
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Trop2 promotes proliferation, invasion and EMT of nasopharyngeal carcinoma cells through the NF-κB pathway
Nan Cheng†
a,
Haixia Li†a and
Junpeng Luo *b
*b
aDepartment of Otolaryngological, Huaihe Hospital of Henan University, Kaifeng, 475000, China
bDepartment of Oncology, Huaihe Hospital of Henan University, No. 8, Baobei Road, Gulou District, Kaifeng, 475000, China. E-mail: pengluoun@163.com; Tel: +86-371-23906055
First published on 17th November 2017
Abstract
Human trophoblast cell surface antigen 2 (Trop2), a cell surface transmembrane glycoprotein receptor, has been demonstrated to be closely associated with increasing tumor aggressiveness, metastasis and unfavorable prognosis. However, the biological roles of Trop2 and its underlying mechanism in nasopharyngeal carcinoma cells (NPC) are still unclear. In this study, we found that Trop2 was up-regulated in NPC tissues and cell lines compared with normal nasopharyngeal epithelial tissues and cells. Trop2 overexpression promoted cell proliferation, invasion and the EMT process and activated the NF-κB signaling pathway, while Trop2 downregulation showed the opposite effects in NPC cells. Moreover, NF-κB inhibitor pyrolidinedithiocarbamate (PDTC) or NF-κB knockdown attenuated Trop2-induced cell proliferation, invasion and EMT in NPC cells. Taken together, Trop2 accelerates cell proliferation, invasion and EMT in NPC via activating the NF-κB pathway, indicating that Trop2 may be exploited in targeted therapy for patients with NPC.
1. Introduction
Nasopharyngeal carcinoma (NPC) is a kind of head and neck squamous cell carcinoma originating from the epithelial cells in the nasopharynx. Although rare in most of the world, NPC frequently occurs in certain geographic areas, such as Southeast Asia and southern China, with a yearly incidence rate of 20 to 50 cases per 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people.1,2 Most NPC patients were found at an advanced stage of the disease when first diagnosed because of the vague symptoms and deep location. Although great achievements have been made in chemotherapy and radiotherapy, the rates of treatment failure in NPC patients still remain high due to local recurrence and distant metastasis due to the high potential of metastasis and invasiveness, which is considered as a main cause for NPC lethality.3–5 Thus, it is urgently required to better understand the molecular mechanisms underlying NPC progression in order to develop effective strategies for NPC therapy.
000 people.1,2 Most NPC patients were found at an advanced stage of the disease when first diagnosed because of the vague symptoms and deep location. Although great achievements have been made in chemotherapy and radiotherapy, the rates of treatment failure in NPC patients still remain high due to local recurrence and distant metastasis due to the high potential of metastasis and invasiveness, which is considered as a main cause for NPC lethality.3–5 Thus, it is urgently required to better understand the molecular mechanisms underlying NPC progression in order to develop effective strategies for NPC therapy.
Human trophoblast cell surface antigen 2 (Trop2, also referred to as TACSTD2, M1S1, EGP-1), a 35 kDa cell surface transmembrane glycoprotein receptor, is encoded by the gene TACSTD2 mapped on chromosome 1p32. Trop2 was initially discovered in human trophoblasts, a highly invasive tissue at the interface between fetal and maternal circulation.6 Subsequently, Trop2 was found significantly overexpressed in the majority of human epithelial carcinomas, such as squamous cell carcinomas,7 cervical cancers,8 and laryngeal carcinomas.9 Overexpression of Trop2 has been demonstrated to be able to accelerate tumor growth and is closely associated with increased tumor aggressiveness, metastasis and unfavorable prognosis.10,11 Moreover, Trop2 has been reported be an independent predictor for poor clinical outcome in NPC.12 However, little is known about the function and mechanism of Trop2 in NPC.
The transcription factor nuclear factor-kappa B (NF-κB), a family of multipotent transcription factors, has been revealed to be implicated in diverse physiological processes.13 Activation of NF-κB was reported to facilitate cell proliferation and invasion, inhibit apoptosis, and promote angiogenesis and metastasis in numerous tumors.14,15 Moreover, a previous study pointed out that NF-κB was commonly activated in NPC and related to NPC carcinogenesis.16
