
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: FAM172A controls endoplasmic reticulum (ER) stress related to NF-κB signaling pathway in hepatocellular carcinoma
Wenfeng Shen†
a,
Zhiqiang Feng†*b,
Ping Wangc and
Jinqian Zhang *d
*d
aDepartment of Ultrasound, The Affiliated Hospital of Inner Mongolia Medical University, Hohhot, 010059, China
bDepartment of Hepatobiliary Surgery, Air Force General Hospital, Beijing 100142, China. E-mail: fengzq1151@163.com; Fax: +86-10-66928312; Tel: +86-10-66928312
cDepartment of Nuclear Magnetic Resonance, Air Force General Hospital, Beijing 100142, China
dDepartment of Laboratory Medicine, Guangdong Second Provincial General Hospital, Southern Medical University, No. 466 Xingang Middle Road, Haizhu District, Guangzhou 510317, Guangdong Province, China. E-mail: jingwanghou@163.com; Fax: +86-20-89168239; Tel: +86-20-89168239
First published on 7th November 2017
Abstract
Our previous investigation suggested that FAM172A shows significantly low expression in hepatocellular carcinoma. However, its mechanism is not very clear. This study showed that the FAM172A proteins are related to ER stress. FAM172A promoted the expression of NF-κB in liver cancer cells and induced ER stress through the PERK–eIF2α pathway in liver cancer cells. FAM172A could be a secreted protein and could restrain the proliferation of hepatoma cells through suppressing the cell cycle. In addition, FAM172A could also promote apoptosis or differentiation of hepatoma cells. FAM172A protein interacted with Notch 1–4 and NF-κB. The FAM172A recombinant proteins suppressed tumor formation in a mouse xenograft model. These results indicated that FAM172A could be an anti-oncogene and plays a vital role in controlling cell proliferation and the cell cycle by inducing the arrest of the G1/S phase through PERK/eIF2α via an ER stress-independent pathway and correlates with NF-κB. FAM172A, which could act as a novel treatment target of HCC.
Introduction
HCC (hepatocellular carcinoma) is a common cause of cancer and is ranked fifth, while it is also the second most common reason for cancer deaths among humans. HCC primarily occurs from the inflammation process and liver cirrhosis induced by aflatoxins, ethanol and chronic hepatitis B or C infection.1 The prognosis of HCC is so poor2 that only a tiny minority of patients could be cured by surgical resection or liver transplantation. Consequently, it is necessary to research and development the effective therapies or medicines for HCC.For many years, different genetic alterations were thought to be the origin of hepatocarcinogenesis and induced malignant transformation.3 Therefore, the development of cancer is not only considered to be the result of genomic or genetic alterations but also due to lipid metabolism.4
Proper functioning of the endoplasmic reticulum and mitochondria is essential for cellular homeostasis and the regulation of metabolic pathways. Perturbation of their function has been linked to pathophysiological states, including metabolic and liver diseases particularly in HCC. In addition to their independent contributions to metabolic and hepatic pathologies, the mitochondria and ER directly interact regulating each other's functions and the ER–mitochondria interface is involved in several molecular pathways, such as the induction of autophagy and triggering of inflammatory cascades. The disturbance in these interactions has already been implicated in different human diseases, and increasing interest has arisen in their role in liver illnesses.5
The results based on our previous studies have indicated that FAM172A shows significantly low expression in the liver tissue of patients with HCC or cirrhosis particularly in HCC. The intracellular localization of FAM172A was investigated in HepG2 cells using confocal microscopy. The binding activity between FAM172A and Ca2+ or active single sugar was investigated using surface plasmon resonance technology. The cell cycle experiments of HepG2 were carried out using flow cytometry. We found that in HepG2 cells, the sub-cellular localization of FAM172A protein was in the endoplasmic reticulum. In normal liver tissue, the protein showed moderate expression. A high concentration of FAM172A recombinant protein induced arrest of the cell cycle at the S-phase and suppression of cell proliferation in co-cultured HepG2 cells. Our study demonstrates that FAM172A could be an anti-oncogene and plays a vital role in controlling the proliferation of hepatoma cells and the cell cycle by inducing the arrest of the G1/S phase by up-regulating the expression of cyclin E and Notch 3.6
However, the functions of FAM172A are not very clear. This study was carried out to clarify the effects of FAM172A on the functions of hepatoma cells. To identify the protein expression level of FAM172A, western blot analysis was performed. FAM172A could regulate cell proliferation, cell cycle and cell apoptosis through the ER stress pathway via NF-κB. This study would be evaluated for the exploration of targeted therapy in HCC.
Results
Low expression levels of FAM172A in liver cancer tissues
To explore the potential regulation mechanism of FAM172A in HCC, we tested the protein expression levels of FAM172A in liver cancer tissues of patients using the western blot method. The results obtained indicated that the expression of FAM172A protein in the tumor samples was decreased when compared to non-cancerous specimens particularly in patients 1–4 (Fig. 1A).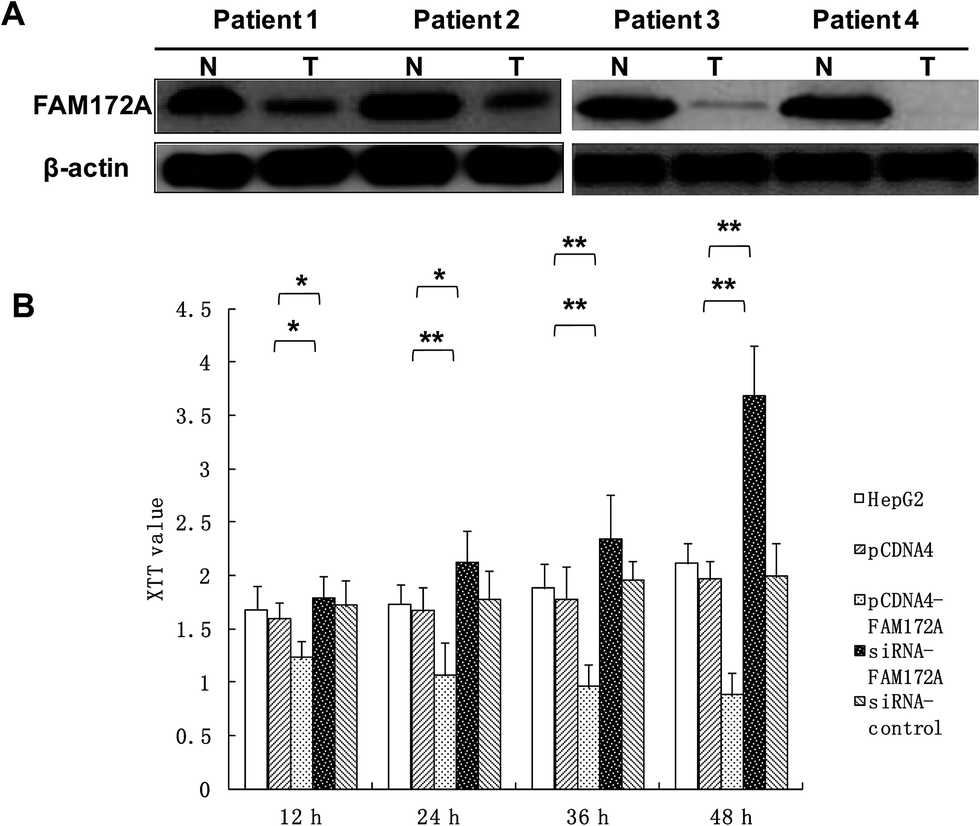 | ||
| Fig. 1 Low expression level of FAM172A in the tissues of patients with HCC. FAM172A inhibited the proliferation of liver cancer cells. (A) The expression of FAM172A protein in tumor samples decreased when compared to non-cancerous specimens particularly in patients 1–4. (B) The XTT assay results showed that FAM172A significantly suppressed the proliferation of the hepatoma cell line HepG2 when compared to the control after treatment for 12, 24, 36, and 48 h. Moreover, FAM172A silencing caused a remarkable increase in the proliferation of hepatoma cell when compared to the control after treatment for 12, 24, 36, and 48 h. *P < 0.05, **P < 0.01; the P value was generated using Kruskal–Wallis ANOVA. | ||
FAM172A inhibited the proliferation of liver cancer cells
Using an XTT assay, the results showed that FAM172A clearly suppressed the proliferation of the hepatoma cell line HepG2 when compared to the control after treatment for 12, 24, 36, and 48 h. Moreover, FAM172A silencing caused a remarkable increase in the proliferation of the hepatoma cell contrast control after treatment for 12, 24, 36 and 48 h (Fig. 1B).FAM172A induced the arrest of S-phase in liver cancer cells
When compared to the control, the cell cycle of HepG2 cells transfected with FAM172A was arrested at the G1/S phase. The S-phase of HepG2 transfected with pcDNA6.2–FAM172A shRNA for 48 h was 0.18%, which was significantly lower than the control groups (Fig. 2A). The changes of cell cycle induced by FAM172A was determined using the supernatant of the cultured cells. The S-phase proportion of HepG2 cultured for 48 h was 40.1% (Fig. 2B). However, after the supernatant was changed and then HepG2 cells were cultured for an additional 48 h, the S-phase was undetectable. The above supernatant was obtained from the cultured HepG2 cells transfected with pEGFP-C1–FAM172A for 48 h (Fig. 2B).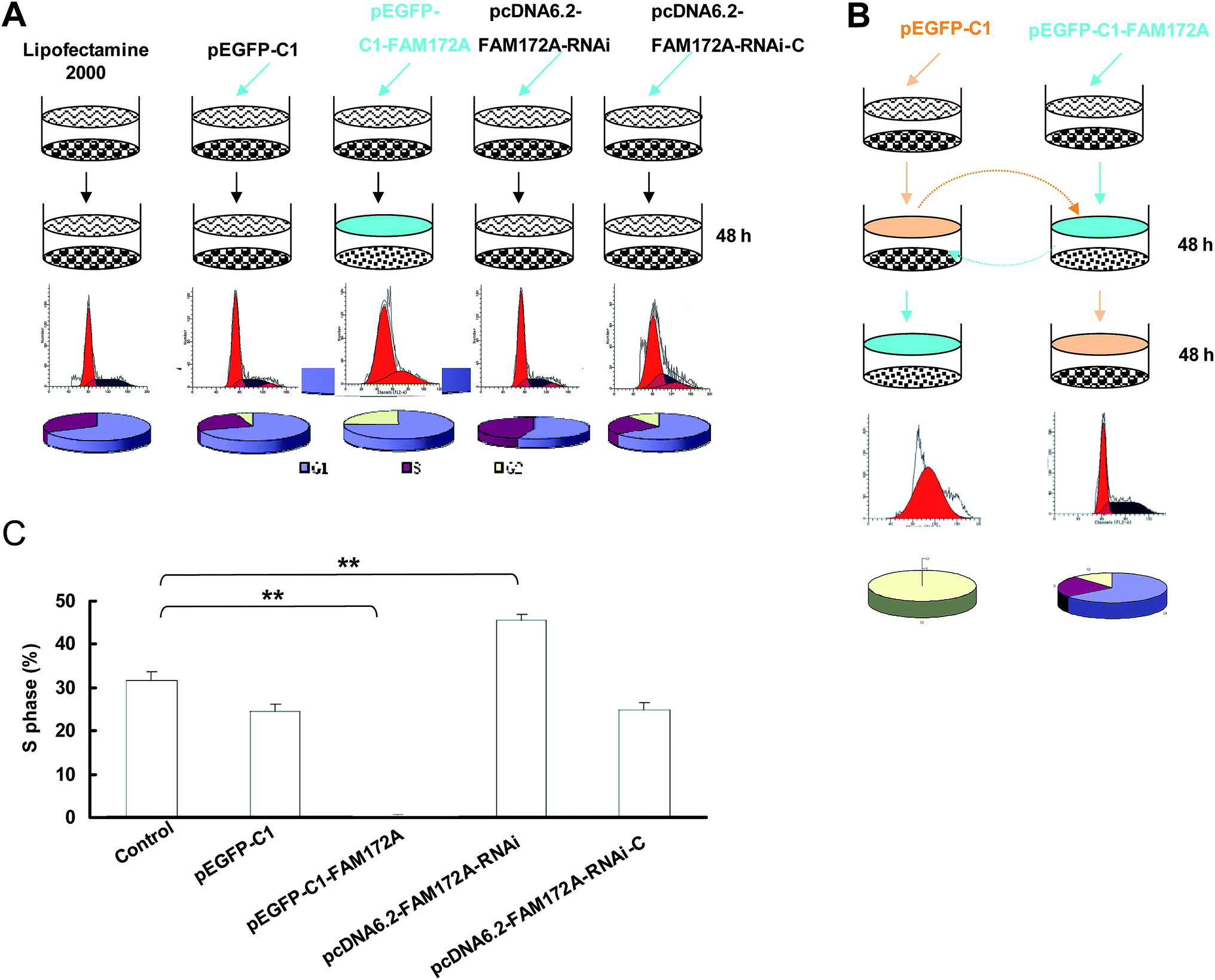 | ||
| Fig. 2 Effects of FAM172A on the cell cycle in HepG2 cells. (A) When compared to the control, the cell cycle of HepG2 cells transfected with FAM172A was arrested at the G1/S phase. The S-phase of HepG2 transfected with pcDNA6.2–FAM172A shRNA for 48 h was 0.18%, which was significantly lower than the control groups. (B) The effects on the cell cycle of HepG2 caused by FAM172A were determined using the supernatant of the cultured cells. The S-phase proportion of HepG2 cultured for 48 h was 40.1%. However, after the supernatant was changed, and then HepG2 cells were cultured for an additional 48 h, the S-phase was undetectable. The above supernatant was obtained from the cultured HepG2 cells transfected with pEGFP-C1–FAM172A for 48 h. (C) After being transfected with FAM172A, the HepG2 was cultured for 48 h and the supernatant of the cells was changed with that obtained from the cultured HepG2 cells for 48 h. The results of the S-phase showed that it had not been arrested. Moreover, in the control group, HepG2 cells transfected with FAM172A were arrested at the G1/S phase. In the negative control groups (pcDNA6.2–RNAi-c-transfected group and pEGFP-C1-transfected group), the cell number of these two groups only slightly decreased. Moreover, the S-phase of the cells transfected with RNAi for FAM172A was higher than the other cells. The data are presented as the mean ± SD from three independent experiments. *P < 0.05, **P < 0.01; the P value was generated using Kruskal–Wallis ANOVA. | ||
After being transfected with FAM172A, HepG2 was cultured for 48 h and the supernatant of these cells was changed with that obtained from the cultured HepG2 cells for 48 h. The results obtained for the S-phase showed that it had not been arrested. Moreover, in the control group, HepG2 cells transfected with FAM172A were arrested at the G1/S phase. In the negative control groups (pcDNA6.2–RNAi-c-transfected group and pEGFP-C1-transfected group), the cell number of these two groups only slightly decreased. Moreover, the S-phase of the cells transfected with RNAi for FAM172A was higher than that observed in the other cells (Fig. 2C). These results indicate that FAM172A could be a secreted protein and could suppress hepatoma cell growth.
FAM172A promoted differentiation and apoptosis of HepG2 cells
Flow cytometry experiments were conducted to determine the influence of FAM172A on apoptosis in hepatoma cells. The rate of apoptosis in HepG2 cells increased (P < 0.01), which was induced by an over-expression of FAM172A. Furthermore, the siRNA of FAM172A led to a significant reduction in the apoptosis rate in HepG2 cells (P < 0.01) (Fig. 3A). The significant differentiation of HepG2 cells was caused by the recombinant protein of FAM172A (Fig. 3C, 100×; and 3E, 400×) when compared with the control (Fig. 3B, 100×; and 3D, 400×).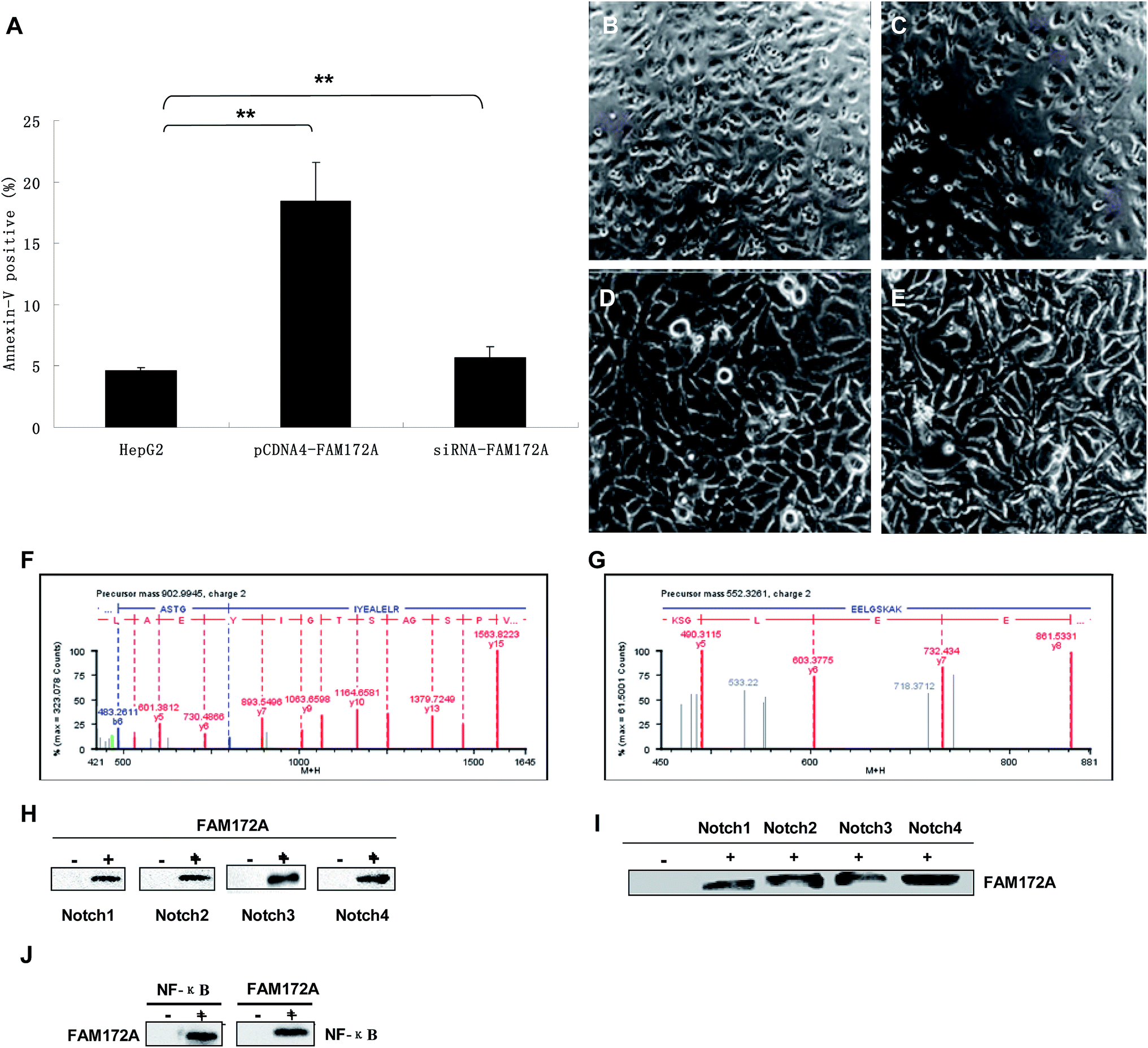 | ||
| Fig. 3 FAM172A promoted differentiation and apoptosis of HepG2. FAM172A proteins interacted with Notch 1–4 and NF-κB. Flow cytometry experiments were conducted to determine the influence of FAM172A on apoptosis in hepatoma cells. The rate of apoptosis in the HepG2 cells increased (P < 0.01), which was induced by the over-expression of FAM172A. Furthermore, the shRNA of FAM172A led to a significant reduction of the apoptosis rate in HepG2 cells (P < 0.01) (A). The remarkable differentiation of HepG2 cells was caused by the recombinant protein of FAM172A (C, 100×; and E, 400×) when compared with the control groups (B, 100×; and D, 400×). The data are presented as the mean ± SD from three independent experiments. *P < 0.05, **P < 0.01; the P value was generated using Kruskal–Wallis ANOVA. (F) The map of ESI-MS/MSMS showing the spectrum of a peptide fragment originating from α-enolase [ASTGIYEALELR, m/z = 902.99, (M + 2H)2+]. (G) The map of the spectrum of another fragment originating from α-enolase [EELGSKAE, m/z = 552.33, (M + 2H)2+]. (H) HepG2 cells were harvested after 48 h in culture, then all proteins of the cells were extracted and CO-IP'd (co-immunoprecipitated) with the antibodies of FAM172A, followed by anti-Notch antibodies. (I) The obtained proteins were then Co-IP'd with anti-Notch antibodies followed by the antibodies of FAM172A. (J) The obtained proteins were CO-IP'd with antibodies of FAM172A or NF-κB p65 and then analyzed with antibodies of NF-κB p65 or FAM172A, respectively. | ||
FAM172A protein interacted with Notch 1–4 and NF-κB
The proteins that interacted with FAM172A were analyzed using UPLC-QTOF/MS and CO-IP. Fig. 3F shows the ESI-MS/MSMS spectrum of a peptide fragment originating from α-enolase [ASTGIYEALELR, m/z = 902.99, (M + 2H)2+]. Fig. 3G shows the spectrum of another fragment originating from α-enolase [EELGSKAE, m/z = 552.33, (M + 2H)2+]. The results demonstrate that FAM172A protein may interact with α-enolase in the hepatoma cell line HepG2.HepG2 cells were harvested after being cultured for 48 h, then all the proteins in the cells were extracted and CO-IP'd (co-immunoprecipitated) with the antibodies of FAM172A followed by anti-Notch antibodies (Fig. 3H). Then, the above proteins were Co-IP'd with anti-Notch antibodies followed with the antibodies of FAM172A (Fig. 3I). Fig. 3C and D show that FAM172A protein could interact with Notch 1–4. The above proteins were CO-IP'd with the antibodies of FAM172A or NF-κB p65 and then analyzed with the antibodies of NF-κB p65 or FAM172A, respectively (Fig. 3J). This identified the role of the interaction between FAM172A and NF-κB p65. These results demonstrated that FAM172A protein could interact with Notch 1–4 and NF-κB in the liver cancer cell line HepG2.
FAM172A promoted the expression of NF-κB in HepG2 cells
In the HepG2 hepatoma cell line, the influence on NF-κB expression induced by FAM172A was determined.6 The over-expression of FAM172A significantly increased the expression of NF-κB. Furthermore, the RNAi of FAM172A decreased the expression of NF-κB significantly (Fig. 4A). After treatment with the recombinant protein FAM172A, the expression of NF-κB in HepG2 also increased in a dose-dependent manner (Fig. 4B).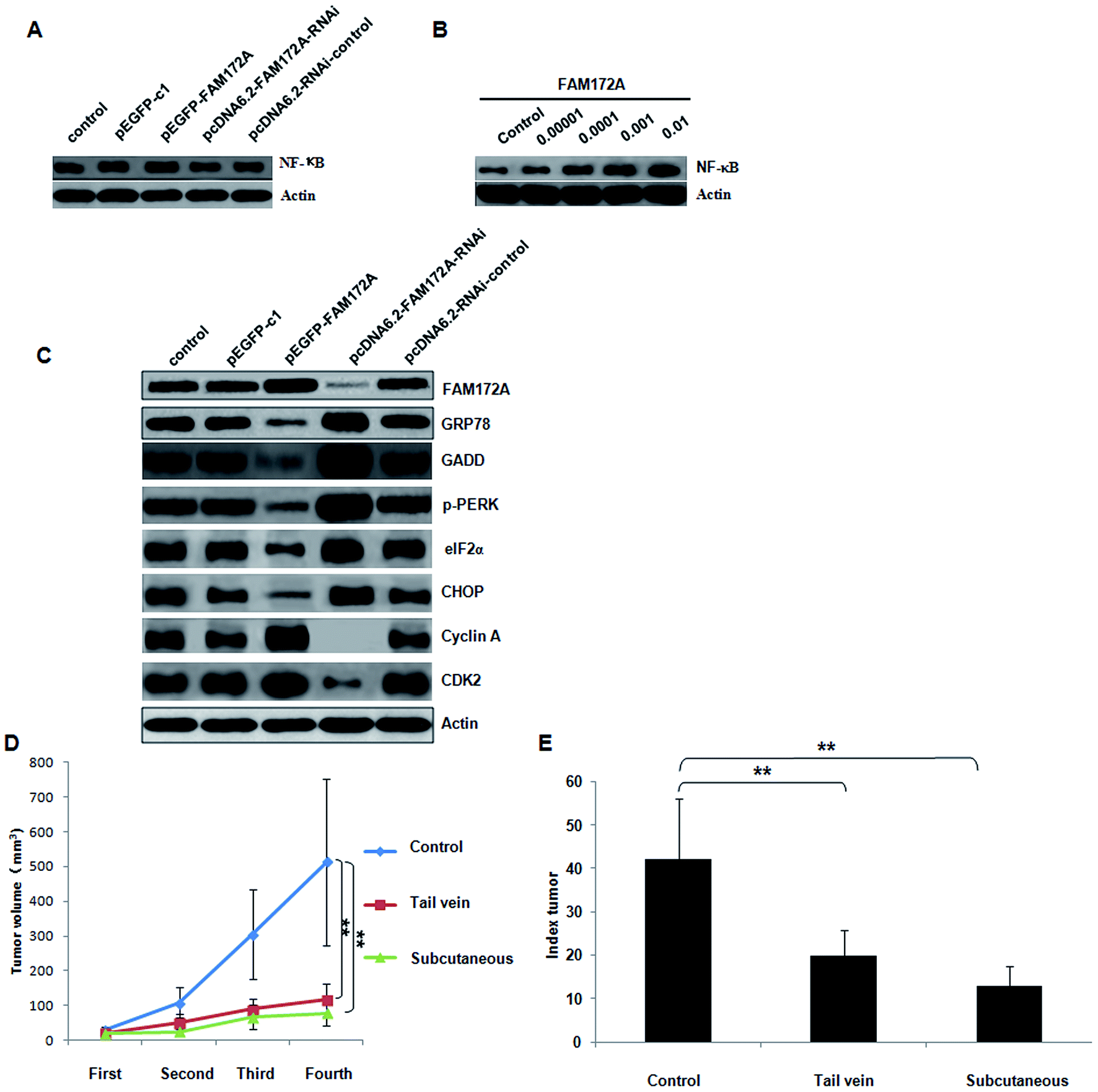 | ||
| Fig. 4 FAM172A promoted the expression of NF-κB and induced ER stress through the PERK–eIF2α pathway in HepG2 cells. FAM172A recombinant proteins inhibited tumor formation and proliferation of liver cancer in vivo. In the HepG2, hepatoma cell line, the influence on NF-κB expression induced by FAM172A was determined. Over-expression of FAM172A significantly increased the expression of NF-κB. Furthermore, the RNAi of FAM172A significantly decreased the expression of NF-κB (A). After treatment with the recombinant protein FAM172A, the expression of NF-κB in HepG2 cells was also increased in a dose-dependent manner (B). Up-regulation of FAM172A expression in liver cancer cells significantly inhibited the expression of GRP78, GADD, p-PERK, eIF2α and CHOP, whilst promoting the expression of cyclin A and CDK 2. Furthermore, treatment with shRNA of FAM172A in liver cancer cells significantly increased the expression of GRP78, GADD, p-PERK, eIF2α and CHOP, but inhibited the expression of cyclin A and CDK 2 (C). The antitumor role induced by FAM172A was evaluated in vivo using a nude mouse xenograft model. After injection with hepatoma cells for fifteen days, the tumor xenografts treated with the recombinant protein of FAM172A presented a decline in tumor growth when compared to the control group. Furthermore, the average volume of the tumor treated with FAM172A recombinant protein significantly reduced when compared with the control group (P < 0.01; D). The tumor proliferation index (TPI) also demonstrated that FAM172A attenuated the growth of the tumor (E) and the weight of the tumor group treated with the FAM172A recombinant proteins was significantly less than that found in the control group. | ||
FAM172A induced ER stress through the PERK–eIF2α pathway in liver cancer cells
The PERK–eIF2α pathway is one of the three key pathways associated with ER.6 The expression of FAM172A, GRP78, GADD, p-PERK, eIF2α, IRE, XBP1, ATF6, cyclin A, B, D, E and CDK 1, 2, 4, 6 proteins was detected using western blot analysis. The up-regulation of FAM172A expression in liver cancer cells significantly inhibited the expression of GRP78, GADD, p-PERK, eIF2α and CHOP, but promoted the expression of cyclin A and CDK 2. Furthermore, treatment with the shRNA of FAM172A in liver cancer cells significantly increased the expression of GRP78, GADD, p-PERK, eIF2α and CHOP, but inhibited the expression of cyclin A and CDK 2 (Fig. 4C). FAM172A had no effect on the expression of IRE, XBP1, ATF6, cyclin B, D, E and CDK 1, 4, 6 (data not shown).FAM172A recombinant proteins inhibited tumor formation and proliferation of liver cancer in vivo
The antitumor role induced by FAM172A was evaluated in vivo using a nude mouse xenograft model. After injection into hepatoma cells for fifteen days, the tumor xenografts treated with the recombinant protein of FAM172A presented a decline in tumor growth when compared to the control group. Furthermore, the average volume of the tumors treated with the FAM172A recombinant protein was significantly reduced when compared with the control group (P < 0.01; Fig. 4D). The tumor proliferation index (TPI) also demonstrated that FAM172A attenuated the progress of tumor growth (Fig. 4E) and the weight of the mice in the tumor group treated with FAM172A recombinant proteins was significantly less than those measured in the control group. These data demonstrated that FAM172A inhibited the growth and formation of tumor xenografts in vivo.Discussion
The results obtained in our previous study indicated that FAM172A could be implicated in the regulation of proliferation of HepG2 cells.6 Interestingly, the results also demonstrated that FAM172A could almost delete the S-phase of HepG2 cells at higher concentrations. This could be associated with a negative supervision in carcinogenesis. In particular, higher concentrations of FAM172A completely eliminated the S-phase of HepG2 cells.6 Therefore, we considered that FAM172A was a tumor-suppressor in liver cancer. However, the mechanism of action of FAM172A on liver cancer cells is not well understood.In this study, our results demonstrated that the up-regulation of FAM172A in liver cancer cells significantly inhibited the expression of GRP78 and GADD, which are markers of ER stress. In contrast, the expression of GRP78 and GADD significantly increased following the RNA interference of FAM172A. As shown in previous studies, tunicamycin can induce ER stress and activate EOR (ER overloading response pathway) and UPR (the unfolded protein response).10,11 When treated with different doses of tunicamycin, the expression levels of GRP78 and GADD were higher than in the control cells, while the expression of FAM172A was significantly down-regulated. Thus, our results suggest that FAM172A could be a survival factor for cells and a novel gene that plays a role in suppressing ER stress.
The ER (endoplasmic reticulum) is an organelle with a dynamic membrane, which participate in protein processing, folding and transport. Some adverse factors such as metabolic disorders, viral proteins, and chemical agents could induce protein accumulation or misfolding in the ER, which resulted in ER stress.12 The UPR pathway contains three components: PERK (ER localized kinase), IRE1 (RNA-processing enzyme) and ATF6 (basic leucine zipper transcription factor). We investigated which signaling pathway was activated to induce apoptosis, cell cycle arrest or differentiation of liver cancer cells treated with FAM172A. The results obtained from UPLC-QTOF/MS and CO-IP indicated that the FAM172A protein interacts with α-enolase, Notch 1–4 and NF-κB in liver cancer cells. In HepG2, the influence on NF-κB expression induced by FAM172A was further investigated. The over-expression of FAM172A significantly increased the expression of NF-κB. Furthermore, the RNAi of FAM172A significantly decreased the expression of NF-κB. After treatment with the recombinant protein FAM172A, the expression of NF-κB in HepG2 also increased in a dose-dependent manner.
Previous studies have indicated that during ER stress, reactive oxygen species products could activate NF-κB through PERK and IRE1 signaling pathways, as well as the release of calcium.13–17 This can regulate more than 100 genes related to different cell processes, such as cell differentiation, apoptosis, proliferation, immune responses and inflammation.18 The results obtained demonstrated that FAM172A promoted the expression of NF-κB, cyclin A and CDK 2 in liver cancer cells, but significantly inhibited the expression of p-PERK, eIF2α and CHOP. These results revealed that FAM172A mediated ER stress through PERK–eIF2α or NF-κB pathways in liver cancer cells.
The hepatoma cell line (HepG2) was treated with the recombinant protein of FAM172A at different concentrations. The results obtained indicated that FAM172A could be a secreted protein and could inhibit the proliferation of liver cancer cells. Moreover, FAM172A could induce the differentiation and apoptosis of HepG2 through activation of NF-κB. The differentiation of liver cancer cells induced by FAM172A was also associated with cyclin A–CDK 2-mediated pathway.
Recently, researchers have identified that apoptosis caused by ER stress was related to several pathways, such as IRE1/PERK/JNK, CHOP/GADD 153 and caspase-12/caspase-4.19–21 However, FAM172A could protect against ER stress related to the CHOP/GADD or PERK–eIF2α pathway and inhibit apoptosis. As a consequence, the FAM172A gene regulates the apoptosis of HepG2 by playing a dual role.
Finally, in vivo, the antitumor role of FAM172A was evaluated using a nude mouse xenograft model. The tumor xenografts treated with the recombinant protein of FAM172A presented a decrease in tumor growth when compared to the control. Furthermore, the average volume of the tumor treated with FAM172A recombinant protein was significantly reduced when compared to the control group. The TPI data and weight of the tumors inferred that FAM172A suppressed the progress of the tumor growth. This demonstrates that FAM172A inhibited the growth and formation of tumor xenograft in vivo.
In conclusion, these data provide a better understanding of the FAM172A gene, which could play a novel role as a tumor suppressor and cell cycle regulator in HCC. FAM172A protein could arrest the G1/S phase in HepG2 mediated by ER stress signaling through the PERK–eIF2α or NF-κB pathway. Further studies are required to explore, preferably, its application in the treatment for HCC.
Materials and methods
The ethics committees of Inner Mongolia Medical University, Air Force General Hospital and Southern Medical University approved our study protocol involving human research. The tissue samples were obtained from the Department of Pathology, Inner Mongolia Medical University, Air Force General Hospital and Southern Medical University. The animal protocols were authorized by the Animal Care and Use Committee in Air Force General Hospital and were implemented based on the approved guidelines.Cell culture
The hepatoma cell line HepG2 was purchased from ATCC (American Type Culture Collection) and stored in liquid nitrogen. At 37 °C, HepG2 in DMEM with 10% FBS (Thermo Fisher Scientific, USA) was cultured under a 5% CO2 atmosphere.Production of recombinant protein and mono-clonal antibody for FAM172A
The cDNA sequence was in accordance to the mRNA of FAM172A obtained from PubMed (NM-032042). The primers (upstream: 5′-GGTACCATGTCTATTTCCTTGAGCTC-3′ and downstream 5′-AAGCTTCAGCTCTTCGTGCTTGATG-3′) were amplified from HepG2 cells and then linked with the pET-32a (+) vector. E. coli cells BL 21 DE 3 were used to express the FAM172A protein and induced with isopropyl β-D-1-thiogalactopyranoside (IPTG) (Sigma-Aldrich, USA). Furthermore, the recombinant protein of FAM172A was purified and then inoculated into mice for the preparation of its mono-colonel antibody.RNA interference of FAM172A
Based on the gene sequence of FAM172A, four miRNA oligonucleotides and one irrelevant sequence were designed and synthesized, which were reconstructed to the GFP-tagged pcDNA™6.2-GW/EmGFPmiR vector with EmGFP and confirmed by sequence analysis. For the primary screening process, a total of 4 × 105 cells in each well of 6-well plates were added to 4 μg of plasmids using Lipofectamine 2000 (Invitrogen, USA). The plates were sealed and incubated for 24 h or 48 h. Then, the transfected cells were separated using a FACS Calibur (Becton Dickinson, USA) flow cytometer based on GFP-tagging. The mRNA of FAM172A was estimated using a RT-PCR method and GAPDH cDNA was used as the control gene. Amplification was achieved using ABI Prism 7500 and the conditions were 95 °C, 10 s; 95 °C, 5 s with 40 cycles and 60 °C, 60 s. The results were analyzed using the ABI7500 system software v2.0.6. The online Roche universal probe library was used to design the oligonucleotide primers. The primers included: FAM172A: upstream: 5′-tcctcactgcttgtgtcgag-3′ and downstream: 5′-tgggctctgggaagaaaac-3′; GAPDH: upstream: 5′-ccagtatgattctacccacggcaa-3′ and downstream: 5′-acagtcttctgagtggcagtgatg-3′.Western blot analysis of FAM172A
The protein expression levels of GRP78, GADD, p-PERK, eIF2α, IRE, XBP1, ATF6, CDK 1, 2, 4, 6, and FAM172A were determined. The proteins were separated using SDS-PAGE gel and then transferred onto the PVDF membrane (Sigma-Aldrich, USA). Furthermore, the membrane was probed with anti-β-actin (sc-81178) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500) (Santa Cruz, USA), anti-FAM172A (1:200), anti-GADD (ab205252), anti-GRp78 (ab140318) (1:500) (Abcam, U.K), anti-p-PERK (sc-377400), anti-eIF2α (sc-365477), anti-IRE (sc-166022), anti-XBP-1s (sc-8015), anti-CHOP (sc-421772) (1:500) (Santa Cruz, USA) and anti-CDK 1, 2, 4, 6 (#9111, 14174, 12790, 13331) (1:500) (CST, USA). Each experiment was repeated three times using a Typhoon imager FLA9500 (General Electric, USA).
:500) (Santa Cruz, USA), anti-FAM172A (1:200), anti-GADD (ab205252), anti-GRp78 (ab140318) (1:500) (Abcam, U.K), anti-p-PERK (sc-377400), anti-eIF2α (sc-365477), anti-IRE (sc-166022), anti-XBP-1s (sc-8015), anti-CHOP (sc-421772) (1:500) (Santa Cruz, USA) and anti-CDK 1, 2, 4, 6 (#9111, 14174, 12790, 13331) (1:500) (CST, USA). Each experiment was repeated three times using a Typhoon imager FLA9500 (General Electric, USA).
Cells count assay and determination of cell cycle
HepG2 cells were co-cultured with the recombinant protein of FAM172A (0 ng mL−1, 0.1 ng mL−1, 1.0 ng mL−1, 10 ng mL−1 and 100 ng mL−1, respectively). Then, the harvested cells were washed with PBS and fixed in 70% ice-cold ethanol at 4 °C overnight. The above cells were resuspended and stained with 7-AAD (Sigma-Aldrich, USA), and then detected using a flow cytometer (BD, USA). Flowjo software was used to analyze the experimental results.Cell proliferation assay
The tetrazolium salt, XTT (Roche, Germany), was used for estimating the cell proliferation. The number of seeding cells was 106 cells per well in each well of 96-well plates and the volume of media before adding the reagents was 2 mL. A 5 mg mL−1 solution of XTT dissolved in PBS was prepared, sterilized, filtered through a 0.2 μM filter and stored at 2–8 °C. Then, 5 h before the end of the incubation, we added 20 μL of the XTT solution into the wells of the 96-well plates containing the cultured cells at 37 °C. The cells were cultured for another 4 h and then 150 μL (or 200 μL) of DMSO was added to each well, pipetted up or down to dissolve the crystals and incubated for 5 min to dissolve air bubbles. The XTT value was measured using a micro-titer plate reader at 450 nm (the absorbance of XTT is at 450 nm).Detection of cell apoptosis
According to protocol,7 the staining with annexin V (BD, USA) was implemented to determine cell apoptosis. Cells (3 × 105) were cultured for 48 h, subsequently washed with PBS and then incubated with an annexin-V solution. The cells were washed again and fixed with 1% paraformaldehyde (Sigma, USA). 7-AAD was used for dual staining of the cells. The total number of 7-AAD + annexin-V double-positive cells was determined using a flow cytometer (BD, USA).8Co-immunoprecipitation
5 μL phenylmethanesulfonyl fluoride (PMSF) (Sigma, USA) was added to the cultured cells and the cells were then lysed. Each supernatant obtained from the cell extraction step was added to 50 μL of protein G plus/protein A Agarose Suspension (Calbiochem, USA). Then, 10 μL of anti-FAM172A or anti-NF-κB antibody was added to the supernatants and rotated gently overnight. Further, 60 μL of protein G plus/protein A Agarose Suspension was added to the samples and rotated gently for 9 h at 4 °C, followed by centrifugation for 5 min. The immunoprecipitates were washed and resuspended with loading buffer before boiling or electrophoresis, then resolved by SDS-PAGE; anti-NF-κB, anti-IκBα and anti-FAM172A antibodies were used for immunoblotting.UPLC-ESI-MS/MSMS analysis
In this study, the proteins, co-immunoprecipitated with FAM172A, were prepared as described above. After digesting with trypsin, the peptides were separated using an analytical column (BEH 130 C18, Waters) and then analyzed using nanoAcquity™ UPLC™/ESI/MS (Micromass Q-Tof Micro, Waters Co. UK). Then, the sample was dissolved in water/formic acid and injected into the above column with linear gradient. After desalting, the samples were determined using a chromatography method performed on a nanoAcquity™ UPLC™. Efficient MS data were obtained for analysis from the MS to MS/MS and the positive peptides were identified using a public database library.Tumor xenograft assay
BALB/c nude mice (4–6 weeks old) were purchased from Beijing Experimental Animal Center; they were injected with HepG2 cells subcutaneously and housed in the Animal Center.9 The L (length) and W (width) of the tumors were measured with a sliding caliper and used to determine the tumor volume using the formula: L × W2 × 0.5. The mice were euthanized after 15 days of treatment with the FAM172A recombinant protein (10 mg kg−1, fourth injection each three days) in the tail vein or subcutaneous injection of tumor cells. After the mice were euthanized, each tumor was weighed and the tumor index was calculated (tumor index = weight of tumor/weight of mice × 1000).Statistical analysis
The data were displayed as the mean ± SD. Moreover, each experiment was replicated at least 6 times. The significance differences between data were estimated using a two-tailed Student's t test or one-way ANOVA analysis. P < 0.05 was considered significant.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 30600524 and 81341067), Special Project of Beijing Health Development and Scientific Research, China (No. 2011-5041-02), Key Projects in the PLA's logistics Program during the Twelfth Five-year Plan Period (No. BWS11J029), Seventh Special Financial Grant from China Postdoctoral Science Foundation (No. 2014T71010) and 54th General Financial Grant from China Postdoctoral Science Foundation (No. 2013M542539). This work was supported by grants from the National Natural Science Foundation of Guangdong Province, China (No. 2017A030313510) and Introduction of Talent Fund of Guangdong Second Provincial General Hospital (No. YY2016-006). The study sponsors had no involvement in the work.References
- C. Herold, T. Reck, P. Fischler, R. Ott, M. Radespiel-Troeger, M. Ganslmayer, W. Hohenberger, E. G. Hahn and D. Schuppan, Prognosis of a large cohort of patients with hepatocellular carcinoma in a single European centre, Liver, 2002, 22, 23–28 CrossRef PubMed.
- K. Okuda, Hepatocellular carcinoma, J. Hepatol., 2000, 32, 225–237 CrossRef CAS PubMed.
- K. N. Bhalla, Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies, J. Clin. Oncol., 2005, 23, 3971–3993 CrossRef CAS PubMed.
- J. K. Dowman, L. J. Hopkins, G. M. Reynolds, N. Nikolaou, M. J. Armstrong, J. C. Shaw, D. D. Houlihan, P. F. Lalor, J. W. Tomlinson, S. G. Hübscher and P. N. Newsome, Development of Hepatocellular Carcinoma in a Murine Model of Nonalcoholic Steatohepatitis Induced by Use of a High-Fat/Fructose Diet and Sedentary Lifestyle, Am. J. Pathol., 2014, 184, 1550–1561 CrossRef CAS PubMed.
- A. Blas-García, N. Apostolova, V. Valls-Bellés and J. V. Esplugues, Endoplasmic reticulum and mitochondria: independent roles and crosstalk in fatty liver diseases and hepatic inflammation, Curr. Pharm. Des., 2016, 22, 2607–2618 CrossRef.
- Z. Feng, H. Li, S. Liu, J. Cheng, G. Xiang and J. Zhang, FAM172A induces S phase arrest of HepG2 cells via Notch 3, Oncol. Rep., 2013, 29, 1154–1160 CrossRef CAS PubMed.
- J. Zhang, Q. Sun, J. Bo, R. Huang, M. Zhang, Z. Xia, L. Ju and G. Xiang, Single-walled carbon nanohorn (SWNH) aggregates inhibited proliferation of human liver cell lines and promoted apoptosis, especially for hepatoma cell lines, Int. J. Nanomed., 2014, 9, 759–773 CrossRef PubMed.
- P. Wu, Y. Tang, J. He, L. Qi, W. Jiang and S. Zhao, ARC is highly expressed in nasopharyngeal carcinoma and confers X-radiation and cisplatin resistance, Oncol. Rep., 2013, 30, 1807–1813 CrossRef CAS PubMed.
- W. J. Wang, Y. Yao, L. L. Jiang, T. H. Hu, J. Q. Ma, Z. J. Liao, J. T. Yao, D. F. Li, S. H. Wang and K. J. Nan, Knockdown of lymphoid enhancer factor 1 inhibits colon cancer progression in vitro and in vivo, PLoS One, 2013, 8, e76596 CAS.
- A. D. Elbein, Inhibitors of the biosynthesis and processing of N-linked oligosaccharide chains, Annu. Rev. Biochem., 1987, 56, 497–534 CrossRef CAS PubMed.
- Y. Misumi, K. Miki, A. Takatsuki, G. Tamura and Y. Ikehara, Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes, J. Biol. Chem., 1986, 261, 11398–11403 CAS.
- W. C. Lin, Y. C. Chuang, Y. S. Chang, M. D. Lai, Y. N. Teng and I. J. Su, Endoplasmic Reticulum Stress Stimulates p53 Expression through NF-κB Activation, PLoS One, 2012, 7, e39120 CAS.
- K. Haze, H. Yoshida, H. Yanagi, T. Yura and K. Mori, Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress, Mol. Biol. Cell, 1999, 10, 3787–3799 CrossRef CAS PubMed.
- J. Ye, R. B. Rawson, R. Komuro, X. Chen, U. P. Davé, R. Prywes, M. S. Brown and J. L. Goldstein, ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs, Mol. Cell, 2000, 6, 1355–1364 CrossRef CAS PubMed.
- H. Yoshida, T. Matsui, A. Yamamoto, T. Okada and K. Mori, BP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor, Cell, 2001, 107, 881–891 CrossRef CAS PubMed.
- B. C. Mak, Q. Wang, C. Laschinger, W. Lee, D. Ron and H. P. Harding, et al., Novel function of PERK as a mediator of force-induced apoptosis, J. Biol. Chem., 2008, 283, 23462–23472 CrossRef CAS PubMed.
- A. H. Lee, N. N. Iwakoshi and L. H. Glimcher, XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response, Mol. Cell. Biol., 2003, 23, 7448–7459 CrossRef CAS PubMed.
- S. Ghosh and M. Karin, Missing pieces in the NF-kappaB puzzle, Cell, 2002, 109, S81–S96 CrossRef CAS PubMed.
- T. Nakagawa and J. Yuan, Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis, J. Cell Biol., 2000, 150, 887–894 CrossRef CAS PubMed.
- S. J. Marciniak, C. Y. Yun, S. Oyadomari, I. Novoa, Y. Zhang, R. Jungreis, K. Nagata, H. P. Harding and D. Ron, CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum, Genes Dev., 2000, 18, 3066–3077 CrossRef PubMed.
- F. Urano, X. Wang, A. Bertolotti, Y. Zhang, P. Chung, H. P. Harding and D. Ron, Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1, Science, 2000, 287, 664–666 CrossRef CAS PubMed.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |