
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergistic effect of upconversion and plasmons in NaYF4:Yb3+, Er3+, Tm3+@TiO2–Ag composites for MO photodegradation†
Yu Gao‡
b,
Cheng Shi‡a,
Jinzhao Fengb,
Guiyan Zhaoa,
He Yub,
Yanfeng Bi *a,
Fu Ding*b,
Yaguang Sunb and
Zhenhe Xu*b
*a,
Fu Ding*b,
Yaguang Sunb and
Zhenhe Xu*b
aCollege of Chemistry, Chemical Engineering and Environmental Engineering, Liaoning Shihua University, Fushun 113001, P. R. China. E-mail: biyanfeng@lnpu.edu.cn
bThe Key Laboratory of Inorganic Molecule-Based Chemistry of Liaoning Province, College of Applied Chemistry, Shenyang University of Chemical Technology, Shenyang 110142, P. R. China. E-mail: xuzh@syuct.edu.cn
First published on 28th November 2017
Abstract
Structure-based rational design of photocatalysts to enable combination of nanocomponents of radically different properties for enhanced solar energy utilization is a very challenging task. Herein, we integrated up-conversion material NaYF4:Yb3+, Er3+, Tm3+ with TiO2 shells and Ag nanoparticles to synthesise a new class of materials with good stability, broadband absorption from ultraviolet to near infrared, and excellent photocatalytic activity. Structure analysis proved that intimate contact between the NaYF4:Yb3+, Er3+, Tm3+ core and the TiO2 shell and between the TiO2 shell and Ag NPs is the reason for the enhanced photocatalytic activity. Furthermore, the materials also showed exceptionally high stability and reusability under similar experimental conditions. All these results reveal that hierarchical core–shell microspheres exhibit great prospects for developing efficient solar photocatalysts.
1. Introduction
Semiconductors, as light harvesters, can transfer solar light to other forms of energy, and much research has been preliminarily achieved in several fields such as photovoltaic cells, hydrogen fuel generation from water, and photocatalysis.1–6 However, the efficiency of solar energy utilization is far from satisfactory thanks to the wide band gap and low quantum efficiency.7–9 For example, TiO2 is a very attractive material as a photocatalyst due to its fascinating features such as nontoxicity, good chemical and thermal stability, excellent electronic and optical properties,8,9 which render it greatly promising in photocatalysis for the removal of inorganic and organic pollutants,10 for hydrogen generation,11 and dye-sensitized solar cells.12 But TiO2 has a band gap of 3.0–3.2 eV, and can only be activated by high-energy ultraviolet (UV) light, which accounts for only 5% of the incoming solar energy.13 Therefore, photons with energy lower than the band gap energy of TiO2, that is, more than 90% of the overall solar spectrum, cannot be utilized to activate this photocatalyst for photocatalysis.14 To resolve this problem, much effort has been devoted to developing next-generation TiO2, including metal-ion, and nonmetal doping,15 dye sensitization,16 and narrow band gap semiconductor coupling,17 and so forth. These methods have been confirmed to be viable ways to extend the absorption of TiO2-based photocatalysts to some extent. However, to date, these materials typically suffer from thermal instability, photocorrosion, fast photoelectrons (e−)/holes (h+) recombination rates, high cost, and environment-harmful problem.18 Therefore, it is still a challenge to use an appropriate way not only to extend the photo absorption but also improve the photocatalytic activity.Recently, noble-metals (Au, Ag) nanoparticles (NPs) strongly absorb visible light due to localized surface plasmon resonance (LSPR), which offered a new opportunity to overcome the limited efficiency of photocatalysts.19–22 For example, the LSPR effect of Ag NPs causes the intense local electromagnetic fields, which can speed the formation rate of h+ and e− with semiconductor.23 Additionally, the favorable Fermi level of Ag NPs facilitates the separation of h+ and e−, which in turn enhances the quantum efficiency of semiconductor.20,24 Furthermore, the efficient utilization of solar light can be realized due to LSPR absorption in the visible light region as well as UV light.21,24 So researchers have explored the applicability of plasmonic processes in the field of photocatalytic chemistry for organic molecule decomposition,25,26 CO oxidation,25 organic synthesis,26 water splitting.27
As is well known, the near-infrared (NIR) light occupies about 44% of solar irradiation, therefore, there is still a large space to be improved for utilizing solar energy. Lanthanide-doped up-conversion materials, which can convert longer wavelength radiation (near infrared, NIR) to shorter wavelength fluorescence (UV or/and visible light) via a two-photon or/and multiphoton mechanism,28–31 are being considered for one of the most promising solutions and have already been studied in photocatalysis and solar cells.32–40 Therefore, the combination of lanthanide-doped up-conversion NPs, TiO2 and Ag NPs may be the more promising way to effectively using the whole solar light (UV, visible, NIR light) for photocatalysis. In this article, a novel hierarchical NaYF4:Yb3+, Er3+, Tm3+@TiO2–Ag (denoted as NYF@TiO2–Ag) core–shell microspheres have been synthesized, which can improve solar energy utilization by harnessing UV, visible, and NIR light. The photocatalytic efficiency of the photocatalysts toward Methyl Orange (MO) is also examined. More importantly, the photocatalytic stability and the effect of amount of Ag NPs on the NYF@TiO2 core–shell microspheres under UV, visible, and NIR light should be the most important in the consideration of effective utilization of solar light.
2. Results and discussion
A simple, facile preparation of hierarchical NYF@TiO2–Ag core–shell microspheres is realized by reproducible functionalization-step-free solution strategy. The NYF@TiO2–x wt% Ag (x = 0.5, 1, 2, and 3) samples show the similar structure and morphology. Herein the NYF@TiO2–2 wt% Ag sample is used as an example to demonstrate the typical microstructure and morphology. The fabrication process of these core–shell microspheres is schematically illustrated in Fig. 1A. To address this destination, three steps were taken: (1) monodisperse NYF microspheres with a diameter of 400 nm were prepared via a hydrothermal reaction (Fig. 1B). (2) The core–shell NYF@TiO2 microspheres were obtained by using the NYF microspheres as the starting template. After the hydrothermal reaction and calcination, a porous TiO2 shell with thickness of 200 nm was grown on the NYF to form the core–shell microsphere, as illustrated in the TEM image of Fig. 1C. Furthermore, the porous shell was made of a lot of nanoparticles as small as 10 nm (Fig. 1C). (3) AgNO3 solution was simply mixed with the core–shell NYF@TiO2 microspheres and was irradiated under a 300 W Xe lamp to assemble Ag NPs onto the microspheres' surfaces. Fig. 1D demonstrates that Ag NPs homogeneously disperse on the surface of the core–shell NYF@TiO2 microspheres, and the size of the Ag NPs is about 10–20 nm. The EDS spectrum of the selected area in Fig. 1E confirms the existence of O, Ti, Y, Yb, F, Na, and Ag elements. In addition, by suitably controlling the amount of AgNO3 solution, we were able to modulate the density of Ag NPs deposited on the surfaces of TiO2.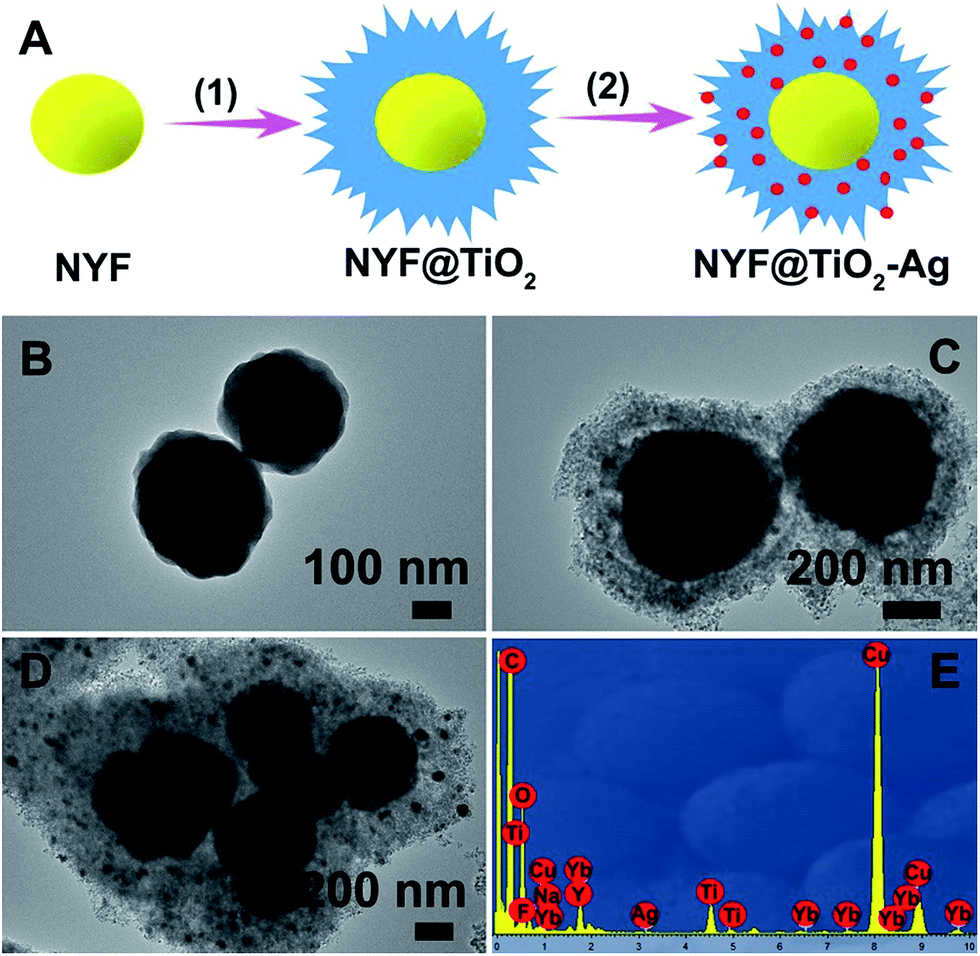 | ||
| Fig. 1 (A) Schematic illustration of the synthesis process of the NYF@TiO2–Ag core@shell microspheres. TEM images of (B) NYF, (C) NYF@TiO2, (D) NYF@TiO2–2 wt% Ag photocatalyst. (E) The EDX spectrum of the NYF@TiO2–2 wt% Ag photocatalyst. | ||
The crystal structures of the NYF, NYF@TiO2, NYF@TiO2–x wt% Ag (x = 1, 2, 3, 4 and 5) samples were analyzed by XRD technique (Fig. 2A). The result shows hierarchical core–shell NYF@TiO2–Ag microsphere is made up by hexagonal β-NaYF4 (JCPDS no. 16-0334) and anatase TiO2 (JCPDS no. 21-1272). No characteristic peaks of Ag NPs can be found in our samples because of the overlap of Ag and NYF diffraction peaks, and a low amount of Ag NPs. In order to compare the optical properties, the UV-vis absorption spectra of NYF, NYF@TiO2, and NYF@TiO2–2 wt% Ag are shown in Fig. 2B. It can be apparently observed that NYF has an absorption in the UV region. After being coated with the TiO2 shell, absorption sharply arises from 400 nm toward shorter wavelengths, corresponding to its band gap absorption of ∼3.2 eV (∼380 nm). After the loading of Ag NPs, an additional shoulder peak apparently appears at around 450 nm, which can be attributed to the SPR excitation of Ag NPs. So the combination of all these absorption features in a single architecture renders the possibility of a broad photoresponse spanning the UV-vis-NIR wavelength ranges of these microspheres. As aforementioned, the NYF is a classic upconversion material, converting the NIR photons into the higher energy emissions in the UV and visible regions. In order to prove whether these emissions are capable of exciting the TiO2 and Ag NPs, the upconversion luminescence spectra of as-synthesized NYF, NYF@TiO2 and NYF@TiO2–2 wt% Ag were measured under the excitation of the 980 nm laser (Fig. 2C). As for the emission spectrum of NYF sample, the emission bands located at 345, 360, 452, 474, 521 and 544 nm can be attributed to the transitions of 1I6 → 3F4, 1D2 → 3H6, 1D2 → 3F4, 1G4 → 3H6, 2H11/2 → 4I15/2, and 4S3/2 → 4I15/2 transition of Tm3+ and Er3+ ions, respectively. After formation of the TiO2 shell, the intensity of overall emissions was largely enhanced because of the increase of crystallinity of NYF during the calcination treatment at 350 °C. Higher crystallinity of NYF generally means less traps and thus stronger luminescence. However, the intensities of the emissions at 345, 360 nm decreased markedly while the intensities of the other emission increased, verifying the energy transfer from the NYF to the TiO2. After further loading Ag NPs, all emissions would be efficiently absorbed by TiO2 and Ag NPs, eventually leading to the significantly reduction of emissions, which indicates that the charge carries recombination in NYF@TiO2–2 wt% Ag was largely suppressed. Therefore, the up-conversion emissions efficiently reabsorbed by TiO2 and Ag NPs are beneficial to photocatalysis.
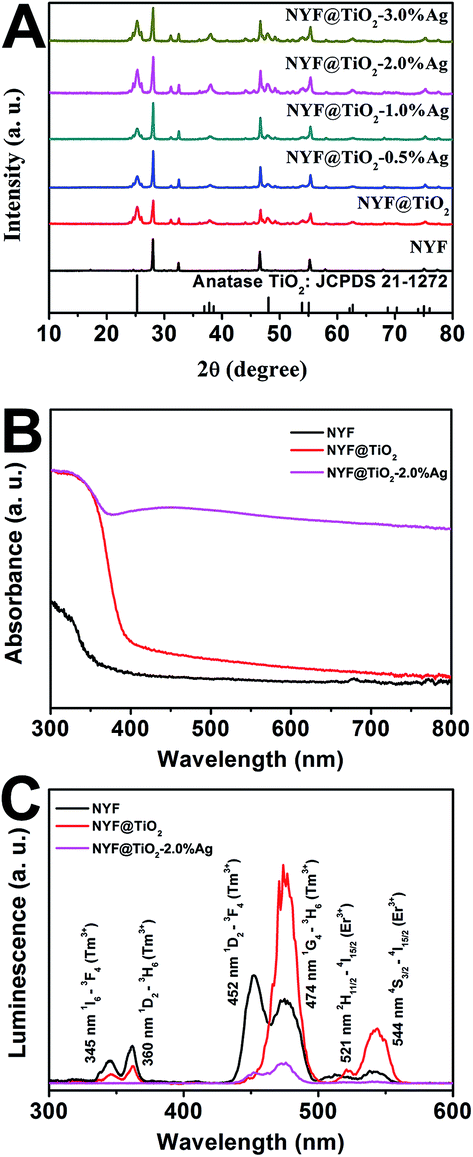 | ||
| Fig. 2 (A) XRD patterns of NYF, NYF@TiO2 and NYF@TiO2–x wt% Ag (x = 0, 0.5, 1, 2, and 3) samples. (B) UV-vis absorption spectra and (C) upconversion luminescence spectra of the NYF, NYF@TiO2 and NYF@TiO2–2 wt% Ag samples. | ||
The photocatalytic activities of samples were investigated by photodegradation MO under the irradiation of UV, visible, and NIR light, respectively (Fig. 3). This photodegradation method is common used for the assessment of the activity of photocatalysts.41 The photodegradation of MO on the photocatalysts loaded with varying Ag NPs amount was studied systematically since the Ag NP loading is an important factor in achieving high photocatalytic activity. For comparison, the photodegradation abilities of Ag NPs, NYF, NYF@TiO2, NYF@TiO2–x wt% Ag (x = 0.5, 1, 2, and 3), and without using a catalyst were used under same experimental condition. There were no obvious photodegradation activities for NYF, Ag NPs and without using a catalyst under UV, visible, 980 nm laser irradiation.
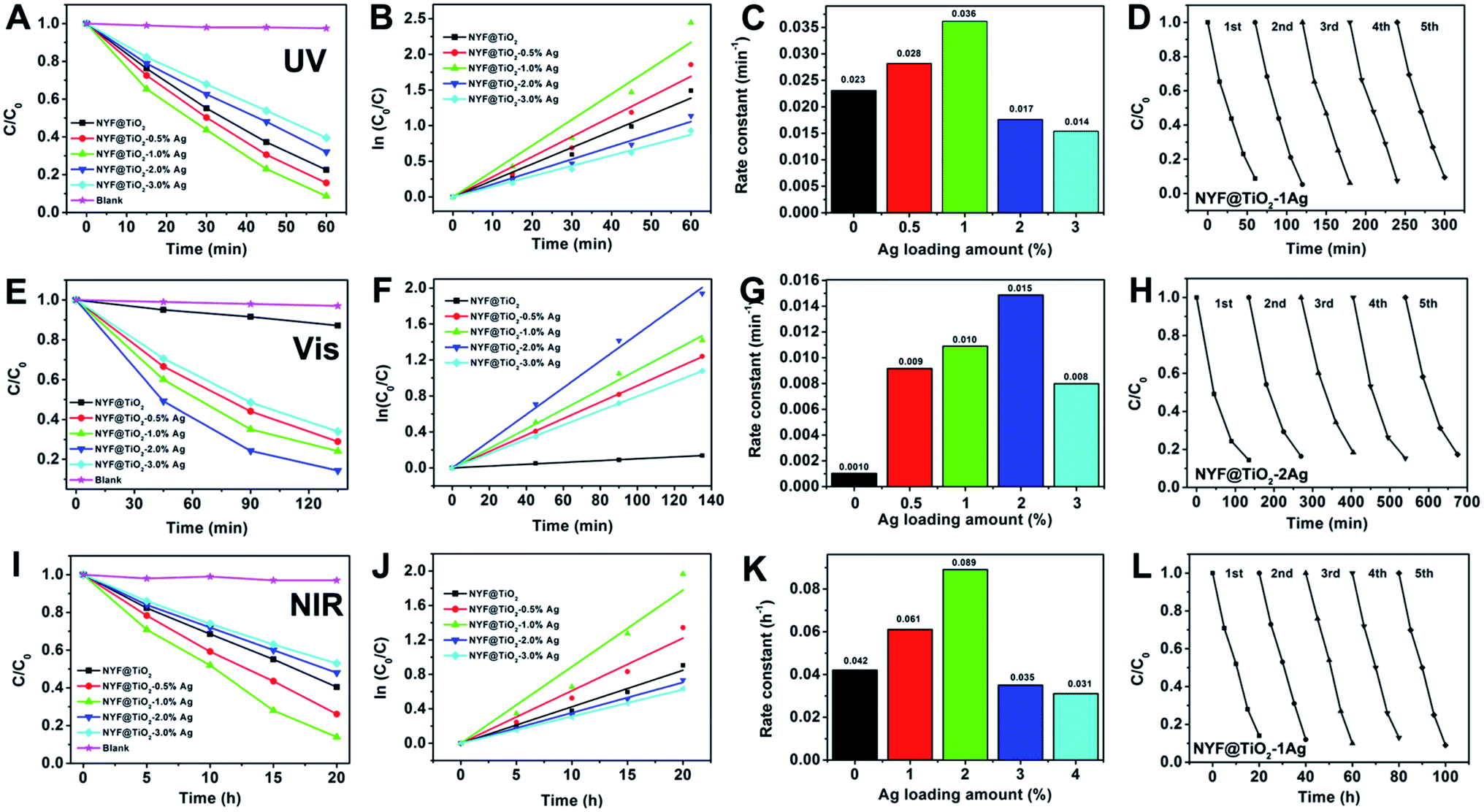 | ||
| Fig. 3 Photocatalytic degradation of MO aqueous solution in the present of the NYF@TiO2–x wt% Ag (x = 0, 0.5, 1, 2, and 3) photocatalysts under (A–D) UV, (E–H) visible (I–L) NIR light irradiation. The plot of (C/C0) vs. reaction time (A, E, I), ln(C0/C) vs. reaction time (B, F, J), the apparent rate constants vs. Ag loading (C, G, K), photocatalytic stability in five successive cycling reactions (D, H, L). | ||
Fig. 3A shows the UV light photocatalytic activity of as-prepared photocatalysts toward the degradation of MO. After 60 min of UV light irradiation, the percentages of MO degraded by NYF@TiO2, NYF@TiO2–0.5 wt% Ag, NYF@TiO2–1.0 wt% Ag, NYF@TiO2–2.0 wt% Ag, and NYF@TiO2–3.0 wt% Ag are 77.5%, 84.4%, 91.3%, 67.9%, and 60.5%, respectively. It is noteworthy to point out that the photodegradation of MO under UV irradiation in the absence of photocatalysts is very slow, confirming that the photocatalytic activity indeed originates from the photocatalyst. So in the photodegradation of MO reaction, the optimum amount for the Ag NPs loaded was 1 wt%, and the order of photocatalytic activity for the samples can be summarized as follows: NYF@TiO2–1.0 wt% Ag > NYF@TiO2–0.5 wt% Ag > NYF@TiO2 > NYF@TiO2–2.0 wt% Ag > NYF@TiO2–3.0 wt% Ag. The corresponding rate constant calculated from the first-order reaction model follows the order: 0.036 min−1 > 0.028 min−1 > 0.023 min−1 > 0.017 min−1 > 0.014 min−1 (Fig. 3B and C). As the UV light cannot excite the SPR effect of Ag NPs, the enhanced UV light activity was not attributed to the SPR effect of Ag NPs. In fact, decreased recombination rate of the charge carries in TiO2, this is the UV light-excited electrons can be transferred from the CB of TiO2 to the Ag NPs, is the reason for improved the UV photocatalytic activity. However, further increased loading of Ag NPs (>1 wt%) causes the reduction in the photocatalytic activity because of the introduced interface defects and the competition in utilizing incident light between the TiO2 and the loaded Ag NPs. All the NYF@TiO2–Ag materials have very excellent photocatalytic performance under UV light. Here, we take the best NYF@TiO2–1.0 wt% Ag sample as a typical example to show the photocatalytic stability experiment. As can be seen from Fig. 3D, after conducting five successive runs under UV light illumination, almost no obvious change in activity is found for the NYF@TiO2–1.0 wt% Ag photocatalyst, and the percentage of degradation of MO still remains as high as 91%
Under visible light illumination (λ > 420 nm), the photocatalytic activity of NYF@TiO2 photocatalyst free of Ag NPs in the photodegradation of MO is quite poor (Fig. 3E). After the introduction of only a small amount Ag NPs, the photoactivity was remarkably enhanced. The photocatalytic degradation efficiency follows the order: NYF@TiO2–2.0 wt% Ag > NYF@TiO2–1.0 wt% Ag > NYF@TiO2–0.5 wt% Ag > NYF@TiO2–3.0 wt% Ag > NYF@TiO2. The above results confirm the important role of SPR of Ag NPs in improving photocatalytic performance under visible light illumination. The corresponding rate constant calculated from the first-order reaction model follows the order: 0.015 h−1 > 0.010 h−1 > 0.009 h−1 > 0.008 h−1 > 0.001 h−1 (Fig. 3F and G). The highest rate constant k (0.015 h−1) achieved by the NYF@TiO2–2.0 wt% Ag sample is 15 times higher than of the NYF@TiO2 sample (0.001 h−1). It is important to point out that the defects introduced by excessive Ag NPs at the interface between TiO2 layer and Ag NPs can act as the recombination centers for charge carriers to decrease the photocatalytic activity under visible light illumination. Furthermore, cycling tests were conducted to test the stability of the as-prepared highest activity sample NYF@TiO2–2.0 wt% Ag under visible light illumination and the results are presented in Fig. 3H. As we can see, the photocatalytic performance of NYF@TiO2–2.0 wt% Ag sample is no obvious change, and more than 82% of MO still can be photodegraded after five successive cycling tests, indicating the good stability of NYF@TiO2–2.0 wt% Ag sample.
The excellent UV and visible light induced photocatalytic activities for these samples were mainly assigned to the excellent UV and visible absorption of TiO2 and Ag NPs, but no NIR light induced photodegradation of MO was performed, because TiO2 and Ag NPs could not be excited by NIR light. It is well known that NYF has been widely recognizing as the most efficient host for up-conversion due to it normally possess a high refractive index and low phonon energy. In this part experiment, NYF can emit UV and visible light under 980 nm light excitation, which are expected to be reabsorbed by the TiO2 and Ag NPs owing to their band gap and SPR excitations, respectively. Fig. 3I gives the MO photodecomposition efficiency curves as a function of the irradiation time with the presence of as-prepared photocatalysts under the NIR irradiation. After 20 h irradiation, almost 80% of the MO was degraded by NYF@TiO2–1.0 wt% Ag sample. The rate constant calculated from the first-order reaction model follows the order: NYF@TiO2–1.0 wt% Ag > NYF@TiO2–0.5 wt% Ag > NYF@TiO2 > NYF@TiO2–2.0 wt% Ag > NYF@TiO2–3.0 wt% Ag (Fig. 3J and K). The highest degradation rate of NYF@TiO2–1.0 wt% Ag sample (0.089 h−1) is more than two times faster than that of the no Ag loading sample (NYF@TiO2). In addition, Fig. 3L shows that the photocatalytic degradation of MO still reach over 86% after 5 successive runs under 980 nm NIR light illumination, indicating that NYF@TiO2–1.0 wt% Ag catalyst is excellent stability in the photocatalytic degradation of MO. The above results indicate that NYF@TiO2–Ag catalysts not only possess excellent UV, visible, and NIR light photocatalytic activity for degradation of MO, but also have a high stability, and therefore they are beneficial to the large-scale application of photocatalysis in the environmental pollution remediation by using abundant solar energy. However, we have to point that the uniform and dense dispersion of Ag NPs may lead to a maximized degree of enhancement of catalytic efficiency. When the density of Ag NPs is further increased, it may as a recombination center, which negatively affects the catalytic efficiency. This may explain why NYF@TiO2–3.0 wt% Ag sample, containing the highest density of Ag NPs in this study, showed comparatively worse photocatalytic efficiency. As mentioned above, the superior catalytic activity is NYF@TiO2–1.0 wt% Ag under UV light irradiation. For other light irradiation, NYF@TiO2–2.0 wt% Ag sample is the best. This is reason that higher content of Ag NPs not only influences the penetration of light but also becomes the recombination center, which negatively affects the catalytic efficiency.
To better understand the photocatalytic process, the trapping experiments of the photocatalytic degradation of MO in the presence of three different types of scavengers, BQ, t-BuOH and Na2EDTA, were designed to identify the active species of hydroxyl radical (OH˙), hole (h+), and superoxide radical anion (O2˙−) involved in this reaction system. Fig. 4 presents the photodegradation of MO catalyzed by NYF@TiO2–2.0 wt% Ag in the presence of these three scavengers under visible light illumination. Compared with the scavenger-free reaction (black line), the system in the presence of the hole scavenger Na2EDTA (red line) is slightly slower. While in the presence of the O2˙− scavenger BQ and OH˙ scavenger t-BuOH, the photocatalytic activities were greatly reduced, with 17% and 58% MO degraded in 135 min, respectively. These results strongly indicate that O2˙− and OH˙ radicals play more important roles than the holes in the photocatalytic degradation of MO, but O2˙− radical is a key intermediate as its trapping results in a greatly suppression of catalytic activity.
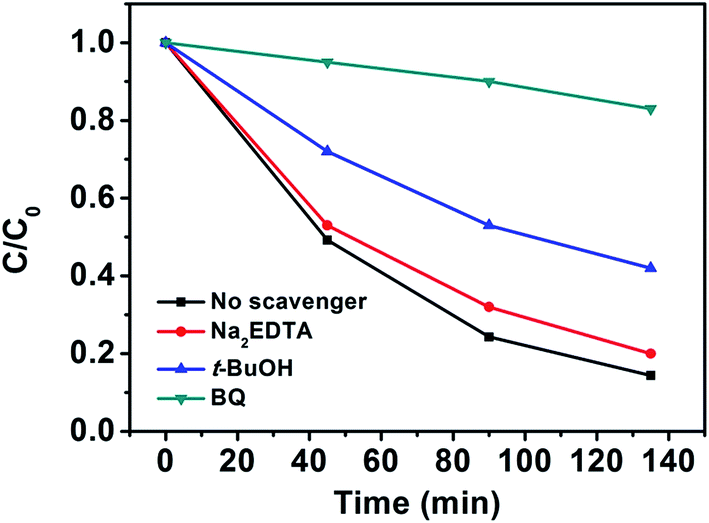 | ||
| Fig. 4 Photocatalytic degradation of MO in the presence of three types of scavengers and NYF@TiO2–2 wt% Ag photocatalyst under visible light irradiation. | ||
For better understanding the underlying mechanisms under different light illumination, three possible schematics for the charge transfer processes are depicted in Fig. 5. Mechanism of degradation of MO for NYF@TiO2–Ag under UV light is shown in Fig. 5A. Deposition of Ag NPs on the surface of TiO2 shells enhances the photocatalytic efficiency under UV light trapping the excitons, which can lead to the interfacial charge transfer and decrease the recombination rate.42 Upon exposure to the UV light, a lot of electrons and holes generated by band gap excitation of TiO2 semiconductor. Then, the excited electrons can be transferred from the conduction band of TiO2 to Ag NPs. This step can prevent the recombination of the electron–hole pairs by capturing the free electrons from TiO2 and transfer them to the adsorbed O2 on the surface of NYF@TiO2–Ag to form superoxide anion radicals (O2−˙).43 Simultaneously, the holes are used for the generation of OH˙ radicals. In addition to the UV light enhanced photocatalytic active, NYF@TiO2–Ag materials have a unique characteristic to interact with visible light through excitation of LSPR. The LSPR effect on the surface of Ag NPs provides electrons to TiO2 by diffusion. Different from the mechanism under UV light, there are two crucial steps that can assist TiO2 to work as visible light photocatalyst (Fig. 5B). In visible light, the photon absorption induces a strong LSPR phenomenon at the TiO2–Ag interface, which enhances the generation of electron–hole pairs. The electrons generated by the LSPR effect in the Ag NPs of NYF@TiO2–Ag move through the TiO2–Ag interface into the CB of TiO2 shell, enhancing the generation of O2−˙ radicals. The holes generated in the Ag NPs are used for the generation of OH˙ radicals. Both O2−˙ and OH˙ radicals play a crucial role in the degradation of organic dyes. It should be pointed that the visible light photocatalytic activity of NYF@TiO2 toward the degradation of MO is attributed to the photosensitized effect of dye.10 Under visible light irradiation, it should be pointed that dye MO molecules rather than TiO2 is excited, which leads to generate the electrons and MO*. Then, the produced electrons inject into the CB of TiO2, and are further scavenged by O2 to form ˙O2−. Both MO* and ˙O2− species are responsible for the photodecomposition of MO. Fig. 5C illustrates the mechanism for the NIR-driven photocatalysis. In essence, the pumping of 980 nm of light only excites the Yb3+ ion, because it possesses a large absorption cross section at 980 nm, and energy transfer occurs as a result of the large spectral overlap between the Yb3+ emission 2F5/2 → 2F7/2 and the Tm3+ absorption 3H5 ← 3H6 or the Er3+ absorption 4I11/2 ← 4I15/2 bands. For the emissions of the sample, up-conversion from Tm3+ and Er3+ ions are due to energy transfer (ET) processes, because it has no ground or excited-state absorption (GSA or ESA) that matches the 980 nm photon. First, the pump light of 980 nm excites only the Yb3+ ions, resulting in population of the long-lived 2F5/2 level in Yb3+. Then, the Tm3+ or Er3+ ions will be excited due to the efficient energy transfer from Yb3+. For the ultraviolet and blue emission, the lights centered at 345, 360, 452, 474 nm, which are totally in the TiO2 absorption range. Then the mechanism is the same as under UV light. For the green light, the emission peaks centered at 521, and 544 nm are totally in the range of the Ag LSPR region. So the TiO2 shells and Ag NPs can absorb the UV and visible emissions from NYF phosphor, respectively, and induce the formation of e− and h+. The other processes are the same as under UV and visible light irradiation.
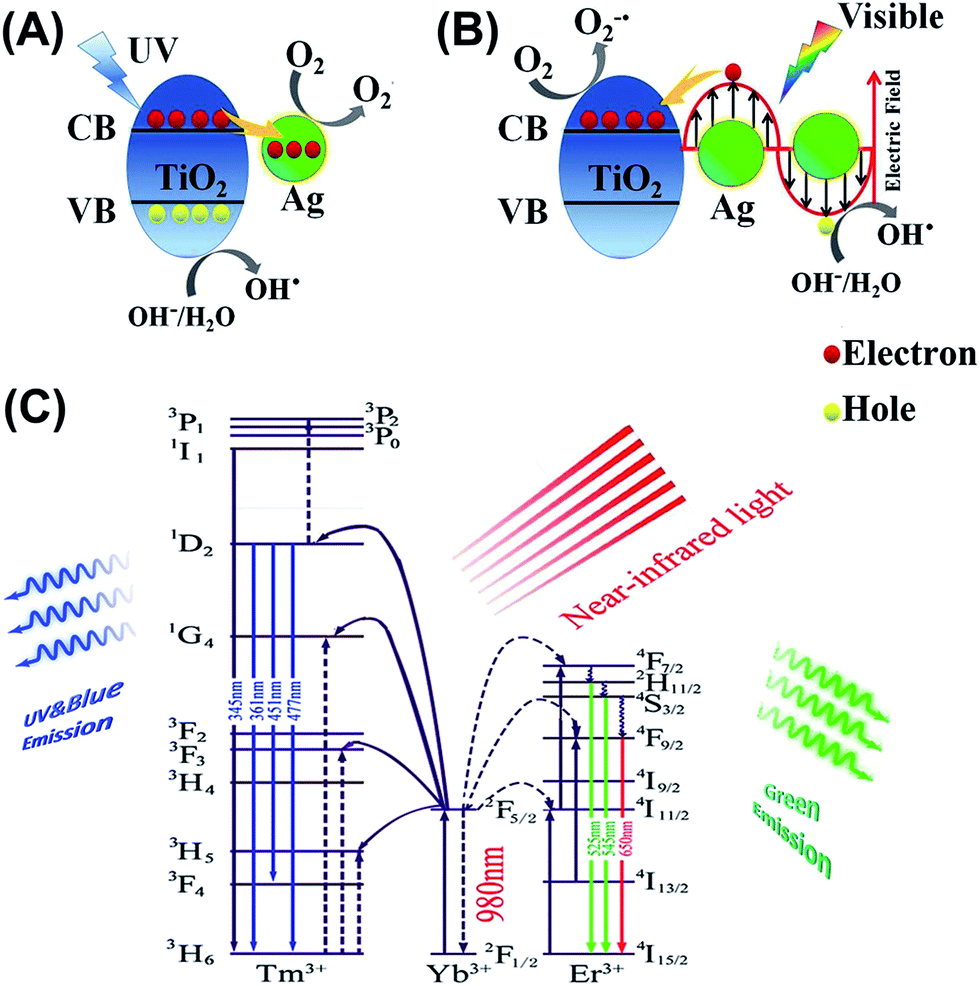 | ||
| Fig. 5 Proposed photocatalytic mechanism of NYF@TiO2–Ag photocatalyst under (A) UV and (B) visible light, (C) schematic energy level diagrams, upconversion excitation and UV and visible light emissions for the NYF microspheres under the NIR light (980 nm laser) illumination. | ||
3. Conclusion
In summary, we have developed a facile approach for the synthesis of hierarchical NYF@TiO2–Ag core–shell microspheres. The morphology and composition of the photocatalysts were investigated thoroughly. Significantly, these photocatalysts display enhanced UV, visible, and NIR photocatalytic activity. Moreover, the photocatalysts showed good stability for the photocatalytic degradation of MO solution in the recycled tests. The strategy of introducing up-conversion material might open an avenue to develop broadband solar light sensitive photocatalysts.4. Experimental section
Materials
Y(NO3)3·4H2O, Yb(NO3)3·5H2O, Er(NO3)3·5H2O, Tm(NO3)3·5H2O, silver nitrate, isopropanol, diethylenetriamine (DETA), titanium isopropoxide (TIP, 97%), anhydrous ethanol, trisodium citrate (Na3Cit), sodium fluoride (NaF), 1,4-benzoquinone (BQ), disodium ethylenediaminetetraacetate (Na2EDTA), and tert-butyl alcohol (t-BuOH) were purchased from Sigma-Aldrich and used without further purification. Water was purified by a Millipore Ultrapure water system and has a resistivity of 18.2 MΩ cm at 25 °C.Preparation of NYF microspheres
In a typical synthesis, 1.488 ml of Y(NO3)3 (0.5 M), 0.5 ml of Yb(NO3)3 (0.5 M), 0.1 ml of Er(NO3)3 (0.01 M), and 0.5 ml of Tm(NO3)3 (0.01 M) stock solutions were added into 15 ml of aqueous solution containing 0.5 mmol of trisodium citrate (labeled as Cit3−) and magnetically stirred for 1 h, forming the Ln3+–Cit3− complex. Then 4 mmol of NaF was introduced into the solution. After additional agitation for 1 h, the as-obtained mixing solution was transferred into Teflon bottle held in a stainless steel autoclave, sealed, and maintained at 180 °C for 12 h. As the autoclave cooled to room temperature naturally, the precipitates were separated by centrifugation, washed with ethanol and deionized water in sequence, and then dried in air at 80 °C for 12 h.Preparation of core–shell NYF@TiO2 microspheres
The as-prepared NYF microspheres (0.05 g) were dispersed in isopropanol (42.00 ml), followed by the addition of DETA (0.04 ml). After stirring for 10 min, TIP (2 ml) was added to the solution. The mixture was then transferred into Teflon bottle held in a stainless steel autoclave with a capacity of 100 ml and kept at 200 °C for 24 h. As the autoclave cooled to room temperature naturally, the precipitates were separated by centrifugation, washed with ethanol, and then dried in air at 80 °C for 12 h. Finally, the product was calcined at 350 °C for 2 h with a heating rate of 1 °C min−1 to obtain the high crystalline phase.Preparation of core–shell NYF@TiO2–Ag microspheres
The as-prepared core–shell NYF@TiO2 microspheres were dispersed into 20 ml of distilled water containing an appropriate amount of AgNO3. The suspension was then transferred to a vessel and irradiated with a 300 w xenon lamp for 2 h. The, the powder was recovered by centrifugation, washed successively with ethanol and deionized water, and dried in air at 60 °C for 12 h. A series of Ag NPs modified NYF@TiO2 core–shell microspheres were prepared using different amount of AgNO3 solution. The weight percentages of Ag NPs in the initial NYF@TiO2 were 0, 0.5, 1.0, 2.0, and 3.0 wt%, labeling as NYF@TiO2 NYF@TiO2–0.5 Ag, NYF@TiO2–1.0 Ag, NYF@TiO2–2.0 Ag, and NYF@TiO2–3.0 Ag, respectively.The evaluation of photocatalytic activity
The photocatalytic activity of the core–shell NYF@TiO2–Ag microspheres was investigated by the photodegradation of MO solution. In a typical measurement, 10 mg of the core–shell NYF@TiO2–Ag microspheres were added to 10 ml of a solution of 20 mg l−1 of MO solution in a 100 ml quartz flask. Prior to photoirradiation, the suspensions were magnetically stirred in the dark for 0.5 h to establish adsorption–desorption equilibrium between the dye and the surface of the catalysts under ambient conditions. A commercial photoreactor (LUZ-4V, Luzchem) equipped with fourteen 8 W ultraviolet (Luzchem LZC-UVA) lamps was used for UV photocatalysis experiments (Fig. S1, ESI†). The visible light photocatalytic reactions were performed by adopting a 300 W xenon lamp (optical filter, λ > 420 nm) as an outer irradiation source (Fig. S2, ESI†). There is a cooling water circulator between the light and quartz flask which is made of quartz to keep the reactor in a constant temperature condition. A 2 W 980 nm diode laser was used as the NIR light source. At varied irradiation time intervals, an aliquot of the mixed solution was collected and centrifuged, and the residual MO concentration in the supernatant was analyzed by UV-vis spectrometer. To evaluate the photocatalytic stability of the core–shell NYF@TiO2–Ag microspheres, three cycles of MO solution photocatalytic degradation were carried out in the presence of the core–shell NYF@TiO2–Ag microspheres. After each degradation cycle, the degraded solution was centrifuged. The obtained precipitate was washed with pure water for several times to get the clean the core–shell NYF@TiO2–Ag microspheres photocatalyst, and then dried at 80 °C for the next degradation cycle. To study the MO degradation kinetics in the presence of NYF@TiO2–Ag microspheres, the apparent reaction rate constant (k) was calculated by using the following first-order reaction model:| ln(C0/C) = kt, |
Characterization
The crystal structure of the as-prepared other samples was characterized by X-ray spectroscopy (XRD, Bruker D8 Advanced Diffractometer, Cu Kα radiation). UV-visible diffuse reflectance spectroscopy was conducted using Varian 5000 spectrometer. The up-conversion luminescent measurements have been carried on under 975 nm excitation using a Thorlabs fiber-coupled laser diode (maximum power 330 mW) focused on the sample using a lens to obtain a spot with Gaussian intensity distribution (0.4 mm diameter). The emission light has been also collected by a lens in a 90 degrees configuration, and then transferred using optical fiber to a spectrophotometer (Avaspec-2048L-USB2). All the emission spectra have been corrected by the response curve of the whole set-up that has been calibrated using an Ocean Optics halogen tungsten lamp. Transmission electron microscopy (TEM, JEOL 2100F) and energy-dispersive X-ray spectroscopy (EDS) were applied for a detailed analysis of microstructure and composition. All experiments were carried out under ambient conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC 51402198, 21671139), the Natural Science Foundation of Liaoning Province (201602592, 20170540715), Educational Bureau of Liaoning Province for the Fundamental Research of Key Lab (Grand No. LZ2014028, LZ2016003, L2017LZD002), the talent scientific research/scientific research cultivation fund of LSHU (No. 2015XJJ-009/2016PY-003).References
- J. Cai, Y. He, X. Wang, L. Zhang, L. Dong, H. Lin, L. Zhao, X. Yi, W. Weng and H. Wan, RSC Adv., 2013, 3, 20862–20868 RSC.
- Z. Xu, S. Bian, T. Liu, L. Wang, Y. Gao, H. Lian and J. Lin, RSC Adv., 2012, 2, 11067–11077 RSC.
- Z. Li, W. Luo, M. Zhang, J. Feng and Z. Zou, Energy Environ. Sci., 2013, 6, 347–370 CAS.
- J. I. L. Chen, E. Loso, N. Ebrahim and G. A. Ozin, J. Am. Chem. Soc., 2008, 130, 5420–5421 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970 CrossRef CAS PubMed.
- Z. Xu, C. Li, D. Yang, W. Wang, X. Kang, M. Shang and J. Lin, Phys. Chem. Chem. Phys., 2010, 12, 11315–11324 RSC.
- Z. Xu, M. Quintanilla, F. Vetrone, A. O. Govorov, M. Chaker and D. Ma, Adv. Funct. Mater., 2015, 25, 2950–2960 CrossRef CAS.
- Z. Xu, Y. Liu, F. Ren, F. Yang and D. Ma, Coord. Chem. Rev., 2016, 320–321, 153–180 CrossRef CAS.
- Y. Cao, Q. Gao, Q. Li, X. Jing, S. Wang and W. Wang, RSC Adv., 2017, 7, 40727–40733 RSC.
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
- J. Lee, S. Mubeen, X. Ji, G. D. Stucky and M. Moskovits, Nano Lett., 2012, 12, 5014–5019 CrossRef CAS PubMed.
- X. Feng, K. Zhu, A. J. Frank, C. A. Grimes and T. E. Mallouk, Angew. Chem., Int. Ed., 2012, 51, 2727–2730 CrossRef CAS PubMed.
- H. Zhang, F. Liu, H. Wu, X. Cao, J. Sun and W. Lei, RSC Adv., 2017, 7, 40327–40333 RSC.
- Y. Tang, W. Di, X. Zhai, R. Yang and W. Qin, ACS Catal., 2013, 3, 405–412 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed.
- W. Zhao, Y. Sun and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 12566–12567 CrossRef CAS PubMed.
- H. Tada, Q. Jin, H. Nishijima, H. Yamamoto, M. Fujishima, S.-i. Okuoka, T. Hattori, Y. Sumida and H. Kobayashi, Angew. Chem., Int. Ed., 2011, 50, 3501–3505 CrossRef CAS PubMed.
- L. Liu, S. Ouyang and J. Ye, Angew. Chem., Int. Ed., 2013, 52, 6689–6693 CrossRef CAS PubMed.
- M. R. Kim, Z. Xu, G. Chen and D. Ma, Chem.–Eur. J., 2014, 20, 11256–11275 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- M. Zang, L. Shi, L. Liang, D. Li and J. Sun, RSC Adv., 2015, 5, 56136–56144 RSC.
- Z. Xu, M. G. Kibria, B. AlOtaibi, P. N. Duchesne, L. V. Besteiro, Y. Gao, Q. Zhang, Z. Mi, P. Zhang, A. O. Govorov, L. Mai, M. Chaker and D. Ma, Appl. Catal., B, 2018, 221, 77–85 CrossRef CAS.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467–472 CAS.
- Y. Gao, J. Lin, Q. Zhang, H. Yu, F. Ding, B. Xu, Y. Sun and Z. Xu, Appl. Catal., B, 2018, 224, 586–593 CrossRef CAS.
- W. H. Hung, M. Aykol, D. Valley, W. Hou and S. B. Cronin, Nano Lett., 2010, 10, 1314–1318 CrossRef CAS PubMed.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC.
- S. C. Warren and E. Thimsen, Energy Environ. Sci., 2012, 5, 5133–5146 CAS.
- Z. Xu, Q. Zhao, Y. Sun, B. Ren, L. You, S. Wang and F. Ding, RSC Adv., 2013, 3, 8407–8416 RSC.
- R. Lv, P. Yang, B. Hu, J. Xu, W. Shang and J. Tian, ACS Nano, 2017, 11, 1064–1072 CrossRef CAS PubMed.
- D. Yang, G. Yang, P. Yang, R. Lv, S. Gai, C. Li, F. He and J. Lin, Adv. Funct. Mater., 2017, 27, 1700371 CrossRef.
- Z. Xu, S. Bian, J. Wang, T. Liu, L. Wang and Y. Gao, RSC Adv., 2013, 3, 1410–1419 RSC.
- Y. Yang, P. Zhou, W. Xu, S. Xu, Y. Jiang, X. Chen and H. Song, J. Mater. Chem. C, 2016, 4, 659–662 RSC.
- Y. Zhang and Z. Hong, Nanoscale, 2013, 5, 8930–8933 RSC.
- Z.-X. Li, F.-B. Shi, T. Zhang, H.-S. Wu, L.-D. Sun and C.-H. Yan, Chem. Commun., 2011, 47, 8109–8111 RSC.
- W. Qin, D. Zhang, D. Zhao, L. Wang and K. Zheng, Chem. Commun., 2010, 46, 2304–2306 RSC.
- Y. Chen, S. Mishra, G. Ledoux, E. Jeanneau, M. Daniel, J. Zhang and S. Daniele, Chem.–Asian J., 2014, 9, 2415–2421 CrossRef CAS PubMed.
- S. Gai, C. Li, P. Yang and J. Lin, Chem. Rev., 2014, 114, 2343–2389 CrossRef CAS PubMed.
- R. Lv, D. Yang, P. Yang, J. Xu, F. He, S. Gai, C. Li, Y. Dai, G. Yang and J. Lin, Chem. Mater., 2016, 28, 4724–4734 CrossRef CAS.
- D.-X. Xu, Z.-W. Lian, M.-L. Fu, B. Yuan, J.-W. Shi and H.-J. Cui, Appl. Catal., B, 2013, 142–143, 377–386 CrossRef CAS.
- M.-Z. Huang, B. Yuan, L. Dai and M.-L. Fu, J. Colloid Interface Sci., 2015, 460, 264–272 CrossRef CAS PubMed.
- J. Zhang, Y. Huang, L. Jin, F. Rosei, F. Vetrone and J. P. Claverie, ACS Appl. Mater. Interfaces, 2017, 9, 8142–8150 CAS.
- S. T. Kochuveedu, Y. H. Jang and D. H. Kim, Chem. Soc. Rev., 2013, 42, 8467–8493 RSC.
- H. Eom, J.-Y. Jung, Y. Shin, S. Kim, J.-H. Choi, E. Lee, J.-H. Jeong and I. Park, Nanoscale, 2014, 6, 226–234 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09368c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |