
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural insights into HIV-1 protease flap opening processes and key intermediates†
Yuqi Yu ab,
Jinan Wanga,
Zhaoqiang Chena,
Guimin Wangab,
Qiang Shao*a,
Jiye Shi*c and
Weiliang Zhu*a
ab,
Jinan Wanga,
Zhaoqiang Chena,
Guimin Wangab,
Qiang Shao*a,
Jiye Shi*c and
Weiliang Zhu*a
aDrug Discovery and Design Center, CAS Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai, 201203, China. E-mail: qshao@mail.shcnc.ac.cn; wlzhu@mail.shcnc.ac.cn
bUniversity of Chinese Academy of Sciences, UCAS, No.19A Yuquan Road, Beijing, 100049, China
cUCB Biopharma SPRL, Chemin du Foriest, Braine-l'Alleud, Belgium. E-mail: Jiye.Shi@ucb.com
First published on 21st September 2017
Abstract
HIV-1 protease (PR) is an effective drug target for antiviral inhibitors. The conformational dynamics in the flaps of HIV-1 PR plays a crucial role in the mechanism of substrate binding. Here, the structural properties of the functionally important intermediate states of the flap opening transition of HIV-1 PR have been characterized by enhanced sampling molecular dynamics simulation as well as long-time conventional non-enhanced simulations at atomic level. Not only crystallographically measured “closed” and “semi-open” structures but also a novel “curled” structure of HIV-1 PR is captured by both kinds of simulations qualitatively and quantitatively. The observation of the “curled” intermediate state helps to connect all other functionally important states to provide an integrated view of the transition pathway of the flap opening of HIV-1 PR (closed → curled → semi-open → fully open). The key residue–residue interactions which are broken or formed in the transition are analyzed to reveal the inherent driving force for the protein conformational transition.
Introduction
HIV-1 (human immunodeficiency virus type 1) protease (PR) is an effective drug target for antiviral inhibitors, which is essential to the replication and invasion of HIV as the protease is responsible for cleaving large polyprotein precursor gag and releasing small structural proteins to help the assembly of infectious viral particles.1–3 As a symmetrically assembled homo-dimer, HIV-1 PR consists of six structural segments (Fig. 1): flap (residues 43–58/43′–58′), flap elbow (residues 35–42/35′–42′), fulcrum (residues 11–22/11′–22′), cantilever (residues 59–75/59′–75′), interface (residues 1–5/1′–5′, 95–99/95′–99′), and active site (residues 23–30/23′–30′).4,5 The flexibility of the flaps is believed to control the access of substrates and inhibitors to the active site and is thus recognized as essential for enzymatic activity. Mutations of flap residues may alter the flexibility and conformation of the flaps. For instance, mutations in flap residues such as 46, 47, 48, 50, 53, and 54 are frequently observed in drug-resistant mutants of HIV-1 PR and show various levels of reduced drug susceptibility to different PR inhibitors.6,7 | ||
| Fig. 1 Superposition of the crystal structures of the closed (3IXO), semi-open (1HHP), and wide-open (1TW7) forms of HIV-1 PR. Arrows indicate the direction of changes during the flap opening. | ||
Altered flap conformations have been reported for wild-type or mutated HIV-1 PR in the unliganded form or with bound inhibitors.8,9 All of the crystal structures of protease–inhibitor complexes in Protein Data Bank (PDB) adopt “closed” conformations, in which the flaps take a downward configuration towards the active site (Fig. 1). The inhibitor-free (apo) HIV-1 PR shows a variety of flap conformations, ranging from closed8,10 to semi-open9,11–14 to wide-open.9,15,16 Accordingly, the flap hairpin moieties could move apart by several angstroms (upon ∼7.7 Å) compared to the closed conformation (Fig. 1). No fully open structure has been captured by currently available experimental techniques yet,3,12,14 which is probably attributed to its short transient lifetime. It is speculated that flaps can fully open to provide access for the substrate and then Asp25 and protonated Asp25′ residues in the active site aid a lytic water to hydrolyze the peptide bond of substrate, producing smaller infectious protein.17,18
The flexibility of the flaps results in not only the ambiguity in the preferred flap conformation of HIV-1 PR in aqueous solution but also the difficulties in tracking the flap opening transition in real time at the level of a single protein molecule. Early sub-nanosecond timescale NMR experiment by Torchia and coworkers19–21 suggested that the semi-open conformation accounts for a major fraction of the equilibrium conformational ensemble for apo HIV-1 PR in solution, and a structural fluctuation is measurable on flap tips which is in a slow equilibrium (∼100 μs) from semi-open to fully open form. Fanucci and Simmerling investigated the dynamics of the inhibitor-bound and unbound flap by the site-directed spin labeling (SDSL) double electron–electron resonance (DEER) electron paramagnetic resonance (EPR) spectroscopy combined with molecular dynamics (MD) simulation.22 They observed that drug pressure selected mutations can alter the flap dynamics.23 In addition, by directly comparing EPR-measured distance distribution to MD simulation results, Simmerling found that the semi-open conformation is dominant for apo HIV-1 PR.24 Very recent NMR experiment, however, indicated that apo HIV-1 PR, on average, adopts a closed conformation which is very similar to the inhibitor-bound state in solution.25 The X-ray diffraction data from Robbins et al., on the other hands, show different flap orientation preference among different subtypes of HIV:8 while apo HIV-1 subtype A has only been crystallized in a closed conformation, apo HIV-1 PR isolated from subtype B has six structures in a semi-open conformation, two in a wide-open conformation, and only one in the closed conformation. However, these data cannot distinguish whether the heterogeneity reflects the preferred flap conformation or results affected by crystal packing condition.8,9
The dynamic properties of HIV-1 PR flaps have been investigated by molecular simulations as well. Early MD simulation suggested that the closed conformation is predominant in solution,26 several more recent all-atom or coarse-grained MD simulations observed the flap opening transition.27–30 For instance, an irreversible opening was observed starting from the semi-open structure in 10 ns MD simulation.27 More recent implicit solvent MD simulation observed the spontaneous opening and reclosing of flaps in the timescale of ∼40 ns.29 One drawback to these prior computational studies is that a very small number of transition events can be sampled in short time conventional MD simulations, making it difficult to quantitatively evaluate the thermodynamic stabilities of the different states and show the statistical significance of the transition pathway(s). Recently, a kinetic network model constructed from detailed atomistic simulations was developed to compute transition pathways of HIV-1 PR.31 A variety of different transition pathways were assessed and their temperature dependence was measured through free energy landscape analysis. In addition, the umbrella sampling simulation along collective “consensus” normal modes allows the calculation of the potential of mean force (PMF) for the conformational transition of HIV-1 PR.32 The detailed structural properties of HIV-1 PR in transition pathway(s) were, however, not deeply studied in the two enhanced sampling molecular simulations. Therefore, despite the progress of simulations, the understanding of the flap conformational transition and its impact on substrate binding still remains incomplete.
Enhanced conformational sampling methods enable one to explore the transition of HIV-1 PR flap opening that would require long conventional MD simulations in order to properly account for its conformational and energetic aspects. Replica exchange molecular dynamics (REMD)33 is probably the most popular enhanced sampling method, which utilizes a series of replicas of system to cover a wide range of temperatures.34–39 Protein configurations with similar energy in neighboring replicas are exchanged on the basis of Metropolis exchange criterion40 at intervals. The requirement in computational resource is, however, huge from standard REMD. We have modified a velocity-scaling optimized hybrid explicit/implicit solvent REMD (hREMD) strategy to reduce the replica number on the premise of maintaining high sampling efficiency: each replica is simulated explicitly and independently while the potential energy of protein is calculated by generalized Born (GB) model implicitly and used as a criterion for exchange attempt.41 Once exchange attempt completes, removed solvent molecules are restored at their previous positions and velocities and REMD simulation resumes in purely explicit solvent regime. The application of such method on the folding of small polypeptide as well as the conformational transition of small protein domain can highly maintain the relative calculation accuracy and meanwhile save massive computation cost.41,42
In this study, we utilized hREMD to explore the conformation space and free energy surface of apo wild-type HIV-1 PR so as to qualitatively and quantitatively identify crucial intermediate states in the pathway of the flap opening transition. In addition, we ran long-time conventional MD simulations on apo wild-type HIV-1 PR as well as two multi-drug resistant variants (Flap+ (L10I/G48V/I54V/V82A) and Act (V82T/I84V)) to further indicate the dynamic properties of flaps. The combination of hREMD and conventional MD simulations connects the functionally important states involved in the flap opening and provides an integrated view of the conformational transition of HIV-1 PR.
Computational methods
Enhanced conformation sampling of HIV-1 protease using hybrid explicit/implicit solvent REMD (hREMD) simulation
The detailed theoretical description of hybrid explicit/implicit solvent REMD (hREMD) method is referred to our previous publications.41,42 To run the molecular simulation, the initial structure of apo HIV-1 PR was obtained from the crystal wide-open form of multi-drug resistant variant (PDB code: 1TW7 (ref. 15)), with the mutated residues of 10, 36, 46,54, 62, 63, 71, 82, 84, and 90 replaced back by the wild-type ones. It is noteworthy that multiple residues take two alternative conformations in the crystal structure (e.g., residues 34, 35, 37, 43, 85 in chain A and residues 7′, 34′, 35′, 41′, 45′, 85′ in chain B). For these residues, the first conformation that shows higher occupancy in PDB file was chosen to construct the initial simulation structure. In addition, Asp25′ in chain B of HIV-1 PR was protonated considering its special role in catalyzing substrate hydrolysis.43,44 Protein was parameterized using AMBER FF03 force field45 and solvated in a cubic box (67.7 Å × 66.2 Å × 82.2 Å) containing 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 153 TIP3P waters.46 Five chloride counterions were added to neutralize the system.
153 TIP3P waters.46 Five chloride counterions were added to neutralize the system.
The constructed system was minimized using the steepest descent minimization of 50000 steps followed by a conjugate gradient minimization of 50000 steps. Then the system was heated to 300 K in 500 ps with a harmonic restraint (force constant = 10 kcal mol−1 Å−2) applied to the heavy atoms of protein. Finally, hREMD simulation was performed for the heated structure. All simulations were carried out with AMBER 10 program.47
The hREMD simulation was run with 14 replicas covering the temperatures from 280 to 450 K (280.00, 290.50, 302.00, 313.00, 324.90, 337.00, 349.40, 362.70, 376.50, 389.80, 404.20, 418.30, 433.87, and 450.00 K). The number of replicas was determined using the webserver (http://folding.bmc.uu.se/remd/),48 and the temperature distribution among the replicas was subsequently optimized using the algorithm of Nadker and Hansmann49 to give a theoretical acceptance probability of 20%. Hydrogen atoms was constrained using SHAKE50 algorithm and time step is set to 2.0 fs. The isotropic periodic sum51,52 was used to calculate the long-range interaction calculation, including electrostatic and van der Waals interactions. The Langevin thermostat with a collision frequency of 3.0 ps−1 was used to keep whole system at corresponding temperatures. Exchanges between neighboring replicas were attempted every 1000 steps. The implicit solvent term Pis, that is used for calculating the exchange move probabilities, was calculated through the generalized-Born model.53 The simulation continued for ∼120 ns per replica.
Conventional molecular dynamics simulation of wild-type protease and two multi-drug resistant variants
Wild-type HIV-1 PR, Flap+ and Act mutants (PDB codes: 3EKV, 3EKP, 1T7J respectively) after deleting inhibitors were used as starting structures.54,55 All crystal waters are kept considering their possible critical role in conformational change of HIV-1 protease. The procedure of system construction is the same with the above descriptions. All simulations were carried out by the program GROMACS version 4.5.3 using the NPT ensemble and periodic boundary condition. Each simulation system was first subjected to energy minimization using the steepest descents algorithm. Subsequently, a 5 ns MD simulation was carried out to heat the system to 300 K with the protein fixed using a harmonic restraint (force constant = 10 kcal mol−1 Å−2), followed by another 5 ns MD simulation with the protein Cα atoms fixed. Finally, production molecular dynamics was run without any constraints for 500 ns. The temperature of each system was maintained at 300 K using V-rescale thermostat56 with response time as 1.0 ps−1. The pressure was kept at 1 bar using the Parrinello–Rahman pressure coupling scheme (τ = 1 ps).57 The cutoff for Lennard-Jones interactions was set as 12 Å and the electrostatic interactions were calculated using the particle Mesh Ewald (PME) algorithm58 with a real-space cutoff of 12 Å. The LINCS59 method was used to restrain bond lengths that including hydrogen atoms, allowing an integration step of 2 fs.Results and discussion
Structural characterization of important states of HIV-1 PR on the free energy surface
Using backbone root-mean-square deviation (RMSD) of the entire protein and the RMSD of the flap region with respect to the crystal closed structure as the reaction coordinates, the two-dimensional free energy landscape at room temperature (300 K) was depicted in Fig. 2. The free energy landscape was calculated with the normalized probability, P(x) = Z−1e−βW(x) from a histogram analysis, where x is any set of reaction coordinate(s). is the relative free energy or potential of mean force.60 Four free energy local minima are presented in the free energy profile. For each local minimum, snapshots which satisfy the criteria according to the RMSDs were selected from 300 K trajectory and hierarchical clustering analyses (kclust algorithm available in MMTSM Toolset61) was performed focused on the selected snapshots to identify the representative (most populated) conformation. The four local minima correspond to closed, curled, semi-open, and fully open states, with the population of 7.32%, 36.5%, 41.1%, and 1.3% in the equilibrium conformational ensemble in 300 K water solution, respectively. To show the convergence of the hREMD simulation, we calculated the time series of the fraction of the four populated structures at 300 K. As shown in Fig. S1 in the ESI,† all the fractions reach the plateau eventually, clearly indicating the convergence of present hREMD simulation. In addition, the representative replica is ergodic for every temperature in the desired temperature range during the simulation, again showing the efficiency of hREMD search (Fig. S2†).
is the relative free energy or potential of mean force.60 Four free energy local minima are presented in the free energy profile. For each local minimum, snapshots which satisfy the criteria according to the RMSDs were selected from 300 K trajectory and hierarchical clustering analyses (kclust algorithm available in MMTSM Toolset61) was performed focused on the selected snapshots to identify the representative (most populated) conformation. The four local minima correspond to closed, curled, semi-open, and fully open states, with the population of 7.32%, 36.5%, 41.1%, and 1.3% in the equilibrium conformational ensemble in 300 K water solution, respectively. To show the convergence of the hREMD simulation, we calculated the time series of the fraction of the four populated structures at 300 K. As shown in Fig. S1 in the ESI,† all the fractions reach the plateau eventually, clearly indicating the convergence of present hREMD simulation. In addition, the representative replica is ergodic for every temperature in the desired temperature range during the simulation, again showing the efficiency of hREMD search (Fig. S2†).
 | ||
| Fig. 2 (A) Two-dimensional free energy landscape at 300 K as the function of the overall backbone RMSD and the RMSD of flap region with respect to the crystal closed structure of HIV-1 PR. (B) Representative conformations of the distinct states in the free energy landscape and their population in the equilibrium conformational ensemble at 300 K. | ||
Previous experiments captured the closed,8,10 semi-open,9,11–14 and wide-open9,15,16 structures for apo wild-type HIV-1 PR and various mutants. We compared the closed and semi-open structures indicated in the free energy profile to the experimentally measured ones and found that both structures are consistent between the present simulation and previous experiments (Fig. 3A and C). In addition, the semi-open structure is the most popular in the equilibrium conformational ensemble, which is consistent with the observation of the sub-nanosecond timescale NMR experiment19–21 and the X-ray diffraction data for HIV-1 subtype B.8 On the other hand, the wide-open structure which is more preferably adopted by mutated HIV-1 PR rather than wild-type (e.g., K7Q, I33L, R41K, I63L in 2R8N, K7Q in 2PC0, I10L, N25D, V36M, L46M, V54I, V62I, P63L, V71A, A82V, V84I, M90L in 1TW7) is randomly sampled in the present simulation, accounting for only 0.55% in the equilibrium conformational ensemble at 300 K. A fully open structure, which is defined as conformations with large flap–flap separations (I50Cα–I50′Cα distance > 13 Å), instead of wide-open structure appears distinctly in the free energy profile: in comparison to the latter structure, the former one has the disposition of the flaps more expanded so that they are ∼1.1 Å more apart (Fig. S3†).
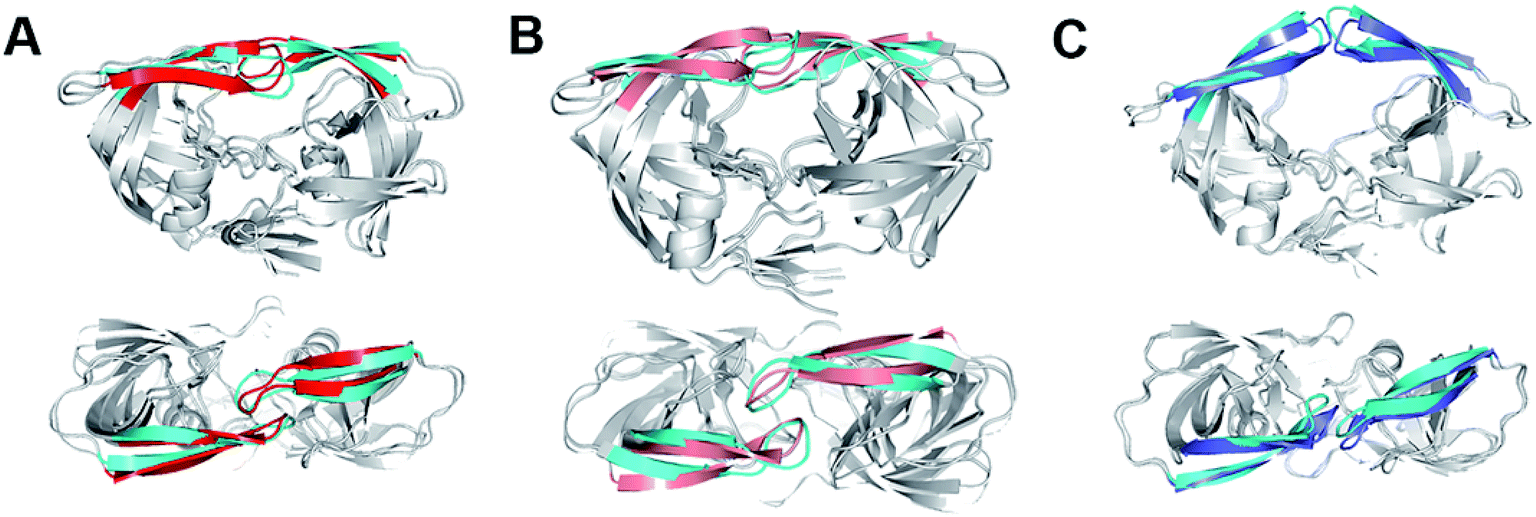 | ||
| Fig. 3 (A) The closed structure from hREMD (cyan flap) superimposed onto crystal closed structure 3IXO (red flap). (B) The curled structure from hREMD (cyan flap) superimposed onto the curled structure from conventional MD (purple flap). (C) The semi-open structure from hREMD (cyan flap) superimposed onto crystal semi-open structure 1TW7 (blue flap). Upper: side view, lower: top view. | ||
An interesting observation in the present study is that another major component of wild-type HIV-1 PR is a “curled” structure which has been not reported by experiments. In comparison to the closed and semi-open structures, the curled structure has a large change in the Φ and Ψ torsion values of the flap tip residues 48–52 (Fig. 4A). It is noteworthy that although multiple previous molecular simulation studies described the flap curling of HIV-1 PR,27,31 the flap curling observed here is certainly different to the previously reported ones. In the previous simulations, a curled-in conformational change occurs in the transition between semi-open and fully open structures of HIV-1 PR: the flap tips are curled in the downward direction to lead a bent “L” structure.27,31 The authors suggested that such curling-in behavior of flap tips buries the initially solvent accessible hydrophobic cluster and stabilizes the open conformation. In the present simulation, the flap tips are curled in lateral orientation of the flaps but not the downward orientation (Fig. 4A). In addition, the curled structure is in between closed and semi-open structures. The main contribution of the flap curling in the closed structure seems to loosen the hydrophobic cluster in HIV-1 PR to trigger the flap opening.
 | ||
| Fig. 4 (A) Comparison of closed (green) and curled (orange) conformations of the flaps (left: side view, right: top view). (B–D) Contact maps of the closed, curled, and semi-open structures of the HIV-1 PR from hREMD. Key hydrophobic contacts are highlighted by pink squares. | ||
Fig. 4B–D indicates the contact maps of the representative conformations of the closed, curled, and semi-open structures of HIV-1 PR. In the closed structure, the residue–residue contacts among the two flaps and 80s (80's) loops are mainly the hydrophobic interactions (see the highlights in Fig. 4B). These hydrophobic clusters are supposed to play an important role in locking the closed conformation. The center distance of the hydrophobic side-chains which are involved in the hydrophobic clusters was averaged for all four states and organized in Table 1. In the curled structure, the hydrophobic interactions between the flaps are weakened due to the curling of flap tips while the strength of the hydrophobic interactions between the flaps and 80s (80's) loops keeps unchanged. More specifically, it is the side-chain distances of the hydrophobic contacts of I50–I50′, I50–F53′, and F53–I50′ that are elongated to weaken the corresponding flap–flap hydrophobic interactions (Table 1). In the semi-open structure, the flap tips convert back to the original uncurled conformation and meanwhile the flaps move upwards. As a result, all the flap-80s (80's) loop hydrophobic interactions are broken and only a portion of flap–flap hydrophobic interactions are remained (I50–I50′, I50–F53′, and F53–I50′).
| States | Flap–flap interaction pairs | Flap–80s (80's) loop interaction pairs | |||||
|---|---|---|---|---|---|---|---|
| 50–50′ | 50–47′ (47–50′) | 50–53′ (53–50′) | 54–50′ (50–54′) | 79–50′ (50–79′) | 81–50′ (50–81′) | 84–50′ (50–84′) | |
| Closed | 7.2 | 6.0(6.1) | 9.3(11.2) | 7.3(5.9) | 8.4(8.7) | 8.5(8.1) | 5.7(7.8) |
| Curled | 10.7 | 6.4(7.5) | 10.6(10.6) | 6.5(7.6) | 8.0(7.6) | 6.5(9.9) | 8.8(8.8) |
| Semi-open | 7.6 | 10.4(9.9) | 6.6(7.0) | 10.4(10.3) | 14.8(15.4) | 12.5(11.8) | 14.6(14.9) |
| Fully open | 16.4 | 17.4(16.7) | 19.7(12.8) | 16.6(20.4) | 21.1(21.0) | 20.6(13.1) | 20.4(14.3) |
TriCa angles (the angles between three adjacent Cα) of residues Gly48-Gly49-Ile50 and residues Gly49-Ile50-Gly51 can be used as reaction coordinates to illuminate the curling motion of flap tips.4,44 We calculated the distribution of the angles in individual states of HIV-1 PR measured by hREMD. One can see from Fig. 5 that the distribution of the TriCa angle of Gly48-Gly49-Ile50 overlaps substantially in all states. On the other hand, the distribution of the TriCa angle of Gly49-Ile50-Gly51 in the curled structure is apparently different to those of the other three states: the angle distribution is centered at ∼110° in the curled structure but is shifted to ∼90° in the closed, semi-open, and fully open structures. The similar distribution of the two TriCa angles can be also seen in the previous MD simulation of closed structure of I50V and I50L/A71V mutants of HIV-1 PR.45
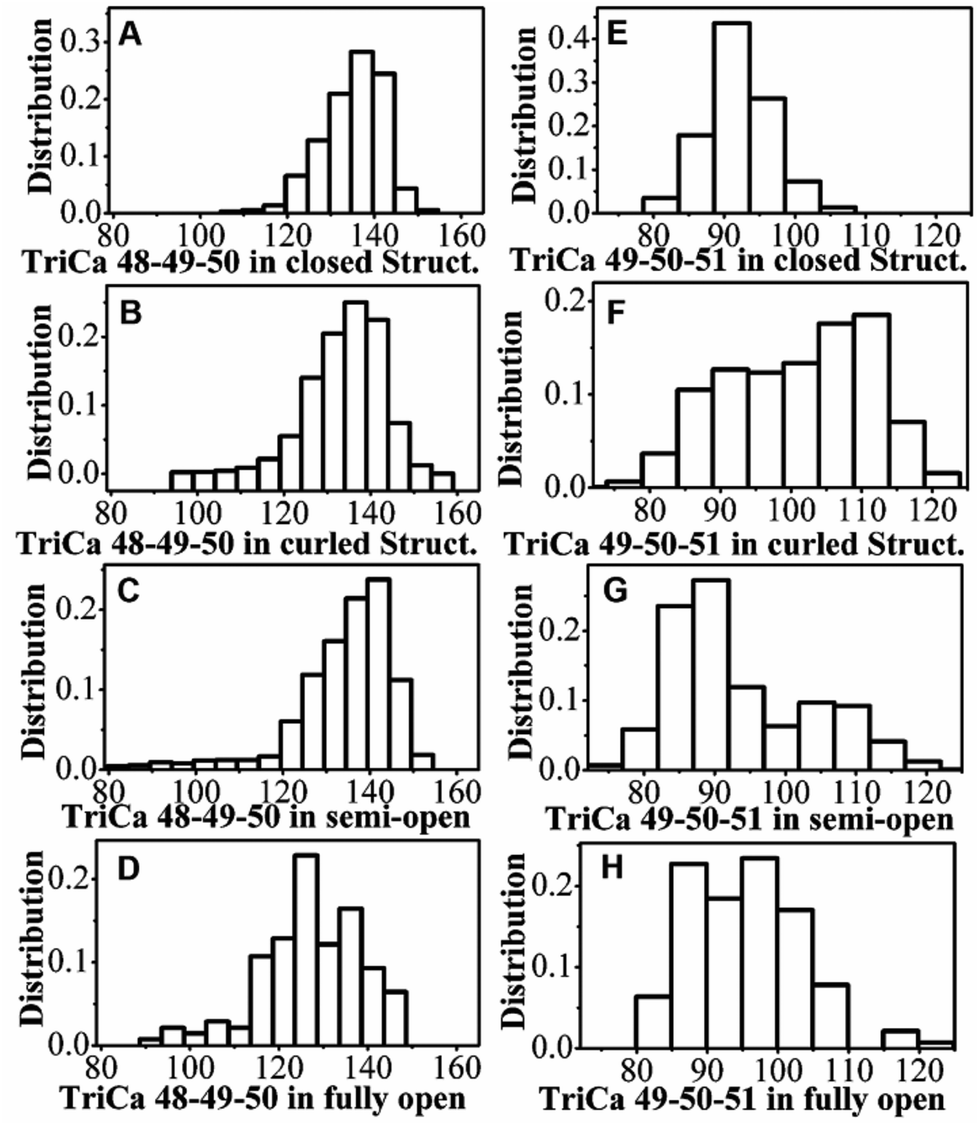 | ||
| Fig. 5 The distribution of TriCa angles of (A–D) Gly48-Gly49-Ile50 and (E–H) Gly49-Ile50-Gly51 in the individual states of HIV-1 PR measured by hREMD. | ||
Comparison of hREMD and conventional MD simulations
While REMD simulation explores the structural and thermodynamic properties of the statistically weighted states on the free energy surface, conventional MD can simulate the protein motion from kinetic perspective. The combination of REMD and conventional MD can essentially provide an integrated view of protein motion. We performed conventional MD simulations on not only the apo wild-type HIV-1 PR but also two variants of Flap+ and Act in aqueous solution. The crystal closed forms of the three proteases were used as initial structures. Fig. 6 indicates that both wild-type and Flap+ proteases show similar motion inferred from the mild fluctuation of the flap RMSD whereas Act has a remarkable increase in the flap RMSD at ∼100 ns, suggesting a motion of flaps only in Act protease. The origin of the amplified flexibility of the flaps in Act variant can be attributed to the two mutations of V82 and I84 in 80s (80's) loop by less hydrophobic residues. The hydrophobic interactions between flaps and 80s (80's) loop residues (e.g., I50–I84′ and I50′–I84) along with the hydrophobic interactions between the two flaps play an important role in maintaining the closed conformation of HIV-1 PR (Table 1); the double mutation of the 80s (80's) loop residues (V82T/I84V) in Act variant weakens the hydrophobic interactions between flap and 80s (80's) loop regions and thus destabilize the closed conformation.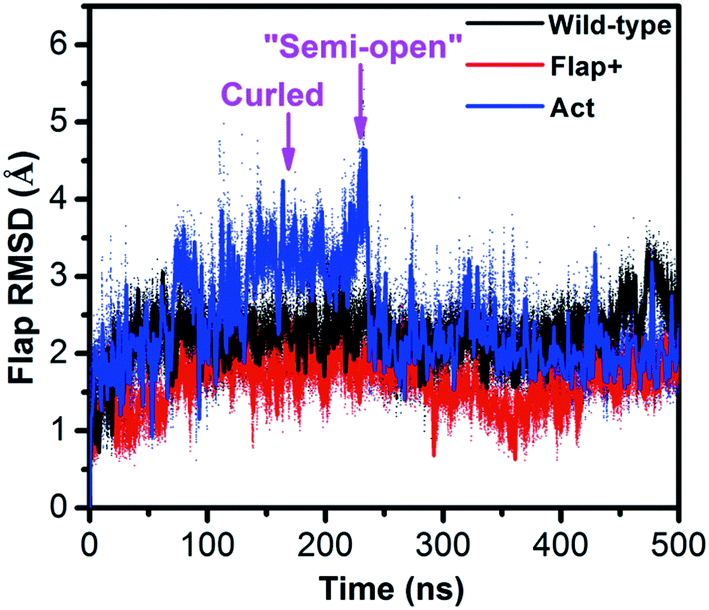 | ||
| Fig. 6 Time series of the RMSD of flap region with respect to the crystal closed structure for the conventional MD simulations of wild-type HIV-1 PR, Flap+ and Act variants. | ||
The flap motion of Act variant leads to a curled structure which is very close to the counterpart on the free energy surface generated by hREMD (see the superposition of the two curled structures in Fig. 3B). Subsequently, the flap residues in the curled structure further move apart until reaching a “semi-open” structure (at ∼220 ns). In comparison to initial closed conformation of flaps, this “semi-open” structure has its flap in one monomer (chain A) moving upward while the flap in the other monomer (chain B) is slightly moving downward. Furthermore, the two flaps also have lateral motions, which is consistent with the observation in previous simulation study.28 However, this “semi-open” structure is deviated from the semi-open structure found in hREMD and experiments: the flap tips still have downward curling conformation and are not completely upward-oriented (Fig. S4†).
The curling motion of flaps in Act variant is delineated by the time series of the TriCa angles of residues Gly48-Gly49-Ile50 and residues Gly49-Ile50-Gly51 (Fig. S5†). One can see that the TriCa angle of Gly49-Ile50-Gly51 increases from ∼90° to ∼105°, fitting with the angle change of flap curling which is quantitatively assessed by hREMD simulation (Fig. 5E–H). In addition, the TriCa angle of Gly48-Gly49-Ile50 also has a remarkable change from ∼130° to 110° along with the flap curling motion in conventional MD simulation, which is not observed in hREMD simulation. This trivial difference between hREMD and conventional MD simulation might be induced by the residue mutation of Act which alters the structural flexibility of flaps. In summary, although the conventional MD simulations cannot reach the experimentally identified semi-open structure of HIV-1 PR due to its low conformation sampling ability, the results clearly demonstrate that the flap curling is an early event in the flap opening process.
Transition pathway of HIV-1 PR flap opening
The flap separation occurs through a combination of upward rotations and lateral movements of the flaps (Fig. 1). The dynamic flap motion described in conventional MD simulation together with the qualitative and quantitative measurement of various states in hREMD shed light into the transition pathway of HIV-1 PR flap opening. Fig. 7 shows the pathway for the flap opening transition, in which the curled and semi-open structures are two crucial intermediates (closed → curled → semi-open → fully open). In the transition from the closed to curled structure, the flap tips are curled so that the hydrophobic interactions between the two flaps are weakened which triggers the opening of flaps. No upward motion of the flaps occurs in this transition step and all the hydrophobic interactions between flaps and 80s (80's) loop are maintained. As the curled structure is transited to semi-open structure, flaps move upwards as a result of a concerted downward motion of the cantilever, fulcrum, and flap elbows. Accordingly, the hydrophobic interactions between flaps and 80s (80's) loop are broken. Meanwhile, the curled flap tips convert back to the original uncurled conformation and the hydrophobic interactions between the flaps are partially maintained (I50–I50′, I50–F53′, and F53–I50′), which may stabilize the semi-open structure. The further upward movement of flaps to the fully open structure needs to break the remaining hydrophobic interactions between the flaps, making the fully open structure thermodynamically unfavorable and kinetically inaccessible. | ||
| Fig. 7 Intermediate conformations in the pathway for closed → fully open transition. Upper: top view (surface representation), lower: side view (cartoon representation). The two flaps are purple and orange colored, respectively. Arrows in curled structure indicate the motion tendency of HIV-1 PR. | ||
Conclusions
The opening of HIV-1 PR flaps controls the access of substrates and inhibitors to the active site and is recognized as essential for enzymatic activity. Drug-resistant mutations in HIV-1 PR can alter the flexibility and conformation of the flaps, leading to the reduced drug susceptibility to different PR inhibitors. Although multiple altered flap conformations including closed, semi-open, and wide-open forms have been reported for wild-type HIV-1 PR and various mutants either in the unliganded form or with bound inhibitors,8–16 the understanding of how HIV-1 PR converts from one structure to another remains incomplete.In this study, we utilized a hybrid explicit/implicit solvent REMD simulation to explore the structural properties of intermediate states in the conformational transition pathway of wild-type HIV-1 PR and determine the conformational equilibrium among functionally important states. In addition, we ran long-time conventional MD simulations on wild-type HIV-1 PR and its Flap+ (L10I/G48V/I54V/V82A) and Act (V82T/I84V) variants to measure the dynamic properties of flaps. Starting from the closed structure in the conventional MD simulations, the flaps in Act protease are moderately opened whereas the flaps in wild-type and Flap+ proteases maintain the closed conformation. Intriguingly, a novel “curled” structure which is heavily weighted in the free energy landscape of wild-type HIV-1 PR generated by hREMD simulation is also observed at the very beginning of the flap opening event of Act variant in the conventional MD simulation. Therefore, consistency can be seen between the enhanced sampling and conventional non-enhanced molecular simulations. The combination of hREMD and conventional MD simulations connects the functionally important states and provides integrated information for the molecular mechanism of the flap opening of HIV-1 PR.
The results presented here demonstrate that the flap opening transition of HIV-1 PR occurs through two important intermediate states: the curled and semi-open states. While the semi-open structure was reported by X-ray crystallography experiments,9,11–14 the curled structure of HIV-1 PR has not been observed in any experiments yet. Although Scott and Schiffer introduced the term flap curling of the TriCa angles involving the residues within the flap tip or nearby region,27 the presently observed flap curling motion and induced curled conformation of flap tips are apparently different to those in previous simulations.27,31 In the multiple–state transition of flap opening from the closed structure, a local structure adjustment occurs initially at flap tips while the main body of the protease keeps unchanged: the flap tips are curled to moderately loosen the hydrophobic clusters which lock the closed conformation of protease, triggering the flap opening. The flaps then move upwards along with a concerted downward motion of the cantilever, fulcrum, and flap elbows. The important role of flap curling in triggering the flap opening is revealed from both thermodynamic and kinetic perspectives for the first time. The hydrophobic contacts affected by flap curling (I50–I50′, I50–F53′, and F53–I50′) could be the resource for residue mutation to effectively alter the flexibility and conformation of flaps.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Key Research and Development Program (Grant 2016YFA0502301), the National Natural Science Foundation of China (Grant no. 21373258, 21403283), National Basic Research Program (Grant no. 2014CB910400). The simulations were run at shanghai Supercomputing Center (SSC) and TianHe-1 supercomputer in Tianjin.References
- F. Clavel and A. J. Hance, N. Engl. J. Med., 2004, 350, 1023–1035 CrossRef CAS PubMed.
- A. Wlodawer and J. Vondrasek, Annu. Rev. Biophys. Biomol. Struct., 1998, 27, 249–284 CrossRef CAS PubMed.
- M. A. Navia, P. M. Fitzgerald, B. M. McKeever, C. T. Leu, J. C. Heimbach, W. K. Herber, I. S. Sigal, P. L. Darke and J. P. Springer, Nature, 1989, 337, 615–620 CrossRef CAS PubMed.
- A. L. Perryman, J. H. Lin and J. A. McCammon, Protein Sci., 2004, 13, 1108–1123 CrossRef CAS PubMed.
- W. E. Harte Jr, S. Swaminathan, M. M. Mansuri, J. C. Martin, I. E. Rosenberg and D. L. Beveridge, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 8864–8868 CrossRef CAS.
- R. W. Shafer, Clin. Microbiol. Rev., 2002, 15, 247–277 CrossRef CAS PubMed.
- R. W. Shafer, S. Y. Rhee, D. Pillay, V. Miller, P. Sandstrom, J. M. Schapiro, D. R. Kuritzkes and D. Bennett, AIDS, 2007, 21, 215–223 CrossRef CAS PubMed.
- A. H. Robbins, R. M. Coman, E. Bracho-Sanchez, M. A. Fernandez, C. T. Gilliland, M. Li, M. Agbandje-McKenna, A. Wlodawer, B. M. Dunn and R. McKenna, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 233–242 CrossRef CAS PubMed.
- H. Heaslet, R. Rosenfeld, M. Giffin, Y. C. Lin, K. Tam, B. E. Torbett, J. H. Elder, D. E. McRee and C. D. Stout, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 866–875 CAS.
- B. Pillai, K. K. Kannan and M. V. Hosur, Proteins, 2001, 43, 57–64 CrossRef CAS.
- A. Wlodawer, M. Miller, M. Jaskolski, B. K. Sathyanarayana, E. Baldwin, I. T. Weber, L. M. Selk, L. Clawson, J. Schneider and S. B. H. Kent, Science, 1989, 245, 616–621 CAS.
- R. Lapatto, T. Blundell, A. Hemmings, J. Overington, A. Wilderspin, S. Wood, J. R. Merson, P. J. Whittle, D. E. Danley, K. F. Geoghegan, S. J. Hawrylik, S. E. Lee, K. G. Scheld and P. M. Hobart, Nature, 1989, 342, 299–302 CrossRef CAS PubMed.
- F. L. Liu, A. Y. Kovalevsky, J. M. Louis, P. I. Boross, Y. F. Wang, R. W. Harrison and I. T. Weber, J. Mol. Biol., 2006, 358, 1191–1199 CrossRef CAS PubMed.
- S. Spinelli, Q. Z. Liu, P. M. Alzari, P. H. Hirel and R. J. Poljak, Biochimie, 1991, 73, 1391–1396 CrossRef CAS PubMed.
- P. Martin, J. F. Vickrey, G. Proteasa, Y. L. Jimenez, Z. Wawrzak, M. A. Winters, T. C. Merigan and L. C. Kovari, Structure, 2005, 13, 1887–1895 CrossRef CAS PubMed.
- R. M. Coman, A. H. Robbins, M. M. Goodenow, B. M. Dunn and R. McKenna, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64, 754–763 CrossRef CAS PubMed.
- N. Okimoto, T. Tsukui, M. Hata, T. Hoshino and M. Tsuda, J. Am. Chem. Soc., 1999, 121, 7349–7354 CrossRef CAS.
- N. Okimoto, K. Kitayama, M. Hata, T. Hoshino and M. Tsuda, J. Mol. Struct., 2001, 543, 53–63 CrossRef CAS.
- D. I. Freedberg, R. Ishima, J. Jacob, Y. X. Wang, I. Kustanovich, J. M. Louis and D. A. Torchia, Protein Sci., 2002, 11, 221–232 CrossRef CAS PubMed.
- R. Ishima, D. I. Freedberg, Y. X. Wang, J. M. Louis and D. A. Torchia, Structure, 1999, 7, 1047–1055 CrossRef CAS PubMed.
- E. Katoh, J. M. Louis, T. Yamazaki, A. M. Gronenborn, D. A. Torchia and R. Ishima, Protein Sci., 2003, 12, 1376–1385 CrossRef CAS PubMed.
- M. B. Luis Galiano and G. E. Fanucc, J. Am. Chem. Soc., 2007, 129, 11004–11005 CrossRef PubMed.
- Y. Cai, W. Myint, J. L. Paulsen, C. A. Schiffer, R. Ishima and N. Kurt Yilmaz, J. Chem. Theory Comput., 2014, 10, 3438–3448 CrossRef CAS PubMed.
- F. Ding, M. Layten and C. Simmerling, J. Am. Chem. Soc., 2008, 130, 7184–7185 CrossRef CAS PubMed.
- J. Roche, J. M. Louis and A. Bax, ChemBioChem, 2015, 16, 214–218 CrossRef CAS PubMed.
- D. M. York, T. A. Darden and L. G. Pedersen, J. Chem. Phys., 1993, 99, 8345–8348 CrossRef CAS.
- W. R. Scott and C. A. Schiffer, Structure, 2000, 8, 1259–1265 CrossRef CAS PubMed.
- V. Tozzini and J. A. McCammon, Chem. Phys. Lett., 2005, 413, 123–128 CrossRef CAS.
- V. Hornak, A. Okur, R. C. Rizzo and C. Simmerling, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 915–920 CrossRef CAS PubMed.
- A. L. Perryman, J. H. Lin and J. A. McCammon, Protein Sci., 2004, 13, 1108–1123 CrossRef CAS PubMed.
- N. J. Deng, W. H. Zheng, E. Gallicchio and R. M. Levy, J. Am. Chem. Soc., 2011, 133, 9387–9394 CrossRef CAS PubMed.
- P. R. Batista, G. Pandey, P. G. Pascutti, P. M. Bisch, D. Perahia and C. H. Robert, J. Chem. Theory Comput., 2011, 7, 2348–2352 CrossRef CAS PubMed.
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 1999, 314, 141–151 CrossRef CAS.
- K. Ostermeir and M. Zacharias, Biochim. Biophys. Acta, 2013, 1834, 847–853 CrossRef CAS PubMed.
- H. Fukunishi, O. Watanabe and S. Takada, J. Chem. Phys., 2002, 116, 9058 CrossRef CAS.
- X. Huang, M. Hagen, B. Kim, R. A. Friesner, R. Zhou and B. J. Berne, J. Phys. Chem. B, 2007, 111, 5405–5410 CrossRef CAS PubMed.
- P. Liu, B. Kim, R. A. Friesner and B. J. Berne, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13749–13754 CrossRef CAS PubMed.
- S. Jang, S. Shin and Y. Pak, Phys. Rev. Lett., 2003, 91, 058305 CrossRef PubMed.
- Y. Meng, D. S. Dashti and A. E. Roitberg, J. Chem. Theory Comput., 2011, 7, 2721–2727 CrossRef CAS PubMed.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, J. Chem. Phys., 1953, 21, 1087–1092 CrossRef CAS.
- J. Wang, W. Zhu, G. Li and U. H. Hansmann, J. Chem. Phys., 2011, 135, 084115 CrossRef PubMed.
- Y. Q. Yu, J. A. Wang, Q. Shao, J. Y. Shi and W. L. Zhu, J. Chem. Phys., 2015, 142, 125105 CrossRef PubMed.
- Y.-X. Wang, D. I. Freedberg, T. Yamazaki, P. T. Wingfield, S. J. Stahl, J. D. Kaufman, Y. Kiso and D. A. Torchia, Biochemistry, 1996, 35, 9945–9950 CrossRef CAS PubMed.
- B. R. Meher and Y. Wang, J. Phys. Chem. B, 2012, 116, 1884–1900 CrossRef CAS PubMed.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo and T. Lee, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- D. Case, T. Cheatham III, C. Simmerling, J. Wang, R. Duke, R. Luo, M. Crowley, R. Walker and W. Zhang, et al., AMBER10, University of California, San Francisco, 2008 Search PubMed.
- A. Patriksson and D. van der Spoel, Phys. Chem. Chem. Phys., 2008, 10, 2073–2077 RSC.
- W. Nadler and U. H. E. Hansmann, J. Phys. Chem. B, 2008, 112, 10386–10387 CrossRef CAS PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- X. Wu and B. R. Brooks, J. Chem. Phys., 2008, 129, 154115 CrossRef PubMed.
- X. Wu and B. R. Brooks, J. Chem. Phys., 2005, 122, 044107 CrossRef PubMed.
- G. D. Hawkins, C. J. Cramer and D. G. Truhlar, Chem. Phys. Lett., 1995, 246, 122–129 CrossRef CAS.
- N. M. King, M. Prabu-Jeyabalan, R. M. Bandaranayake, M. N. Nalam, E. A. Nalivaika, A. Ozen, T. Haliloglu, N. K. Yilmaz and C. A. Schiffer, ACS Chem. Biol., 2012, 7, 1536–1546 CrossRef CAS PubMed.
- D. L. N. G. Surleraux, A. Tahri, W. G. Verschueren, G. M. E. Pille, H. A. de Kock, T. H. M. Jonckers, A. Peeters, S. De Meyer, H. Azijn, R. Pauwels, M. P. de Bethune, N. M. King, M. Prabu-Jeyabalan, C. A. Schiffer and P. B. T. P. Wigerinck, J. Med. Chem., 2005, 48, 1813–1822 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- A. E. Garcia and K. Y. Sanbonmatsu, Proteins, 2001, 42, 345–354 CrossRef CAS.
- M. Feig, J. Karanicolas and C. L. Brooks, MMTSB Tool Set; MMTSB NIH Research Resource, The Scripps Research Institute, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09691g |
| This journal is © The Royal Society of Chemistry 2017 |