
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing surface oxide formations on SiO2-supported platinum nanocatalysts under CO oxidation†
Sunyoung Ohab,
Seoin Backa,
Won Hui Doh b,
Song Yi Moonab,
Jeongjin Kimab,
Yousung Jung*a and
Jeong Young Park*ab
b,
Song Yi Moonab,
Jeongjin Kimab,
Yousung Jung*a and
Jeong Young Park*ab
aGraduate School of EEWS, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: ysjn@kaist.ac.kr; Tel: +82-42-350-1712
bCenter for Nanomaterials and Chemical Reactions, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea. E-mail: jeongypark@kaist.ac.kr; Fax: +82-42-350-1710; Tel: +82-42-350-1713
First published on 21st September 2017
Abstract
Formations of an ultrathin oxide layer on noble metal catalysts affect the characteristics of fundamental molecular behaviours such as adsorption, diffusion, and desorption on their surfaces. That is directly correlated to enhancement of catalytic activity under operating conditions because the kinetics of catalytic reactions are also simultaneously influenced. Especially, a sub-monolayered surface oxide is known as having a key role for improving catalytic activity, but revealing its existence in catalysis is challenging due to their fast chemical conversion. Herein, we report the first evidence of surface oxide formations on platinum (Pt) nanocatalysts under CO oxidation probed with a diffuse reflectance infrared Fourier transform (DRIFT) technique. Spectroscopic information demonstrates that the abrupt blue shift of adsorbed CO molecules vibrational frequencies of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching mode on the reduced Pt nanocatalyst surface is initiated prior to aggressive CO conversion to CO2 gas molecules. Site-specific replacements of the adsorbed CO molecule with dissociative oxygen occur just before the ignition temperature that is supposed to be an important reaction step for CO oxidation over a Pt nanocatalyst. Density functional theory (DFT) calculation results support this phenomenon as a function of relative atomic fractions between CO and O on a Pt model surface and consistently show a similar trend with experimental evidence.
O stretching mode on the reduced Pt nanocatalyst surface is initiated prior to aggressive CO conversion to CO2 gas molecules. Site-specific replacements of the adsorbed CO molecule with dissociative oxygen occur just before the ignition temperature that is supposed to be an important reaction step for CO oxidation over a Pt nanocatalyst. Density functional theory (DFT) calculation results support this phenomenon as a function of relative atomic fractions between CO and O on a Pt model surface and consistently show a similar trend with experimental evidence.
1. Introduction
Effects of oxide layers formation on a metal surface have been investigated due to their remarkably different catalytic activities with corresponding metallic surfaces for carbon monoxide (CO) oxidation. Molecular behaviours on a platinum (Pt) model surface at low temperature visualise that CO molecules are efficiently converted to CO2 gas molecules over a dissociative oxygen (O) covered Pt surface.1 In addition, Over et al.2,3 explained with theoretical calculations that the origin of high catalytic activity of RuO2 catalysts, with the highly active phase at high pressure for CO oxidation, is not on metallic Ru(0001) but rather on a RuO2 film surface. Pt-group metals under high O2 partial pressure environments could yield the “surface oxide” as an active phase in industrial catalytic reaction processes such as CO oxidation4–6 and oxygen reduction reaction (ORR).7 On the other hand, bulk oxides formation of Pt-group metals are known as inactive for CO oxidation according to well-defined model catalyst studies in ultra-high vacuum (UHV).8–10 To investigate effects of oxidised components on the Pt catalysts during CO oxidation, extensive studies have been performed with a variety of pressures and CO/O2 ratios.11–14 The nature of a surface oxide in altering its catalytic activity for CO oxidation on noble metal nanoparticles (NPs) was studied.15 For instance, a surface oxide appears to have chemically active properties, which is consistent with oxidised Pt NPs after an ultraviolet-ozone treatment.16 Moreover, the rate of CO conversion is influenced not only by the size of metallic rhodium (Rh) NPs, but also by the thickness of Rh oxide layers, which are measured using ambient-pressure X-ray photoelectron spectroscopy (AP-XPS).17,18 In this way, investigating surface oxides is significantly important because they play a key role in enhancing catalytic activity under catalytic reactions that have quite different features from their corresponding metallic surfaces.19,20As a matter of fact, revealing the existence of surface oxides in catalysis is considered to be a great challenge because of their chemically fast conversion. Even though many efforts using a high-pressure flow reactor integrated with scanning tunnelling microscopy (STM) and AP-XPS have discovered formation of a surface oxide having a dramatic effect for CO2 evolution,21–23 unfortunately, no evidence for vibrational motions of adsorbate molecules on the surface during CO oxidation has not been observed yet. To fully distinguish this fascinating phenomenon, and to design a rational nanocatalyst structure with a high-performance, well-characterised evidences for surface oxides using surface-sensitive techniques are indispensably needed. Fourier-transform infrared spectroscopy (FT-IR) is particularly suitable for investigating fundamental molecular behaviours such as adsorption and desorption at an active site24–27 without limitations of the pressure gap issue.28,29
In this study, we probed an instantly formed surface oxide phase on SiO2-supported Pt nanocatalysts under CO oxidation by employing an in situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) technique. It is important to have a thorough understanding of how catalytic activity is affected by the surface oxide phase in catalysis. Observation of an abrupt blue shift of adsorbed CO molecules vibrational frequencies of the CO stretching mode [v(CO)] on the reduced Pt nanocatalyst surface is obviously related to the formations of ultrathin oxide layered Pt NPs under the catalytic reaction condition at an elevated temperature. The ultrathin oxide layer on the Pt NPs surface is initiated prior to the aggressively occurring CO conversion to CO2 gas molecules. Furthermore, the identified modes of vibrational frequency shifts on the ultrathin surface oxide layers are supported by density functional theory (DFT) calculations as a function of relative CO and O atomic fractions on the Pt model surface.
2. Experimental and model methods
2.1 Synthesis of SiO2-supported Pt nanocatalysts
A colloidal spherical silica (with diameters of 25–30 nm) solution was prepared by a modified Stöber method.30 Briefly, 600 mL of anhydrous ethanol and 9.3 mL of ammonium aqueous solution (NH3·H2O, 27%) were mixed with 24 mL of deionised (DI) water; the solution was stirred for 20 min at room temperature. Then, 30 mL of tetraethyl orthosilicate (TEOS, ≥97%) was added, followed by stirring for 24 h at room temperature. After stirring, the mixed solution suddenly turned a muddy white colour, and then it was centrifuged to collect the spherical silica NPs followed by washing with ethanol. To prepare an amino group functionalised surface, the collected silica NPs were dispersed in a mixture of isopropanol (84 mL) and (3-aminopropyl)triethoxysilane (APTES, 300 μL) using ultrasonication for 10 min. After the amino functionalisation, the dispersed silica NPs were heated at 80 °C for 2 h.31 Finally, the obtained amine-functionalised spherical silica NPs (f-SiO2) were centrifuged and washed several times with ethanol, and then re-dispersed in 1.5 mL of ethanol.The PVP [poly(N-vinylpyrrolidone)] stabilised Pt NPs were synthesised using the following procedure. A mixture solution (i.e., 100 mg of Pt precursor (H2PtCl6·6H2O, >99.9%), 10 mL of ethylene glycol, and 5 mL of 0.5 M NaOH) was refluxed at 160 °C for 2 h. When the reflux was finished, the Pt NPs were dispersed in ethanol after adding 20.3 mg of PVP (Mw = 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) under sonication for 20 min.32 The PVP-stabilised Pt NPs colloid (3 mL) dispersed in 7 mL of ethanol was mixed with 1.5 mL of the functionalised silica (f-SiO2) solution followed by sonication for 30 min. The resultant Pt/SiO2 colloids were centrifuged and washed three times with ethanol to remove any Pt NPs not bound to the silica particles. The Pt NPs anchored on the prepared f-SiO2 were then dispersed in 1.5 mL of ethanol. Following the washing process, the as-synthesised SiO2-supported Pt nanocatalysts (Pt/SiO2) were prepared with free-of-Cl− residues (ESI, Fig. S1†).
000) under sonication for 20 min.32 The PVP-stabilised Pt NPs colloid (3 mL) dispersed in 7 mL of ethanol was mixed with 1.5 mL of the functionalised silica (f-SiO2) solution followed by sonication for 30 min. The resultant Pt/SiO2 colloids were centrifuged and washed three times with ethanol to remove any Pt NPs not bound to the silica particles. The Pt NPs anchored on the prepared f-SiO2 were then dispersed in 1.5 mL of ethanol. Following the washing process, the as-synthesised SiO2-supported Pt nanocatalysts (Pt/SiO2) were prepared with free-of-Cl− residues (ESI, Fig. S1†).
2.2 Transmission electron microscopy (TEM) measurements
Morphology and size distribution of the prepared Pt/SiO2 catalysts were visualised using TEM operated at an acceleration voltage of 300 kV (Tecnai TF30 ST, FEI). Prior to the measurements, the catalysts were well-dispersed in ethanol solution for 30 min, followed by drop-casting on an amorphous carbon-coated TEM grid, and then dried for a few minutes.2.3 X-ray photoelectron spectroscopy (XPS) measurements
Surface oxidation states of the prepared Pt NPs were examined by XPS (K-alpha, Thermo VG Scientific) equipped with an Al Kα X-ray source (1486.6 eV). Each XPS spectrum was measured at a resolution of 0.05 eV per step with a pass energy of 50 eV. The core-level photoelectron peaks in the measured XPS spectra were calibrated in accordance with adventitious carbon (i.e., C 1s core-level peak at 284.8 eV of binding energy) on the substrate.2.4 Vibrational frequency shift analysis of Pt/SiO2 catalysts
DRIFTS analysis of the Pt/SiO2 catalysts were carried out using a Fourier-transform infrared spectrometer (Carry 660, Agilent) equipped with diffuse reflectance optics (Harrick), a mercury cadmium telluride (MCT) detector (every 32 scans with a resolution of 4 cm−1), and ZnSe windows integrated into the reaction cell. The inside of the spectrometer was purged by purified nitrogen (N2) gas during all measurements. The DRIFTS cell was connected to a heater and gas lines that were controlled by mass flow controllers (BROOKS Instrument).All synthesised powder samples were mechanically pressed onto a cleanly prepared stainless-steel mesh to prevent potentially exposed contamination in the reactor cell. For preparation of the O2-treated Pt/SiO2 catalysts, as-prepared Pt/SiO2 catalysts were treated under 100 sccm of O2 flow condition at 350 °C for 3 h in the DRIFTS reaction cell. Before the CO oxidation reaction, the reducing or oxidising pre-treatment of the nanocatalysts was performed in the reaction cell before the CO oxidation reaction and was carried out in the following steps: the samples were (i) purged with helium (He) gas at 27 °C for 30 min, (ii) heated at 210 °C in 5% H2/Ar or in O2 for 1 h, and finally (iii) cooled to room temperature (27 °C) under He flow conditions.
To investigate the vibrational frequency shifts of adsorbate molecules on the Pt/SiO2 catalysts during CO oxidation, the samples were exposed to two different gas feeding environments before the DRIFTS measurements at an elevated temperature. 10 mL min−1 of 10% CO/He was added to the He balance gas flow (CO feed) to provide a CO environment in the reaction cell. Then, 10 mL min−1 of 10% CO/He and 4 mL min−1 of O2 flow (CO + O2 feed, 1:4 v/v) were simultaneously introduced into the reaction cell for the CO oxidation environment.
Total flow rate of the gas was kept at 50 mL min−1 using He gas as the balance. A FTIR spectrum using potassium bromide (KBr) was recorded at atmospheric N2 flow condition for background subtraction for all spectra. The acquired background spectrum was also measured in N2 at each reaction temperature that had no particular changes in the obtained spectra (ESI, Fig. S2†). All spectra were obtained in Kubelka–Munk (KM) units. Following the washing process, the obtained SiO2-supported platinum nanocatalysts (Pt/SiO2) were synthesised with the free-of-Cl− residues (ESI, Fig. S1†).
2.5 Characterisations of Pt/SiO2 catalysts
The morphologic shapes, with size distributions and surface oxidation states of the anchored Pt NPs on spherical silica, were characterised to examine the effect of oxygen annealing. A TEM image of as-synthesised Pt/SiO2 showed that the Pt NPs were almost uniformly dispersed on the silica spheres (i.e., approximate size was 25–30 nm), and they had an average size of 1.9 ± 0.2 nm as displayed in Fig. 1a. After an annealing process of the samples in atmospheric air at 300 °C, the size and shape of Pt NPs showed negligible changes with a mean diameter of 2.1 ± 0.3 nm in Fig. 1b. This indicated that the size and shape of Pt NPs were quite resistive to the heating process at atmospheric pressure with oxygen maintained for their own nanoscale morphology.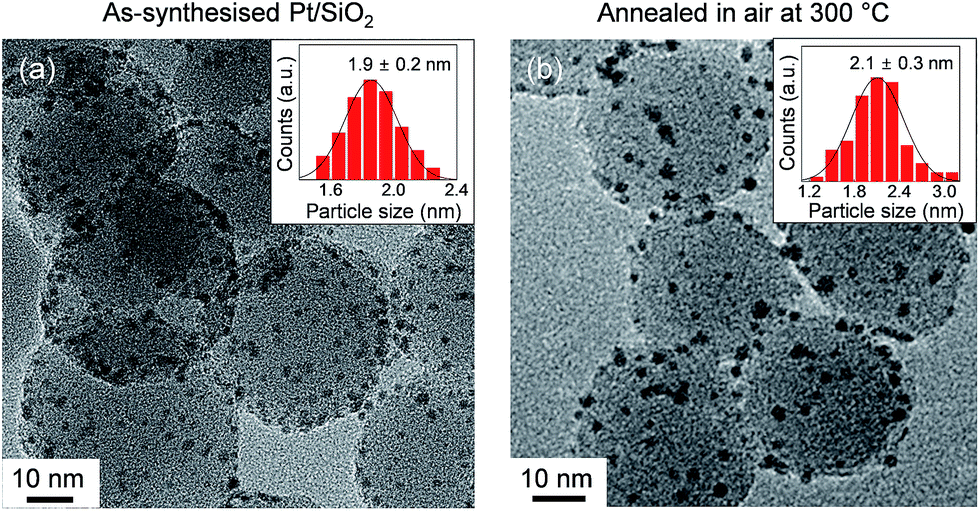 | ||
| Fig. 1 TEM images and size distribution histograms of anchored Pt NPs on the spherical silica supports for (a) as-synthesised and (b) oxidised Pt/SiO2 catalysts in air at 300 °C. | ||
In other conditions, the surface oxidation states of the Pt/SiO2 catalysts showed dramatic changes after a highly purified O2 (99.999%) annealing treatment. In Fig. 2a, as-synthesised Pt NPs surface consists of metallic Pt for Pt 4f7/2 at 71.1 eV of binding energy and a small amount of PtO species at 72.4 eV of binding energy; however, their compositional ratio is clearly changed as demonstrated in Fig. 2b. PtO species becomes a major species by the oxidation of metallic Pt NPs on the surface; in addition, a fully oxidised bulk α-PtO2 species at 75.0 eV is also characterised as a small amount. Our characterisations agree with earlier literatures for each species in the XPS spectra, respectively.33–35
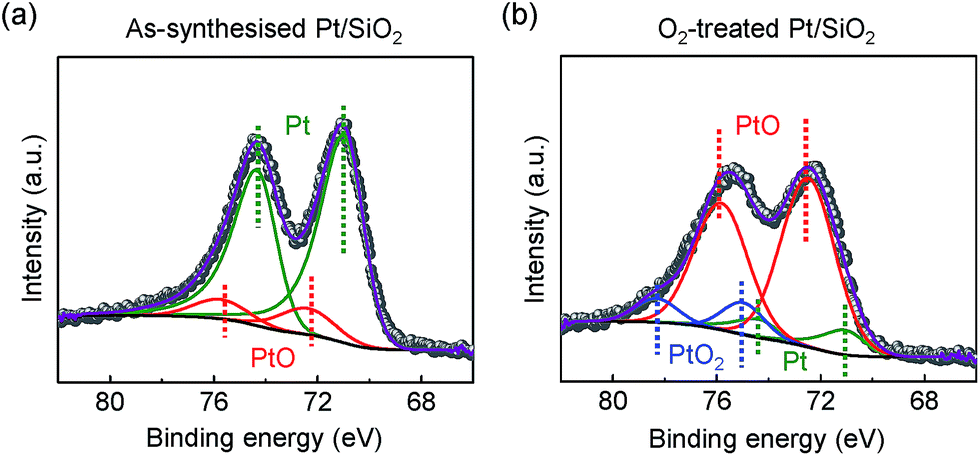 | ||
| Fig. 2 XPS spectra of (a) as-synthesised and (b) O2-treated Pt/SiO2 catalysts. The oxidation state of each assigned peak corresponds to metallic Pt0 (green colour), Pt2+ (red colour), and Pt4+ (blue colour), respectively. | ||
We note that as-synthesised Pt/SiO2 catalysts inevitably contain the negligible amount of PtO species because the spherical Pt NPs are composed of a majority of face-centre cubic (fcc) structured {111} facets at terrace sites and a small amount of {100} facets at step sites.36,37 Unlike investigations on a well-defined model surface, the probed PtO species of as-synthesised Pt/SiO2 catalysts in the Pt 4f core-level spectrum is associated with slightly formed oxide at the step sites of Pt NPs caused by spontaneously dissociated oxygens on the surface23 so the oxygen blocked sites of Pt atoms can be excluded from potential formations of surface oxide in catalysis.16,38
2.6 Computational model calculation methods
Structural relaxation and vibrational frequency calculations were performed using the Vienna ab initio Simulation Package (VASP)39,40 with the Perdew–Burke–Ernzerhof (RPBE) exchange functional41,42 and projector-augmented wave pseudopotential.43 The cut-off energy for the plane wave basis set was set at 500 eV and k-points were sampled using a 3 × 3 × 1 Monkhorst–Pack mesh.44 A three-layered Pt(111) slab was designed using a (3 × 3) surface unit cell containing 27 atoms in total. The bottom two Pt layers were fixed with their bulk positions and all other Pt atoms and adsorbates were relaxed until the forces on them were less than 0.01 eV Å−1. The convergence criterion for total energy was set to 10−8 eV. Vibration frequencies were obtained by a finite difference approximation to the Hessian Matrix, where the adsorbed CO was displaced by 0.01 Å in the x-, y-, and z-directions.3. Results and discussion
3.1 CO molecules adsorption on the Pt NPs
Molecular CO adsorption on the Pt/SiO2 catalysts (i.e., as-synthesised and O2-treated) surfaces were investigated by spectral changes of the vibrational modes. The employed DRIFTS technique shows a surface-sensitive feature for site-specific CO adsorption on Pt atoms. According to molecular orbital (MO) theory, the adsorbed CO peak [v(CO)] positions in DRIFT spectra are directly related to the bond strength between the carbon (C) and oxygen (O) atom of the intramolecular structure of a CO molecule. The bond strength between C and O is weakened when a CO molecule is adsorbed on a transition metal (e.g., Pt) because the empty 2π* antibonding orbital of the CO plays the role of electron acceptor from the d-orbital of Pt (i.e., the so-called as π-backbond effect).45,46 Thus, the v(CO) peak could be shifted to a higher or a lower frequency in DRIFT spectra depending on the oxidation states of the Pt NPs owing to the reduced back donation effect that originates from the difference of a small quantity of d-electrons for adsorbate-induced Pt sites compared with metallic Pt arrays on the surface.47
Fig. 3a exhibits the shift of adsorbed CO molecules of the v(CO) mode peak at the lower position of a wavenumber (i.e., red shift) on the as-synthesised Pt/SiO2 catalysts with increasing temperature under a CO gas flow condition. Otherwise, the observed vibration frequency changes for the O2-treated Pt/SiO2 catalyst show the recorded peak position at a higher wavenumber (i.e., blue shift) than that of the as-synthesised Pt/SiO2 catalysts with apparently lower signal intensity in the linearly vibrating mode. It is speculated that the bulk oxide formations on the metallic Pt surface after the O2 pre-treatment process affects a hindrance of chemically active sites.33,34 In earlier studies, the peak position of the v(CO) mode, which is also correlated with ensembles of dipole–dipole interactions between CO molecules and the metallic Pt atoms,38 the degree of Pt surface reduction,48 and CO-induced island formations appeared.49,50 It is also generally understood that partially oxidised Pt NPs could accept more transferred electrons from the CO molecules. As a result, the Pt NPs become more reduced under the CO gas flow condition,51,52 and the red shift of the v(CO) peak of CO at elevated temperature means that gradually increasing numbers of reduced Pt sites form due to CO molecule adsorptions on the Pt NPs surface.
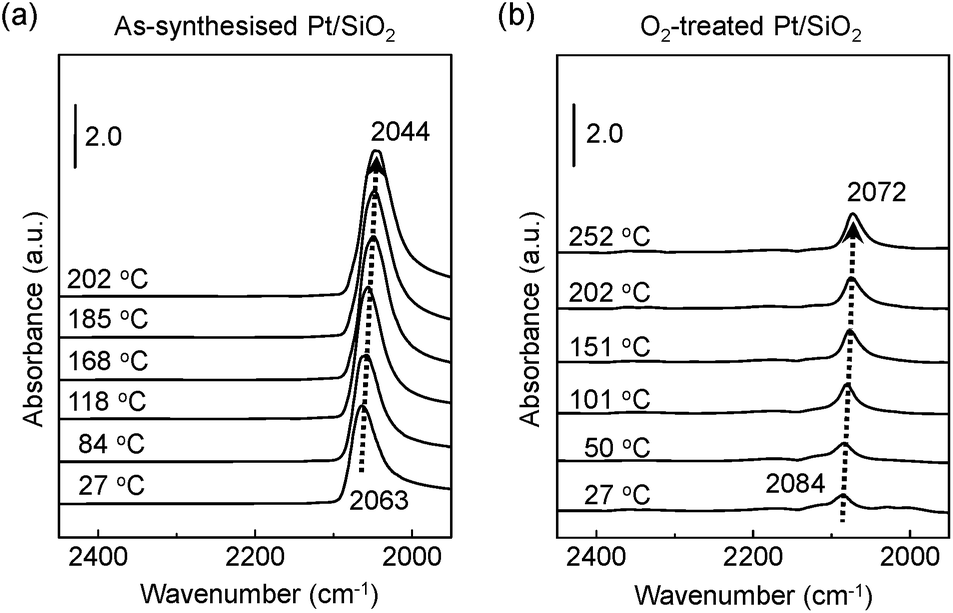 | ||
| Fig. 3 Vibration frequency changes of adsorbed CO molecules on the catalyst surfaces at elevated temperature. DRIFT spectra of (a) as-synthesised and of (b) O2-treated Pt/SiO2 catalysts under CO gas flow condition. | ||
3.2 Relation of chemical adsorptions and frequency shifts
Fig. 4a and b show obviously different characteristics of the linearly bound CO absorption behaviour between as-synthesised and O2-treated Pt/SiO2 catalysts. The position and full width at half maximum (FWHM) of the v(CO) peak rely on the number of Pt NPs sites or sizes, and how the CO molecules exist on the nanoparticle's surfaces.37 Typically, the crystalline nature of the Pt NPs is confirmed by X-ray diffraction (XRD) corresponding to a presence of the crystalline Pt fcc phase, which belongs to (111), (200), and (220) crystalline planes compared with a Pt single crystal.53 So, the pronounced band splitting peaks in Fig. 4a indicate that there are two main CO bonded sites such as (111) and (200), corresponding to 2065 cm−1 and 2043 cm−1 at 27 °C, respectively. All the peaks of fitting parameters and IR analysis are shown in ESI, Table S1.† The observed CO adsorption peaks positions are similar to previously reported works.46,49
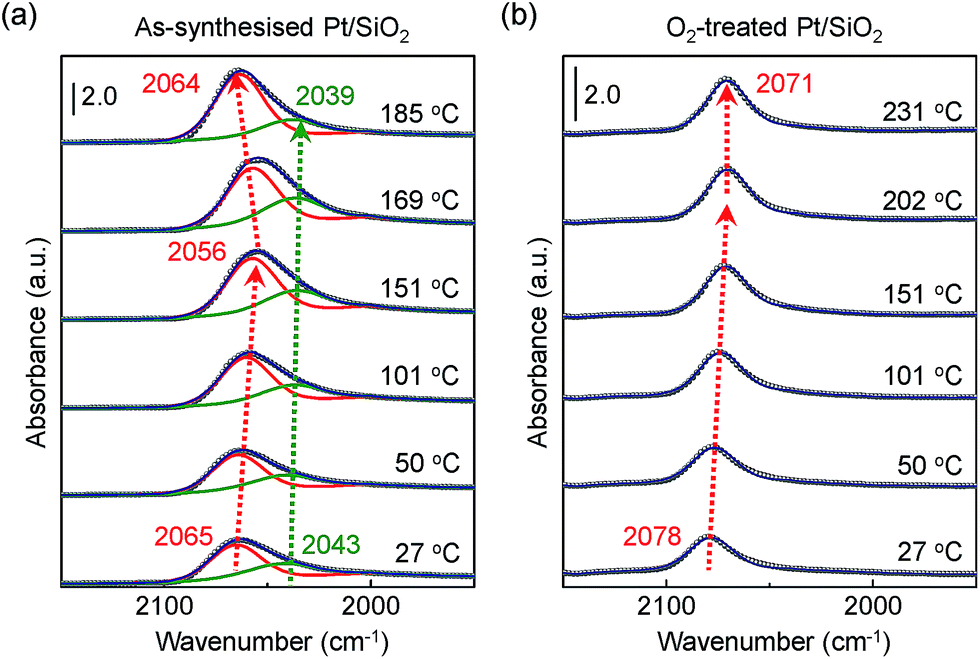 | ||
| Fig. 4 DRIFT spectra of (a) as-synthesised and (b) O2-treated Pt/SiO2 catalysts at elevated temperature under the mixed CO/O2 gas flow condition. Deconvolution peaks correspond to vibrational modes for adsorbed CO molecules on atop sites (red colour) and on the bridge sites (green colour) of Pt atoms on a surface, respectively. | ||
When the as-synthesised Pt/SiO2 catalysts are exposed to the gas mixture (CO + O2 feed) for a CO oxidation reaction, the linearly adsorbed CO on Pt for (111) is shifted from 2065 cm−1 to 2056 cm−1 as the temperature increases to 151 °C. Then, the consecutively adsorbed CO peak for (111) is shifted to a higher wavenumber at 2064 cm−1 as temperature increases. In general, the density of the excess charges decreases with increasing nanoparticle size, resulting in a blue shift of the CO absorption band due to the reduced amount of π-back-donation into the CO.38 After heating at 300 °C in air, the Pt/SiO2 is thermally stable, as shown in the TEM image in Fig. 1b, because the initial morphology of the structure and Pt dispersion are preserved; thus, the blue shift observed in Fig. 4a is definitely not the result of Pt nanoparticle agglomeration. The peak position of v(CO) for O2-treated Pt/SiO2 is higher than the as-synthesised Pt/SiO2 catalyst, indicating an oxidised Pt surface because of oxygen treatment, which is in an agreement with the XPS results (Fig. 2a and b). Interestingly, the blue shifted v(CO) peak is not observed on the O2-treated Pt/SiO2 catalyst during the CO oxidation reaction in Fig. 4b, whereas the red shifted v(CO) peak is only observed with increasing temperature. When the bare SiO2 spheres were exposed to the CO oxidation condition, the frequency of linearly adsorbed CO at about 2060 cm−1 was not observed (ESI, Fig. S3†).
3.3 Surface oxide formations on Pt NPs for CO oxidation
As demonstrated in Fig. 4a, the abruptly occurring wavenumber changes in the v(CO) mode are clearly related to the formation of oxygen species on the Pt NPs surface. To confirm the interplay between surface oxide and catalytic activity during CO oxidation, we performed further DRIFTS investigations for the as-synthesised and O2-treated Pt/SiO2 catalysts operated in time-resolved fast scanning mode.
In Fig. 5a, a contour-map visualises an intriguing behaviour of the adsorbed CO molecules on the Pt NPs surface with the evolution of CO2 gas molecules. A plot of the wavenumber for the linearly adsorbed CO molecules and the integrated area of the evolved CO2 gas molecules indicates apparently that there is a similar trend between the aggressive CO2 evolutions and the blue shift of the v(CO) mode as shown in Fig. 5b. CO adsorption sites on the Pt NPs are possibly generated as the number of adsorbed CO molecules increases until the temperature reaches 151 °C because the Pt NPs are more reduced by accepting more non-bonding electrons from the CO molecules. A sudden change in the linearly adsorbed CO [v(CO)] peak, such as a blue shift in a mild heating temperature region (151–185 °C), is observed along with produced CO2 gas at approximately 2350 cm−1, which is attributed to the formation of an ultrathin oxide layered on the Pt surface under the O2-rich condition. Finally, the peak of the asymmetric stretching mode [vas(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)] for the evolved CO2 gas phase increases dramatically at 190 °C while the v(CO) peak disappears, as clearly shown in Fig. 5a. It can be demonstrated that the Pt surface is not fully covered by CO molecules because adsorption and desorption of CO molecules take place simultaneously at an elevated temperature. The measured CO oxidation reaction initiated and eventually accelerated at around ignition temperature once the ultrathin oxide layer was formed on the Pt surface. The Pt surface needs to be partially oxidised to initiate and to accelerate CO oxidation. These results clearly reveal that formation of surface oxide on the Pt surface involves the CO oxidation to CO2 that proceeds via a Mars–van Krevelen mechanism.
O)] for the evolved CO2 gas phase increases dramatically at 190 °C while the v(CO) peak disappears, as clearly shown in Fig. 5a. It can be demonstrated that the Pt surface is not fully covered by CO molecules because adsorption and desorption of CO molecules take place simultaneously at an elevated temperature. The measured CO oxidation reaction initiated and eventually accelerated at around ignition temperature once the ultrathin oxide layer was formed on the Pt surface. The Pt surface needs to be partially oxidised to initiate and to accelerate CO oxidation. These results clearly reveal that formation of surface oxide on the Pt surface involves the CO oxidation to CO2 that proceeds via a Mars–van Krevelen mechanism.
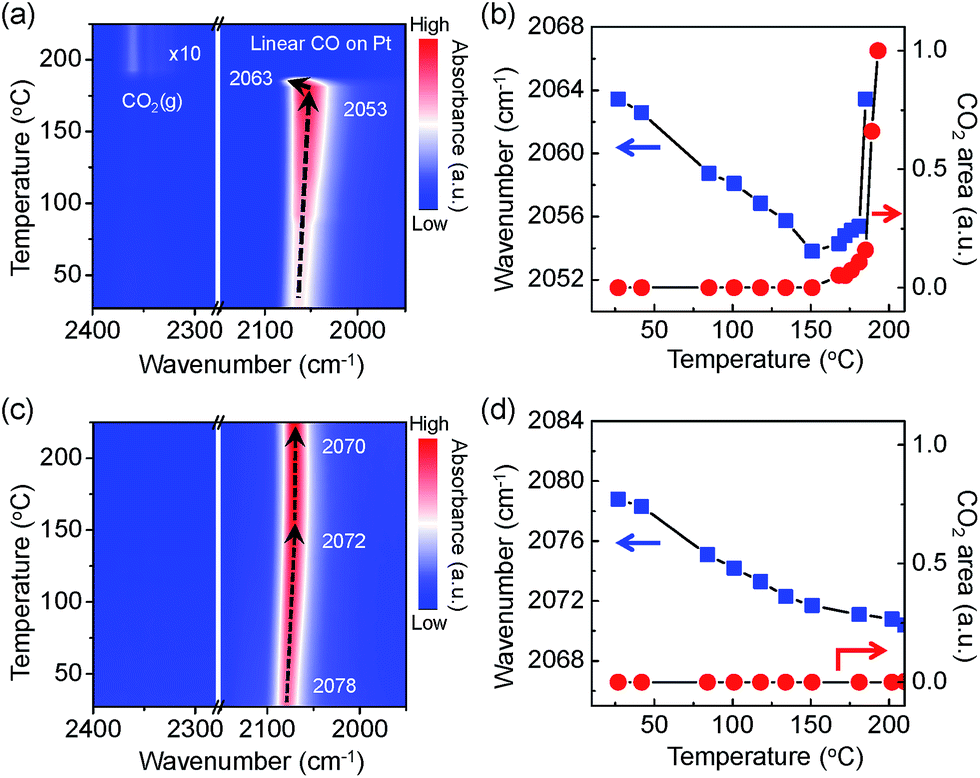 | ||
| Fig. 5 Contour-maps of the in situ measured DRIFT spectra during the CO oxidation over Pt/SiO2 catalysts and the plots of vibration frequency shifts of v(CO) mode with integrated areas of the peaks for a vas(CO) band. (a) A contour-map and (b) the plots for as-synthesised Pt/SiO2 catalysts, and (c) a contour-map and (d) the plots for O2-treated Pt/SiO2 catalysts, respectively. | ||
The trend of the v(CO) peak and formation of produced gaseous CO2 on the O2-treated Pt/SiO2 catalysts are shown in Fig. 5c and d as a function of temperature. The observed red shift of the v(CO) peak has a similar trend as the results of as-synthesised Pt/SiO2 catalysts at elevated temperatures. The evolution of gas phase CO2 does not occur until 225 °C as shown in Fig. 5c. Since bulk Pt oxides are formed during oxygen treatment, the Pt NPs surface has fewer active sites than a metallic Pt surface, which is responsible for decreased catalytic activity.34 It can be expected that the adsorption probability of CO molecules on the Pt NPs catalysts is relatively lower than for as-synthesised Pt NPs catalyst due to fewer reaction sites on the Pt NPs surfaces; even the oxidised Pt NPs are reduced as the CO coverage increases. Thus, the ignition temperature of the CO oxidation reaction on the O2-treated catalysts is slightly higher than that on the as-synthesised Pt/SiO2. The metallic Pt sites can be completely deactivated to non-active phases by the O2-treatment. The energy barrier for the CO oxidation reaction could decrease with an adequate metal oxide layer on the catalysts, as mentioned above. This would be considered as strong evidence that initiation and ignition of the CO oxidation reaction was significantly affected by the presence of an ultrathin oxide layer on the Pt NPs.
Overall, the results of in situ DRIFT analysis show that catalytic activity of the supported Pt NPs catalyst increases by the existence of an ultrathin oxide layer on the Pt NPs; meanwhile the fully oxidised Pt NPs are responsible for a deactivation phenomenon.
3.4 Theoretical calculation for vibration frequency shifts
To further understand the relationship between the measured wavenumber and surface oxide formation in the DRIFT spectra, we performed theoretical calculations for the vibration frequency of the v(CO) mode with relative atomic fractions (AF) of several different configurations of C and O on a model Pt(111) surface using density functional theory (DFT). We assumed that CO molecules adsorb on the top sites of the Pt(111) surface in our proposed models, as described in previously reported experimental and theoretical results.37,54,55 Adsorbed atoms were arranged as far as possible from each other on a model surface with a (3 × 3) unit cell of Pt atoms to prevent potential molecular interferences at a maximum total coefficient value of 3/9. Fig. 6 shows that the calculated wavenumber of the v(CO) mode is considerably changed as a function of the configurations for AF between C and O. The wavenumber of the v(CO) mode is shifted to 2033 cm−1 with an increasing coefficient value of AF(CO) up to 3/9, whereas the red shifted wavenumber is constantly blue shifted up to 2060 cm−1 with an increasing coefficient value of AF(O).
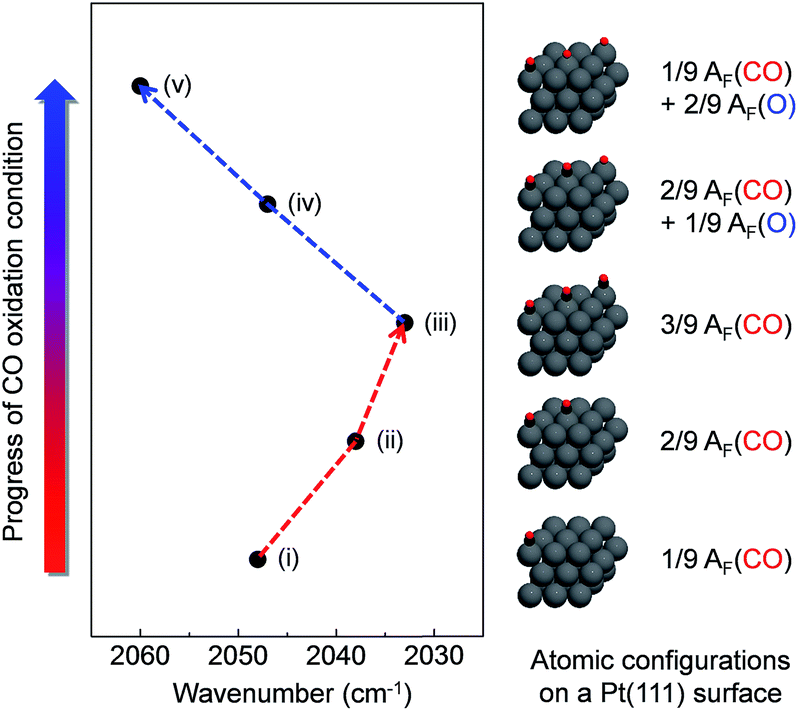 | ||
| Fig. 6 Simulation results of vibration frequency shifts depending on different configurations of CO/O atomic fractions on the Pt model surface. | ||
The expected theoretical wavenumber changes demonstrate a similar trend with our DRIFTS results showing that an underlying mechanism for the red shift of the v(CO) mode is a weakening of the CO bond via enhanced back-bonding of the Pt–CO bond upon the adsorption of additional CO molecules. Likewise, weakening of the Pt–CO bond under the more oxidised conditions (i.e., due to less back-bonding) yields a shortening of the CO bond, which gives rise to a blue shift of the CO stretching mode. The calculated Pt–C and CO bond lengths also qualitatively support these trends (ESI, Table S2†). These results indicate that formation of surface oxide on the terrace sites of a Pt NPs surface is necessary to have a chemically active phase during CO oxidation. The probed intriguing phenomenon would affect controlling catalytic reactivity on the Pt nanocatalysts surfaces.
4. Conclusions
In summary, we reveal the apparent formation of surface oxide on SiO2-supported Pt nanocatalyst surfaces under CO oxidation conditions by in situ investigations employing a DRIFTS technique. The probed vibrational frequency changes for the v(CO) mode on the prepared Pt/SiO2 catalysts surface could be sensitively influenced by the adsorbed molecules chemical binding compositions of nearest-neighbourhood Pt atoms. Under the CO oxidation condition, peak position of the v(CO) mode is abruptly blue shifted because of the formation of an ultrathin oxide layer on the reduced Pt nanocatalyst's surface just below the ignition temperature. This process occurs gradually with the site-specific chemical binding replacements between CO and O on the Pt atoms at an elevated temperature. Otherwise, the O2-pretreated Pt/SiO2 catalysts do not show a similar trend in the process because they have chemically inactive Pt oxides (i.e., multi-layered PtO and PtO2) rather than a surface oxide that already fully covers the Pt nanocatalyst surfaces. DFT calculations suggest reasonable results showing a similar trend of experimental observations where a vibrational frequency change in the v(CO) mode is dependent on the relative atomic fractions of CO and O on the Pt nanocatalyst surfaces. This is inferred to be a key role for the oxide layer in the catalytic activity for CO oxidation, and these investigations provide strong evidence for the existence of the surface oxide as an active species in catalysis.
Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work was supported by Institute for Basic Science (IBS) [IBS-R004-A2-2017-a00]. Y. J. acknowledge the support through the National Research Foundation of Korea from the Korean Government (NRF-2015R1A2A1A15055539 & NRF-2017R1A2B3010176).References
- J. Wintterlin, S. Völkening, T. V. W. Janssens, T. Zambelli and G. Ertl, Science, 1997, 278, 1931–1934 CrossRef CAS PubMed.
- H. Over, Y. D. Kim, A. P. Seitsonen, S. Wendt, E. Lundgren, M. Schmid, P. Varga, A. Morgante and G. Ertl, Science, 2000, 287, 1474–1476 CrossRef CAS PubMed.
- H. Over and M. Muhler, Prog. Surf. Sci., 2003, 72, 3–17 CrossRef CAS.
- A. L. Gerrard and J. F. Weaver, J. Chem. Phys., 2005, 123, 224703 CrossRef PubMed.
- J. G. Wang, W. X. Li, M. Borg, J. Gustafson, A. Mikkelsen, T. M. Pedersen, E. Lundgren, J. Weissenrieder, J. Klikovits, M. Schmid, B. Hammer and J. N. Andersen, Phys. Rev. Lett., 2005, 95, 256102 CrossRef CAS PubMed.
- M. D. Ackermann, T. M. Pedersen, B. L. M. Hendriksen, O. Robach, S. C. Bobaru, I. Popa, C. Quiros, H. Kim, B. Hammer, S. Ferrer and J. W. M. Frenken, Phys. Rev. Lett., 2005, 95, 255505 CrossRef CAS PubMed.
- D. Friebel, D. J. Miller, C. P. O'Grady, T. Anniyev, J. Bargar, U. Bergmann, H. Ogasawara, K. T. Wikfeldt, L. G. M. Pettersson and A. Nilsson, Phys. Chem. Chem. Phys., 2011, 13, 262–266 RSC.
- T. Engel and G. Ertl, Adv. Catal., 1979, 28, 1–78 CAS.
- M. Salmerón, L. Brewer and G. A. Somorjai, Surf. Sci., 1981, 112, 207–228 CrossRef.
- P. J. Berlowitz, C. H. F. Peden and D. W. Goodman, J. Phys. Chem., 1988, 92, 5213–5221 CrossRef CAS.
- M. S. Chen, Y. Cal, Z. Yan, K. K. Gath, S. Axnanda and D. W. Goodman, Surf. Sci., 2007, 601, 5326–5331 CrossRef CAS.
- S. M. McClure and D. W. Goodman, Chem. Phys. Lett., 2009, 469, 1–13 CrossRef CAS.
- F. Gao, Y. Wang, Y. Cai and D. W. Goodman, J. Phys. Chem. C, 2009, 113, 174–181 CAS.
- F. Gao, S. M. McClure, Y. Cai, K. K. Gath, Y. Wang, M. S. Chen, Q. L. Guo and D. W. Goodman, Surf. Sci., 2009, 603, 65–70 CrossRef CAS.
- J. Aβmann, D. Crihan, M. Knapp, E. Lundgren, E. Löffler, M. Muhler, V. Narkhede, H. Over, M. Schmid, A. P. Seitsonen and P. Varga, Angew. Chem., Int. Ed., 2005, 44, 917–920 CrossRef PubMed.
- J. Y. Park, C. Aliaga, J. R. Renzas, H. Lee and G. A. Somorjai, Catal. Lett., 2009, 129, 1–6 CrossRef CAS.
- M. E. Grass, Y. W. Zhang, D. R. Butcher, J. Y. Park, Y. M. Li, H. Bluhm, K. M. Bratlie, T. F. Zhang and G. A. Somorjai, Angew. Chem., Int. Ed., 2008, 47, 8893–8896 CrossRef CAS PubMed.
- K. Qadir, S. H. Joo, B. S. Mun, D. R. Butcher, J. R. Renzas, F. Aksoy, Z. Liu, G. A. Somorjai and J. Y. Park, Nano Lett., 2012, 12, 5761–5768 CrossRef CAS PubMed.
- J. Gustafson, R. Westerström, A. Mikkelsen, X. Torrelles, O. Balmes, N. Bovet, J. N. Andersen, C. J. Baddeley and E. Lundgren, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 045423 CrossRef.
- D. A. J. M. Ligthart, R. A. van Santen and E. J. M. Hensen, Angew. Chem., Int. Ed., 2011, 50, 5306–5310 CrossRef CAS PubMed.
- B. L. M. Hendriksen and J. W. M. Frenken, Phys. Rev. Lett., 2002, 89, 046101 CrossRef CAS PubMed.
- D. R. Butcher, M. E. Grass, Z. H. Zeng, F. Aksoy, H. Bluhm, W. X. Li, B. S. Mun, G. A. Somorjai and Z. Liu, J. Am. Chem. Soc., 2011, 133, 20319–20325 CrossRef CAS PubMed.
- Z. W. Zhu, F. Tao, F. Zheng, R. Chang, Y. M. Li, L. Heinke, Z. Liu, M. Salmeron and G. A. Somorjai, Nano Lett., 2012, 12, 1491–1497 CrossRef CAS PubMed.
- J. T. Yates, T. M. Duncan, S. D. Worley and R. W. Vaughan, J. Chem. Phys., 1979, 70, 1219–1224 CrossRef CAS.
- I. X. Green, W. J. Tang, M. Neurock and J. T. Yates, Science, 2011, 333, 736–739 CrossRef CAS PubMed.
- F. Zaera, ChemCatChem, 2012, 4, 1525–1533 CrossRef CAS.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- G. A. Somorjai and J. Y. Park, Chem. Soc. Rev., 2008, 37, 2155–2162 RSC.
- G. A. Somorjai and J. Y. Park, J. Chem. Phys., 2008, 128, 182504 CrossRef PubMed.
- Q. Zhang, I. Lee, J. P. Ge, F. Zaera and Y. Yin, Adv. Funct. Mater., 2010, 20, 2201–2214 CrossRef CAS.
- K. Gude and R. Narayanan, J. Phys. Chem. C, 2010, 114, 6356–6362 CAS.
- R. M. Rioux, H. Song, J. D. Hoefelmeyer, P. Yang and G. A. Somorjai, J. Phys. Chem. B, 2005, 109, 2192–2202 CrossRef CAS PubMed.
- D. J. Miller, H. Öberg, S. Kaya, H. Sanchez Casalongue, D. Friebel, T. Anniyev, H. Ogasawara, H. Bluhm, L. G. M. Pettersson and A. Nilsson, Phys. Rev. Lett., 2011, 107, 195502 CrossRef CAS PubMed.
- D. Miller, H. Sanchez Casalongue, H. Bluhm, H. Ogasawara, A. Nilsson and S. Kaya, J. Am. Chem. Soc., 2014, 136, 6340–6347 CrossRef CAS PubMed.
- S. K. Calderón, M. Grabau, L. Óvári, B. Kress, H.-P. Steinrück and C. Papp, J. Chem. Phys., 2016, 144, 044706 CrossRef PubMed.
- M. J. Kale and P. Christopher, ACS Catal., 2016, 6, 5599–5609 CrossRef CAS.
- C. Lentz, S. P. Jand, J. Melke, C. Roth and P. Kaghazchi, J. Mol. Catal. A: Chem., 2017, 426, 1–9 CrossRef CAS.
- G. Wu, Y. S. Chen and B. Q. Xu, Electrochem. Commun., 2005, 7, 1237–1243 CrossRef CAS.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- G. Blyholder, J. Phys. Chem., 1964, 68, 2772–2777 CrossRef CAS.
- M. A. Albiter and F. Zaera, Langmuir, 2010, 26, 16204–16210 CrossRef CAS PubMed.
- R. A. Shigeishi and D. A. King, Surf. Sci., 1976, 58, 379–396 CrossRef CAS.
- Y. Borodko, H. S. Lee, S. H. Joo, Y. W. Zhang and G. Somorjai, J. Phys. Chem. C, 2010, 114, 1117–1126 CAS.
- I. Lee, Q. Zhang, J. P. Ge, Y. D. Yin and F. Zaera, Nano Res., 2011, 4, 115–123 CrossRef CAS.
- F. Sen, Y. Karatas, M. Gulcan and M. Zahmakiran, RSC Adv., 2014, 4, 1526–1531 RSC.
- F. Stoop, F. J. C. M. Toolenaar and V. Ponec, J. Catal., 1982, 73, 50–56 CrossRef CAS.
- X. Lin, B. Yang, H. M. Benia, P. Myrach, M. Yulikov, A. Aumer, M. A. Brown, M. Sterrer, O. Bondarchuk, E. Kieseritzky, J. Rocker, T. Risse, H. J. Gao, N. Nilius and H. J. Freund, J. Am. Chem. Soc., 2010, 132, 7745–7749 CrossRef CAS PubMed.
- Y. Xing, J. Phys. Chem. B, 2004, 108, 19255–19259 CrossRef CAS.
- H. Froitzheim, H. Hopster, H. Ibach and S. Lehwald, Appl. Phys., 1977, 13, 147–151 CAS.
- H. Steininger, S. Lehwald and H. Ibach, Surf. Sci., 1982, 123, 264–282 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08952j |
| This journal is © The Royal Society of Chemistry 2017 |