In the present study, we focused on the function of Trop2 in NPC development, as well as its potential molecular basis. The results showed that Trop2 was up-regulated in NPC tissues and cell lines, and Trop2 promoted NPC cell proliferation, invasion and epithelial mesenchymal transition (EMT). Further mechanistic analysis demonstrated that Trop2 exerted its biological roles in NPC cells by regulating NF-κB pathway.
2. Materials and methods
2.1. Cell lines and tissue samples
Human nasal epithelial cells HNEpC were obtained from PromoCell (Heidelberg, Germany) and maintained in an airway epithelial growth medium (PromoCell) in a 5% CO2 humidified incubator at 37 °C. Human NPC cell lines including C666-1, CNE1, CNE2, HONE1 and its subclone 5-8F, of which C666-1 is the only well-known Epstein–Barr virus (EBV) positive NPC cell line and CNE1, CNE2, 5-8F, and HONE1 are EBV negative, were purchased from the China Type Culture Collection and Cancer Research Center (Wuhan, China) and cultured in RPMI 1640 medium (Hyclone, Waltham, MA, USA) supplemented with 10% heated-inactivated fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA), 1% penicillin and streptomycin (P/S) (Invitrogen, Grand Island, NY, USA) in a humidified chamber at 37 °C with 5% CO2. A total of 29 primary fresh NPC biopsies and 8 normal nasopharyngeal epithelial biopsy tissues were collected from the Huaihe Hospital of Henan University, Kaifeng, China between March 2014 and March 2015. The prior consents from the patients and approval from the Research Ethics Committee of Huaihe Hospital of Henan University were obtained.2.2. Cell transfection and treatment
siRNA targeting Trop2 (si-Trop2), siRNA targeting NF-κB (si-NF-κB), siRNA negative control (si-control), pcDNA3.1-Trop2 and pcDNA3.1-control were all purchased from GenePharma (Shanghai, China). HONE1 and CNE2 cells were seeded into 6-well plates and grown to 80% confluency, followed by transfected with 100 pmol l−1 of si-Trop2, si-NF-κB, si-control, pcDNA3.1-Trop2 or pcDNA3.1-control using Lipofectamine 2000 (Invitrogen). siRNA sequences were listed as below: si-Trop2 forward: 5′-GCACGCUCAUCUAUUACCUTT-3′, si-Trop2 reverse: 5′-AGGUAAUAGAUGAGCGUGCTT-3′; si-NF-κB forward: 5′-GGAGUACCCUGAAGCUAUATT-3′, si-NF-κB reverse: 5′-UAUAGCUUCAGGGUACUCCAT-3′; si-control forward: 5′-UUCUCCGAACGUGUCACGUTT-3′, si-control reverse: 5′-ACGUGAC ACGUUCGGAGA ATT-3′. NF-κB inhibitor, pyrolidine dithiocarbamate (PDTC, SC-203224, 50 μM, Santa Cruz Biotechnology, Santa Cruz, CA, USA), was also applied to observe the involvement of NF-κB in Trop2-mediated modulation in NPC development.2.3. Quantitative real-time PCR (qRT-PCR)
Total RNA were extracted from cells or tissues by using Trizol Reagent (TaKaRa, Dalian, China) and reversely transcribed to cDNA by using the Super Script III Reverse Transcriptase (Invitrogen). RT-qPCR examination was performed using ABI Step-One plus (Applied Biosystems, Foster City, CA, USA) with SYBR Green qRT-PCR master mix (TaKaRa). The procedure for PCR were carried out as below: denaturation at 95 °C for 5 min, followed by 40 cycles of 95 °C for 10 s and annealing at 60 °C for 30 s. The relative amount of Trop2 mRNA was calculated using 2−ΔΔCt method with β-actin serving as the internal normalization control. The primers were listed as follows: Trop2, F: 5′-CTCTTGAAGCCCCCTTAGAG-3′, R: 5′-TAGTTCCTTGACCCCCTCAA-3′; β-actin, F: 5′-GGCGGCAACACCATGTACCCT-3′, R: 5′-AGGGGCC GGACTCGTCATACT-3′.2.4. Cell proliferation assay
Cell viability was evaluated by MTT assay as previously described.17 Briefly, HONE1 and CNE2 cells (5 × 103 per well) were seeded into 96-well plates and cultured for 24 h. Then cells were transfected with different siRNAs or pcDNA plasmids. Following incubating for another 24 h, 48 h and 72 h at 37 °C with 5% CO2, 20 μl MTT (2 mg ml−1; Sigma, St. Louis, MO, USA) was added to each well for 4 h incubation. Then the supernatant was discarded and 200 μl of dimethyl sulfoxide (DMSO) was added to dissolve the MTT formazan precipitate for 10 min. Absorbance at 490 nm were measured by a microtitre plate reader (Bio-Rad, Hercules, CA, United States). Experiments were performed at least three times.2.5. Transwell invasion assays
Cell invasion assay were carried out in Bio-Coat Invasion Transwell chambers (with 8.0 μm pores; Corning Costar, Cambridge, MA, USA) coated with Matrigel (BD Biosciences, San Diego, CA, USA). 1 × 105 transfected HONE1 and CNE2 cells were incubated into the upper chamber filled with serum free medium. RPMI 1640 with 10% FBS was loaded to the lower chamber. Following incubated for 24 h, the non-invading cells on the upper surface of membranes were removed with a cotton-tipped swab. The invasive cells adhering to the lower surface were fixed with 100% precooling methanol for 5 min, stained with Giemsa (Beyotime Institute of Biotechnology, Shanghai, China) for 30 min and counted from 5 randomly selected optical fields by a microscope (Nikon, Tokyo, Japan) at 100× magnification.2.6. Western blot assay
HONE1 and CNE2 cells were collected and lysed in RIPA buffer (Beyotime, Jiangsu, China) to extract the total protein 48 h post transfection. The concentrations of extracted proteins were determined by the bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA). Equal amounts of protein were mixed with Laemmle buffer, subjected to 10% SDS-PAGE and blotted to a polyvinylidenedifluoride (PVDF) membrane (Millipore, Billerica, MA, USA). After blocked with 5% nonfat dry milk for 2 h at room temperature, the membrane were probed with the primary antibodies against COX-2 (SC-19999), iNOS (SC-58399), cyclin D1 (SC-70899), Ki-67 (SC-23900), E-cadherin (SC-71009), N-cadherin (SC-59987), vimentin (SC-73258), Twist1 (SC-81417), AKT (SC-81434), p-AKT (SC-7985-R), ERK1/2 (SC-514302), p-ERK1/2 (SC-81492) (1:1000 dilution; Santa Cruz Biotechnology) and β-actin (SC-58673) (1:2000 dilution; Santa Cruz Biotechnology) at 4 °C overnight, followed by incubation with the appropriate peroxidase-labeled secondary IgG (SC-516142, 1:10000; Santa Cruz Biotechnology) antibody for 1 h at room temperature. The hybridization signal was visualized with an enhanced chemiluminescence detection reagent (Cwbiotech, China).
2.7. NF-κB DNA-binding analysis
Nuclear extracts were obtained from HONE1 and CNE2 cells using a Nuclear Extract kit (Active Motif, Carlsbad, CA, USA). The DNA binding activities of NF-κB were quantified using an ELISA-based colorimetric Trans-AM kit purchased from Active Motif 48 h post transfection. Briefly, 5 μg nuclear extracts were added into 96-well plate coated with immobilized oligonucleotides containing the NF-κB consensus DNA-binding site (5′-GGGACTTTCC-3′) and incubated for 1 h. Following three washes, NF-κB DNA-binding activity from nuclear extracts was quantified by probing with primary antibody against NF-κB p65 (a main NF-κB subunit accumulated in the nucleus after NF-κB activation) followed by incubated with a horseradish peroxidase-conjugated secondary antibody in an ELISA-like assay (Active Motif) at 450 nm.2.8. Statistical analysis
Results were expressed as mean ± standard deviation (SD) from at least three independent experiments. Statistical differences were analyzed by Student's t test between two groups and one-way multivariate analysis of variance (ANOVA) among three or more groups using GraphPad Prism version 5.0 (GraphPad Software, La Jolla, CA, USA). P values less than 0.05 was considered as statistically significant.3. Results
3.1. Trop2 was aberrantly up-regulated in NPC tissues and cell lines
To explore the effect of Trop2 on NPC, qRT-PCR was first performed in 29 primary fresh NPC biopsies and 8 normal nasopharyngeal epithelial biopsy tissues. The results showed that Trop2 was obviously upregulated in NPC tissues samples compared with normal nasopharyngeal epithelial biopsy tissues (Fig. 1A). Subsequently, we further evaluated Trop2 expression in different NPC cell lines (C666-1, CNE1, CNE2, HONE1, and 5-8F) and normal nasal cells (HNEpC). As expected, Trop2 expression was significantly increased in varying degrees in 4 NPC cells (C666-1, CNE1, CNE2 and HONE1) compared with HNEpC cells, especially in CNE2 and HONE1 (Fig. 1B). The results indicated that abnormal expression of Trop2 may be associated with NPC tumorigenesis.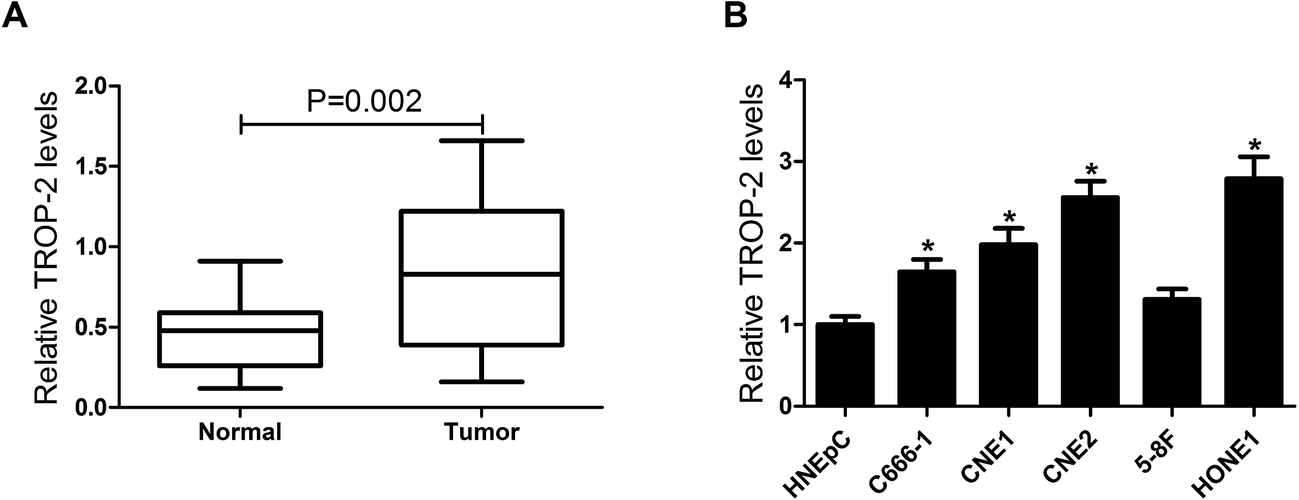 | ||
| Fig. 1 Aberrant expression of Trop2 in NPC tissues and cell lines. (A) The mRNA expression of Trop2 was detected by qRT-PCR in 29 primary fresh NPC biopsies and 8 normal nasopharyngeal epithelial biopsy tissues. (B) The mRNA expression of Trop2 was determined by qRT-PCR in NPC cell lines (C666-1, CNE1, CNE2, HONE1, and 5-8F) and normal nasal epithelial cells HNEpC. The expression of Trop2 was normalized to β-actin. *P < 0.05 vs. normal tissue or HNEpC cells. | ||
3.2. Trop2 promoted cell proliferation and invasion in NPC cells
MTT assay, western blot and Transwell invasion assay were carried out to investigate the effect of Trop2 on NPC cell proliferation and invasion, respectively. Trop2 overexpression by pcDNA3.1-Trop2 significantly promoted growth and Ki-67 expression in HONE1 (Fig. 2A) and CNE2 (Fig. 2B) cells compared with the control groups, whereas Trop2 downregulation by si-Trop2 exhibited the opposite effect on the proliferation and Ki-67 expression in HONE1 (Fig. 2C) and CNE2 (Fig. 2D) cells. Moreover, Trop2 up-regulation dramatically enhanced cell invasive capability in HONE1 (Fig. 2E) and CNE2 (Fig. 2G) cells compared with the control groups, while the invasive ability was suppressed following Trop2 depletion (Fig. 2F and H). Taken together, these results indicated that overexpression of Trop2 facilitated proliferation and invasion in NPC cells.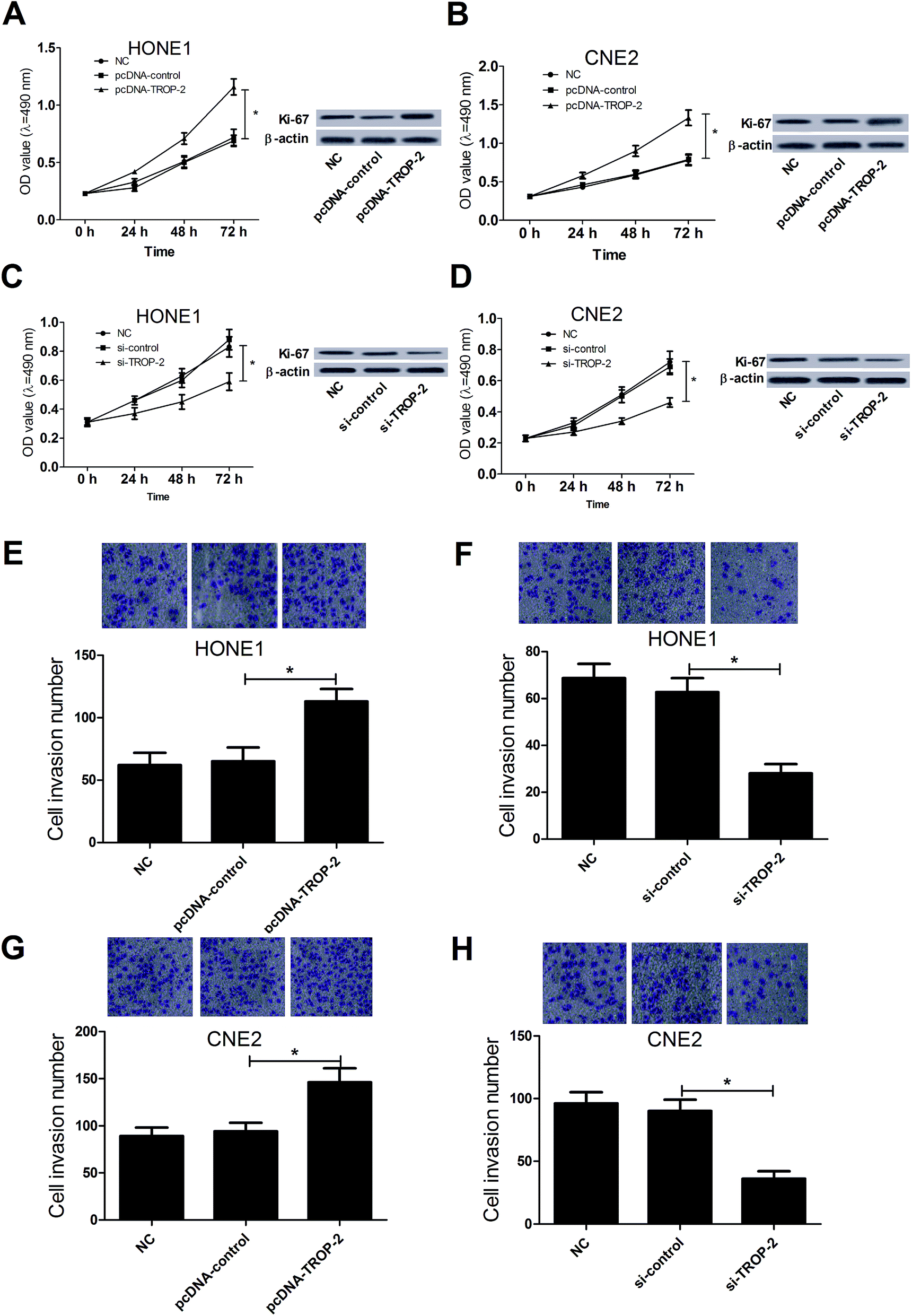 | ||
| Fig. 2 Effects of Trop2 on proliferation and invasion in NPC cells. (A and B) MTT and western blot analysis was performed to detect the effect of Trop2 overexpression on the proliferation and Ki-67 expression in HONE1 and CNE2 cells. (C and D) MTT and western blot was carried out to determine the effect of Trop2 knockdown on the growth and Ki-67 expression in HONE1 and CNE2 cells. (E and G) Transwell invasion assay was performed to evaluate the effect of Trop2 up-regulation on invasion of HONE1 and CNE2 cells at 24 h after transfection. (F and H) Transwell invasion assay was applied to assess the effect of Trop2 silence on invasion of HONE1 and CNE2 cells at 24 h after transfection. *P < 0.05 vs. control group. | ||
3.3. Trop2 induced epithelial mesenchymal transition (EMT) in NPC cells
E-cadherin is a cell–cell adhesion molecule which often served as a hallmark of EMT. The loss of E-cadherin expression is consistently observed at sites of EMT in cancer and its expression level is often inversely correlated to the tumor grade and stage. Transcription factor Twist has been implicated in the transcriptional repression of E-cadherin and could be responsible for the invasion and metastasis of many carcinomas.18,19 N-cadherin is up-regulated in epithelial malignant tumors and closely related to EMT. Vimentin is closely linked to tumorigenesis and metastasis, and acted as a target for anti-metastatic therapies and research.20,21 As EMT is an indispensable step to obtain the metastatic and invasive capacity in tumors, western blot was performed to detect the levels of EMT-related epithelial marker E-cadherin, transcription factor Twist, and mesenchymal marker N-cadherin and vimentin. Western blot analysis showed that the levels of N-cadherin, vimentin and Twist were markedly increased while E-cadherin levels were dramatically decreased in HONE1 (Fig. 3A and B) and CNE2 (Fig. 3E and F) cells transfected with pcDNA-Trop2 when compared to control groups. Conversely, Trop2 downregulation greatly inhibited the expressions of N-cadherin, vimentin and Twist but enhanced E-cadherin expression compared with si-control groups in HONE1 (Fig. 3C and D) and CNE2 (Fig. 3G and H) cells. These results indicated that Trop2 upregulation promoted EMT in NPC cells. | ||
| Fig. 3 Effects of Trop2 on EMT in NPC cells. Western blot analysis of E-cadherin, N-cadherin, vimentin and Twist in HONE1 (A and B) and CNE2 (E and F) cells transfected with pcDNA-control or pcDNA-Trop2. Western blot analysis of E-cadherin, N-cadherin, vimentin and Twist in HONE1 (C and D) and CNE2 (G and H) cells transfected with si-control or si-Trop2. β-Actin was served as a loading control. *P < 0.05 vs. control group. | ||
3.4. Trop2 activated NF-κB signaling pathway in NPC cells
DNA-binding ELISA-based assay was performed to investigate the effect of Trop2 overexpression or downregulation on the DNA-binding activity of NF-κB protein complex. The results revealed that Trop2 overexpression significantly increased the DNA-binding activity of NF-κB in HONE1 (Fig. 4A) and CNE2 (Fig. 4C) cells, while downregulation of Trop2 decreased the DNA-binding activity of NF-κB (Fig. 4B and D). Furthermore, the effect of Trop2 on the NF-κB downstream target gene including COX-2, iNOS, and cyclin D1 (ref. 22–24) was detected by western blot. The results displayed that Trop2 overexpression resulted in an evident increase in the levels of COX-2, iNOS and cyclin D1 in HONE1 (Fig. 4E and F) and CNE2 (Fig. 4I and J) cells, and si-Trop2-mediated Trop2 knockdown strikingly reduced the levels of COX-2, iNOS, and cyclin D1 in HONE1 (Fig. 4G and H) and CNE2 (Fig. 4K and L) cells. These results demonstrated that Trop2 stimulated NF-κB signaling pathway in NPC cells.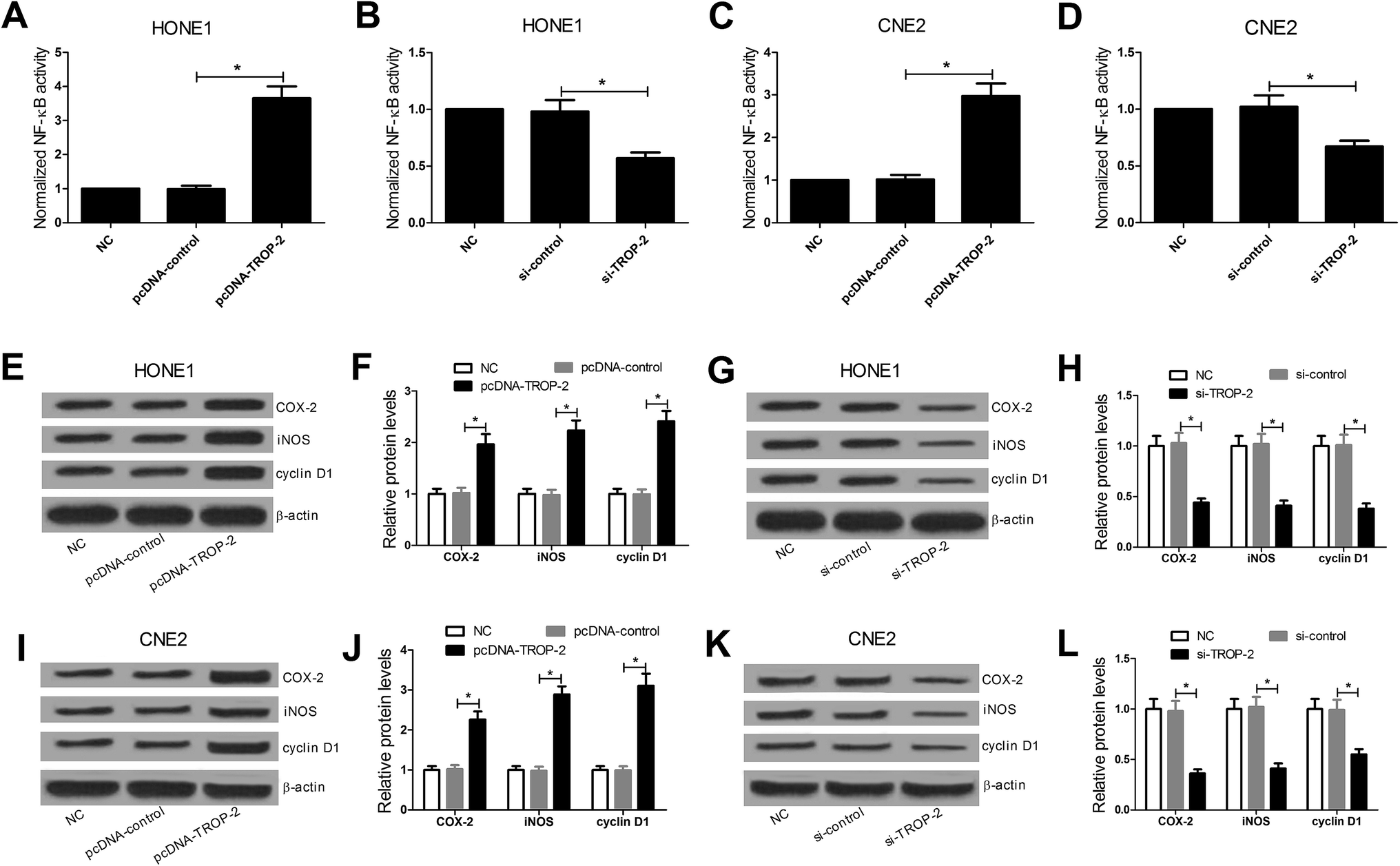 | ||
| Fig. 4 Effects of Trop2 on NF-κB signaling pathway. (A and C) ELISA-based colorimetric assay was carried out to assess the effect of pcDNA-Trop2 on the DNA-binding activity of NF-κB in HONE2 and CNE2 cells at 48 h after transfection. (B and D) ELISA-based colorimetric assay was carried out to assess the effect of si-Trop2 on the DNA-binding activity of NF-κB in HONE2 and CNE2 cells at 48 h after transfection. Western blot analysis of COX-2, iNOS and cyclin D1 in HONE1 (E and F) and CNE2 (I and J) cells transfected with pcDNA-Trop2 48 h post transfection. Western blot analysis of COX-2, iNOS and cyclin D1 in HONE1 (G and H) and CNE2 (K and L) cells transfected with si-Trop2 48 h post transfection. β-Actin was used as an internal control. *P < 0.05 vs. control group. | ||
3.5. Inactivation of NF-κB pathway attenuated Trop2-induced proliferation, invasion and EMT in NPC cells
The NF-κB inhibitor, PDTC,25 was used to further confirm whether Trop2 promoted cell proliferation, invasion and EMT in NPC cells by NF-κB pathway. HONE1 and CNE2 cells transfected with pcDNA-control or pcDNA-Trop2 were pretreated with or without PDTC for 1 h, followed by measurement of cell proliferation, invasion and EMT. The results demonstrated that compared with pc-DNA control group, PDTC treatment obviously suppressed the growth (Fig. 5A and B), invasion (Fig. 5C and D) and EMT (Fig. 5E and F) of NPC cells. Moreover, Trop2 overexpression induced increase in proliferation (Fig. 5A and B), invasion (Fig. 5C and D) and EMT (Fig. 5E and F) was greatly abated following PDTC treatment. These data suggested that Trop2 induced proliferation, invasion and EMT via activating NF-κB pathway in NPC cells.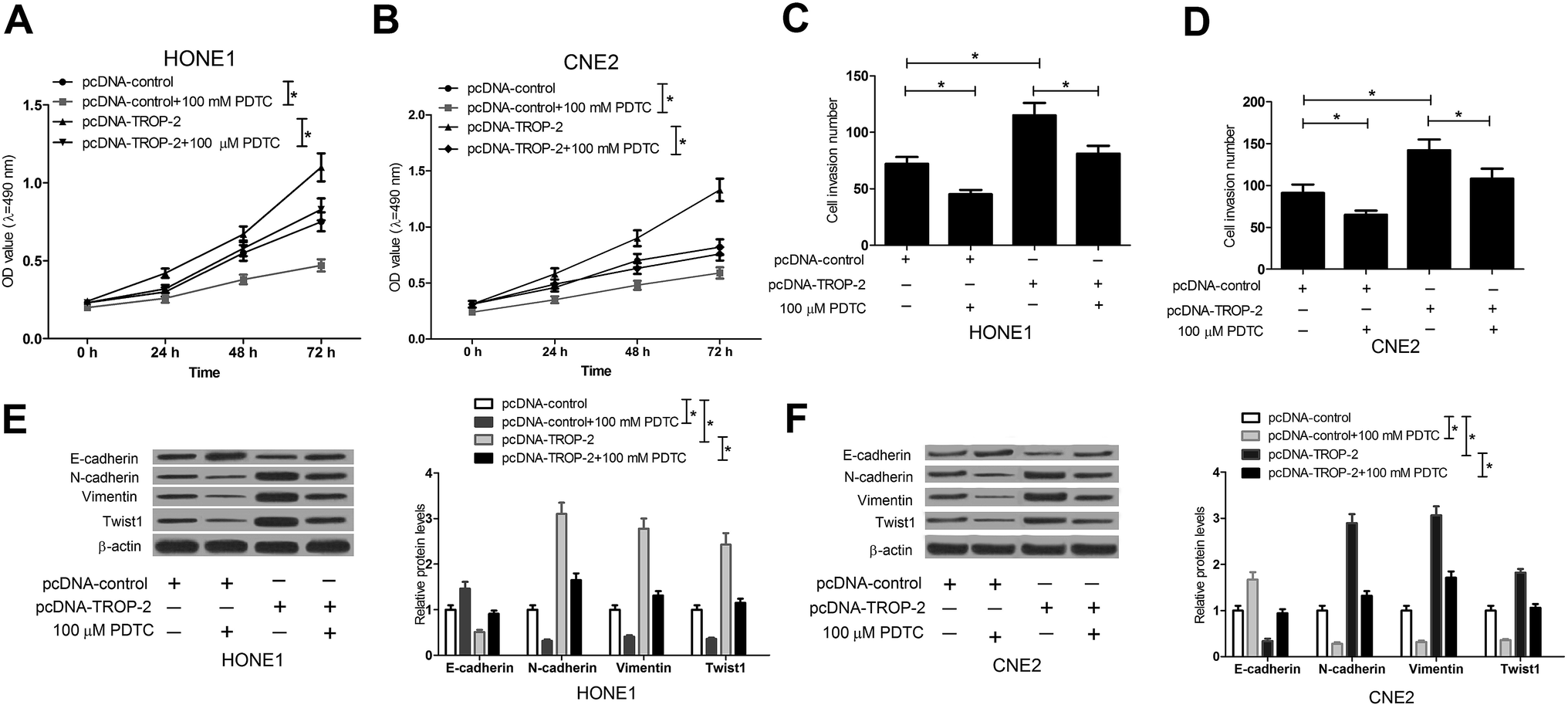 | ||
| Fig. 5 NF-κB inhibitor abated the effect of Trop2 on NPC cell proliferation, invasion and EMT. HONE1 and CNE2 cells transfected with pcDNA-control or pcDNA-Trop2 were treated with or without 100 μM PDTC for 1 h. (A and B) The viability of HONE1 and CNE2 cells was evaluated by MTT assay at 24 h, 48 h and 72 h. (C and D) The invasion numbers of HONE1 and CNE2 cells were detected by Transwell invasion assay 24 h post transfection. Western blot was used to determine the levels of E-cadherin, N-cadherin, vimentin and Twist in HONE1 (E) and CNE2 (F) cells at 48 h after transfection. The protein level was normalized to β-actin. *P < 0.05 vs. control group. | ||
3.6. NF-κB knockdown suppressed Trop2-induced proliferation, invasion and EMT in NPC cells
To further confirm that Trop2 promotes cancer aggressiveness through the NF-κB activation, NF-κB expression was inhibited by introducing si-NF-κB in pcDNA-Trop2-transfected HONE1 and CNE2 cells. The results manifested that NF-κB knockdown substantially weakened Trop2-triggered enhancement in proliferation (Fig. 6A and B), invasion (Fig. 6C and D) and EMT (Fig. 6E and F) in HONE1 and CNE2 cells. All these data indicated that Trop2 promoted NPC progression by activation of NF-κB pathway. | ||
| Fig. 6 NF-κB knockdown inhibited the effect of Trop2 on NPC cell proliferation, invasion and EMT. HONE1 and CNE2 cells were transfected with pcDNA-control, pcDNA-Trop2, pcDNA-Trop2 + si-control, orpcDNA-Trop2 + si-NF-κB. (A and B) The viability of HONE1 and CNE2 cells was measured by MTT assay at 24 h, 48 h and 72 h. (C and D) The invasion ability of HONE1 and CNE2 cells were analyzed by Transwell invasion assay 24 h post transfection. Western blot was employed to detect the expression of E-cadherin, N-cadherin, vimentin and Twist in HONE1 (E) and CNE2 (F) cells at 48 h after transfection. The protein level was normalized to β-actin. *P < 0.05 vs. respective control group. | ||
3.7. Trop2 activated ERK1/2 and Akt pathway in NPC cells
We further explored the possible effect of Trop2 overexpression on ERK1/2 and Akt pathway. As presented in Fig. 7A and B, compared with control groups, p-ERK1/2 and p-Akt protein level were both upregulated in CNE2 and HONE1 cells introduced with pcDNA-Trop2, indicating that Trop2 may exert role in NPC progression via ERK1/2 and Akt pathway.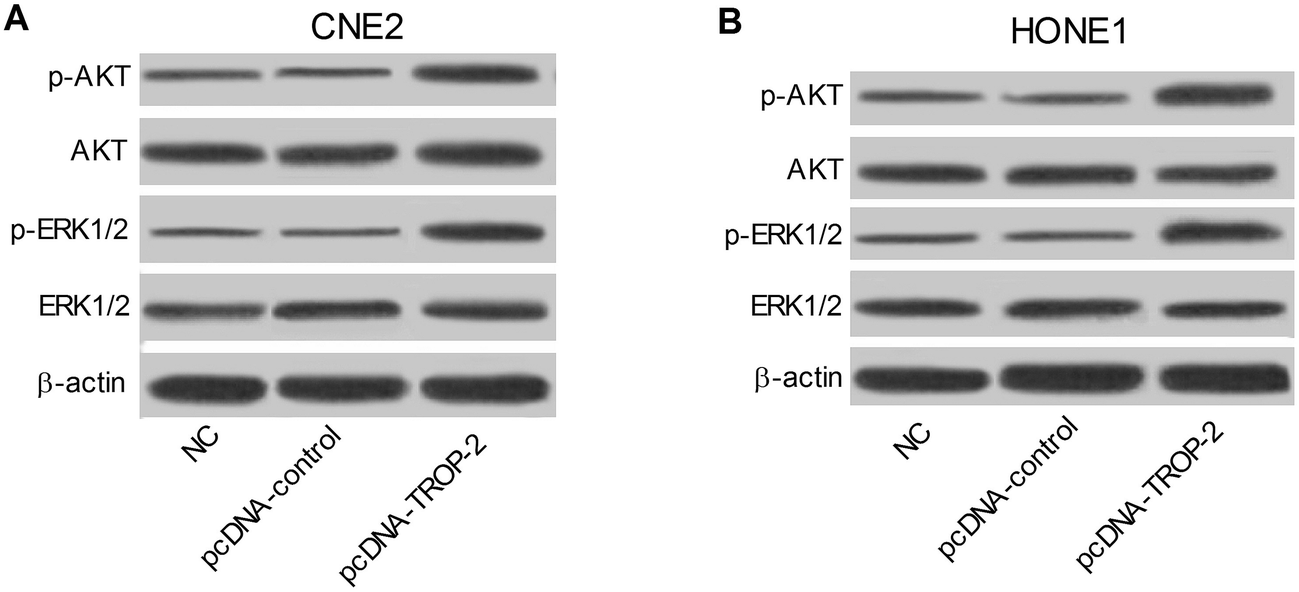 | ||
| Fig. 7 Effects of Trop2 on MAPK/ERK and PI3K/AKT signaling pathway. Western blot analysis of p-ERK1/2, ERK1/2, p-AKT and AKT in CNE2 (A) and HONE1 (B) cells transfected with pcDNA-control or pcDNA-Trop-2. | ||
4. Discussion
In the present study, we found that Trop2, an anti-adhesive and pro-migratory transmembrane protein, was upregulated in NPC tissues and cell lines. Overexpression of Trop2 contributed to proliferation, invasion and EMT in HONE1 and CNE2 cells. To further explore the underlying molecular mechanism of Trop2 in NPC progression, the relationship between Trop2 and NF-κB signaling pathway was investigated.Trop2, a tumor-associated calcium signal transducer, was found to be differently expressed in various tumors. For example, Trop2 was overexpressed in human epithelial tumor types including squamous cell carcinoma,7 gastric,26 and pancreatic cancer11 and downregulated in some cancers such as breast cancer.27 Mounting evidence suggests that abnormal expression of Trop2 is significantly correlated with cancer development and poor prognosis in numerous cancers and confers the capacity for proliferation and invasion to cancer cells.28,29 For example, Trop2 overexpression enhanced cell proliferation, migration and invasion in non-small cell lung carcinoma cells, while Trop2 knockdown presented an opposite effect.30,31 In laryngeal squamous cell carcinoma (LSCC), si-Trop2-mediated Trop2 silencing suppressed the proliferation and invasion by ERK/MAPK signaling pathway.32 In the current study, we found that Trop2 was highly expressed in NPC tissues and cell lines compared with normal nasopharyngeal epithelial tissues and nasal epithelial cells HNEpC, consistent with the previous study.12 Moreover, overexpression of Trop2 facilitated proliferation and invasion, whereas downregulation of Trop2 suppressed proliferation and invasion in HONE1 and CNE2 cells, suggesting that Trop2 was involved in proliferation and invasion in NPC.
Tumor metastasis is the one of the major cause of NPC deaths and understanding the potential molecular mechanism in cancer invasion and metastasis may provide a novel therapeutic approaches against NPC.33 EMT is a dynamical process by which cell changes the original epithelial morphology with disappearance of inter-cellular connection, thereby acquiring motility and invasiveness.34 Therefore, EMT is considered as a common molecular mechanism of tumor metastasis.35 E-cadherin, N-cadherin, vimentin and Twist1 are the typical regulators of EMT. In the current study, lower expression of E-cadherin and higher expressions of vimentin, Twist1, and N-cadherin were observed in NPC cells with Trop2 overexpression, whereas silencing endogenous Trop2 expression showed the contrary effect on EMT-related proteins including E-cadherin, N-cadherin, vimentin and Twist1, indicating that Trop2 played a key role in triggering EMT process in NPC.
Guan et al. have reported that overexpression of Trop2 appears to be an independent predictor for poor clinical outcome in NPC.12 They investigated the correlation of Trop2 expression with clinicopathologic features through a large number of statistical data. However, the detailed function and precise mechanism of Trop2 in NPC remains unclear. In our research, we confirmed the carcinogenicity of Trop2 in NPC via measuring the effects of Trop2 on proliferation, invasion and EMT by gain- and loss-of function analysis in HONE1 and CNE2 cells. On this basis, we further discussed the possible molecular basis involved in the regulation of Trop2 in NPC progression.
NF-κB is responsible for the regulation of a variety of cellular biological process, including proliferation, apoptosis and metastasis and angiogenesis by targeting its downstream genes.36 Activation of NF-κB signaling pathway was found in NPC and dysregulation of the pathway was implicated with NPC carcinogenesis.37 The biomarkers of tumor promotion and inflammation including iNOS and COX-2, frequently upregulated in majority of human tumor cells,38,39 are regulated by NF-κB pathway at the gene transcriptional or translational levels and are associated with the pathogenesis of inflammation and cancer.40,41 Cyclin D1, an NF-κB target gene, is a cell cycle-dependent regulatory protein and has been known as markers of cell growth.42 DHMEQ, a small molecular inhibitor of NF-κB, has been demonstrated to significantly inhibit NPC cell proliferation, invasion, migration, as well as anchorage-independent growth.42 Trop2 is an intracellular calcium signal transducer and it is composed of 323 amino acids. A serine phosphorylation site at S303 could be phosphorylated by protein kinase C (PKC). When position S303 is activated, more PKC is activated in a positive feedback mechanism, which could lead to more phosphorylated Trop2 and the activation of NF-κB pathway.43 In the present study, we observed that Trop2 overexpression increased NF-κB activity, and Trop2 silencing decreased NF-κB activity compared with that in corresponding control groups. Trop2 overexpression promoted the expression of NF-κB downstream target proteins including COX-2, iNOS, and cyclin D1, whereas Trop2 downregulation inhibited their expressions. Additionally, further studies found that NF-κB inhibitor PDTC or NF-κB knockdown attenuated Trop2-induced proliferation, invasion and EMT in NPC cells. All these results indicated that Trop2 promoted cell proliferation, invasion and EMT by activating NF-κB pathway in NPC. Prior to our study, some studies reported the effects of trop2 on NPC, while other reports showed that NF-κB signaling pathway was involved in NPC.12,44 However, few reports demonstrated that the effect of trop2 on NPC via activating NF-κB signaling pathway. Therefore, the innovative point in our research is to identify the regulation of trop2 in NPC cells through the activation of NF-κB pathway.
In our research, we also analyzed the effect of Trop2 on other important signaling proteins for EMT and metastasis, such as MAPK/ERK and PI3K/AKT pathway. A previous study reported that Trop2 regulated cell cycle via MAPK/ERK pathway in cervical cancer.45 Also, Trop2 was revealed to modulate the growth, invasion and migration of laryngeal carcinoma cells through the ERK/MAPK signaling pathway.32 Additionally, Trop2 was demonstrated to be a mandatory activator of AKT in breast cancer cells.46 Consistently, our results discovered that the protein level of p-ERK1/2 in MAPK/ERK pathway and p-AKT in PI3K/AKT pathway was up-regulated in CNE2 and HONE1 cells transfected with pcDNA-Trop2, indicating that Trop2 may affect NPC development by activating MAPK/ERK and PI3K/AKT signaling pathway. However, the detailed molecular mechanism still needs to be further analyzed in the future work.
In addition, we also found some inconsistencies between our experimental data and the previous results. In our research, over-expression of Trop2 suppressed the expression of E-cadherin in NPC cells, which is consistent with the results in gallbladder and cervical cancer cells.45,47 However, over-expression of Trop2 promoted E-cadherin expression in squamous cell carcinoma, which exhibited a loss-of-function role for Trop2 in tumorigenesis and mesenchymal transdifferentiation.48 These results indicate the complexity of Trop2 function in invasion and proliferation of cancer cells in different tissues. Further studies are needed to verify the role of Trop2 in cancer cells of different tissues.
EMT is known to be associated with chemoresistance of various cancers. It was documented that down-regulation of Trop2 resulted in enhanced sensitivity to cisplatin in cervical cancer cells.45 Nevertheless, the effect of Trop2 on NPC cells in the presence of cisplatin has not been reported, which is also the focus of our follow-up study.
5. Conclusions
Taken together, our findings demonstrated that Trop2 was up-regulated in NPC tissues and cell lines. Moreover, Trop2 overexpression contributed to the progression of NPC by promoting cell proliferation, invasion and EMT via NF-κB pathway. Therefore, Trop2 may be developed as a novel therapeutic target for the treatment of NPC.Authors' contributions
N. C. conceived the concept and designed the experiments. N. C. and H. L. conducted the experiments and analyzed the data. J. L. reviewed and approved the final manuscript.Conflicts of interest
The authors have no conflict of interest to declare.Funding
None.Acknowledgements
Thanks for all participants involved in our study.References
- S. M. Cao, M. J. Simons and C. N. Qian, Chin. J. Cancer, 2011, 30, 114–119 CrossRef PubMed.
- J. T. Wee, T. C. Ha, S. L. Loong and C. N. Qian, Chin. J. Cancer, 2010, 29, 517–526 CrossRef CAS PubMed.
- S. Y. Wang, J. L. Lou, J. Chen, S. Z. Zhang and L. Guo, World J. Surg. Oncol., 2016, 14, 89 CrossRef PubMed.
- I. W. Tham, S. W. Hee, R. M. Yeo, P. B. Salleh, J. Lee, T. W. Tan, K. W. Fong, E. T. Chua and J. T. Wee, Int. J. Radiat. Oncol., Biol., Phys., 2009, 75, 1481–1486 CrossRef PubMed.
- W. I. Wei and J. S. Sham, Lancet, 2005, 365, 2041–2054 CrossRef.
- E. Ripani, A. Sacchetti, D. Corda and S. Alberti, Int. J. Cancer, 1998, 76, 671–676 CrossRef CAS PubMed.
- D. Fong, G. Spizzo, J. M. Gostner, G. Gastl, P. Moser, C. Krammel, S. Gerhard, M. Rasse and K. Laimer, Mod. Pathol., 2008, 21, 186–191 CAS.
- T. Liu, Y. Liu, X. Bao, J. Tian, Y. Liu and X. Yang, PLoS One, 2013, 8, e75864 CAS.
- H. Wu, H. Xu, S. Zhang, X. Wang, H. Zhu, H. Zhang, J. Zhu and J. Huang, Head Neck, 2013, 35, 1373–1378 Search PubMed.
- M. Trerotola, P. Cantanelli, E. Guerra, R. Tripaldi, A. L. Aloisi, V. Bonasera, R. Lattanzio, R. de Lange, U. H. Weidle, M. Piantelli and S. Alberti, Oncogene, 2013, 32, 222–233 CrossRef CAS PubMed.
- D. Fong, P. Moser, C. Krammel, J. M. Gostner, R. Margreiter, M. Mitterer, G. Gastl and G. Spizzo, Br. J. Cancer, 2008, 99, 1290–1295 CrossRef CAS PubMed.
- G. F. Guan, D. J. Zhang, L. J. Wen, D. J. Yu, Y. Zhao, L. Zhu, Y. Y. Guo and Y. Zheng, Int. J. Clin. Exp. Pathol., 2015, 8, 10995–11004 Search PubMed.
- S. Ghosh and M. S. Hayden, Immunol. Rev., 2012, 246, 5–13 CrossRef PubMed.
- S. K. Sandur, A. Deorukhkar, M. K. Pandey, A. M. Pabon, S. Shentu, S. Guha, B. B. Aggarwal and S. Krishnan, Int. J. Radiat. Oncol., Biol., Phys., 2009, 75, 534–542 CrossRef CAS PubMed.
- A. B. Kunnumakkara, P. Diagaradjane, S. Guha, A. Deorukhkar, S. Shentu, B. B. Aggarwal and S. Krishnan, Clin. Cancer Res., 2008, 14, 2128–2136 CrossRef CAS PubMed.
- W. Shi, C. Bastianutto, A. Li, B. Perez-Ordonez, R. Ng, K. Y. Chow, W. Zhang, I. Jurisica, K. W. Lo, A. Bayley, J. Kim, B. O'Sullivan, L. Siu, E. Chen and F. F. Liu, Int. J. Cancer, 2006, 119, 2467–2475 CrossRef CAS PubMed.
- W. L. Chen, Y. Qian, W. F. Meng, J. Y. Pang, Y. C. Lin, Y. Y. Guan, S. P. Chen, J. Liu, Z. Pei and G. L. Wang, Biochem. Pharmacol., 2009, 78, 941–950 CrossRef CAS PubMed.
- X. Li, J. Yang, X. Wang, X. Li, J. Liang and H. Xing, Eur. J. Gynaecol. Oncol., 2016, 37, 100–108 CrossRef CAS PubMed.
- A. E. Stepan, M. Ciuca, C. Simionescu, D. Stepan and C. Margaritescu, Curr. Health Sci. J., 2015, 41, 219–226 Search PubMed.
- C. C. Fan, T. Y. Wang, Y. A. Cheng, S. S. Jiang, C. W. Cheng, A. Y. Lee and T. Y. Kao, J. Cancer Res. Clin. Oncol., 2013, 139, 1735–1744 CrossRef CAS PubMed.
- X. Zhai, H. Zhu, W. Wang, S. Zhang, Y. Zhang and G. Mao, Med. Oncol., 2014, 31, 970 CrossRef PubMed.
- M. Hermanova, J. Trna, R. Nenutil, P. Dite and Z. Kala, Eur. J. Gastroenterol. Hepatol., 2008, 20, 732–739 CrossRef CAS PubMed.
- Z. Feng, J. Chen, H. Wei, P. Gao, J. Shi, J. Zhang and F. Zhao, Exp. Mol. Pathol., 2011, 91, 569–577 CrossRef CAS PubMed.
- P. Letelier, P. Garcia, P. Leal, C. Ili, K. Buchegger, I. Riquelme, A. Sandoval, O. Tapia and J. C. Roa, Appl. Immunohistochem. Mol. Morphol., 2014, 22, 530–536 CrossRef CAS PubMed.
- M. Z. Cao, W. Z. Mao, G. L. Ma and Y. Li, Chinese Journal of Gastrointestinal Surgery, 2013, 16, 578–582 Search PubMed.
- G. Muhlmann, G. Spizzo, J. Gostner, M. Zitt, H. Maier, P. Moser, G. Gastl, M. Zitt, H. M. Muller, R. Margreiter, D. Ofner and D. Fong, J. Clin. Pathol., 2009, 62, 152–158 CrossRef CAS PubMed.
- H. Huang, J. Groth, K. Sossey-Alaoui, L. Hawthorn, S. Beall and J. Geradts, Clin. Cancer Res., 2005, 11, 4357–4364 CrossRef CAS PubMed.
- M. Trerotola, D. L. Jernigan, Q. Liu, J. Siddiqui, A. Fatatis and L. R. Languino, Cancer Res., 2013, 73, 3155–3167 CrossRef CAS PubMed.
- E. Bignotti, L. Zanotti, S. Calza, M. Falchetti, S. Lonardi, A. Ravaggi, C. Romani, P. Todeschini, E. Bandiera and R. A. Tassi, BMC Clin. Pathol., 2012, 12, 22 CrossRef PubMed.
- Z. Li, X. Jiang and W. Zhang, Biochem. Biophys. Res. Commun., 2016, 470, 197–204 CrossRef CAS PubMed.
- X. Y. Gao, Y. H. Zhu, L. X. Zhang, H. Y. Lu and A. G. Jiang, Exp. Ther. Med., 2015, 10, 429–434 CrossRef CAS PubMed.
- X. D. Wang, Q. Wang, X. L. Chen, J. F. Huang, Y. Yin, P. Da and H. Wu, Mol. Med. Rep., 2015, 12, 865–870 CrossRef CAS PubMed.
- J. Zhang, X. Wen, X. Y. Ren, Y. Q. Li, X. R. Tang, Y. Q. Wang, Q. M. He, X. J. Yang, Y. Sun, N. Liu and J. Ma, J. Exp. Clin. Cancer Res., 2016, 35, 109 CrossRef PubMed.
- J. Yang and R. A. Weinberg, Dev. Cell, 2008, 14, 818–829 CrossRef CAS PubMed.
- P. Li, Y. F. Jia, X. L. Ma, Y. Zheng, Y. Kong, Y. Zhang, S. Zong, Z. T. Chen and Y. S. Wang, Am. J. Cancer Res., 2016, 6, 1741–1757 Search PubMed.
- S. C. Miller, R. Huang, S. Sakamuru, S. J. Shukla, M. S. Attene-Ramos, P. Shinn, D. Van Leer, W. Leister, C. P. Austin and M. Xia, Biochem. Pharmacol., 2010, 79, 1272–1280 CrossRef CAS PubMed.
- D. D. Zhu, J. Zhang, W. Deng, Y. L. Yip, H. L. Lung, C. M. Tsang, W. T. Law, J. Yang, V. M. Lau, W. H. Shuen, M. L. Lung, A. L. Cheung and S. W. Tsao, Int. J. Cancer, 2016, 138, 1175–1185 CrossRef CAS PubMed.
- R. E. Harris, Subcell. Biochem., 2007, 42, 93–126 Search PubMed.
- C. Yi, Y. Zhang, Z. Yu, Y. Xiao, J. Wang, H. Qiu, W. Yu, R. Tang, Y. Yuan, W. Guo and W. Deng, PLoS One, 2014, 9, e99943 Search PubMed.
- C. H. Pan, E. S. Kim, S. H. Jung, C. W. Nho and J. K. Lee, Arch. Pharmacal Res., 2008, 31, 1447–1456 CrossRef CAS PubMed.
- F. Nose, T. Ichikawa, M. Fujiwara and I. Okayasu, Am. J. Clin. Pathol., 2002, 117, 546–551 CrossRef CAS PubMed.
- J. H. Wong, V. W. Lui, K. Umezawa, Y. Ho, E. Y. Wong, M. H. Ng, S. H. Cheng, C. M. Tsang, S. W. Tsao and A. T. Chan, Cancer Lett., 2010, 287, 23–32 CrossRef CAS PubMed.
- R. Cubas, M. Li, C. Chen and Q. Yao, Biochim. Biophys. Acta, 2009, 1796, 309–314 CAS.
- D. D. Zhu, J. Zhang, W. Deng, Y. L. Yip, H. L. Lung, C. M. Tsang, W. T. Law, J. Yang, V. M. Lau, W. H. Shuen, M. L. Lung, A. L. Cheung and S. W. Tsao, Int. J. Cancer, 2016, 138, 1175–1185 CrossRef CAS PubMed.
- T. Liu, Y. Liu, X. Bao, J. Tian, Y. Liu and X. Yang, PLoS One, 2013, 8, e75864 CAS.
- E. Guerra, M. Trerotola, R. Tripaldi, A. L. Aloisi, P. Simeone, A. Sacchetti, V. Relli, A. D'Amore, R. L. Sorda, R. Lattanzio, M. Piantelli and S. Alberti, Clin. Cancer Res., 2016, 22, 4197–4205 CrossRef CAS PubMed.
- X. Li, S. Teng, Y. Zhang, W. Zhang, X. Zhang, K. Xu, H. Yao, J. Yao, H. Wang, X. Liang and Z. Hu, Oncotarget, 2017, 8, 47052–47063 Search PubMed.
- J. B. Wang, K. H. Zhang, D. Grabowska, A. M. Li, Y. Y. Dong, R. Day, P. Humphrey, J. Lewis, D. Kladney, M. Arbeit, D. Weber, H. Chung and S. Michel, Mol. Cancer Res., 2011, 9, 1686–1695 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |