
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of an amphiphilic graft copolymer bearing a hydrophilic poly(acrylate acid) backbone for drug delivery of methotrexate†
Wei Wang‡
a,
Xin Zhou‡a,
Min Weia,
Zude Liu*a,
Guolin Lub and
Xiaoyu Huang *b
*b
aDepartment of Orthopaedic Surgery, Renji Hospital, School of Medicine, Shanghai Jiaotong University, 1630 Dongfang Road, Shanghai 200127, People's Republic of China. E-mail: rjliuzd@126.com; Tel: +86-21-68383706
bKey Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, People's Republic of China. E-mail: xyhuang@mail.sioc.ac.cn; Fax: +86-21-64166128; Tel: +86-21-54925310
First published on 28th November 2017
Abstract
A well-defined amphiphilic graft copolymer consisting of a hydrophilic poly(acrylic acid) (PAA) backbone and hydrophobic poly(lactic acid) side chains was synthesized by the combination of reversible addition–fragmentation chain transfer (RAFT) polymerization, ring open polymerization (ROP), and the grafting-from strategy. RAFT homopolymerization of an OH-containing tert-butyl 2-((4-hydroxy-butanoyloxy)methyl)acrylate (tBHBMA) monomer was first conducted to give a well-defined PtBHBMA homopolymer bearing pendant hydroxyls in every repeat unit. The pendant hydroxyls of PtBHBMA directly initiated the ROP of lactide without post-polymerization functionality transformation so as to provide a well-defined poly(tert-butyl acrylate)-g-poly(lactic acid) (PtBA-g-PLA) graft copolymer via the grafting-from strategy. The hydrophobic PtBA backbone was transformed into a hydrophilic PAA backbone to afford the target well-defined PAA-g-PLA amphiphilic graft copolymer (Mw/Mn = 1.17) containing equally-distributed carboxyls along the backbone. The PAA-g-PLA amphiphilic graft copolymer shows pH-responsive micellization behavior and it can self-assemble into non-toxic large compound micelles under certain conditions for loading methotrexate (MTX). The in vitro accumulative release characteristics of MTX-loaded micelles were investigated by UV/vis spectroscopy. In comparison with free MTX, the MTX-loaded nanoparticles display higher cytotoxicity against MG-63 in 24, 48 and 72 h.
Introduction
Biodegradable polymers have received increased attention and have been seen as a promising solution to the plastic waste issue because they can be broken down to environmentally-friendly low molecular weight products such as water and carbon oxide.1,2 Among the many known biodegradable polymers, polylactides (PLAs) occupy a prominent position due to their biodegradability, biocompatibility, and ready availability from inexpensive renewable resources.3–8 PLAs can be fabricated into sutures, catheters, and prosthetic devices, and recently their application in tissue engineering has also been quite attractive.9 In addition, employing PLAs as carriers for drug delivery is now a hot research topic.10–12 However, PLAs possess certain drawbacks including intrinsic hydrophobicity. Therefore, PLAs are usually copolymerized to improve their physical, chemical, and mechanical properties.13,14It is well known that the architecture of copolymers has great impact on their properties.15 Amphiphilic block copolymers with linear architecture are known to form different morphologies in specific solvents, which brings lots of potential applications including drug delivery,16–19 bioreactor,20,21 and catalysis.22,23 However, it has been showed that non-linear copolymers display fundamentally different properties compared to the linear copolymers.24,25 Amphiphilic graft copolymers, one of those polymers with non-linear structure, are endowed with some unusual properties such as exhibiting more stable aggregation in solution due to their confined and complicated structure.26 PLA-based amphiphilic graft copolymers in which PLA (or derivatives) is either backbone or side chain, could bring interesting properties such as increased hydrophilicity,27–30 higher degradability,28,29,31–33 and better histocompatibility.33,34 Although many works are reported on the synthesis of PLA-based amphiphilic block copolymers,13 it is still unusual to see reports concerned with the synthesis of PLA-based graft copolymer because of synthetic obstacles including complicated topology and highly steric congestion. PLA could be used as the backbone of graft polymer, which could be realized by incorporating functionalities on PLA backbone. For instance, Riva et al. reported PLA-g-PEG amphiphilic graft copolymer by copolymerizing lactide with α-chloro-ε-caprolactone, through which the halogen was introduced into the backbone, followed by grafting PEG onto the backbone via click chemistry.35 However, this approach employing PLA as the backbone is not so convenient since that on one hand, it is not an easy task to incorporate functionalities such as halogen and hydroxyl into the backbone; on the other hand, because of the PLA-based backbone, many characteristics of PLA including crystallinity and mechanical properties might disappear.36,37 It is more common to introduce PLA as side chains while preparing PLA-based graft copolymers.38–41 But it is worthy noting that it is difficult to accomplish the synthesis of PLA-based amphiphilic graft polymers with a hydrophilic backbone whose every repeat unit is hydrophilic. Poly(2-hydroxyethyl methacrylate) (PHEMA), possessing a number of biomedical applications due to its biocompatibility and hydrophilicity, is often introduced as the backbone of PLA-based amphiphilic graft polymers. Atom transfer radical polymerization (ATRP) of HEMA is used to yield the backbones on which a portion of hydroxyls could initiate ring open polymerization (ROP) of lactide to give the desired amphiphilic graft copolymer.42 Poly(vinyl alcohol) (PVA) is also often chosen as the backbone because of its intrinsic hydrophilicity and biogradability. Kissel et al. prepared PLA-based amphiphilic graft polymer using the pendant hydroxyls on PVA backbone to initiate ROP of lactide for affording PVA-g-PLA graft copolymer.43 In order to avoid to use stannous octoate as catalyst of ROP which would hinder the medical applications for the toxicity, Schue et al. synthesized PVA-g-PLA by employing MgH2 as catalyst of ROP of lactide initiated by PVA.44 Akashi et al. also accomplished the synthesis of PLA-based amphiphilic graft copolymers with poly(γ-glutamic acid) (PGA) containing carboxyls as hydrophilic backbone, through a coupling reaction between γ-PGA and PLA activated by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide.45
It is clear that only some parts of the backbone of PLA-based graft copolymers are hydrophilic42–45 since a portion of hydrophilic functionalities including hydroxyls and carboxyls is converted to hydrophobic ester linkages for connecting PLA side chains. Meanwhile, the distribution of those residual hydrophilic groups along the backbone is also random, which means that the precise structure of amphiphilic graft polymers could not be guaranteed. To obtain the amphiphilic graft copolymers with precise structure, especially the hydrophilic functionalities of the backbone are equally distributed, the simplest way is to make every repeat unit of the backbone hydrophilic. To our best knowledge, none has ever reported the synthesis of PLA-based amphiphilic graft copolymers bearing a hydrophilic backbone whose every repeat unit is all hydrophilic for ensuring the equaled distribution of hydrophilic functionalities.
Herein, we report the synthesis of poly(acrylic acid)-g-poly(lactic acid) (PAA-g-PLA) amphiphilic graft copolymer as shown in Scheme 1, in which every repeat unit of hydrophilic backbone possesses a carboxyl and hydrophobic PLA side chains are connected to every repeat unit of backbone. In order to accomplish the synthesis of this new polymer, it is viable to use the monomer containing a ROP initiation group. As mentioned above, PLA side chains can be formed by ROP of lactide initiated by the pendant hydroxyls of the backbone. To achieve the construction of a fully hydrophilic backbone, PAA, a hydrophilic polymer with pH responsiveness, is chosen due to convenient and complete conversion from the hydrolysis of the corresponding polyacrylate. Based on these facts and previous work of our group on the synthesis of diverse amphiphilic graft polymers46–50 and a new trifunctional inimer,51 tert-butyl 2-((4-hydroxybutanoyloxy)methyl)acrylate (tBHBMA) which bears a polymerizable double bond, a hydroxyl as initiating group for ROP, and a tert-butoxycarbonyl as potential carboxyl, the well-defined PtBA-based backbone with pendant hydroxyl in every repeat unit is formed by RAFT homopolymerization of tBHBMA followed by initiating ROP of lactide to form PLA side chains. The desired well-defined amphiphilic graft copolymer, PAA-g-PLA, is finally achieved by selective acidic hydrolysis of PtBA-based backbone using trifluoroacetic acid. PAA-g-PLA graft copolymer could self-assemble into large compound micelles in aqueous media and is used to load methrotrexate (MTX). The morphology, size distribution, in vitro drug accumulative release, and cytotoxicity against MG-63 cells of the MTX-loading nanoparticles are investigated in details.
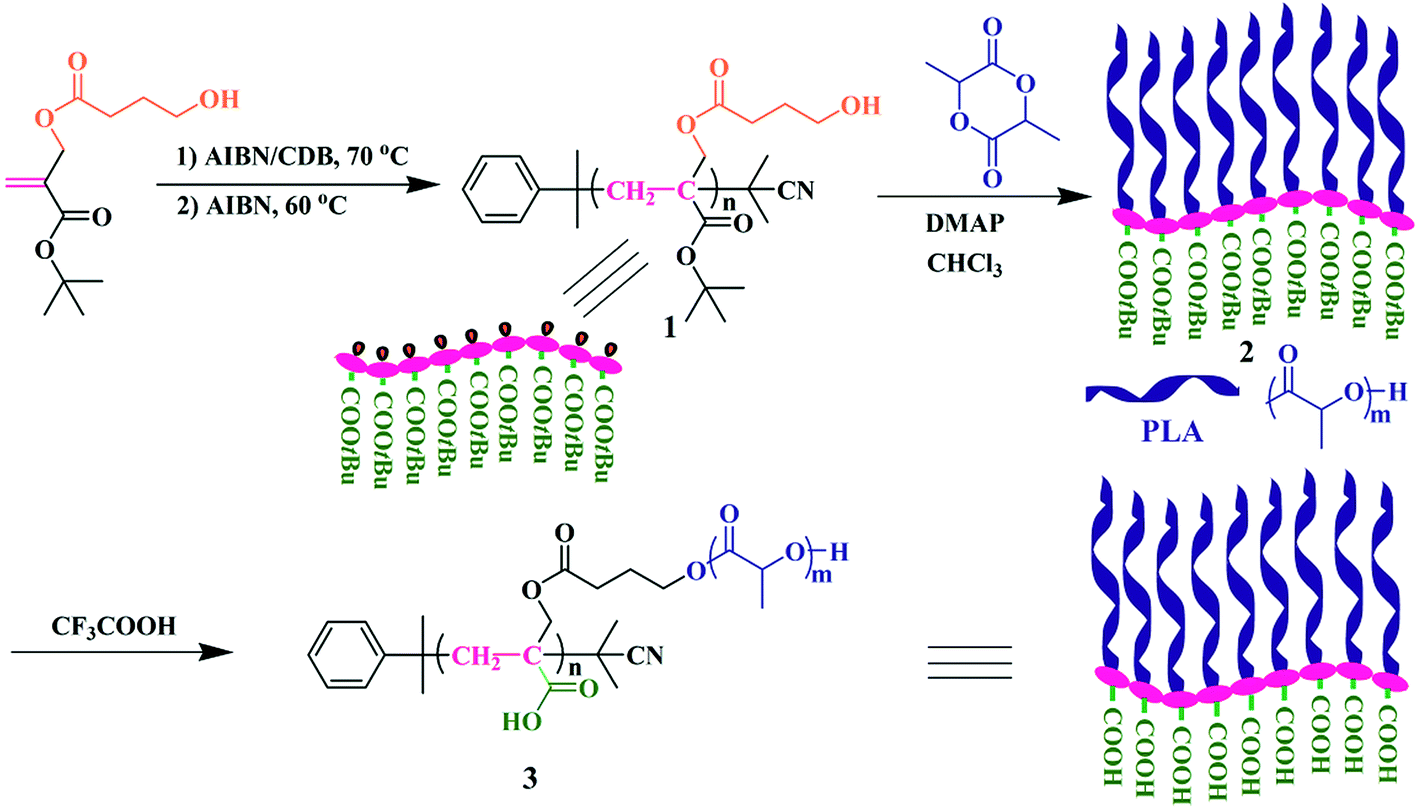 | ||
| Scheme 1 Synthesis of PAA-g-PLA well-defined amphiphilic graft copolymer. | ||
Results and discussion
Synthesis of PtBA-g-PLA well-defined graft copolymer
In 2013, our group developed a new trifunctional acrylate monomer of tBHBMA,51 which consists of a polymerizable double bond, a hydroxyl as initiating group for ROP, and a tert-butoxycarbonyl as potential carboxyl. As it is well-known that hydroxyl survives the reversible-deactivation radical polymerization (RDRP) such as atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain transfer (RAFT) polymerization, therefore we can expect that the pendant hydroxyls of the homopolymer of tBHBMA monomer can directly initiate ROP of lactone monomer including lactide to form the desired graft copolymer without polymeric functionality transformation, and tert-butoxycarbonyl can be readily converted to carboxyl for transforming hydrophobicity into hydrophilicity.RAFT homopolymerization of tBHBMA using AIBN as initiator and CDB as chain transfer agent was performed in DMF at 70 °C with a similar procedure51 to afford PtBHBMA 1 homopolymer bearing pendant hydroxyls in every repeat unit so as to achieve the synthesis of PLA-based amphiphilic graft copolymer with equally distributed carboxyls in the backbone. Fig. 1 shows GPC trace of PtBHBMA 1 homopolymer with a relative molecular weight (Mn,GPC) of 4400 g mol−1 and a narrow molecular weight distribution (Mw/Mn) of 1.26. The chemical structure of PtBHBMA 1 homopolymer is conformed by FT-IR (Fig. S1A†) and 1H NMR (Fig. 2A) and detailed discussion can be found in our previous report.51 The ‘absolute’ molecular weight of PtBHBMA 1 homopolymer (Mn,NMR = 6400 g mol−1, DP of tBHBMA = 25.3) is determined from 1H NMR of PtBHBMA 1 homopolymer using the same method described in our previous report.51 The minute amount of dithiobenzoate moiety was removed by AIBN according to a previous report52 for avoiding any possible effect of dithiobenzoate end group on the following ROP of lactide and facilitating the observation of polymerization, which was evidenced by the color change from pink to white after the treatment with AIBN.
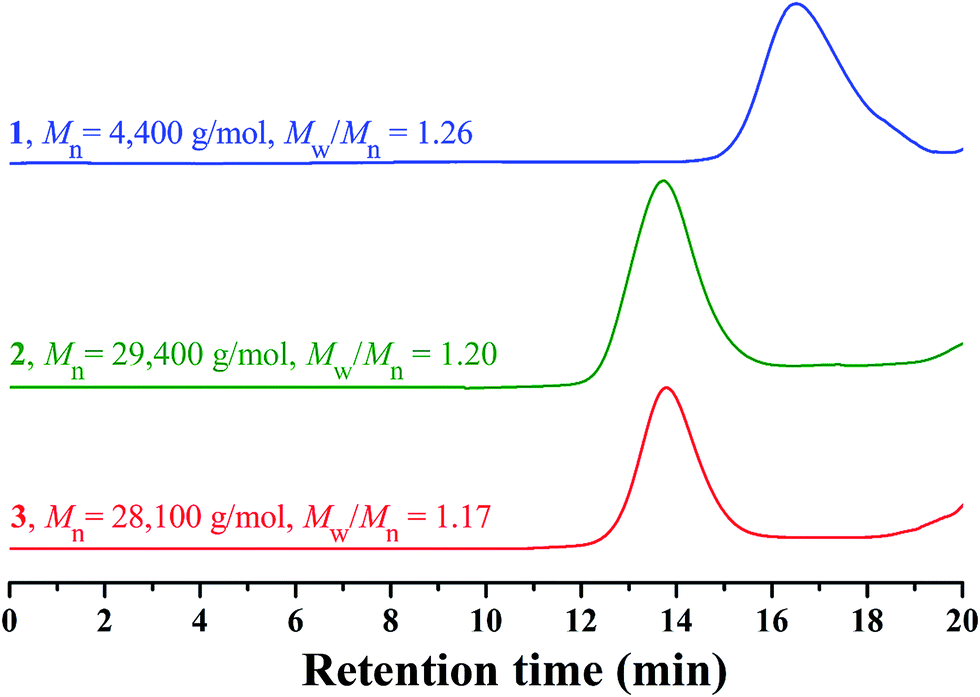 | ||
| Fig. 1 GPC traces of PtBHBMA 1, PtBA-g-PLA 2 and PAA-g-PLA 3 in THF. | ||
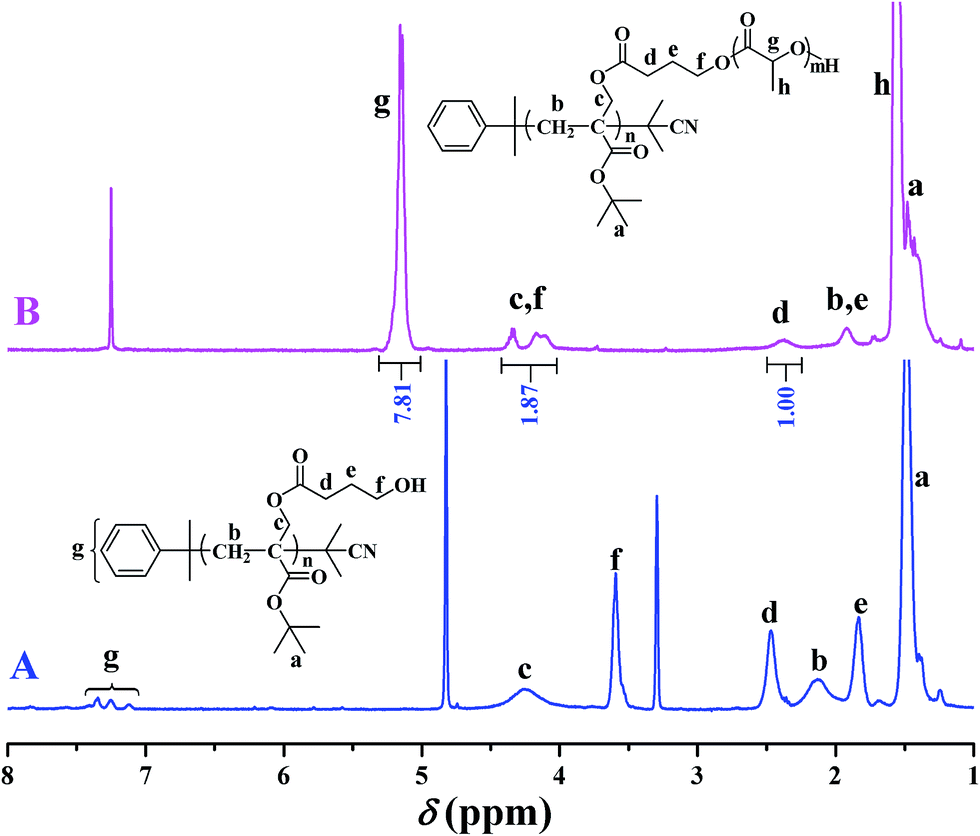 | ||
| Fig. 2 1H NMR spectra of PtBHBMA 1 in CD3OD (A) and PtBA-g-PLA 2 in CDCl3(B). | ||
Recently, organocatalytic ROP of lactones including lactide has been developed very fast since this strategy could eliminate the usage of transition metal as catalyst which could prevent the application of corresponding polymers in biomedical and microelectronic fields.53,54 Therefore, the grafting-from strategy is employed for the construction of PtBA-g-PLA 2 graft copolymer, and ROP of lactide was initiated by the pendant hydroxyls of PtBHBMA 1 homopolymer using DMAP as organocatalyst in this report so as to attach PLA side chains onto every repeat unit of PtBA-based backbone. The purified product shows a unimodal and symmetrical elution peak (Fig. 1) with a narrow molecular weight distribution below 1.20 and its molecular weight (Mn,GPC = 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1) is much higher than that of the macroinitiator, which indicates the proceeding of ROP of lactide.
400 g mol−1) is much higher than that of the macroinitiator, which indicates the proceeding of ROP of lactide.
The chemical structure of PtBA-g-PLA 2 graft copolymer is characterized by FT-IR and 1H NMR. The weak peak at 3510 cm−1 in FT-IR spectrum (Fig. S1A†) is assigned to the terminal hydroxyl of PLA side chains while the signal originating from the pendant hydroxyls of PtBHBMA 1 homopolymer at 3435 cm−1 (Fig. S1B†) is absent in Fig. S1A,† which implies all pendant hydroxyls are consumed to link PLA side chains. Moreover, the signal of carbonyl appears at 1756 cm−1 after ROP of lactide (Fig. S1A†) in comparison with that located at 1740 cm−1 before ROP of lactide (Fig. S1B†), this also witnessing the formation of PLA side chains. The peaks of both backbone and side chain are visible in 1H NMR spectrum (Fig. 2B). Typical signals of PLA side chains appear at 1.56 (peak ‘h’) and 5.15 (peak ‘g’) ppm, which belong to three protons of CHCH3 and one proton of CHCH3, respectively. In particular, the signal originating from two protons of –CH2 group adjacent to pendant hydroxyls of PtBHBMA 1 homopolymer (peak ‘f’ in Fig. 2A) disappears after ROP of lactide, which also clearly demonstrates that all pendant hydroxyls of PtBHBMA 1 homopolymer initiated ROP of lactide, i.e. the number of grafted PLA side chain after ROP of lactide equals to that of –CH2CH2CH2OH initiating group in tBBPMA repeat unit before ROP of lactide, that is 25.3. The peaks at 16.7, 69.1, and 169.4 ppm in 13C NMR spectrum (Fig. S2†) could be assigned to carbons of methyl, methylidyne, and carbonyl in PLA side chains, respectively, which further illustrates the grafting of PLA side chains onto the backbone. This fact indicates that only the terminal hydroxyl of PLA side chains exists in the graft copolymer after ROP of lactide, which is consistent with the result of FT-IR. Thus, we can affirm the chemical structure of PtBA-g-PLA 2 graft copolymer.
Previous work showed that the molecular weight of graft copolymer determined by GPC might differ from the “real” value because of its branched structure.55 So, 1H NMR was used to determine the ‘absolute’ molecular weight of PtBA-g-PLA 2 graft copolymer in the current case. The length of PLA side chain (nLA) and the molecular weight of the graft copolymer (Mn,NMR) were calculated according to eqn (1) (Sg and Sd are the integration area of peak ‘g’ at 5.15 ppm (1 proton of CHCH3) of PLA side chain and peak ‘d’ at 2.37 ppm (2 proton of CH2CH2CH2O) of the backbone in Fig. 2B, respectively) and eqn (2) (25.3 is the number of tBHBMA repeated unit, 6400 and 72 are the molecular weights of PtBHBMA 1 macroinitiator and lactide monomer), respectively. The length of PLA side chain is estimated to be 15.6 and Mn,NMR is evaluated to be 34800 g mol−1, obviously higher than that obtained from GPC.
| nLA = 2Sg/Sd | (1) |
| Mn,NMR = 6400 + 25.3 × 72 × nLA | (2) |
Thus, it can be concluded that a well-defined PtBA-g-PLA 2 graft copolymer bearing a PtBA-based backbone (25.3 repeat units) and equally-distributed PLA side chains (15.6 repeat units per chain) connected to every repeat unit of the backbone has been successfully synthesized by ROP of lactide initiated by PtBHBMA 1.
Conversion of PtBA-g-PLA to PAA-g-PLA
It is well-known that hydrophobic PtBA can be easily transformed into hydrophilic PAA via acidic hydrolysis under mild conditions without affecting polyacrylate skeleton and other functionality.56,57 Therefore, PAA-g-PLA amphiphilic graft copolymer with a PAA backbone can be obtained from the precursor, PtBA-g-PLA. Herein, CF3COOH was used to hydrolyze tert-butyoxycarbonyls of PtBA backbone in PtBA-g-PLA graft copolymer into carboxyls in CH2Cl2 at room temperature according to previous reports,56,57 in which excessive feeding of CF3COOH and long reaction time were employed to facilitate the hydrolysis of tert-butyoxycarbonyls. PLA side chains would keep inert during the hydrolysis process using CF3COOH.58It can be seen from FT-IR spectra before (Fig. S1A†) and after (Fig. S3†) the hydrolysis that a stronger peak at 3317 cm−1 attributed to the newly formed carboxyls appears in FT-IR spectrum after the hydrolysis in comparison with that before the hydrolysis. This fact apparently evidences the formation of PAA backbone. Fig. S3† also shows the remaining of typical signals of PLA (1760 and 1133 cm−1) segment in comparison with Fig. S1A,† which means that PLA segments indeed survive the hydrolysis. The hydrolyzed product also shows a unimodal and symmetrical elution peak (Fig. 1) with a similar molecular weight and molecular weight distribution compared to polymer 2, which affirms that polymeric skeleton was not affected during the hydrolysis. All these evidences confirm that PtBA-g-PLA 2 graft polymer has been selectively hydrolyzed in acidic environment for affording PAA-g-PLA 3 graft copolymer without affecting PLA side chains.
pH-responsive micellization of PAA-g-PLA amphiphilic graft copolymer
The amphiphilic nature of PAA-g-PLA 3 graft polymer, consisting of hydrophilic PAA backbone and hydrophobic PLA side chains, endows the polymer with capability to self-assemble in aqueous media. It has been reported that the solubility of PAA segment is related with the pH value of aqueous solution, i.e. PAA segment can become less soluble in acidic aqueous phase with a low pH due to the protonation of carboxyls in PAA segment.59 By lowering pH value of aqueous solution, PAA chain can turn from hydrophilic to hydrophobic and correspondingly, PAA-g-PLA 3 amphiphilic graft copolymer will also become completely hydrophobic. Therefore, fluorescence spectroscopy with N-phenyl-1-naphthylamine (PNA) as probe was employed to examine the pH-responsiveness of PAA-g-PLA 3 graft copolymer in aqueous media.Fluorescence spectrum of PNA is sensitively affected by the environment and the polarity of its surrounding.60 In the presence of micelles, PNA is solubilized within the interior of the hydrophobic part showing high fluorescence intensity. Fig. 3 shows the relationship of fluorescence intensity of PNA emission band at 418 nm with pH of the aqueous solution of PAA-g-PLA 3 graft copolymer and we can notice that the fluorescence intensity of PNA increases quickly when pH is above 4.36 because of the deprotonation of carboxyls to raise the hydrophilicity of the backbone for solubilizing PNA better, i.e. causing the rising of fluorescence intensity. When pH is above 5.77, the fluorescence intensity of PNA reaches the highest value, suggesting the formation of compact and stable micelles. Fig. 4A shows that the average particle size of polymeric micelles formed by PAA-g-PLA 3 graft copolymer is about 144.7 nm obtained from DLS. Fig. 4B is TEM image of polymeric micelles, which exhibits the spherical micelles with a diameter of ca. 54–78 nm. The smaller size observed by TEM compared to the result of DLS might be attributed to the shrinking of nanoparticles during the drying for the preparation of TEM specimen. Here, “spherical micelles” formed by PAA-g-PLA 3 amphiphilic graft copolymer should be large compound micelles, not common spherical micelles with only hydrophobic core covered by hydrophilic corona. That is, hydrophilic PAA segments form the corona of micelles and the core is consisted of numerous reverse micelles with islands of PAA segments in continuous phase of hydrophobic PLA segments.61,62 All these facts demonstrate that the self-assembly of PAA-g-PLA 3 amphiphilic graft copolymer in aqueous media is pH-dependent.
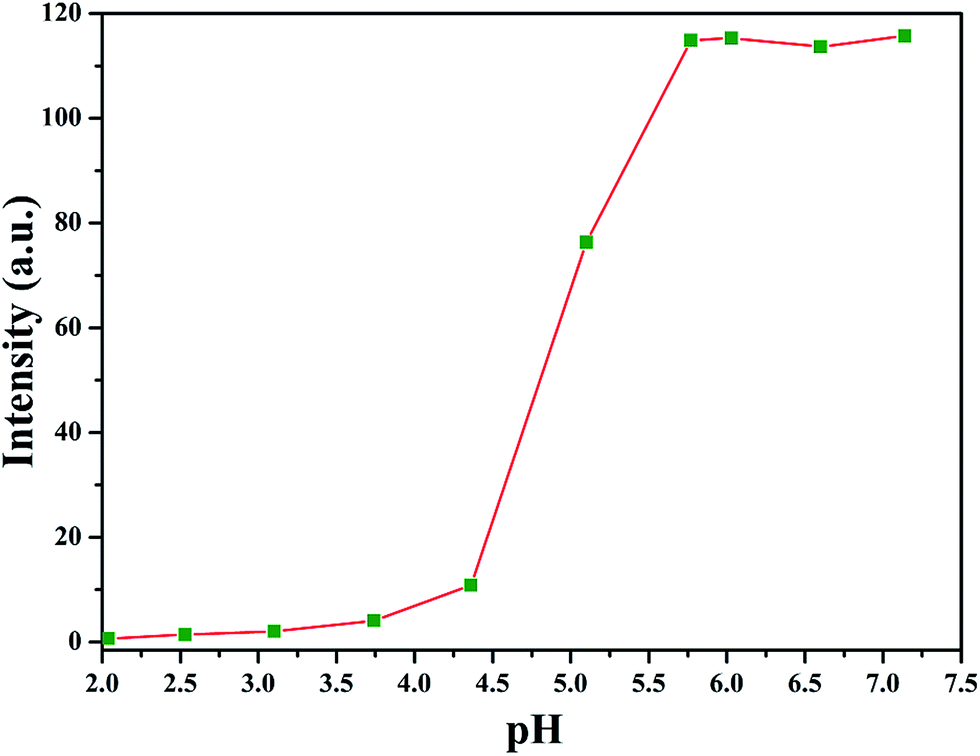 | ||
| Fig. 3 Dependence of fluorescence intensity of PNA emission band at 418 nm on pH of the aqueous solution of PAA-g-PLA 3 graft copolymer. | ||
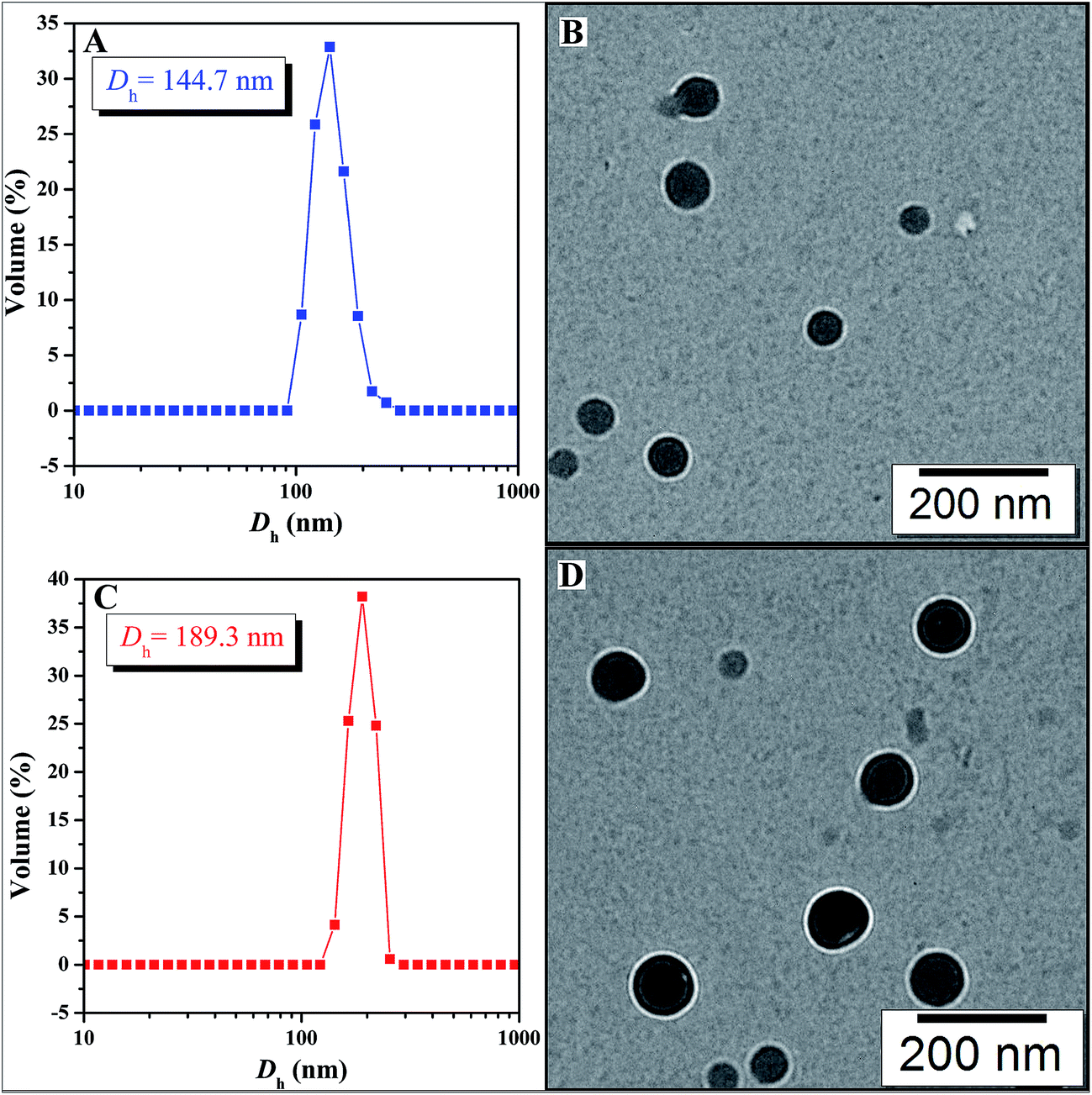 | ||
| Fig. 4 Hydrodynamic diameter distributions of bare (A) and MTX-loading (C) micelles prepared from PAA-g-PLA 3; TEM images of bare (B) and MTX-loading (D) micelles prepared from PAA-g-PLA 3. | ||
Drug delivery of MTX using PAA-g-PLA graft polymer as carrier
On the basis of aforementioned results, PAA-g-PLA 3 amphiphilic graft copolymer could assemble into large compound micelles with a diameter of about 144.7 nm, which greatly benefits the application for drug delivery due to the so-called EPR effect.63,64 Therefore, we used PAA-g-PLA 3 amphiphilic graft copolymer as a carrier to prepare MTX-loading nanoparticles by dialysis approach (Mn,cut-off = 3500 g mol−1) against double-distilled water and the resulting solutions were then subjected to UV/vis spectroscopy for determining the drug loading content (DLC, 5.3%) and drug loading efficiency (DLE, 20.6%). The average particle size and size distribution of MTX-loading micelles were determined by DLS and the size is about 189.3 nm (Fig. 4C), which is among the range of 50–200 nm suitable for EPR effect. TEM image reveals spherical shapes and a good dispersity with nanoscale sizes of about 83–98 nm (Fig. 4D).In vitro release profiles of free MTX and MTX-loading micelles were assessed using the dialysis technique in PBS (0.01 M, pH = 5.0) at 37 °C. As shown in Fig. 5, free MTX almost completely released during 24 h while in the case of MTX-loading micelles, about 33.1% of MTX released during the same time, and the accumulative release amount of MTX increases gradually with the extending of time, which indicates a significantly reduced burst release effect and an obvious sustained drug release behavior.
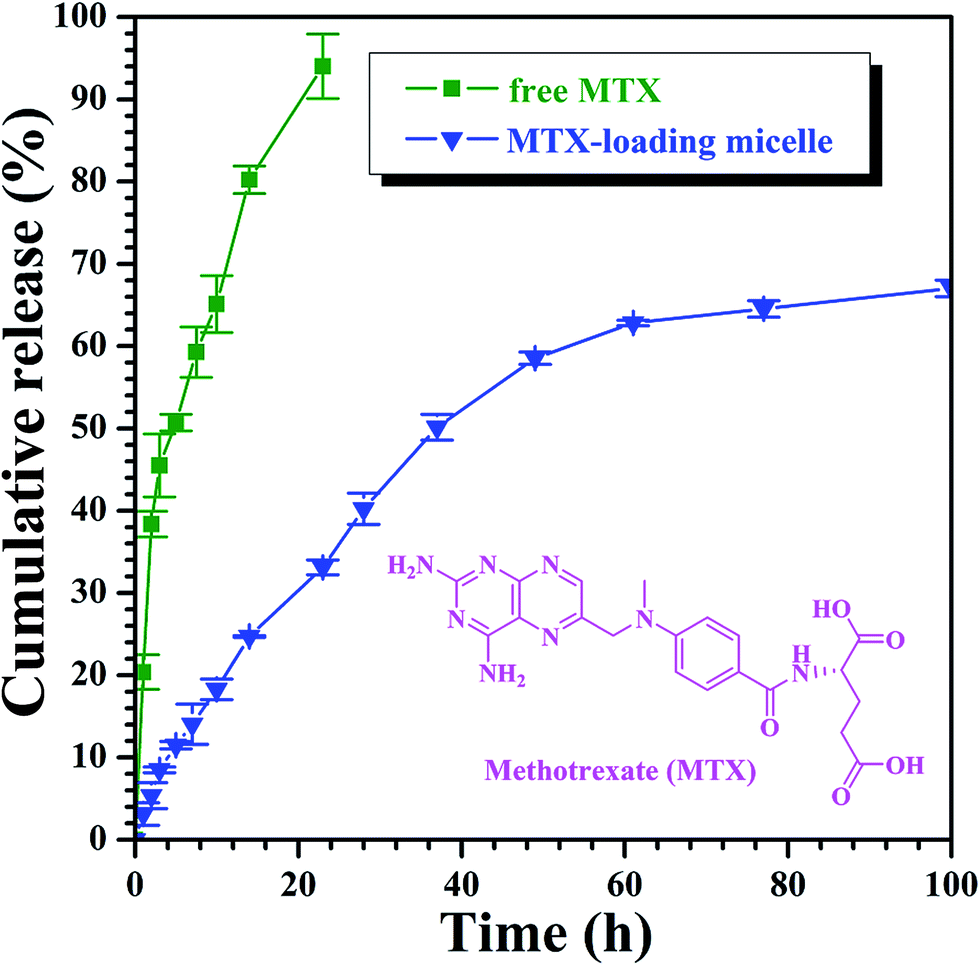 | ||
| Fig. 5 In vitro MTX release profiles of free MTX and MTX-loading nanoparticles at 37 °C under pH = 5.0. | ||
The in vitro cytotoxicity of free MTX, MTX-loading nanoparticles, and the bare PAA-g-PLA carrier were evaluated with MG-63 (human osteogenic sarcoma cell) using WST-8 based colorimetric assay as shown in Fig. 6. High cell viability for MG-63 (>93%) cells are found for various concentrations (up to 16 μM) of PAA-g-PLA 3 micelles after incubating for 72 h, this indicating the very low cytotoxicity of PAA-g-PLA 3 micelles and the suitability to be used as drug carrier. Both MTX-loading nanoparticles and free MTX are found to inhibit MG-63 cells and the relative cell viability declines monotonously with ascending the concentration of MTX. Obviously, MTX-loading nanoparticles show a higher cytotoxic effect on MG-63 cells in comparison with free MTX. For example, the viabilities of MG-63 cells are 47.7% for free MTX and 36.4% for MTX-loading nanoparticles with a settled MTX concentration of 4 μM. Furthermore, it is worthy pointing out that MTX-loading nanoparticles with a low concentration of MTX exhibit a high cytotoxicity similar to free MTX with a high concentration. For instance, the viability of MG-63 cells is 40.6% for free MTX with a concentration of 8 μM, while it is 40.8% for MTX-loading nanoparticles with just 1 μM of MTX, which clearly indicates that PAA-g-PLA could be used to load a low concentration of MTX for achieving the same treatment using a higher concentration of free MTX. Besides, the effect of incubation time on the cytotoxicity of MTX-loading nanoparticles and free MTX with a constant concentration of MTX (16 μM) was investigated. It can be seen from Fig. 6B that MG-63 cells are inhibited by both free MTX and MTX-loading nanoparticles in a time-dependent mode. MTX-loading nanoparticles obviously exhibit a higher cytotoxicity effect on MG-63 cells compared to free MTX at every time interval. These above-mentioned results indicate that micelles prepared from PAA-g-PLA 3 amphiphilic graft copolymer could load MTX to improve the efficacy of MTX.
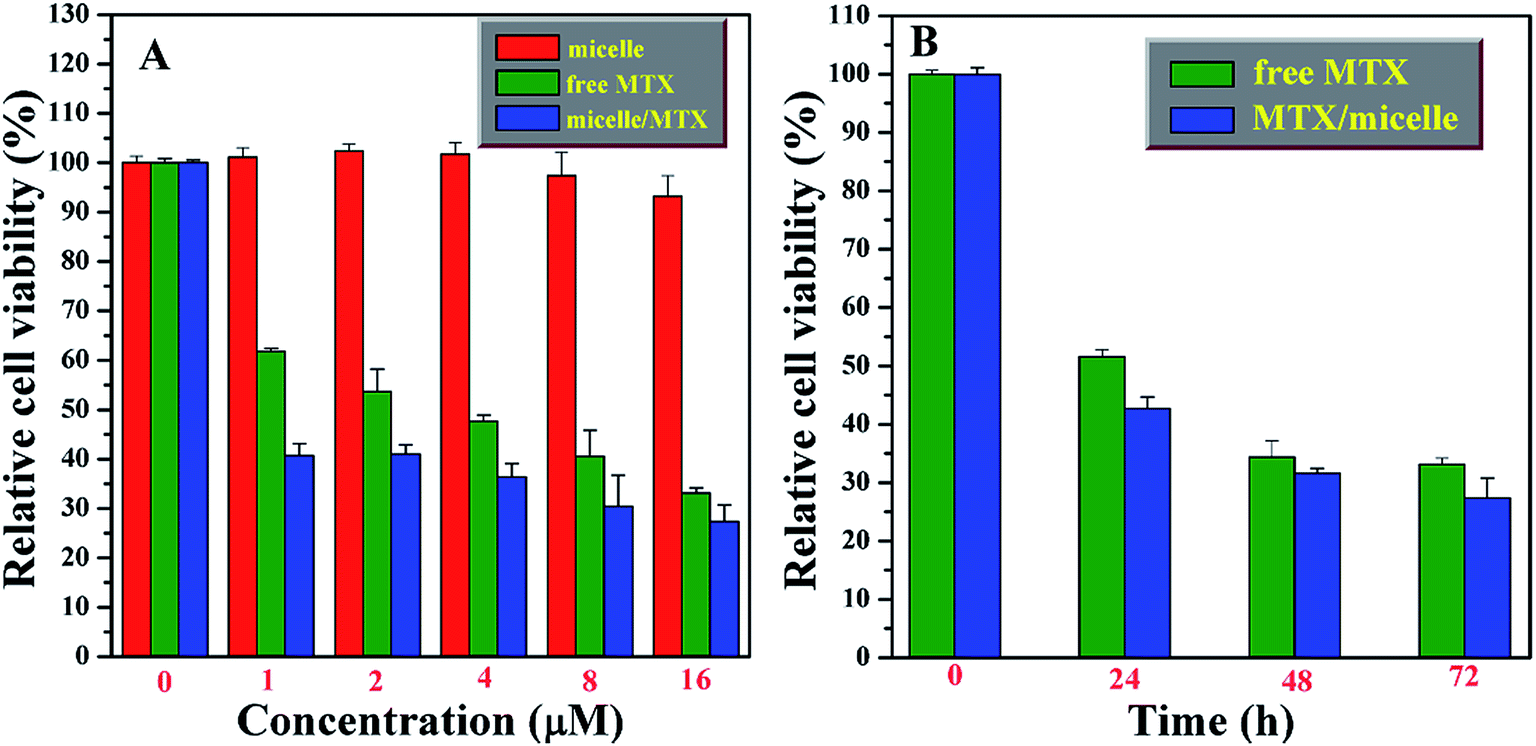 | ||
| Fig. 6 Relative viabilities of MG-63 cells after treatment with different concentrations of PAA-g-PLA 3 micelles, MTX-loading micelles and free MTX for 72 h (A) and with different time at a constant MTX concentration of 16 μM (B). The experiments were repeated three times, all with similar results. The data are presented as the mean ± S. D. (for each group, n = 3). | ||
Conclusions
This report provides the synthesis and self-assembly studies on a new PLA-based amphiphilic graft copolymer, PAA-g-PLA. Target amphiphilic graft copolymer with equally distributed carboxyls in backbone was prepared by the acidolysis of hydrophobic backbone of a well-defined PtBA-g-PLA graft copolymer with a narrow molecular weight distribution (Mw/Mn = 1.20), which was obtained by the combination of RAFT polymerization of tBHBMA monomer and ROP of lactide, into hydrophilic PAA backbone. This amphiphilic graft copolymer is pH-responsive and can aggregate into large compound micelles in aqueous media. MTX was loaded by this kind of micelles self-assembled from PAA-g-PLA amphiphilic graft copolymer to provide MTX-loading nanoparticles with an average size of ca. 189.3 nm (DLS). It is illustrated that MTX-loading nanoparticles show sustained drug release profiles and inhibit MG-63 cells in a concentration- and time-dependent manner so as to exhibit a higher cytotoxicity effect compared to free MTX. All these results suggest that PAA-g-PLA graft copolymer may be a good candidate as carrier for MTX or other anti-tumor drugs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support from National Natural Science Foundation of China (21474127), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000), and Shanghai Scientific and Technological Innovation Project (16JC1402500 and 16520710300).Notes and references
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- M. Akarami, I. Ghasemi, H. Azizi, M. Karrabi and M. Sayedabadi, Carbohydr. Polym., 2016, 144, 254–262 CrossRef PubMed.
- G. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082–2099 CrossRef CAS PubMed.
- L. Yu, K. Dean and L. Li, Prog. Polym. Sci., 2006, 31, 576–602 CrossRef CAS.
- A. Raup, H. Wang, C. V. Synatschke, V. Jérôme, S. Agarwal, D. V. Pergushov, A. H. E. Müller and R. Freitag, Biomacromolecules, 2017, 18, 808–818 CrossRef CAS PubMed.
- M. Vert, Eur. Polym. J., 2015, 68, 516–525 CrossRef CAS.
- B. Gupta, N. Revagade and J. Hilborn, Prog. Polym. Sci., 2007, 32, 455–482 CrossRef CAS.
- Y. J. Xi, T. Song, S. Y. Tang, N. S. Wang and J. Z. Du, Biomacromolecules, 2016, 17, 3922–3930 CrossRef CAS PubMed.
- I. Manavitehrani, A. Fathi, Y. W. Wang, P. K. Maitz, F. Mirmohseni, T. L. Cheng, L. Peacock, D. G. Little, A. Schinder and F. Dehghani, Biomacromolecules, 2017, 18, 1736–1746 CrossRef CAS PubMed.
- V. Lassalle and M. L. Ferreira, Macromol. Biosci., 2007, 7, 767–780 CrossRef CAS PubMed.
- Y. Zhang, S. Liu, X. Wang, Z. Y. Zhang, X. B. Jing, P. Zhang and Z. G. Xie, Chin. J. Polym. Sci., 2014, 32, 1111–1118 CrossRef CAS.
- C. F. Yang, F. Sun, L. J. Zhang, G. D. Zhu, C. Y. Zhang and Y. Qian, Chin. J. Chem., 2012, 30, 1980–1986 CrossRef CAS.
- J. K. Oh, Soft Matter, 2011, 7, 5096–5108 RSC.
- S. Corneillie and M. Smet, Polym. Chem., 2015, 6, 850–867 RSC.
- X. M. Sun, J. X. Xu, J. B. Tang, M. H. Sui and Y. Q. Shen, Chin. J. Polym. Sci., 2011, 29, 427–430 CrossRef CAS.
- F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37–53 CrossRef CAS PubMed.
- Y. Q. Zhu, B. Yang, S. Chen and J. Z. Du, Prog. Polym. Sci., 2017, 64, 1–22 CrossRef CAS.
- Y. F. Xiao, H. Sun and J. Z. Du, J. Am. Chem. Soc., 2017, 139, 7640–7647 CrossRef CAS PubMed.
- Q. M. Liu, L. W. Song, S. Chen, J. Y. Gao, P. Y. Zhao and J. Z. Du, Biomaterials, 2017, 114, 23–33 CrossRef CAS PubMed.
- P. Broz, S. Driamov, J. Ziegler, N. Ben-Haim, S. Marsch, W. Meier and P. Hunziker, Nano Lett., 2006, 6, 2349–2353 CrossRef CAS PubMed.
- C. Y. Yang, Q. Li, C. H. Cai and J. P. Lin, Langmuir, 2016, 32, 6917–6927 CrossRef CAS PubMed.
- S. B. Clendenning, S. Fournier-Bidoz, A. Pietrangelo, G. C. Yang, S. J. Han, P. M. Brodersen, C. M. Yip, Z. H. Lu, G. A. Ozin and T. Manners, J. Mater. Chem., 2004, 14, 1686–1690 RSC.
- K. Nakajima, T. Fukui, H. Kato, M. Kitano, J. N. Kondo, S. Hayashi and M. Hara, Chem. Mater., 2010, 22, 3332–3339 CrossRef CAS.
- Y. L. Cai and S. P. Armes, Macromolecules, 2005, 38, 271–279 CrossRef CAS.
- Y. L. Cai, Y. Q. Tang and S. P. Armes, Macromolecules, 2004, 37, 9728–9737 CrossRef CAS.
- Y. Wang and S. M. Grayson, Adv. Drug Delivery Rev., 2012, 64, 852–865 CrossRef CAS PubMed.
- C. Nouvel, J. Raynaud, E. Marie, E. Dellacherie, J. L. Six and A. Durand, J. Colloid Interface Sci., 2009, 330, 337–343 CrossRef CAS PubMed.
- Y. Wan, X. Cao, Q. Wu, S. Zhang and S. Wang, Polym. Adv. Technol., 2008, 19, 114–123 CrossRef CAS.
- Y. Wan, X. Cao, S. Zhang, S. Wang and Q. Wu, Acta Biomater., 2008, 4, 876–886 CrossRef CAS PubMed.
- S. J. Carlotti, O. Giani-Beaune and F. Schue, J. Appl. Polym. Sci., 2001, 80, 142–147 CrossRef CAS.
- Y. Ohya, S. Maruhashi and T. Ouchi, Macromol. Chem. Phys., 1998, 199, 2017–2022 CrossRef CAS.
- Y. Ohya, S. Maruhashi and T. Ouchi, Macromolecules, 1998, 31, 4662–4665 CrossRef CAS.
- M. Li, Q. Huang and Y. Wu, Pest Manage. Sci., 2011, 67, 831–836 CrossRef CAS PubMed.
- A. Lutzke, J. B. Tapia, M. J. Neufeld and M. M. Reynolds, ACS Appl. Mater. Interfaces, 2017, 9, 2104–2113 CAS.
- R. Riva, S. Schmeits, F. Croisier, P. Lecomte and C. Jerome, Clin. Hemorheol. Microcirc., 2015, 60, 65–70 CrossRef CAS PubMed.
- C. H. Kim, K. Y. Cho and J. K. Park, Polym. Eng. Sci., 2001, 41, 542–553 CAS.
- M. Labet and W. Thielemans, Polym. Chem., 2012, 3, 679–684 RSC.
- X. F. Niu, F. Tian, L. Z. Wang, X. M. Li, G. Zhou and Y. B. Fan, Chin. J. Polym. Sci., 2014, 32, 43–50 CrossRef CAS.
- J. Li, M. Kong, X. J. Cheng, Q. F. Dang, X. Zhou, Y. N. Wei and X. G. Chen, Int. J. Biol. Macromol., 2012, 51, 221–227 CrossRef CAS PubMed.
- T. Okuda, K. Ishimoto, H. Ohara and S. Kobayashi, Macromolecules, 2012, 45, 4166–4174 CrossRef CAS.
- H. Shinoda and K. Matyjaszewski, Macromolecules, 2001, 34, 6243–6248 CrossRef CAS.
- C. Zhao, D. Wu, N. A. Huang and H. Zhao, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 589–598 CrossRef CAS.
- A. Breitenbach and T. Kissel, Polymer, 1998, 39, 3261–3271 CrossRef CAS.
- N. Guerrouani, A. Mas and F. Schue, J. Appl. Polym. Sci., 2009, 113, 1188–1197 CrossRef CAS.
- Y. Zhu, T. Akagi and M. Akashi, Polym. J., 2013, 45, 560–566 CrossRef CAS.
- C. Feng, Y. J. Li, D. Yang, J. H. Hu, X. H. Zhang and X. Y. Huang, Chem. Soc. Rev., 2011, 40, 1282–1295 RSC.
- S. J. Zhai, J. Shang, D. Yang, S. Y. Wang, J. H. Hu, G. L. Lu and X. Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 811–820 CrossRef CAS.
- S. J. Zhai, X. M. Song, C. Feng, X. Y. Jiang, Y. J. Li, G. L. Lu and X. Y. Huang, Polym. Chem., 2013, 4, 4134–4144 RSC.
- X. Y. Jiang, X. Jiang, G. L. Lu, C. Feng and X. Y. Huang, Polym. Chem., 2014, 5, 4915–4925 RSC.
- G. L. Lu, H. Liu, H. F. Gao, C. Feng, Y. J. Li and X. Y. Huang, RSC Adv., 2015, 5, 39668–39676 RSC.
- X. M. Song, W. Q. Yao, G. L. Lu, Y. J. Li and X. Y. Huang, Polym. Chem., 2013, 4, 2864–2875 RSC.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- S. A. Berg, H. Zuilhof and T. Wennekes, Macromolecules, 2016, 49, 2054–2062 CrossRef.
- Z. F. Jia, Q. Fu and J. L. Huang, Macromolecules, 2006, 39, 5190–5193 CrossRef CAS.
- P. P. Li, Z. Y. Li and J. L. Huang, Macromolecules, 2007, 40, 491–498 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, K. L. Wooley and C. J. Hawker, Chem. Mater., 2005, 17, 5976–5988 CrossRef.
- H. H. Liu, S. X. Li, M. J. Zhang, W. Shao and Y. L. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4705–4716 CrossRef CAS.
- S. C. Lee, K. J. Kim, Y. K. Jeong, J. H. Chang and J. Choi, Macromolecules, 2005, 38, 9291–9297 CrossRef CAS.
- L. C. You, F. Z. Lu, Z. C. Li, W. Zhang and F. M. Li, Macromolecules, 2003, 36, 1–4 CrossRef CAS.
- L. F. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- Y. S. Yu and A. Eisenberg, J. Am. Chem. Soc., 1997, 119, 8383–8384 CrossRef CAS.
- L. L. Li, F. Q. Tang, H. Y. Liu, N. J. Hao, D. Chen, X. Teng and J. Q. He, ACS Nano, 2010, 4, 6874–6882 CrossRef CAS PubMed.
- J. M. W. Chan, R. J. Wojtechki, H. Sardon, A. L. Z. Lee, C. E. Smith, A. Shkumatov, S. J. Gao, H. Kong, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2017, 6, 176–180 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11975e |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |