these peaks compared to the corresponding standard pattern, suggesting the formation of the separated phase of PdAu with a core–shell structure.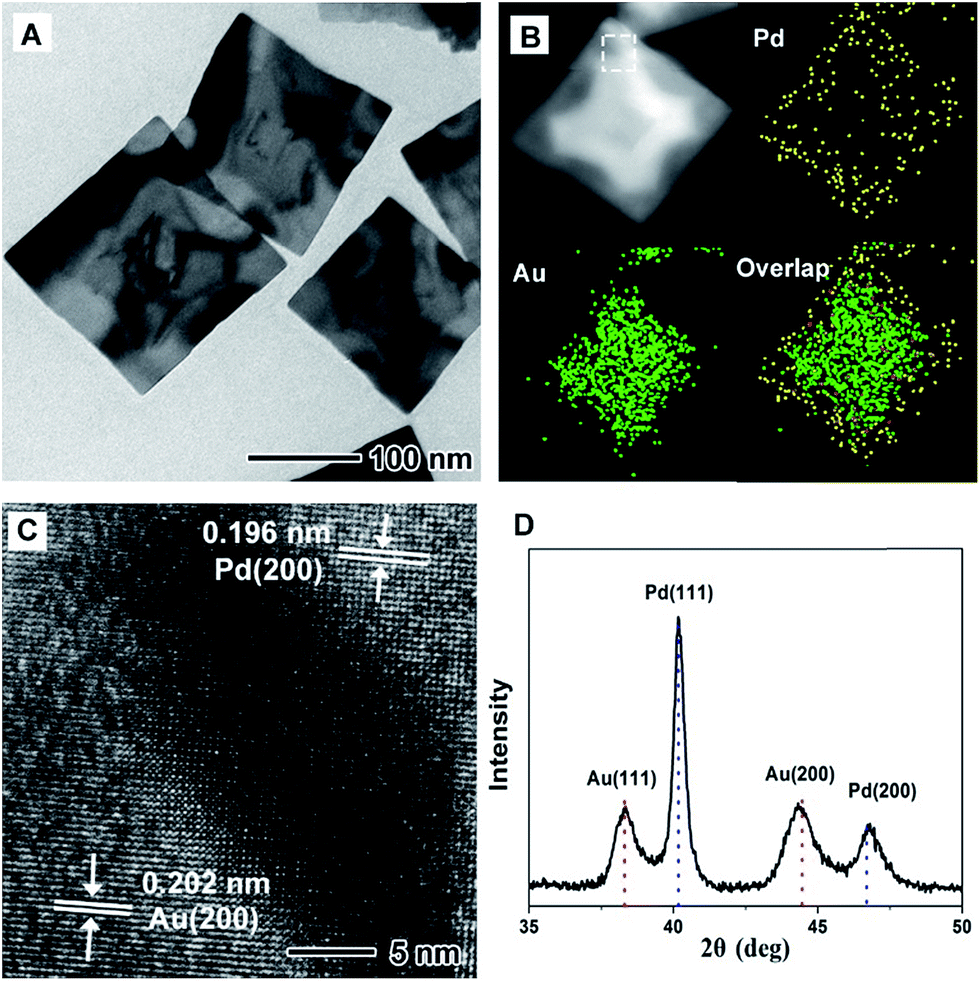 | ||
| Fig. 1 (A) TEM image, (B) HAADF-STEM-EDX mapping image, (C) HRTEM image and (D) XRD pattern of the PdAu@rGO-1. The white dashed frame in HAADF-STEM image is the selected area for HRTEM analysis. The red and blue dash lines in (D) correspond to the standard data for Au (PDF: 65-8601) and Pd (PDF: 65-6174), respectively. | ||
By slowing down the injection rate of HAuCl4 solution (0.5 mL min−1) with other conditions being the same as in the standard procedure, PdAu nanoplates with a different structure on rGO are obtained, which are marked as PdAu@rGO-2. From TEM images in Fig. 2A and S1B,† concave square nanoplates instead of regular square nanoplates are produced. HAADF-STEM-EDX mapping images (Fig. 2B) indicate that Au prefers to be deposited on the surface of the nanoplates with a concentration at the corners, while Pd is distributed throughout the nanoplates, implying a core–shell and alloy integrating structure. HRTEM image in Fig. 2C shows that there are two kinds of lattice fringes in PdAu@rGO-2. The fringe of 0.194 nm can be attributed to {200} facets of Pd while another fringe of 0.199 nm is indexed to {200} facets of PdAu alloy. From XRD pattern in Fig. 2D, one can observe that both of {200} and {111} peaks of Au shift to higher degrees due to the alloying of Pd with a smaller atom size while the peaks of Pd stay unchanged. These results further suggest the formation of a core–shell and alloy integrating structure in which the PdAu alloy shell is generated around a pure Pd nanoplate.
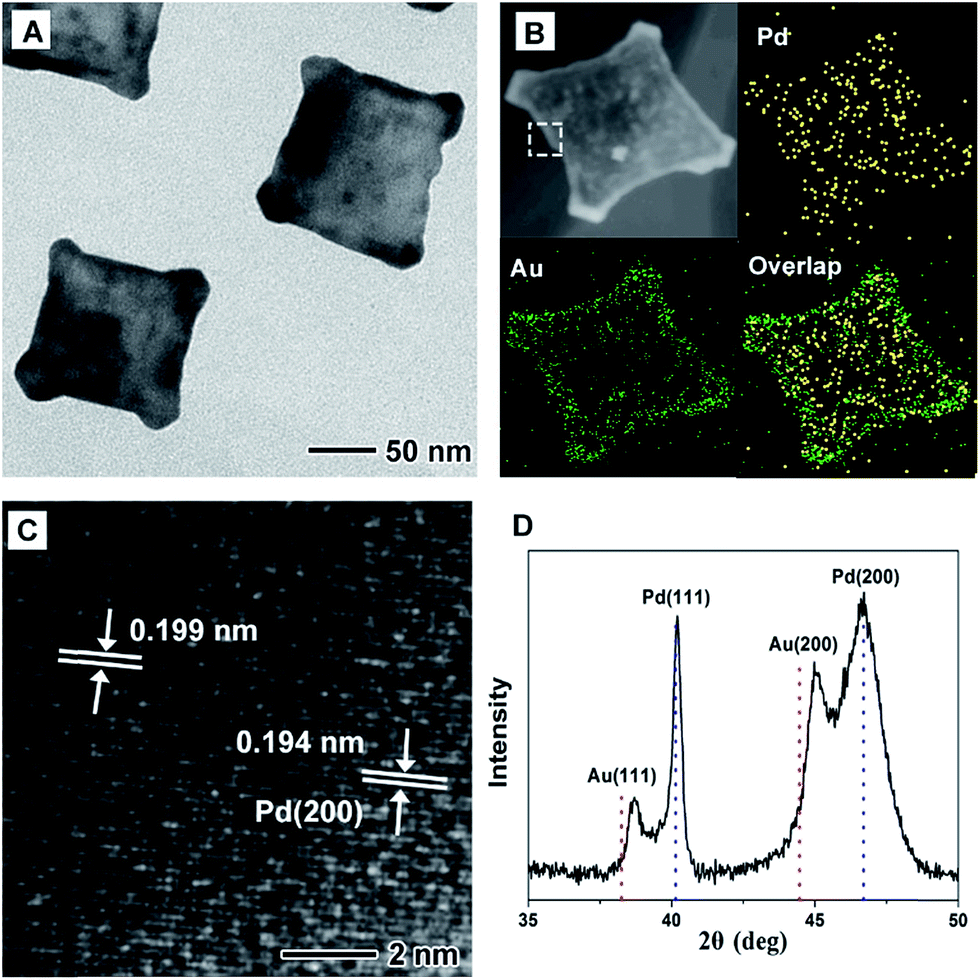 | ||
| Fig. 2 (A) TEM image, (B) HAADF-STEM-EDX mapping image, (C) HRTEM image and (D) XRD pattern of the PdAu@rGO-2. The white dashed frame in STEM image was the selected area for HRTEM analysis. The red and blue dash lines in (D) correspond to the standard data for Au (PDF: 65-8601) and Pd (PDF: 65-6174), respectively. | ||
The synthesis of such PdAu nanoplates involves subsequent addition of Na2PdCl4 and HAuCl4 into an aqueous solution containing AA and KBr. In this synthesis, PdSPs are initially produced before adding HAuCl4 solution, as shown in Fig. S2,† which was demonstrated in our previous report.38 To decipher the growth mechanism of the PdAu nanoplates, TEM images are taken from a series of samples for the synthesis of PdAu@rGO-1 at different reaction times after the injection of HAuCl4 solution at a high rate. In the initial stage (Fig. S3A,† t = 1 min), the in situ generated PdSPs evolve into the cross-shaped nanoplates mainly made of Au. With increasing the reaction time to 5 min (Fig. S3B†), the concave nanoplates with a clear difference in contrast are generated through the deposition of Pd on the side of the cross-shaped Au nanoplates. By prolonging the reaction time to 10 min, the PdAu core–shell nanoplates on rGO are obtained, as shown in Fig. S3C.† Fig. 3a summarizes the main steps for the formation of the PdAu core–shell nanoplates on rGO. In the presence of excess Br− ions, the complexes of AuBr4− and PdBr42− ions are formed through replacement of Cl− in the precursors by Br− owing to the stronger binding between Br− and metal ions.40,41 Arising from the difference in their redox potential (0.49 eV for Pd/PdBr42− versus 0.85 eV for Au/AuBr4−), the galvanic replacement between Pd nanoplates and AuBr4− is spontaneously conducted as soon as the Au precursor is added at a high injection rate. On the account of selective absorption of Br− ions on Pd {100} facets, the concentration of AuBr4− is much higher neighboring the {100} facets according to our previous report.42 As such, the galvanic replacement preferentially takes place from the four {100} side faces of the Pd nanoplates (Fig. 3a, step 1). Pd atoms on the {100} facets are gradually oxidized and dissolved into the solution along four 〈100〉 directions. In the meantime, the newly formed Au atoms by the reduction of AuBr4− preferentially deposit at the corner of the nanoplates since {100} facets are strongly capped by Br− ions, leading to the cross-shaped Au nanoplates (Fig. 3a, step 2). Due to the excess amount of the Pd precursor than Au precursor with a molar ratio of 4:1, the Au precursor is used up during the galvanic replacement. After that, the remnant Pd precursor and oxidized Pd ions are reduced by AA, leading to the subsequent deposition of Pd atoms on the side faces of the cross-shaped Au nanoplates due to the selective adsorption of Br− ions on {100} facets and eventually the formation of the PdAu core–shell nanoplates (Fig. 3a, step 3).
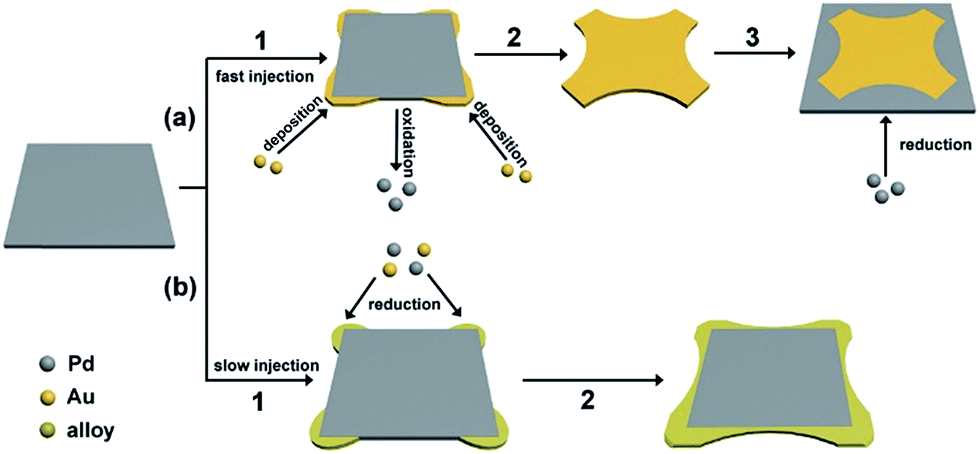 | ||
| Fig. 3 Schematic illustration for the formation of (a) PdAu core–shell nanoplates and (b) core–shell and alloy integrating PdAu nanoplates. | ||
As well-known, injection rate of precursors has a great influence on reaction kinetics, which is usually employed to control shape and structure of metal nanocrystals.43–45 In this work, slowing down the injection rate of the Au precursor makes the PdAu nanoplates transit from Au core@Pd shell to Pd core@PdAu alloy shell. This transition can be attributed to the inhibition of the galvanic replacement between Pd nanoplates and the Au precursor. It is clear that there is a competition between galvanic replacement and reduction reaction in the presence of AA.46 The driving force for galvanic replacement is expected to be remarkably weakened due to the low concentration of the Au precursor in the reaction at a slow injection rate. In this case, the galvanic replacement is severely blocked and the synthesis is dominated by the parallel reduction arising from AA. As such, the Au precursor as well as the remnant Pd precursor are co-reduced by AA, and the PdAu alloy is preferentially deposited at the corners of the Pd nanoplates, leading to a core–shell and alloy integrating structure (Fig. 3b). This demonstration is further confirmed by the control experiments with different injection rates of the Au precursor, as shown in Fig. S4.† At a fast injection rate of 1 mL min−1, the square PdAu core–shell nanoplates on rGO similar to PdAu@rGO-1 are generated (Fig. S4A†). Nevertheless, the PdAu nanoplates with a core–shell and alloy integrating structure on rGO similar to PdAu@rGO-2 are produced when the injection rate is decreased to 0.25 (Fig. S4B†) and 0.125 (Fig. S4C†) mL min−1. The XRD patterns (Fig. S4D†) confirm the formation of PdAu alloy in the latter two samples. The atomic ratio between Pd and Au of PdAu@rGO-1 and -2 was 4.1:1 and 3.95:1, which were closed to the feed ratio, determined by inductively coupled plasma mass spectrometry (ICP-MS). In other controlled experiments in which the molar ratio of Pd and Au precursors are decreasing, the synthesized PdAu bimetallic nanoplates are much less uniform than those prepared using the standard procedure as shown in Fig. S5†
X-ray photoelectron spectroscopy (XPS) was used to characterize the chemical and electronic states of C, Pd and Au. Fig. S6† depicts the XPS spectra of C 1s from the PdSP@rGO, PdAu@rGO-1 and PdAu@rGO-2. In the C 1s spectrum, there are double peaks with binding energies at 284.4 and 286.3 eV, which can be attributed to C–C and C–O, respectively. In the Pd 3d spectrum of the PdSP@rGO (Fig. 4A), the two peaks associated with Pd 3d5/2 and Pd 3d3/2 are located at 334.61 and 339.87 eV, respectively, which have a clear negative shift relative to the standard ones (dash lines in Fig. 4A). The shift of the binding energies provides the strong evidence of the electrons transfer from rGO to nanoplates arising from the difference in their redox potentials (0.38 V of rGO, 0.95 V of Pd2+/Pd, and 1.52 V of Au3+/Au vs. RHE). Compared to the PdSP@rGO, there are more negative shift of Pd 3d5/2 and Pd 3d3/2 for both the PdAu-based samples with an order of PdAu@rGO-1 < PdAu@rGO-2 possibly owing to the enhanced interaction between Pd and Au in the alloyed shell. The same trend is also observed in the Au 4f spectrum of such two samples (Fig. 4B). These XPS results indicate more electrons assembled on the surface of the PdAu@rGO-2, which may be beneficial to the enhancement in activity towards HER.
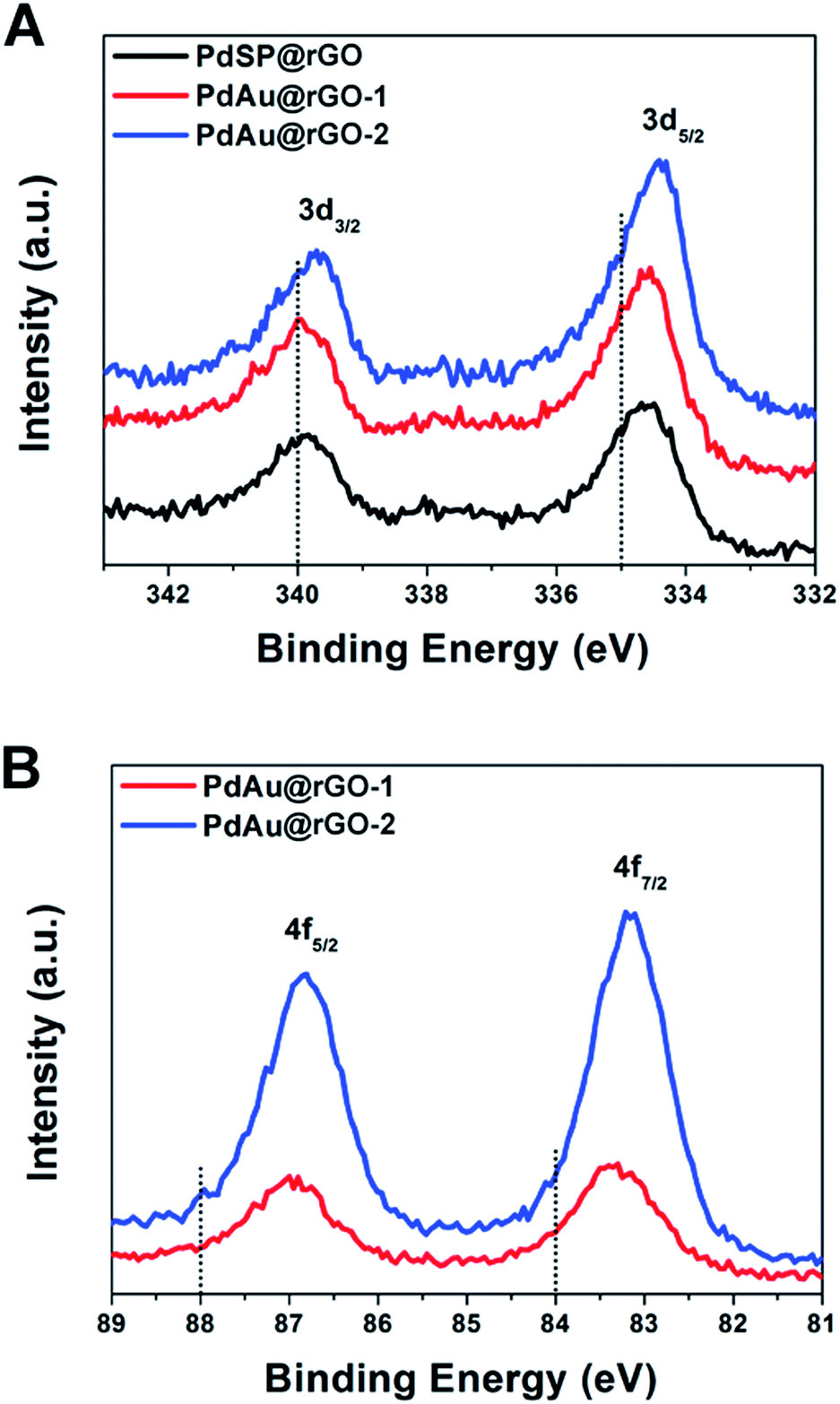 | ||
| Fig. 4 XPS spectra of (A) Pd 3d and (B) Au 4f orbitals of the PdSP@rGO, PdAu@rGO-1, and PdAu@rGO-2. | ||
The PdSP@rGO, PdAu@rGO-1 and PdAu@rGO-2 are evaluated as electrocatalysts towards HER on a three-electrode set-up with commercial Pt/C as a reference. The mass loading of catalysts was determined by inductively coupled plasma mass spectrometry (ICP-MS), which were 25.15%, 30.65% and 31.04%, respectively. The measurements were conducted in a 0.5 M H2SO4 solution after 100 cycles of cyclic voltammetry (CV) for surface cleaning. Fig. 5A shows the polarization curves of these four samples. All current densities are normalized by the corresponding electrochemically active surface areas (ECSAs), which were obtained by measuring the charge of CO oxidation in the CO-stripping curves (Fig. S7A†). Clearly, the steeper slope of the current density against the potential indicates an increased kinetics and higher cathodic currents, namely better catalytic activity for HER. Therefore, the HER activities of these four catalysts are following the sequence: PdSP@rGO < PdAu@rGO-1 < PdAu@rGO-2 < commercial Pt/C. A key estimate of an excellent catalyst for HER is low onset potential in electrolysis. As shown in Table S1,† the onset potential of PdAu@rGO-2 is 13.06 mV, which is next below to commercial Pt/C (6.68 mV). To further decipher their HER kinetics, the Tafel plots are derived from the corresponding polarization curves. The linear regions of the Tafel plots are fitted according to the Tafel equation (η = a + blogj), in which the Tafel slope (b = 2.3RT/αnF) represents the rate determining step and the possible HER reaction pathway. As can be seen from Fig. 5B and Table S1,† the PdAu@rGO-2 processes a smaller Tafel slope (44.35 mV dec−1) compared to PdSP@rGO (86.97 mV dec−1), PdAu@rGO-1 (55.77 mV dec−1), indicating the superior HER performance. The smaller Tafel slope suggests that the Volmer step is quicker on PdAu@rGO-2 relative to other two samples, which is determined by the electrochemical desorption reaction.
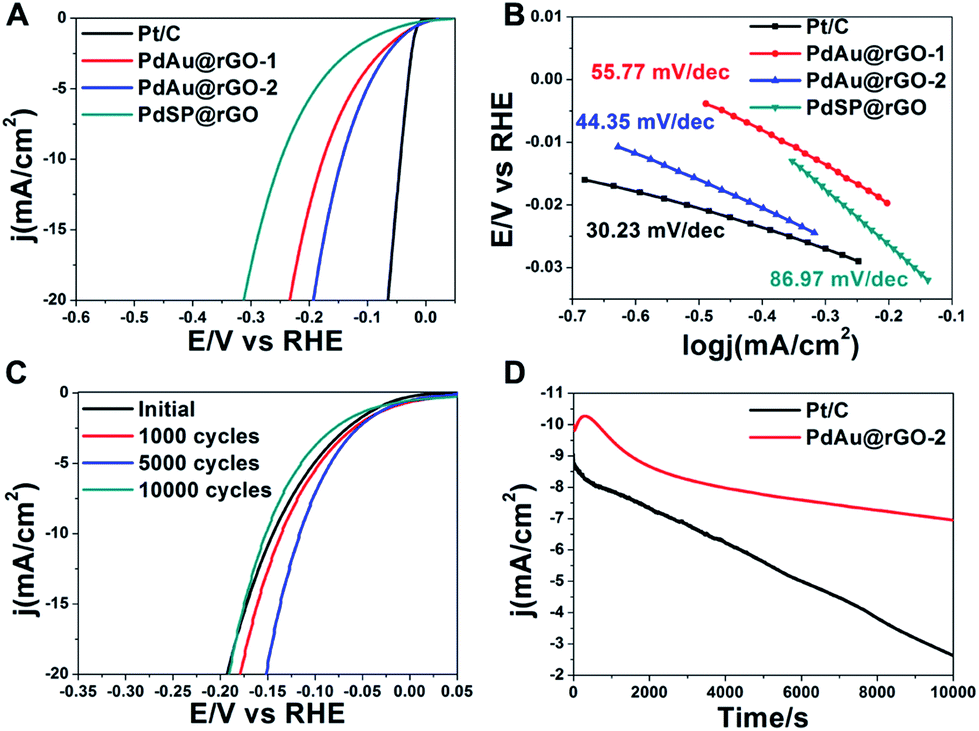 | ||
| Fig. 5 HER performance of several catalysts as shown. (A) Polarization curves after i–R correction recorded in a Ar-saturated 0.5 M H2SO4 solution at a sweep rate of 10 mV s−1. (B) Tafel plots corresponding to the tested catalysts derived from (A). (C) Polarization curves of PdAu@rGO-2 recorded after 1000, 5000, and 10000 CV cycles in a range of −0.3 to 0.1 V vs. RHE including the initial one. (D) Chronoamperometry measurements of commercial Pt/C and PdAu@rGO-2 at an overpotential with the current density being 10 mA cm−2 for 10000 s. | ||
The HER catalytic properties of these samples are further evaluated by exchange current density (j0), which is defined as the current density when overpotential is zero and describes the kinetics of the electrochemical charge transfer reaction at the metal–solution interface. From Table S1,† the PdAu@rGO-2 shows a larger j0 of 0.52 mA cm−2 than PdAu@rGO-1 (0.38 mA cm−2), and PdSP@rGO (0.24 mA cm−2). The large value of j0 in combination with a low Tafel slope and a small onset potential highlight the excellent HER performance of the PdAu@rGO-2, which is close to those of commercial Pt/C. The same conclusion is drawn when comparing their overpotentials at an apparent cathodic current density of 1.0 mA cm−2 (η1.0, Table S1†). The enhanced catalytic activity of PdAu@rGO-2 towards HER is probably owing to the concentration of electrons through the incorporation of Au into the Pd nanoplates, which is supported by the XPS data (Fig. 4). In addition, the alloying of Au with Pd on the surface of the Pd nanoplates can modulate the adsorption of atomic hydrogen to a suitable strength according to the volcano plot, and thus improve the enhancement in the HER activity.19,32
Besides catalytic activity, durability is also an important criterion for an excellent catalyst. The electrochemical stability is measured by continuous CVs between −0.3 and 0.1 V vs. RHE at a sweep rate of 100 mV s−1 in 0.5 M H2SO4 solution for 10000 cycles. As can be seen from Fig. S7B,† the catalytic performance towards HER of commercial Pt/C is gradually decreasing during CV cycles. The same trend is also observed in CV cycles of the PdAu@rGO-1 (Fig. S7C†). On the contrary, there is a significant improvement in HER performance after several thousand CV cycles for the PdAu@rGO-2 (Fig. 5C), indicating an activation process. Even after 10000 cycles of CV, a catalytic performance similar to the origin one is obtained. This result indicates that the PdAu@rGO-2 has an excellent long-term stability. Compared to the PdAu@rGO-1 with monometallic Pd shell being prone to form hydride phase, alloying with Au of the PdAu@rGO-2 brings some changes to lattice and electronic structures of Pd, which is confirmed experimentally and theoretically.29–32 As such, these changes suppress the formation of palladium hydride and improve the cycling stability. The enhanced stability of Au@Pt core–shell catalysts towards ORR by incorporation of Au was also demonstrated in our previous report.47 The superior stability of the PdAu@rGO-2 towards HER is further confirmed by chronoamperometry measurements. The I–t plots are carried out at a constant potential with the initial current density being nearly 10 mA cm−2 for 10000 seconds, as shown in Fig. 5D. As observed, the current density of commercial Pt/C keeps decreasing with time, while the PdAu@rGO-2 has an increase in current density at the first 1000 seconds and there is no obvious decline over the next hours, which is consistent with the CV stability results. Specifically, in comparison with nearly 71% activity loss of commercial Pt/C, the PdAu@rGO-2 is more resistive to degrading in the long term with only 30% activity loss after 10000 seconds. TEM images of the PdAu@rGO-2 after 1000 (Fig. S8A†) and 5000 cycles (Fig. S8B†) of CV show the well-preserved plate-like nanostructures, which may be responsible for the enhanced catalytic stability.
It is important to highlight that the PdAu@rGO-2 exhibits a comparable catalytic performance to Pt/C after the activation process (Fig. S7D†). Their onset potential, η1.0, and Tafel slope are significantly decreased to 5.02 mV, 22.07 mV and 32.27 mV dec−1 from the initial value of 13.06 mV, 31.87 mV and 44.35 mV dec−1, respectively. In order to figure out the reasons for the enhanced properties, CV plots of the PdAu@rGO-2 are recorded in a range of 0–1 V vs. RHE after 1000 and 5000 cycles (Fig. S8C†). As observed, the electric double layer is broadening during thousands of CV cycles. The reason for this result is probably the expansion of the stacking graphene nanosheets by constantly evolving hydrogen, leading to increase in active sites and thus catalytic properties. Moreover, the resistance of the sample decreases after continuous CV cycles between −0.3 to 0.1 V vs. RHE, which is characterized by electrochemical impedance spectroscopy (EIS, Fig. S8D†). Although GO can be reduced by AA during the synthesis, a lot of carbonyl and hydroxyl groups still remain on the surface of rGO, causing a poor conductivity. During the electrochemical measurement, rGO is more thoroughly reduced by the current at a negative bias voltage and the conductivity is significantly improved, which is beneficial to electron transfer during the hydrogen generation. Since the carbon electrode is used as the counter electrode, the factor of deposition of Pt for the enhanced catalytic performance can be eliminated. Taken together, the activation in catalytic performance of the PdAu@rGO-2 can be attributed to the enriched active sites and enhanced conductivity in combination with the superior stability of the PdAu alloyed nanoplates during the electrochemical test.
Conclusion
We have developed a facile method to synthesize PdAu nanoplates on rGO with different core–shell structures. The key to the success of this synthesis is to control the injection rate of HAuCl4 solution with an aim to tune the galvanic replacement and co-reduction in the presence of Br− ions and AA. Core–shell and alloy integrating PdAu@rGO-2 exhibits a substantially enhanced catalytic activity towards HER relative to PdSP@rGO and PdAu@rGO-1, which is close to that of commercial Pt/C. In addition, such PdAu@rGO-2 shows a much superior stability compared to other three catalysts including commercial Pt/C. Such enhancement in activity and stability can be attributed to the synergistic effect between PdAu alloy and graphene as well as the unique 2D nanostructures. This work may shed a new light to the development of advanced materials serving as efficient catalysts towards other catalytic reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work on electron microscopy was carried out in the Center for Electron Microscopy of Zhejiang University. We acknowledged financial support by the National Science Foundation of China (51372222 and 51522103), National Program for Support of Top-notch Young Professionals and Program for Innovative Research Team in University of Ministry of Education of China (IRT13R54).Notes and references
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- J. A. Turner, Science, 1999, 285, 687–689 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao and S. Qiao, Adv. Mater., 2015, 27, 5372–5378 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- C. G. Morales-Guio, L. A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- J. K. Nørskov and C. H. Christensen, Science, 2006, 312, 1322–1323 CrossRef PubMed.
- T. Liao, Z. Sun, C. Sun, S. X. Dou and D. J. Searles, Sci. Rep., 2014, 4, 6256 CrossRef PubMed.
- J. R. McKone, E. L. Warren, M. J. Bierman, S. W. Boettcher, B. S. Brunschwig, N. S. Lewis and H. B. Gray, Energy Environ. Sci., 2011, 4, 3573–3583 CAS.
- C. Kong, S. Min and G. Lu, Chem. Commun., 2014, 50, 9281–9283 RSC.
- J. Durst, C. Simon, F. Hasche and H. A. Gasteiger, J. Electrochem. Soc., 2015, 162, F190–F203 CrossRef CAS.
- L. Zhang, Q. Chang, H. Chen and M. Shao, Nano Energy, 2016, 29, 198–219 CrossRef CAS.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- M. Gong, D. Wang, C. Chen, B. Hwang and H. Dai, Nano Res., 2016, 9, 28–46 CrossRef CAS.
- B. B. Li, S. Z. Qiao, X. R. Zheng, X. J. Yang, Z. D. Cui, S. L. Zhu, Z. Y. Li and Y. Q. Liang, J. Power Sources, 2015, 284, 68–76 CrossRef CAS.
- T. Bhowmik, M. K. Kundu and S. Barman, ACS Catal., 2016, 6, 1929–1941 CrossRef CAS.
- S. Bai, C. Wang, M. Deng, M. Gong, Y. Bai, J. Jiang and Y. Xiong, Angew. Chem., Int. Ed., 2014, 53, 12120–12124 CrossRef CAS PubMed.
- M. Johansson, E. Skulason, G. Nielsen, S. Murphy, R. M. Nielsen and I. Chorkendorff, Surf. Sci., 2010, 604, 718–729 CrossRef CAS.
- M. Zeng and Y. Li, J. Mater. Chem. A, 2015, 3, 14942–14962 CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
- C. Ray, S. Dutta, Y. Negishi and T. Pal, Chem. Commun., 2016, 52, 6095–6098 RSC.
- R. Jana, A. Bhim, P. Bothra, S. K. Pati and S. C. Peter, ChemSusChem, 2016, 9, 2922–2927 CrossRef CAS PubMed.
- J. Li, P. Zhou, F. Li, R. Ren, Y. Liu, J. Niu, J. Ma, X. Zhang, M. Tian, J. Jin and J. Ma, J. Mater. Chem. A, 2015, 3, 11261–11268 CAS.
- F. Ren, H. Lu, H. Liu, Z. Wang, Y. Wu and Y. Li, J. Mater. Chem. A, 2015, 3, 23660–23663 CAS.
- L. Jiao, F. Li, X. Li, R. Ren, J. Li, X. Zhou, J. Jin and R. Li, Nanoscale, 2015, 7, 18441–18445 RSC.
- J. Li, F. Li, S. Guo, J. Zhang and J. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 8151–8160 CAS.
- Y. Liu, S. Liu, Z. Che, S. Zhao, X. Sheng, M. Han and J. Bao, J. Mater. Chem. A, 2016, 4, 16690–16697 CAS.
- L. Chen, N. Chen, Y. Hou, Z. Wang, S. Lv, T. Fujita, J. Jiang, A. Hirata and M. Chen, ACS Catal., 2013, 3, 1220–1230 CrossRef CAS.
- Y. Park, Y. Lee, S. Kang and S. Han, Nanoscale, 2014, 6, 9798–9805 RSC.
- P. Quaino, E. Santos, H. Wolfschmidt, M. A. Montero and U. Stimming, Catal. Today, 2011, 177, 55–63 CrossRef CAS.
- M. Okube, V. Petrykin, J. E. Mueller, D. Fantauzzi, P. Krtil and T. Jacob, ChemElectroChem, 2014, 1, 207–212 CrossRef.
- Z. Zhuang, F. Wang, R. Naidu and Z. Chen, J. Power Sources, 2015, 291, 132–137 CrossRef CAS.
- G. Darabdhara, M. A. Amin, G. A. M. Mersal, E. M. Ahmed, M. R. Das, M. B. Zakaria, V. Malgras, S. M. Alshehri, Y. Yamauchi, S. Szunerits and R. Boukherroub, J. Mater. Chem. A, 2015, 3, 20254–20266 CAS.
- X. Huang, H. Li, S. Li, S. Wu, F. Boey, J. Ma and H. Zhang, Angew. Chem., Int. Ed., 2011, 50, 12245–12248 CrossRef CAS PubMed.
- X. Huang, S. Tang, X. Mu, Y. Dai, G. Chen, Z. Zhou, F. Ruan, Z. Yang and N. Zheng, Nat. Nanotechnol., 2011, 6, 28–32 CrossRef CAS PubMed.
- H. Duan, N. Yan, R. Yu, C. R. Chang, G. Zhou, H. S. Hu, H. Rong, Z. Niu, J. Mao, H. Asakura, T. Tanaka, P. J. Dyson, J. Li and Y. Li, Nat. Commun., 2014, 5, 3093 Search PubMed.
- Y. Yan, H. Shan, G. Li, F. Xiao, Y. Jiang, Y. Yan, C. Jin, H. Zhang, J. Wu and D. Yang, Nano Lett., 2016, 16, 7999–8004 CrossRef CAS PubMed.
- Y. Li, W. Wang, K. Xia, W. Zhang, Y. Jiang, Y. Zeng, H. Zhang, C. Jin, Z. Zhang and D. Yang, Small, 2015, 11, 4745–4752 CrossRef CAS PubMed.
- Y. Jiang, Y. Yan, W. Chen, Y. Khan, J. Wu, H. Zhang and D. Yang, Chem. Commun., 2016, 52, 14204–14207 RSC.
- J. Li, P. Zhou, F. Li, J. Ma, Y. Liu, X. Zhang, H. Huo, J. Jin and J. Ma, J. Power Sources, 2016, 302, 343–351 CrossRef CAS.
- M. Jin, H. Liu, H. Zhang, Z. Xie, J. Liu and Y. Xia, Nano Res., 2011, 4, 83–91 CrossRef CAS.
- J. E. Millstone, W. Wei, M. R. Jones, H. Yoo and C. A. Mirkin, Nano Lett., 2008, 8, 2526–2529 CrossRef CAS PubMed.
- H. Zhang, M. Jin, J. Wang, W. Li, P. H. C. Camargo, M. J. Kim, D. Yang, Z. Xie and Y. Xia, J. Am. Chem. Soc., 2011, 133, 6078–6089 CrossRef CAS PubMed.
- Y. Yan, F. Zhan, J. Du, Y. Jiang, C. Jin, M. Fu, H. Zhang and D. Yang, Nanoscale, 2015, 7, 301–307 RSC.
- S. Xie, S. Choi, N. Lu, L. T. Roling, J. A. Herron, L. Zhang, J. Park, J. Wang, M. J. Kim, Z. Xie, M. Mavrikakis and Y. Xia, Nano Lett., 2014, 14, 3570–3576 CrossRef CAS PubMed.
- H. Zhang, W. Li, M. Jin, J. Zeng, T. Yu, D. Yang and Y. Xia, Nano Lett., 2011, 11, 898–903 CrossRef CAS PubMed.
- J. Li, J. Liu, Y. Yang and D. Qin, J. Am. Chem. Soc., 2015, 137, 7039–7042 CrossRef CAS PubMed.
- T. Bian, H. Zhang, Y. Jiang, C. Jin, J. Wu, H. Yang and D. Yang, Nano Lett., 2015, 15, 7808–7815 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08517f |
| This journal is © The Royal Society of Chemistry 2017 |