
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication of electroactive poly(vinylidene fluoride) through non-isothermal crystallization and supercritical CO2 processing†
Ji Eun Lee,
Yanting Guo,
Richard Eungkee Lee and
Siu Ning Leung *
*
Department of Mechanical Engineering, Lassonde School of Engineering, York University, Toronto, ON M3J 1P3, Canada. E-mail: sunny.leung@lassonde.yorku.ca
First published on 17th October 2017
Abstract
Poly(vinylidene fluoride) (PVDF) with enhanced β phase content has conventionally been manufactured by mechanical stretching and electric poling of PVDF films. More recently, scientific research has been conducted to investigate the use of electrospinning to promote the piezoelectric properties of PVDF. In this work, we developed a novel processing strategy using a combination of non-isothermal crystallization and supercritical carbon dioxide (ScCO2) processing to promote the electroactive (i.e., β and γ) phase content of PVDF. Differential scanning calorimetric, X-ray diffraction, and infrared spectroscopy results revealed that the preferential formation of β and γ crystal phases were induced by the ScCO2 foaming step and the non-isothermal crystallization step, respectively. The sequential crystallization of these electroactive phases were coupled by their relationships with the ScCO2-induced foam expansion. Furthermore, both ScCO2's plasticization effect and annealing helped the promotion of PVDF's degree of crystallinity. Overall, this work offers a new industrially viable methodology to tailor PVDF's crystalline structures. This represents a useful technique to fabricate piezoelectric PVDF film for sensors, actuators, and energy harvesting applications.
Introduction
Poly(vinylidene) fluoride (PVDF) is a thermoplastic fluoropolymer synthesized from the polymerization of vinylidene difluoride. It consists of an alternating arrangement of two fluorine atoms followed by two hydrogen atoms along the carbon backbone. Due to its piezoelectric properties,1,2 it has raised tremendous interest for the fabrication of miniaturized and novel organic electronic devices. These include nanogenerators,3 tactile and infrared imaging sensors,3 and acoustic sound transducers.4 While polycrystalline ferroelectric ceramic materials such as lead zirconate titanate (PZT) have been the dominant materials as the active elements of transducers and arrays, its lead toxicity has raised concerns over its use. A restriction on the lead contents in consumer products has been placed and it will lead to elimination of PZT use, eventually. In this context, PVDF has been considered as an inexpensive, environmentally benign, and conformable alternative to PZT in sensors, actuators, and energy harvesting applications.2 Moreover, conventional piezoceramics have grain sizes in the order of 5–10 μm, which are not suitable for high frequency transducer applications.5 In contrast, piezoelectric polymers such as PVDF and its copolymer with trifluoroethylene (TrFE) have been found to be far more useful for producing high frequency transducers.6,7As a semi-crystalline polymer, PVDF is commonly known to have five distinct polymorphs (i.e. α, β, γ, δ, and ε).8 α and δ have trans-gauche-trans-gauche (TGTG′) chain conformation, β has zigzag all-trans (TTT) chain confirmation, and γ and ε have T3GT3G′ chain conformation. Among these five polymorphs, the three mostly researched and applied PVDF phases are α, β, and γ phases, the two latter being electroactive. In particular, the β phase has the highest dipole moment (i.e., 8 × 10−30 cm) per unit cell among the three polymorphs,9 and thereby possessing the best piezoelectric, pyroelectric, and ferroelectric properties.10 This has led to extensive and on-going research on developing new processing strategies to promote the contents of electroactive crystal phases within the polymer.
With the best piezoelectric properties among all polymorphs, the β phase of PVDF has been the most extensively researched. There are many different processing methods proposed to achieve high β phase content, each with their own advantages and disadvantages. The most commonly used method is through uniaxial mechanical stretching of PVDF films or filaments. α to β phase transformation is achieved by applying stress to the PVDF samples to align the polymer chains and to induce the TTT chain conformation.11–13 Gomes et al. reported that both the draw ratio and the stretching temperature influenced the effectiveness of this crystal phase transformation.11 Various studies also suggested that the maximum β phase content was achieved at a stretch ratio of approximately 5 and a stretching temperature between 80 °C to 100 °C.12–14 Salimi et al. revealed that the stretch ratio had more significant influence than the stretching temperature on the crystal phase transformation.13
Another strategy to promote β phase formation is through melt crystallization at high pressures or high cooling rates.15–17 Increasing the quenching pressure to 800 MPa yielded PVDF with 100% β phase crystals.16 Moreover, Gradys et al. observed that increasing the cooling rate above 150 K s−1 enhanced the formation of β phase. Further increase of the cooling rate to 2000 K s−1 would result in the formation of only β phase.17 Oka and Koizumi proposed that the nucleation rate of β phase was promoted over that of α phase at low temperatures.18 Therefore, an ultra-high cooling rate would suppress the nucleation of the α phase, and subsequently promote the nucleation of β phase. Annealing PVDF was also shown to promote its β phase formation by increasing PVDF's molecular chain group mobility. However, excessive increase in chain mobility beyond an optimal level would cause the β phase chain groups to revert back to α phase.19 Electric poling is commonly used to align the dipoles of PVDF.20 Such post-processing step after mechanical stretching would also enhance PVDF's β phase content.12
A more recently reported approach to promote β phase formation in PVDF is electrospinning, which can simultaneously induce mechanical stretching and electric poling in a single processing step.21,22 In this approach, a high voltage supply is used to induce a high electric field between a small nozzle head and a metal collecting plate. The morphologies and the crystal forms are controlled through the applied voltage, spinning temperature, flow rate, nozzle size, and the distance between the nozzle and the collecting plate. Zheng et al. revealed that lowering the spinning temperature, decreasing the flow rate, and reducing the separation distance between the nozzle and the collecting plate promoted the formation of β phase crystal in the electrospun PVDF fibers.22 While one key advantage of electrospinning is that it does not require any post-processing step, the electrostatically driven jet of polymer solution can only yield electroactive PVDF fibres in submicron to nano-scale.
While most research focused their efforts to promote the formation of the β phase crystals in PVDF, some researchers have also attempted to identify potential methods to facilitate the formation of the electroactive γ phase crystals. Various studies revealed that a possible processing method to obtain γ phase crystals is through melt crystallization at extremely high temperatures (i.e., close to the melting temperature of γ phase crystals) and at very slow cooling rates.23 Prest and Luca reported that isothermal crystallization of PVDF at 158 °C for 15 hours and 95 hours resulted in the formation of 7.2% and 34.8% of γ phase crystals, respectively.24,25 They also revealed that a higher γ phase content could be obtained by increasing the isothermal crystallization temperature to about 170 °C. The presence of α crystal nuclei during the isothermal crystalization would also enhance the formation of γ spherulites.26–28
The aforementioned fabrication methods have focused on the processing of pure PVDF but there are also researches that investigated the addition of ferrite nanofillers to promote the nucleation of β phase29 and γ phase30 crystals. It has been observed that the interaction between the negatively charged surfaces of the nanoparticles and the positive CH2 bonds of the PVDF allowed the molecular chain groups to rearrange themselves into the electroactive phase conformations.29–31 With the use of ferrite nanofillers, β phase content of up to 90% had been achieved.32
Analyses of polymorphs of PVDF can be done by Fourier transform infrared spectroscopy (FTIR),33 X-ray diffraction (XRD),34 and differential scanning calorimetry (DSC).35 In a recent review literature, Martins et al. suggested that a combination of two or more techniques were needed to clearly identify the specific crystal phases in PVDF.8 For example, FTIR allowed researchers to clearly distinguish α phase (i.e., at a wavenumber of 763 cm−1) from the other two electroactive phases (i.e., β and γ phases); however, the common bands for the two electroactive phases at 840 cm−1 led to some ambiguity to differentiate them. In this context, XRD diffraction peaks could be used to confirm the presence of the electroactive phases from the peaks at 2θ = 20.6° and 20.3° for β and γ phases, respectively.14,36 On the other hand, DSC also represents a complementary technique for the identification of PVDF's crystalline phases. In particular, the γ phase has a distinct melting peak at around 179–180 °C, which is higher than those of the α and β phases (i.e., 167–172 °C). While the crystallization exotherms of α and β phases overlap, the one for the β phase demonstrates a low temperature shoulder.17 Similarly, with a wider melting peak for the β phase, a low temperature shoulder would be observed when both α and β phases coexist.8 Recently, Cai et al. suggested that the unique bands at 1275 cm−1 and 1234 cm−1 for the β phase and the γ phase, respectively, could be employed in a universal phase identification procedure using only the FTIR results.37 In the first step, the 763 cm−1 and 840 cm−1 bands would help to quantify the amount of α phase and that of the electroactive phase, respectively. Consequently, the peak-to-valley height ratio between the peaks at 1275 cm−1 and 1234 cm−1 to their nearest valleys can be used to quantify the fractions of individual β and γ phases.
Thermal analyses of PVDF and their composites conducted by DSC and thermogravimetric analysis (TGA) were reported by various researchers. DSC can be used to obtain the degree of crystallinity and melting peaks of polymers,38 while TGA can be used to analyze the thermal stability of polymers.39 With the inclusion of fillers, it is possible to promote the thermal stability of PVDF.39
This paper investigates the uses of a combination of non-isothermal crystallization and supercritical carbon dioxide (ScCO2) processing to promote the formation of electroactive crystal phases (i.e., β and γ) in PVDF. Non-isothermal crystallization at a temperature range above 160 °C and different temperature profiles were studied in attempt to promote the formation of γ phase crystals. Physical foaming of PVDF by ScCO2 was explored to mimic the strain caused by mechanical stretching and thereby to enhance the formation of β crystals. Parametric studies were conducted to study the effects of processing temperatures during non-isothermal crystallization and ScCO2 processing conditions (e.g., pressure and temperature) on PVDF's volume expansion ratio, foam morphology, degree of crystallinity, and electroactive phase (i.e., β and γ) content. The interrelationship among the formation of γ phase, the volume expansion of PVDF in ScCO2 foaming, and the subsequent formation of β phase were also elucidated through a series of FTIR, XRD, DSC, and scanning electronic microscopy (SEM) analyses. The findings in this study provide insight to future research work on foaming to induce the crystallization of electroactive phases of PVDF, advancing its development in piezoelectric applications.
Experimental
Materials
Commercially available PVDF (Kynar 741, from Arkema, with molecular weight of 282![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was utilized. Carbon dioxide (CO2, from Linde Gas Inc.) of 99.8% purity was used as the physical blowing agent and/or the plasticizing medium of PVDF. Both PVDF and CO2 were used as received without further modification. The supercritical CO2 (ScCO2) medium was obtained by injecting a pressurized CO2 into the PVDF polymer melt in a pressure vessel at an elevated temperature.
000 g mol−1) was utilized. Carbon dioxide (CO2, from Linde Gas Inc.) of 99.8% purity was used as the physical blowing agent and/or the plasticizing medium of PVDF. Both PVDF and CO2 were used as received without further modification. The supercritical CO2 (ScCO2) medium was obtained by injecting a pressurized CO2 into the PVDF polymer melt in a pressure vessel at an elevated temperature.
Film preparation
Weighed amounts of PVDF powder were loaded into a circular disc mold of 120 mm in diameter and 500 μm in thickness, and subsequently compression molded into film samples by the following procedures:000, and 15000 lbs-force for 30 s at each pressure (i.e., a total of 90 s).Non-isothermal crystallization and supercritical CO2 processing
PVDF films were pre-cut into square-shaped samples with a side length of 0.5 inches, and then the samples were transferred to a batch foaming system as illustrated in Fig. 1. ScCO2 processing was conducted using two different temperature profiles, as shown in Fig. 2. The procedures conducted at temperature profile 1 were as follows: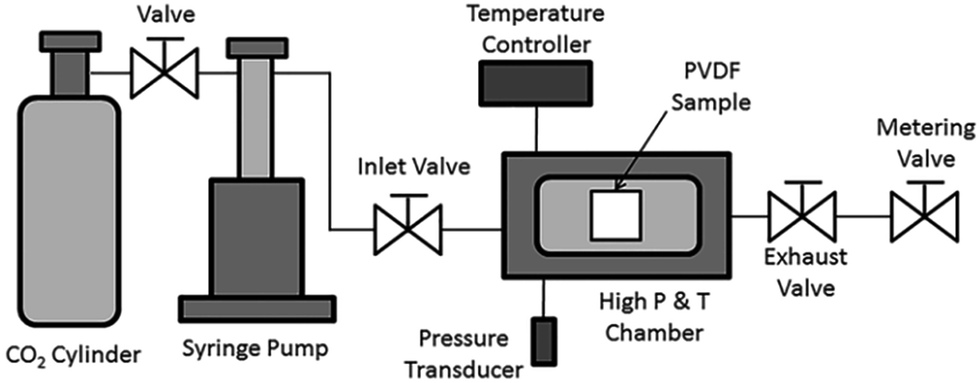 | ||
| Fig. 1 Schematic illustration of a batch foaming system. | ||
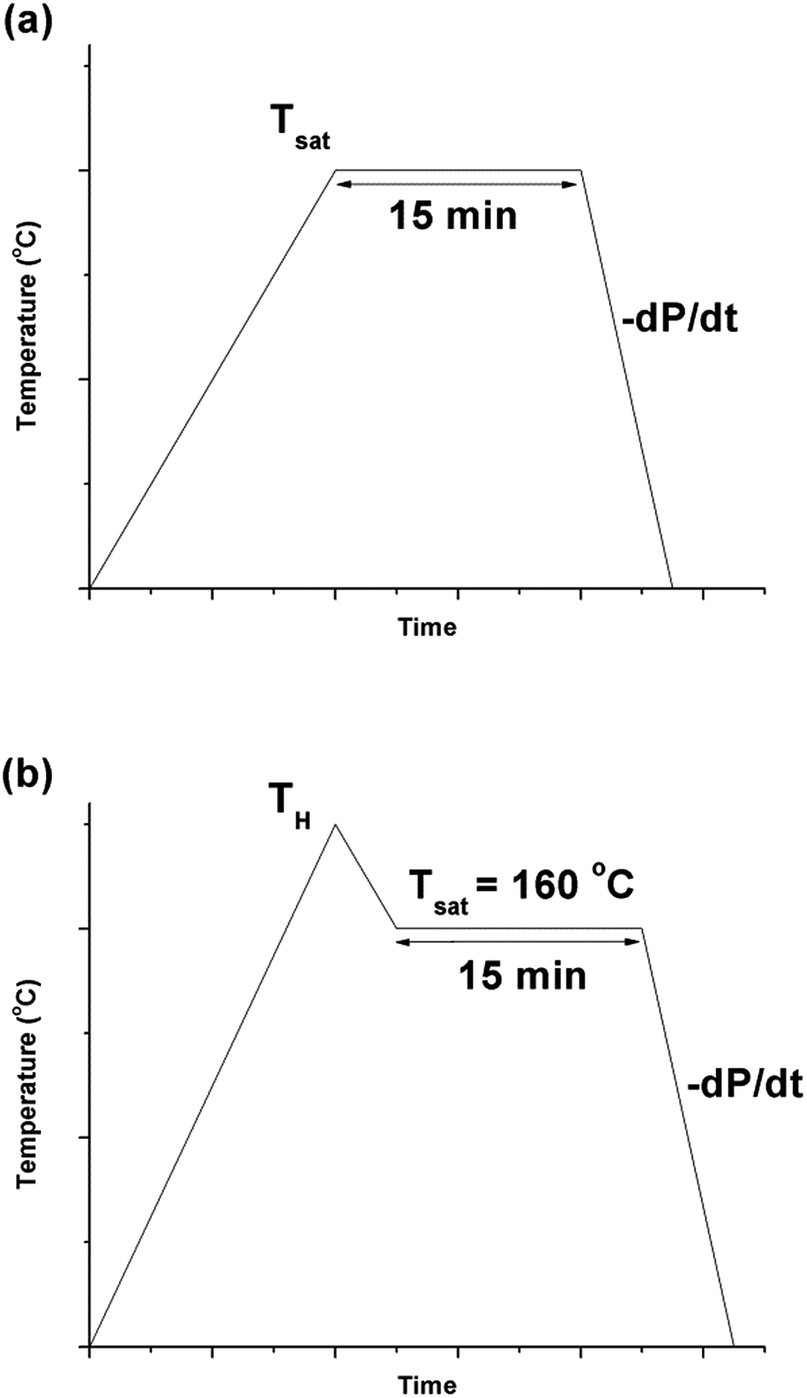 | ||
| Fig. 2 Graphs depicting (a) temperature profile 1 and (b) temperature profile 2, over time. | ||
The procedures for processing PVDF at temperature profile 2 were similar to that conducted at temperature profile 1 with the exception of Step 2. Instead of heating the chamber and sample to Tsat after Step 1, they were heated to a desire heating temperature (TH), which was higher than Tsat. Once TH was reached, the temperature was lowered to Tsat and continued onto Step 3. By varying the opening of the metering valve, two different pressure drop rates (−dP/dt) were achieved during the ScCO2 release. During the temperature decrease from TH to Tsat, non-isothermal crystallization occurred. The crystal structures formed during this step would affect PVDF's crystallization behaviours during the later ScCO2 processing. For experiments conducted at temperature profile 2, the saturation temperature and pressure were fixed at 160 °C and 2000 psi. The selection was based on the results obtained from experiments conducted at temperature profile 1. Details are further explained later in this paper. For comparison purposes, experiments of PVDF samples fabricated at temperature profiles 1 and 2 without the presence of CO2 were also conducted. The experimental conditions for both temperature profiles 1 and 2 are summarized in Table 1.
| Variable | Temperature profile 1 | Temperature profile 2 |
|---|---|---|
| Saturation pressure, Psat (psi) | 1200, 2000, 2500 | 2000 |
| Heating temperature, TH (°C) | N/A | 180, 200 |
| Saturation temperature, Tsat (°C) | 100, 120, 140, 160, 180 | 160 |
| Saturation time, t (min) | 15 | 15 |
| Pressure drop rate, −dP/dt (MPa s−1) | 190.6 | 190.6, 55.2 |
Determination of volume expansion ratio
The volume expansion of ScCO2-processed PVDF samples were determined using ASTM D792 standard. After measuring its weight in air and in water, the apparent density (ρf) and the volume expansion ratio (VER) can be determined by eqn (1) and (2), respectively.
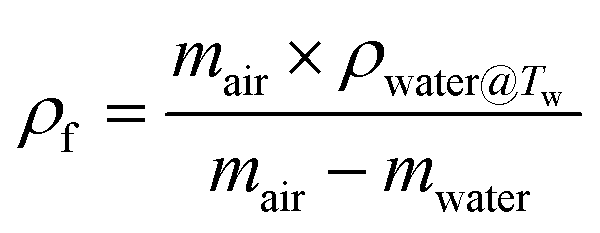 | (1) |
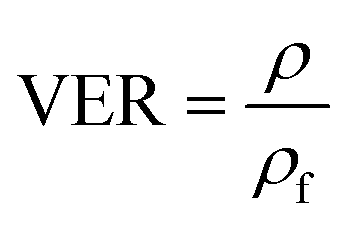 | (2) |
Scanning electron microscopy (SEM)
The morphologies of PVDF samples were characterized by SEM (Quanta 3D FEG, from FEI Company) at different magnifications. The cross-sections were exposed by cryo-fracturing the samples under liquid nitrogen. The fractured surfaces were sputter-coated with gold (Desk V Sputter, Denton Vacuum) before SEM analysis.Differential scanning calorimetry (DSC)
DSC (Q20, from TA Instruments) was used to characterize the melting temperatures and the degree of crystallinity of PVDF samples. Thermal analyses were conducted from 40 °C to 220 °C at a ramping rate of 10°C min−1. The degree of crystallization (Xc) was calculated using eqn (3).
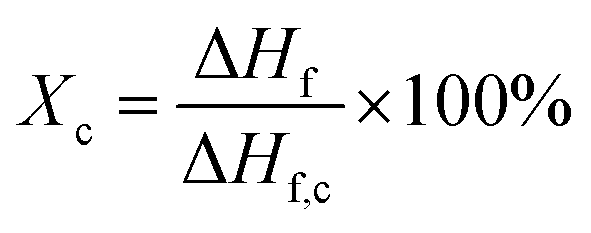 | (3) |
Fourier transform infrared spectroscopy (FTIR)
Infrared spectra of PVDF samples were obtained by a FTIR spectrometer (Alpha-P, from Bruker) by averaging signals from 32 scans in the range from 400 cm−1 to 1500 cm−1 at a resolution of 1 cm−1. As previously mentioned, using FTIR absorbance peaks to analyse the presence of the three main (i.e., α, β, and γ) polymorphs of PVDF is challenging because of the presence of common peaks that appear in multiple crystal phases. In this context, the peak-to-valley height ratio method reported by Cai et al.37 was used to differentiate the three main polymorphs.If the β band at 1275 cm−1 was present but not the γ band at 1234 cm−1, eqn (4) was used. This would denote the 763 cm−1 band as the α phase and the 840 cm−1 electroactive (EA) band as entirely the β phase.
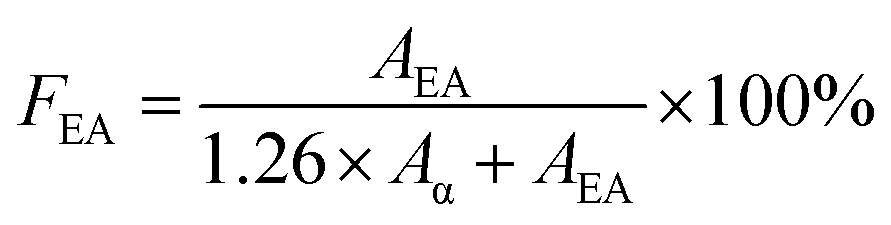 | (4) |
If both 1275 and 1234 cm−1 bands existed, FEA would be subdivided into fractions of β and γ crystals. This could be done using eqn (5) and (6).
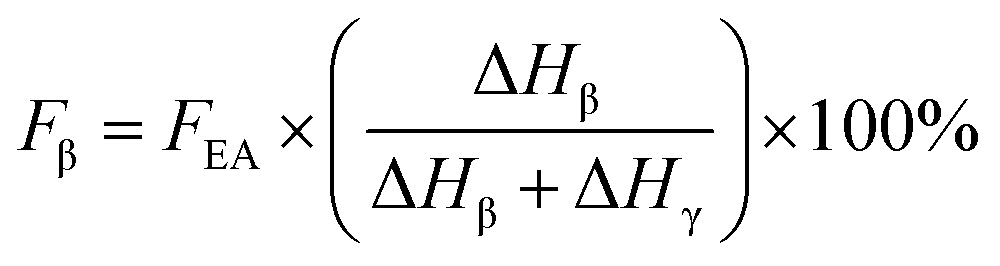 | (5) |
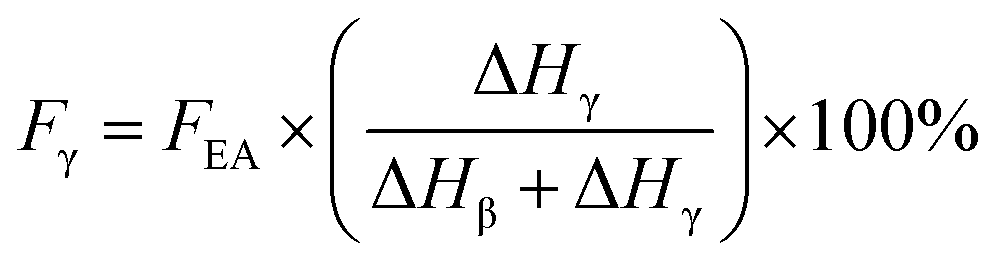 | (6) |
X-ray diffractometer (XRD)
Diffraction peaks of PVDF were obtained using a XRD (Rigaku MiniFlex 600) over a scan range from 10° to 30°, with a scan step of 0.02 at a speed of 2 s per step. Common α peaks were observed at 2θ = 18.4° and 19.9°, β peak at 20.6°, and γ peaks at 20.3°.14,35Results and discussion
Effects of ScCO2 processing conditions on the morphology of PVDF
Fig. 3 shows the effects of ScCO2 processing using temperature profile 1 on the volume expansion ratio (VER) of PVDF. Regardless of the saturation pressure, it can be observed that Tsat of 160 °C led to the highest VER. At other Tsat, the PVDF samples either did not expand or had negligible expansion. When Tsat was too low (i.e., ≤140 °C), the PVDF matrix was too stiff to generate cellular structures. However, when Tsat was too high (i.e., 180 °C), the foam structure could not sustain, resulting in excessive gas loss and decrease in VER.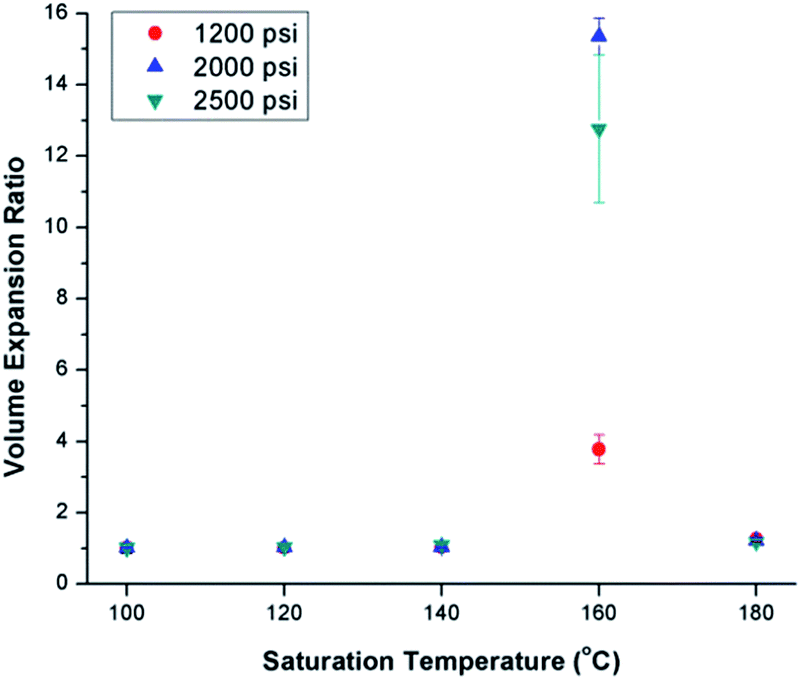 | ||
| Fig. 3 Effect of ScCO2 on the volume expansion ratio of PVDF prepared using temperature profile 1 at various saturation temperatures and pressures. | ||
The foam morphologies of PVDF samples processed at 2000 psi illustrated in Fig. 4 further confirmed these theories. The morphology changed from solid polymer matrix (at 100 and 140 °C), to uniform cellular structure (at 160 °C), and finally to collapsed foam structures with big voids and gas pockets (at 180 °C). It was also observed that when the saturation pressure increased from 1200 psi to 2500 psi at Tsat of 140 °C, tiny voids started to form, as depicted in Fig. 5(a). These voids were only visible under very high magnification (10000×); therefore, they only had negligible effect on the VER (i.e., changing from VER = 1.03 at 1200 psi to VER = 1.08 at 2500 psi). At Tsat of 160 °C, the VER decreased from 15.35 to 11.36 as the saturation pressure increased from 2000 psi to 2500 psi (Part II in ESI†). Such decrease in VER is evidenced by the thin cell walls and the pin holes on them, respectively, shown on Fig. 5(b) and (c), in the sample prepared at 2500 psi. The rupture of cell walls could attribute to the significant plasticization effect induced by ScCO2 and the rapid foam expansion at this high level of saturation pressure.
 | ||
| Fig. 4 SEM micrographs (at 500× magnification) of PVDF samples processed with ScCO2 using temperature profile 1 at 2000 psi and various saturation temperatures: (a) 100 °C; (b) 140 °C; (c) 160 °C; and (d) 180 °C. | ||
 | ||
| Fig. 5 SEM micrographs of PVDF samples processed with ScCO2 using temperature profile 1 at 2500 psi and saturation temperatures: (a) 140 °C at 10000×; (b) 160 °C at 500×; and (c) 160 °C at 5000×. | ||
For samples prepared at temperature profile 1, it seems that PVDF could only foam near Tsat of 160 °C, regardless of the saturation pressure. The highest VER of PVDF was achieved at a saturation pressure of 2000 psi. To elucidate the effect of bubble growth-induced strain on the formation of PVDF's electroactive phases and to explore the potential of ScCO2 processing to tailor PVDF's crystalline structures, the saturation temperature and pressure for samples prepared under temperature profile 2 were kept at 160 °C and 2000 psi, respectively, for all conditions.
Effect of ScCO2 processing conditions on the degree of crystallinity in PVDF
Fig. 6(a) illustrates the degree of crystallinity (Xc) of PVDF samples prepared using temperature profile 1 without the presence of ScCO2. It was observed that Xc decreased as the processing temperature increased. This trend can be explained by the thermogram of compression-molded PVDF sample shown in Fig. 6(b). The melting of PVDF's crystals started at around 140 °C and ended at approximately 180 °C. When the PVDF samples prepared using temperature profile 1 were processed at a temperature lower than its onset of melting (i.e., <140 °C), it yielded a higher Xc due to the annealing effect on the pre-existing crystals.19 Above 140 °C, the annealing effect was less pronounced due to the partial melting of pre-existing crystals; therefore, the overall Xc reduced. When the processing temperature increased to 180 °C, all of the existing crystals had been melted completely. The absence or very limited amount of crystal nuclei would limit the annealing effect to promote PVDF's crystallization. Being processed at this temperature, Xc of PVDF was similar to that of the compression-molded sample, which was approximately 43% (Part III in ESI†).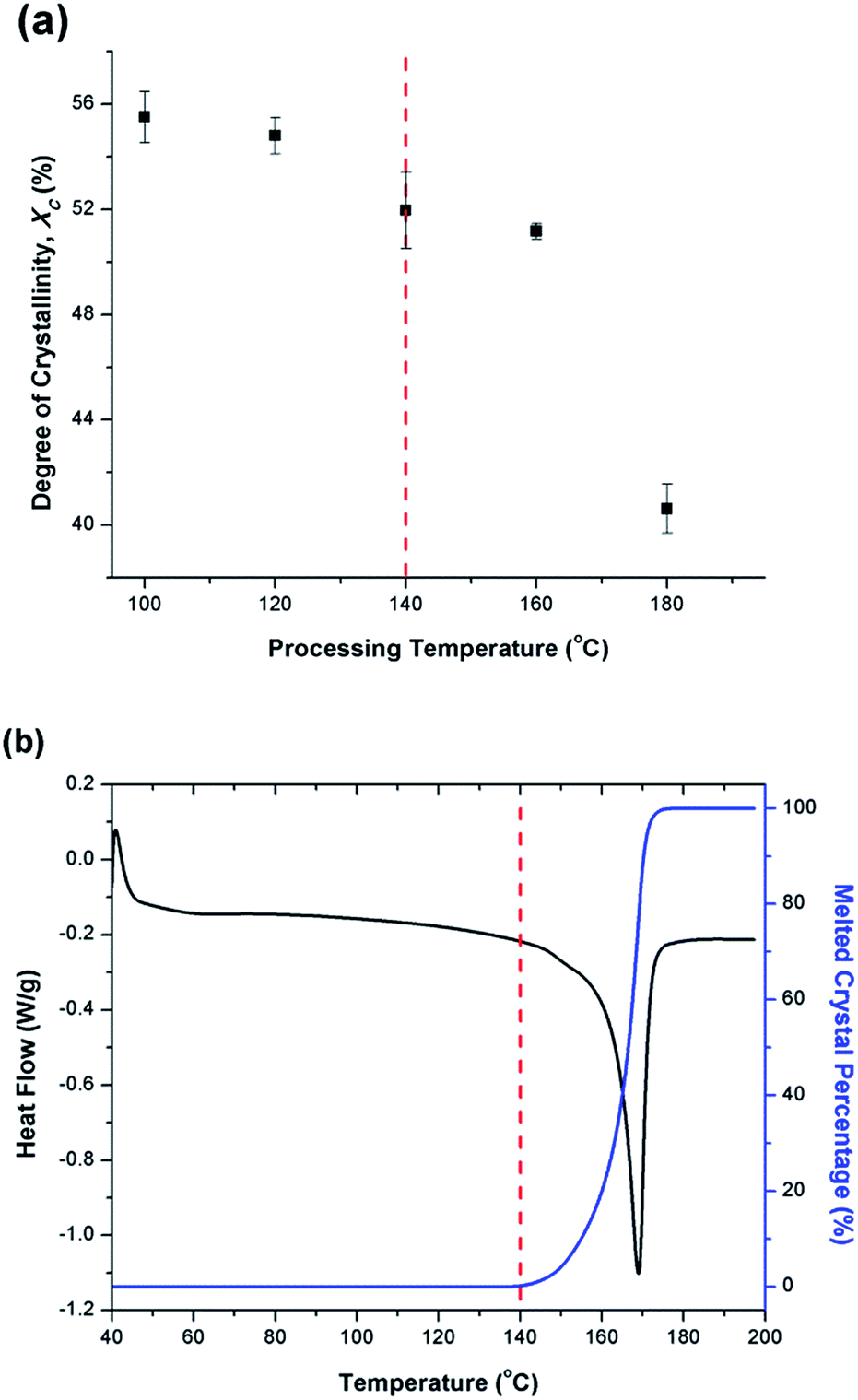 | ||
| Fig. 6 Thermal characteristics of PVDF: (a) degree of crystallinity of samples fabricated using temperature profile 1 without the presence of CO2; and (b) melted crystal percentage and thermogram for compression molded sample. | ||
By preparing PVDF samples using temperature profile 1 with the presence of ScCO2, the plasticization effect of ScCO2 would influence the PVDF's Xc. The results were shown in Fig. 7. The plasticization effect of ScCO2 would increase PVDF's molecular chain mobility and decrease its melting temperature. The level of plasticization effect varied with saturation pressure and temperature. At 140 °C, due to partial melting of crystals, there were pre-existing crystals in the PVDF matrix that were used as seeds for crystal growth and perfection. An optimal level of plasticizing effect would enhance the chain mobility around the crystal nuclei and thereby promote crystal perfection during gas saturation. This increased the Xc of the PVDF matrix. However, when Tsat increased to 160 °C, the suppression of the melting point by ScCO2 plasticizing effect would disrupt the pre-existing crystal structure. This led to a scarce number of seeds for crystal perfection despite the high mobility of PVDF's molecular chain groups. Moreover, the fast CO2 consumption during the foaming process rapidly increased the melting temperature back to the original level. This did not allow sufficient time for the crystals to grow. As a result, the degree of crystallinity decreased despite the high volume expansion ratio observed at 160 °C. Each saturation pressure achieved a maximum degree of crystallinity of approximately 54% at their own distinct saturation temperatures (Part III in ESI†).
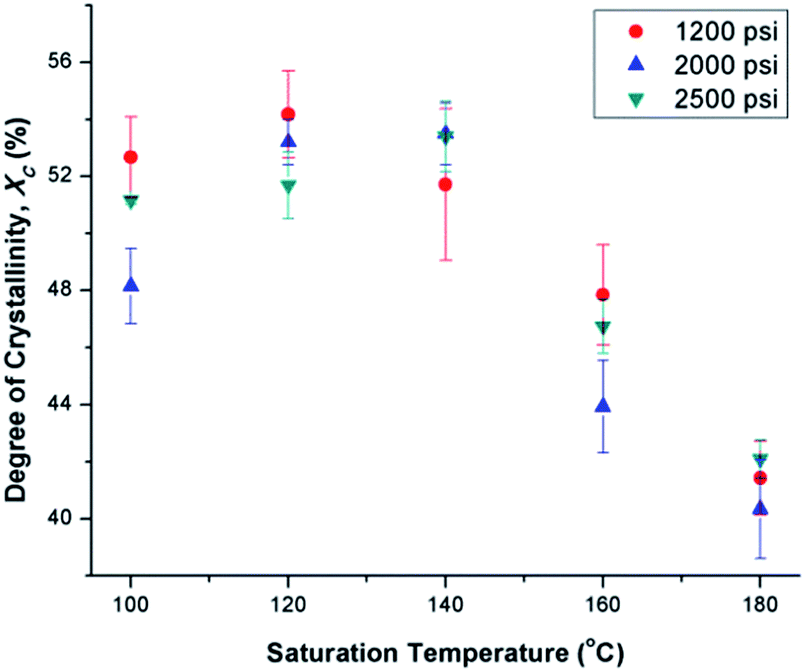 | ||
| Fig. 7 Comparison of the effect of ScCO2 using temperature profile 1 on the degree of crystallinity of PVDF at various saturation temperatures and pressures. | ||
At the same saturation temperature but lower saturation pressure, there is a lower degree of plasticization effect.40 This resulted in a larger amount of existing crystals prior to foaming. This hindered PVDF's foamability because CO2 could only dissolve in its amorphous domains. The non-uniform cell structure and thick cell walls shown in Fig. 8(a) had further confirmed its high Xc. As the saturation pressure increased from 1200 psi to 2000 at a steady Tsat of 160 °C, Xc decreased from 48% to 44%. This was due to the higher degree of plasticization effect, which reduced the amount of crystals and yielded uniform cell structures as observed in Fig. 8(b).
 | ||
| Fig. 8 SEM micrographs of PVDF samples processed with ScCO2 using temperature profile 1 at Tsat = 160 °C and Psat of (a) 1200 psi; and (b) 2000 psi. | ||
Effects of ScCO2 processing conditions on the electroactive phases of PVDF
FTIR analysis was conducted for PVDF samples prepared using temperature profile 1 with the presence of ScCO2 to observe its distinct three main polymorphs (i.e., α, β, and γ). Fig. 9 shows the absorbance curves for the compression molded sample as well as ScCO2 processed samples at 2000 psi and various Tsat (i.e., 140 °C, 160 °C, and 180 °C). Peaks at wavenumbers of 763 cm−1, 840 cm−1, and 1275 cm−1 are present in all FTIR spectra but there is no peak at 1234 cm−1. This indicates that only α and β phases were present, and thereby Fβ was equal to FEA. It can also be seen that the β phase peak at 840 cm−1 increased as Tsat increased to 160 °C, and decreased when it reached 180 °C. At the same time, the α phase peak decreased when Tsat was set at 180 °C. As the Tsat increased beyond PVDF's melting temperature of 168 °C, the crystalline structure transformed to the unorderly amorphous phase due to the excessive chain group mobility. As the quantity of the distinct polymorphs decreased, so did the overall degree of crystallinity. The same trend was also observed in Fig. 7. Moreover, the increase in β phase peak at Tsat of 160 °C could deduce that the formation of β phase was related to the foaming-induced strain and/or fast cooling during the rapid depressurization of the foaming chamber.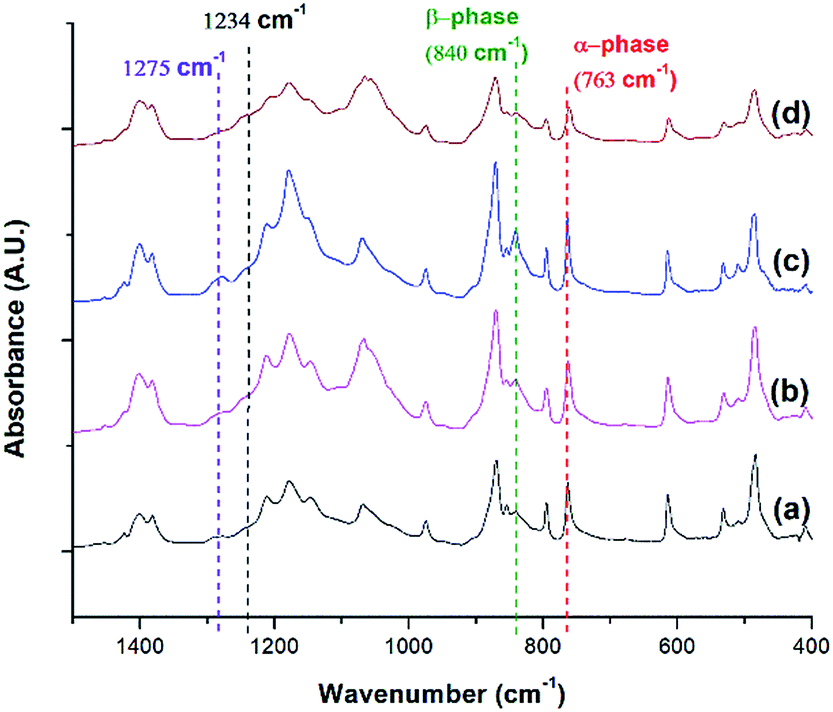 | ||
| Fig. 9 FTIR absorbance graph of PVDF: (a) compression molded sample; ScCO2 processing using temperature profile 1 at Psat = 2000 psi and Tsat of (b) 140 °C; (c) 160 °C; and (d) 180 °C. | ||
Effects of non-isothermal crystallization and ScCO2 processing on the electroactive phases of PVDF
FTIR spectra shown in Fig. 10 indicates that there were drastic changes in the composition of PVDF crystalline phases between the compression molded sample and the samples undergone non-isothermal crystallization and ScCO2 processing under temperature profile 2. An additional heating step, which heated the PVDF sample to an elevated heating temperature (i.e., 180 °C) and then lowering and equilbriating it at Tsat (i.e., 160 °C) generated a peak at 1234 cm−1 (i.e., a wavenumber attributed to the presence of γ phase). In contrast, γ phase was not present in the samples prepared under temperature profile 1. The peak-to-valley height ratio method32 was used to separate the content of the electroactive phase into Fβ and Fγ. Furthermore, non-isothermal crystallization had led to an increase in the height of the electroactive peak at 840 cm−1 and a decrease in the α phase content. This phenomena is more pronounced when the sample was processed with the presence of ScCO2. According to literature,30 the γ phase has a melting temperature at around 179–180 °C. It is believed that non-isothermal crystallization occurred when the temperature decreased from 180 °C to 160 °C. This led to the formation of the γ phase using the existing α phase as seeds. The subsequent step of processing PVDF with ScCO2 decreased the α phase peak and increased the electroactive peak at 840 cm−1 and the β phase peak at 1275 cm−1. The fast consumption of CO2 during foaming suppressed the plasticizing effect and thereby increased the melting temperature of PVDF back to its original level. Together with the fast cooling came with the rapid pressure drop, this favored the growth of β phase crystals.17,18 Moreover, it is also believed that the 4.5 times volume expansion of PVDF would mimic the effects of mechanical stretching on crystal phases transformation. This was evidenced by the increase of β phase content from 12%, from the compression molded sample in Fig. 10(a), to 23% in Fig. 10(b) due to the foam expansion under a ScCO2 environment. Together with the 52% of γ phase, this sample possessed a total of 75% electroactive crystal phase.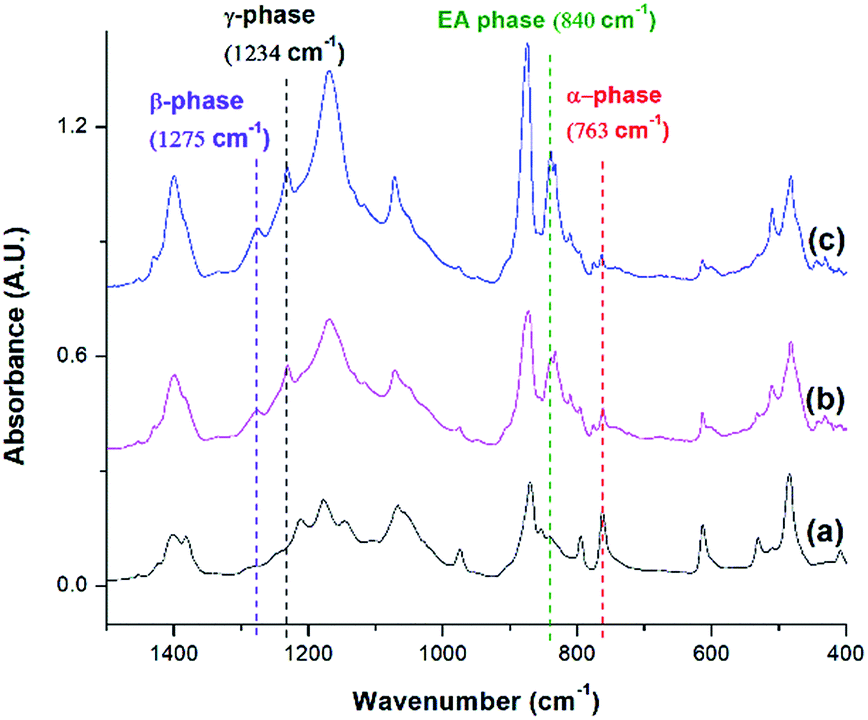 | ||
| Fig. 10 FTIR absorbance graph of PVDF: (a) compression molded sample; using temperature profile 2 at TH = 180 °C (b) without the presence of CO2; and (c) with ScCO2 at −dP/dt = 190.6 MPa s−1. | ||
Fig. 11 compares the three conditions investigated using temperature profile 2: TH = 180 °C, TH = 200 °C, and decreasing the pressure drop rate (−dP/dt) from 190.6 MPa s−1 to 55.2 MPa s−1 at TH = 180 °C. Similar to the previous experiments, the saturation pressure and temperature were set at 2000 psi and 160 °C, respectively. When −dP/dt rate decreased from 190.6 MPa s−1 to 55.2 MPa s−1 at TH = 180 °C, there was a slight decrease in the electroactive phase peak (840 cm−1) and the β phase (1275 cm−1) peak. The reduced gas removal rate caused a slower return of PVDF's melting temperature to is original level. This could have promoted α phase growth and prohibited β phase formation.17,18 The sample prepared at a lower −dP/dt had a lower VER compared than the sample prepared a higher −dP/dt. It is noteworthy that there was negligible the effect of −dP/dt on γ phase content, indicating that the effect of −dP/dt on the electroactive phase content was mainly through its influence on the β phase.
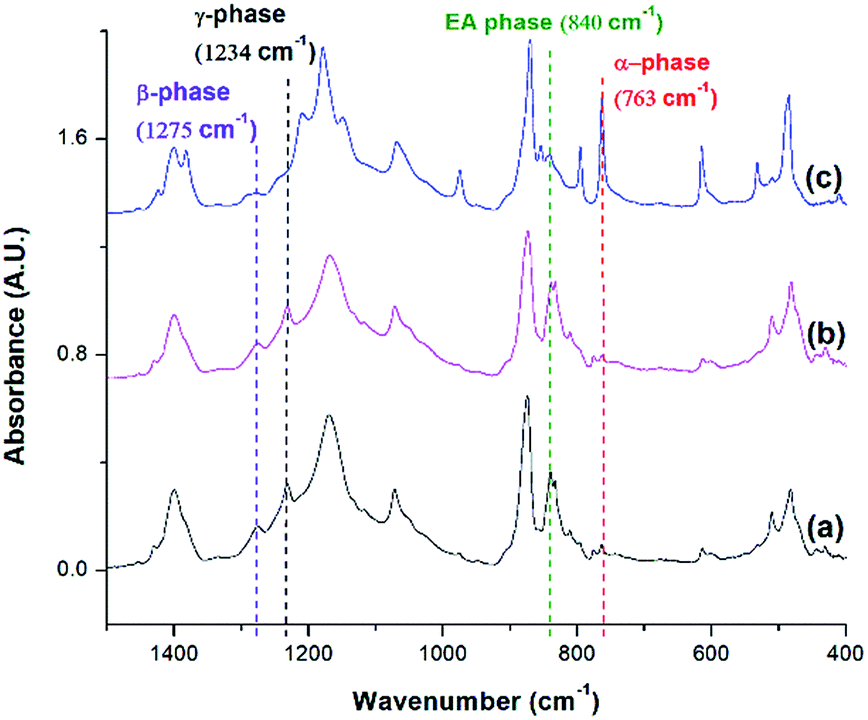 | ||
| Fig. 11 FTIR absorbance graph of ScCO2 processing using temperature profile 2 at Psat = 2000 psi, Tsat = 160 °C, and (a) TH = 180 °C, −dP/dt = 190.6 MPa s−1; (b) TH = 180 °C, −dP/dt = 55.2 MPa s−1; and (c) TH = 200 °C, −dP/dt = 190.6 MPa s−1. | ||
When the TH increased to 200 °C, the γ phase peak at 1234 cm−1 disappeared and there was an increase in the α phase peak. At this high TH, the majority of the α phase crystals could have melted, leaving insufficient amount of α seeds in the PVDF matrix to facilitate the formation γ phase during the non-isothermal crystallization step. Subsequently, cooling the PVDF sample from 200 °C to 160 °C would have caused a reformation of α phase crystals. This led to the absence of the γ phase peak and the increase in the α phase peak compared to the other conditions.
It is important to note that the FTIR spectrum for PVDF prepared at TH = 200 °C showed no γ phase peak at 1234 cm−1 but a β phase peak at 1275 cm−1. Therefore, the electroactive phase peak at 840 cm−1 represented entirely the β phase. As seen in Table 2, the PVDF sample prepared at TH = 200 °C revealed to have the highest β phase content of 29.10%. Interestingly, this condition also had the highest VER of 11 times (Part IV in ESI†) among those prepared using temperature profile 2. By comparing all the results obtained from samples prepared under temperature profiles 1 and 2, it is possible to solidify our earlier speculation that the formation of β phase crystals was due to the strain generated by bubble growth. Higher VER would lead to a higher β phase content. In the meantime, the fast cooling induced by the rapid pressure drop during foaming also played a positive role in β phase formation. It is also worth mentioning that the promotion of γ crystals in the non-isothermal crystallization step was detrimental to foaming (i.e., lower VER) and hence indirectly affected the β phase.
| Psat (psi) | TH (°C) | Tsat (°C) | −dP/dt (MPa s−1) | Average Fβ (%) | Average Fγ (%) |
|---|---|---|---|---|---|
| No CO2 | 180 | 160 | 190.6 | 12.29 | 51.09 |
| 2000 | 180 | 160 | 190.6 | 22.81 | 51.75 |
| 2000 | 180 | 160 | 55.2 | 16.32 | 57.53 |
| 2000 | 200 | 160 | 190.6 | 28.93 | 0.00 |
In addition to FTIR, XRD and DSC analyses were used to confirm the FTIR results. From the XRD diffraction peaks indicated in Fig. 12, the presence of α and the electroactive phases are observed. The α phase peaks at 18.4° and 19.9° are the highest at TH = 200 °C, consistent with the FTIR peaks seen in Fig. 11. The electroactive β and γ phases have boardening peaks at 20.6° and 20.3°, respectively. Due to their close proximity, the peak at 2θ around 20.3° to 20.6° was denoted as the electroactive (EA) phase. This peak is more pronounced at TH of 180 °C, for both pressure drop rates, due to the presence of both β and γ phases. This peak is abruptly decreased at TH of 200 °C, a condition that showed no γ phase and a smaller 840 cm−1 peak in Fig. 11. This qualitative XRD analysis provides additional evidence to the findings from the FTIR spectra on the effects of the various processing parameters on the promotion of the crystallization of the electroactive phases.
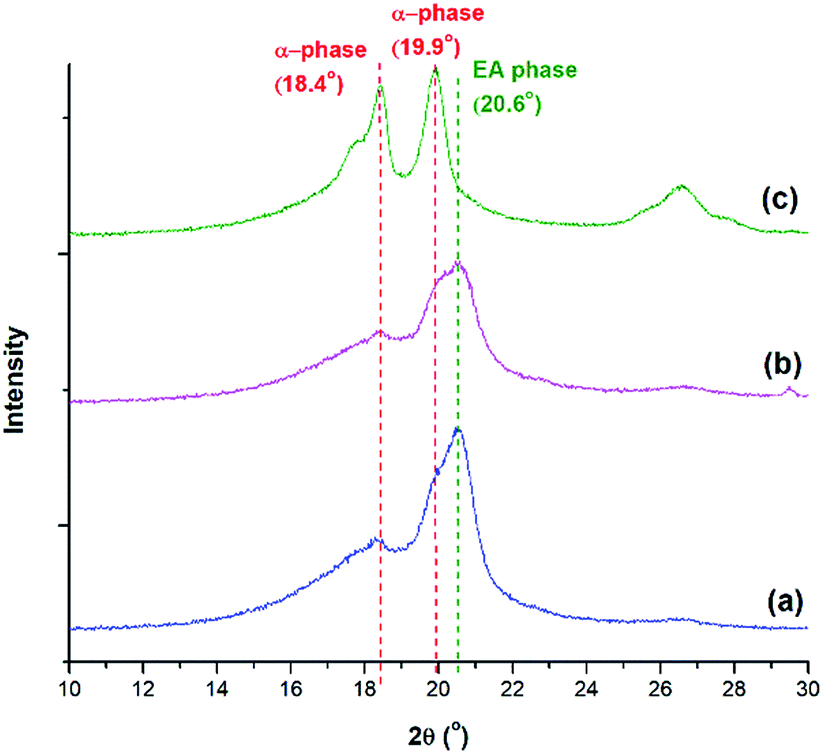 | ||
| Fig. 12 XRD diffraction peaks of PVDF using temperature profile 2 at Psat = 2000 psi, Tsat = 160 °C and (a) TH = 180 °C, −dP/dt = 190.6 MPa s−1; (b) TH = 180 °C, −dP/dt = 55.2 MPa s−1; and (c) TH = 200 °C, −dP/dt = 190.6 MPa s−1. | ||
The γ phase is known to have a melting peak at 179–180 °C.30 DSC thermographs shown in Fig. 13 reveal that γ phase existed in all samples except the one prepared at TH of 200 °C. The α phase, which is known to have a narrower melting peak ranging from 165–172 °C,25,41 is the sharpest for the condition TH = 200 °C. The β phase is known to have a melting peak overlapping with the one for the α phase; however, it is a broader melting peak.8 Such characteristic peak can be seen in all conditions as the initial shoulders and/or part of the wide and lower temperature melting peak. The presence of these distinct traits provide additional evidence to the findings discussed based on the FTIR analyses.
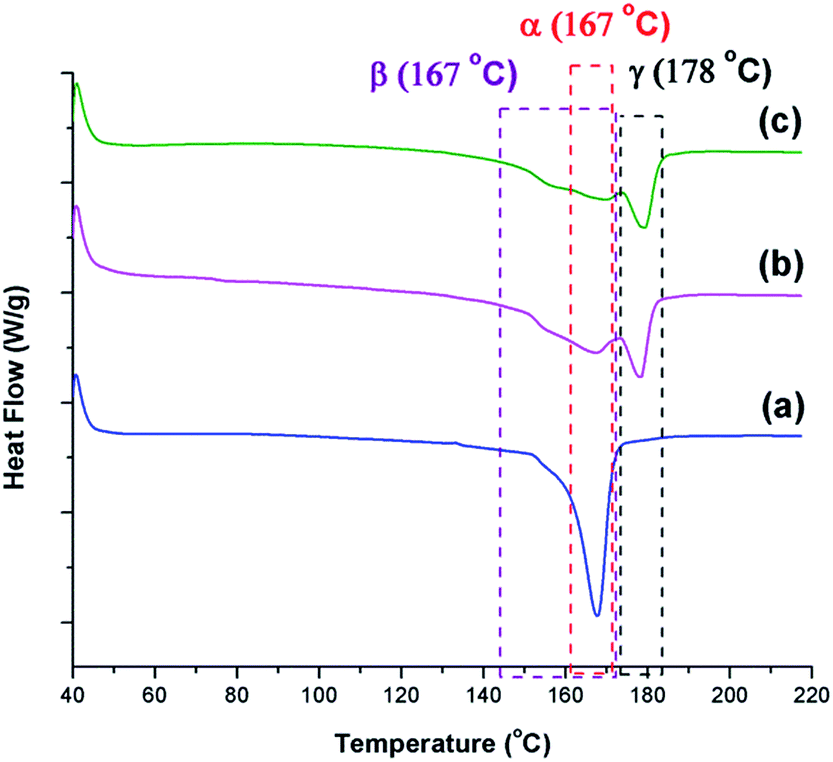 | ||
| Fig. 13 DSC thermograms for samples prepared under temperature profile 2 at Psat = 2000 psi, Tsat = 160 °C, and (a) TH = 200 °C, −dP/dt = 190.6 MPa s−1; (b) TH = 180 °C, −dP/dt = 190.6 MPa s−1; and (c) TH = 180 °C, −dP/dt = 55.2 MPa s−1. | ||
The non-isothermal crystallization and ScCO2 processing did not change PVDF's total degree of crystallinity, which maintained at around 52% (Part V in ESI†). The single exception was the minor reduction in the degree of crystallinity in PVDF sample fabricated at a lower pressure drop rate. As biaxial stretching along the cell walls during foam expansion may assist PVDF crystallization, this decrease in crystallinity could be attributed to the low pressure drop rate and the reduced foam expansion.
Effects of non-isothermal crystallization and ScCO2 processing on the foam morphology of PVDF
The relationship between β phase content and PVDF's VER has become evident by studying various parameters, including the presence of ScCO2, pressure drop rate, and finally, the heating temperature, as discussed in the earlier sections. High VER is beneficial to the β phase formation, which can be attributed to the high local strain introduced on the cell wall of PVDF foam during bubble expansion. Moreover, it can be identified that the mechanism for β and γ phase formations are independent but indirectly affect each other through the ScCO2 foaming step. γ phase formation occurred during the non-isothermal crystallization before the CO2 injection, and the presence of existing α phase crystals was a necessary condition. Experimental results reveal that PVDF foams would have lower VER when there existed a high content of γ crystals prior to foaming. The naturally compact chain group conformation of the γ phase limited the amount of ScCO2 dissolved into the polymer matrix. Therefore, the amount of CO2 available for bubble nucleation and growth decreased, causing a lower VER of the PVDF foam. This ultimately resulted in a reduced β phase content due to the lower degree of local strain within the cell walls. Fig. 14 clearly shows the opposing trends of β phase and γ phase content as the VER varied. In other words, the γ phase content indirectly influences the β phase content through its impact on the foam expansion.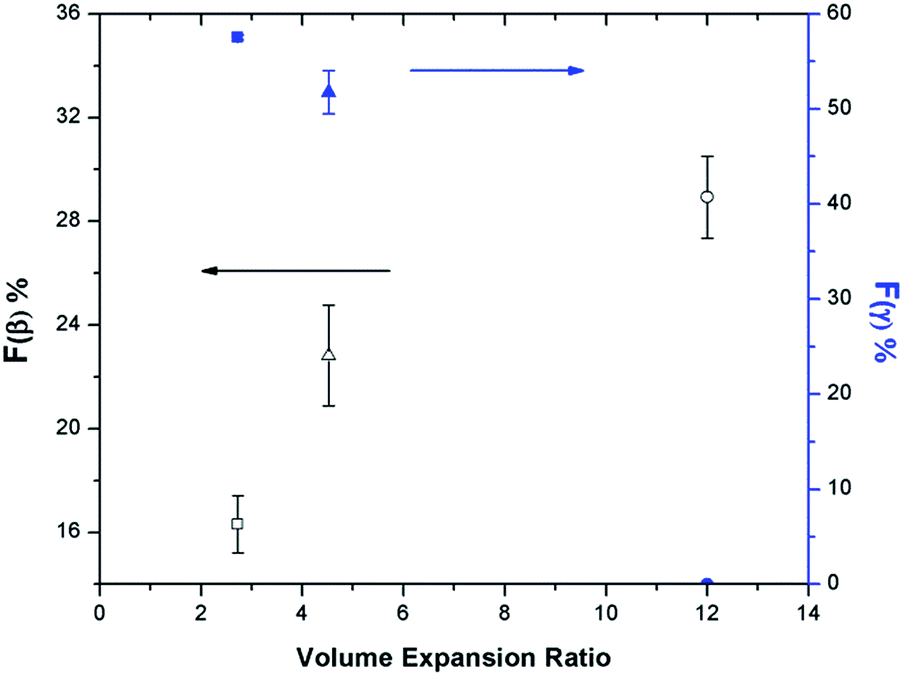 | ||
| Fig. 14 Relationship between Fγ vs. VER vs. Fβ of samples prepared under temperature profile 2 at Psat = 2000 psi, Tsat = 160 °C, and: (square) TH = 180 °C, −dP/dt = 55.2 MPa s−1; (triangle) TH = 180 °C, −dP/dt = 190.6 MPa s−1; (circle) TH = 200 °C, −dP/dt = 190.6 MPa s−1. | ||
The morphologies of the samples prepared using temperature profile 2 are illustrated in Fig. 15. At TH = 180 °C, samples had non-uniform cell structures due to high γ phase content (Fig. 15(a) and (b)). In contrast, when TH increased to 200 °C, it resulted in uniform cell structure without the presence of γ phase crystals (Fig. 15(c) and (d)). Furthermore, the crazes in the cell walls illustrated in Fig. 15(d) indicated that TH was too high, and it led to the rupture of cell walls. The microstructures of PVDF samples observed from SEM are consistent with what have been reported in literature by Dutta et al., and Meng et al. for all the samples processed using temperature profile 2 (Part VI in ESI†).42,43
 | ||
| Fig. 15 SEM micrographs (at 500× magnification) of PVDF samples using temperature profile 2 at 2000 psi, Tsat = 160 °C and TH of: (a) 180 °C, −dP/dt = 190.6 MPa s−1; (b) 180 °C, −dP/dt = 55.2 MPa s−1; (c) 200 °C with −dP/dt = 190.6 MPa s−1; and (d) 200 °C with −dP/dt = 190.6 MPa s−1 at 5 000×. | ||
Conclusion
This paper explored the effects of non-isothermal crystallization and supercritical carbon dioxide (ScCO2) processing on the degree of crystallinity and the formation of electroactive phases of polyvinylidene fluoride (PVDF) through two different temperature profiles. Heating PVDF to 180 °C before conducting ScCO2 foaming at 160 °C yielded PVDF that consisted of 75% electroactive phases (i.e., β and γ). The integrated processing strategy proposed in this work represents a significant technology advancement due to its industrial viability when comparing to other existing technology.Experimental results reveal that ScCO2 plasticized PVDF. This would suppress the melting point of PVDF and influence the pre-existing crystal structures as well as the crystallization behaviours. In particular, if there were no or very limited amount of pre-existing crystals to act as seeds, crystal perfection was absent in the annealing step. Evidenced by this, the decrease in the degree of crystallinity of PVDR decreased as the temperature increased beyond it’s onset temperature of melting. Parametric studies on ScCO2 processing of PVDF revealed that saturation pressure and temperature at 2000 psi and 160 °C, respectively, were the optimal conditions to generate a uniform cellular structure, without excessive gas loss, and yield the highest VER (i.e., 15.35). Therefore, these conditions were adopted for all samples prepared under temperature profile 2 to investigate the effect of bubble growth induced strain on the content of PVDF's electroactive phases.
Temperature profile 2 utilized a combination of non-isothermal crystallization and ScCO2 processing to promote the formation of PVDF's electroactive phases (i.e., β and γ). Parametric studies were conducted to investigate the effects of the presence of ScCO2, pressure drop rate (−dP/dt), and elevated temperature (TH) prior to ScCO2 processing step on the formation of the electioactive crystal phases. Experimental results revealed that the mechanism for β and γ phase formations occurred in independent processing steps but indirectly influenced each other through foam expansion. It was observed that γ phase crystals formed in the non-isothermal crystallization process (i.e., the temperature decrease from TH to Tsat) utilized the existing α phase crystal seeds. This phenomena was absent when TH increased from 180 °C to 200 °C. This was caused by removal of almost all α phase crystals before the non-isothermal crystallization. The increase in the formation of γ crystals during the non-isothermal crystallization hindered foaming and limited VER. The lower degree of local strain within the cell walls decreased the β phase content. It is believed that a high VER would promote the formation of β phase crystals through two mechanisms. Firstly, the strain induced by bubble expansion would mimic the effect of mechanical stretching on transforming α phase crystals into β phase crystals. Secondly, results obtained from samples prepared at different pressure drop rates indicated that the faster cooling rate caused by a more rapid pressure drop also enhanced β phase formation. The presence and the contents of the distinct polymorphs were confirmed through FTIR analysis, XRD diffraction peaks, and DSC thermograms.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada [Grant No. RGPIN-2015-04148].References
- H. Kawai, Jpn. J. Appl. Phys., 1969, 8, 975–976 CrossRef CAS.
- A. J. Lovinger, Developments in Crystalline Polymers, ed. D. C. Basset, Elsevier Applied Science, London, 1982, vol. 1 Search PubMed.
- D. Mandal, S. Yoon and K. J. Kim, Macromol. Rapid Commun., 2011, 32, 831–837 CrossRef CAS PubMed.
- Q. X. Chen and P. A. Payne, Meas. Sci. Technol., 1995, 6, 249–267 CrossRef CAS.
- Q. Zhou, S. Lau, D. Wu and K. K. Shung, Prog. Mater. Sci., 2011, 56, 139–174 CrossRef CAS PubMed.
- M. D. Sherar and F. S. Foster, Ultrason. Imaging, 1989, 11, 75–94 CrossRef CAS PubMed.
- J. A. Ketterling, O. Aristizábal, D. H. Turnbull and F. L. Lizzi, IEEE Trans. Sonics Ultrason., 2005, 52, 672–681 CrossRef.
- P. Martins, A. C. Lopes and S. Lanceros-Mendez, Prog. Polym. Sci., 2014, 39, 683–706 CrossRef CAS.
- H. M. G. Correia and M. M. D. Ramos, Comput. Mater. Sci., 2005, 33, 224–229 CrossRef CAS.
- S. Lanceros-Méndez, J. F. Mano, A. M. Costa and V. H. Schmidt, J. Macromol. Sci., Part B: Phys., 2001, 40, 517–527 CrossRef.
- J. Gomes, J. S. Nunes, V. Sencadas and S. Lanceros-Méndez, Smart Mater. Struct., 2010, 19, 065010 CrossRef.
- V. Sencadas, R. Gregorio Jr and S. Lanceros-Méndez, J. Macromol. Sci., Part B: Phys., 2009, 48, 514–525 CrossRef CAS.
- A. Salimi and A. A. Yousefi, Polym. Test., 2003, 22, 699–704 CrossRef CAS.
- L. Li, M. Zhang, M. Rong and W. Ruan, RSC Adv., 2014, 4, 3938–3943 RSC.
- K. Matsushige, K. Nagata and T. Takemura, Jpn. J. Appl. Phys., 1978, 17, 467–472 CrossRef CAS.
- J. Scheinbeim, C. Nakafuku, B. A. Newman and K. D. Pae, J. Appl. Phys., 1979, 50, 4399–4405 CrossRef CAS.
- A. Gradys, P. Sajkiewicz, S. Adamovsky, A. Minakov and C. Schick, Thermochim. Acta, 2007, 461, 153–157 CrossRef CAS.
- Y. Oka and N. Koizumi, Bull. Inst. Chem. Res., Kyoto Univ., 1985, 63, 192–206 CAS.
- S. Satapathy, S. Pawar, P. K. Gupta and K. B. R. Varma, Bull. Mater. Sci., 2011, 34, 727–733 CrossRef CAS.
- S. K. Mahadeva, J. Berring, K. Walus and B. Stoeber, J. Phys. D: Appl. Phys., 2013, 46, 285035 CrossRef.
- C. Ribeiro, V. Sencadas, J. L. G. Ribelles and S. Lanceros-Méndez, Soft Mater., 2010, 8, 274–287 CrossRef CAS.
- J. Zheng, A. He, J. Li and C. C. Han, Macromol. Rapid Commun., 2007, 28, 2159–2162 CrossRef CAS.
- A. C. Lopes, C. M. Costa, C. J. Tavares, I. C. Neves and S. Lanceros-Méndez, J. Phys. Chem. C, 2011, 115, 18076–18082 CAS.
- W. M. Prest Jr and D. J. Luca, J. Appl. Phys., 1975, 46, 4136–4143 CrossRef.
- W. M. Prest Jr and D. J. Luca, J. Appl. Phys., 1978, 49, 5042–5047 CrossRef.
- Y. Li, J. Xu, L. Zhu, H. Xu, M. Pan, G. Zhong and Z. Li, Polymer, 2014, 55, 4765–4775 CrossRef CAS.
- M. P. Silva, V. Sencadas, G. Botelho, A. V. Machado, A. G. Rolo, J. G. Rocha and S. Lanceros-Mendez, Mater. Chem. Phys., 2010, 122, 87–92 CrossRef CAS.
- A. J. Lovinger, Polymer, 1980, 21, 1317–1322 CrossRef CAS.
- P. Martins, C. Costa, M. Benelmekki, G. Botelho and S. Lanceros-Méndez, CrystEngComm, 2012, 14, 2807–2811 RSC.
- A. C. Lopes, S. A. C. Carabineiro, M. F. R. Pereira, G. Botelho and S. Lanceros-Méndez, ChemPhysChem, 2013, 14, 1926–1933 CrossRef CAS PubMed.
- M. S. Sebastian, A. Larrea, R. Gonçalves, T. Alejo, J. L. Vilas, V. Sebastian, P. Martins and S. Lanceros-Méndez, RSC Adv., 2016, 6, 113007–113015 RSC.
- P. Martins, C. Caparros, R. Gonçalves, P. M. Martins, M. Benelmekki, G. Botelho and S. Lanceros-Méndez, J. Phys. Chem. C, 2012, 116, 15790–15794 CAS.
- M. A. Bachmann, W. L. Gordon, J. L. Koenig and J. B. Lando, J. Appl. Phys., 1979, 50, 6106–6112 CrossRef CAS.
- R. Gregorio Jr, J. Appl. Polym. Sci., 2006, 100, 3272–3279 CrossRef.
- R. Imamura, A. B. Silva and R. Gregorio Jr, J. Appl. Polym. Sci., 2008, 110, 3242–3246 CrossRef CAS.
- P. Thakur, A. Kool, B. Bagchi, N. A. Hoque, S. Das and P. Nandy, RSC Adv., 2015, 5, 62819–62827 RSC.
- X. Cai, T. Lei, D. Sun and L. Lin, RSC Adv., 2017, 7, 15382–15389 RSC.
- P. Thakur, A. Kool, B. Bagchi, N. A. Hoque, S. Das and P. Nandy, Phys. Chem. Chem. Phys., 2015, 17, 13082–13091 RSC.
- P. Thakur, A. Kool, N. A. Hoque, B. Bagchi, S. Roy, N. Sepay, S. Das and P. Nandy, RSC Adv., 2016, 6, 26288–26299 RSC.
- Z. Zhang and Y. P. Handa, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 977–982 CrossRef CAS.
- R. Gregorio Jr and M. Cestari, J. Polym. Sci., Part B: Polym. Phys., 1994, 32, 859–870 CrossRef.
- B. Dutta, E. Kar, N. Bose and S. Mukherjee, RSC Adv., 2015, 5, 105422–105434 RSC.
- N. Meng, X. Zhu, R. Mao, M. J. Reece and E. Bilotti, J. Mater. Chem. C, 2017, 5, 3296–3305 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09162a |
| This journal is © The Royal Society of Chemistry 2017 |