DOI:
10.1039/C7RA09975D
(Paper)
RSC Adv., 2017,
7, 54603-54609
Poly(ethylene glycol) modified Mn2+ complexes as contrast agents with a prolonged observation window in rat MRA†
Received
7th September 2017
, Accepted 20th November 2017
First published on 28th November 2017
Abstract
A novel rigid Mn2+ complex (MnL) with pyridine and pyrrolidine rings was designed and synthesized. The complex was further modified with alkyne, and conjugated to azide-terminated PEG by click chemistry. PEGylated MnL shows considerably higher relaxivity (MnL-PEG2k-MnL: r1 = 6.38 mmol−1 s−1; MnL-PEG4.6k-MnL: r1 = 5.63 mmol−1 s−1) and much longer blood circulation time than free Mn2+ complexes (MnL: r1 = 3.73 mmol−1 s−1), providing a prolonged time window to acquire longitudinal images for rat contrast enhanced magnetic resonance angiography.
Introduction
Manganese(II) based complexes have been studied as MRI contrast agents for many years, because of their excellent magnetic properties including five unpaired electrons and long electronic relaxation time, which can offer good T1 relaxation enhancement in MRI.1–3 Meanwhile, manganese is also a natural cellular constituent, acts as a cofactor for enzymes and receptors in cell biology, and has its own metabolic pathways in vivo.3,4 Compared to gadolinium complex based contrast agents, manganese complexes can be good alternatives in terms of biosafety, and possibly having lower chances in developing nephrogenic systemic fibrosis (NSF) in patients with kidney dysfunctions.5–7
Chelation ligands are crucial for the stabilization of metal ions and enhancement of relaxivity of manganese complexes.8–10 In our previous study, aza-semi-crown pentadentate Mn2+ complexes rigidified with pyridine and piperidine rings were developed.11 Several other Mn2+ complexes based on pentagonal picolinate ligands were also reported recently.12,13 The special rigid structure has shown a relatively higher T1 relaxivity (3.6 Mn mmol−1 s−1, 1.5 T, 20 °C) comparing to commercial agent Teslascan (Mn-DPDP, 2.1 Mn mmol−1 s−1, 1.5 T, 20 °C). However, the time window for contrast enhanced magnetic resonance angiography (MRA) is usually short because of the fast kidney clearance. In many clinical cases, longer MRI observation window can provide more information to the radiologists for better analysis. In order to overcome the limitation, conjugation of small molecule contrast agents to various frameworks such as micelles, liposomes, dendrimers, proteins, bioactive peptides were chosen.14–18 However, complicacy of nano-systems, higher cost of fabrication, and potential toxicity slowed down their clinical translation process.
Polymeric conjugation is a feasible method to prepare macromolecular contrast agents. As a water-soluble, non-antigenic and biocompatible polymer, poly(ethylene glycol) (PEG) has been approved by the Food and Drug Administration (FDA) for human dermal, oral and intravenous applications.19–23 For instance, PEG conjugated interferon (Pegasys®, Roche) provides long circulation time and less side effects than free interferons.24,25 PEGylated small molecule contrast agents can slow down kidney clearance rate while have less RES uptake comparing to nanoparticles, presenting a more feasible solution for enhanced MRA imaging.26,27
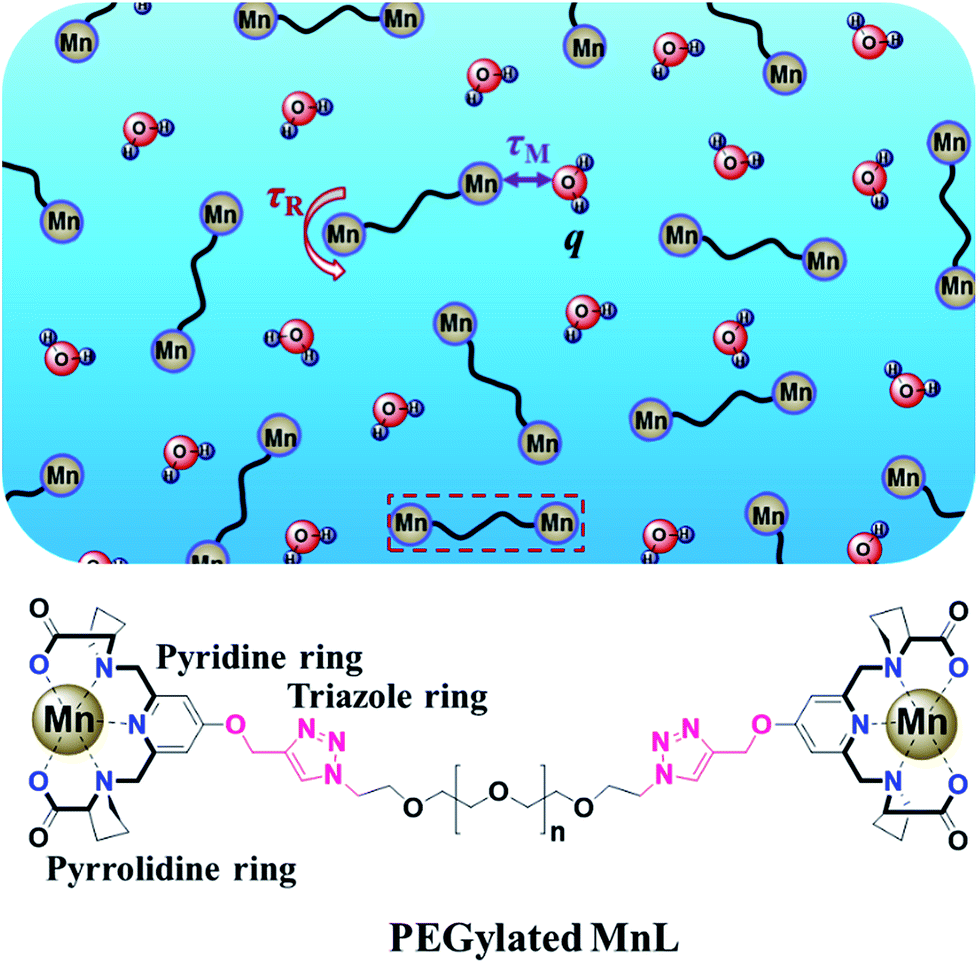
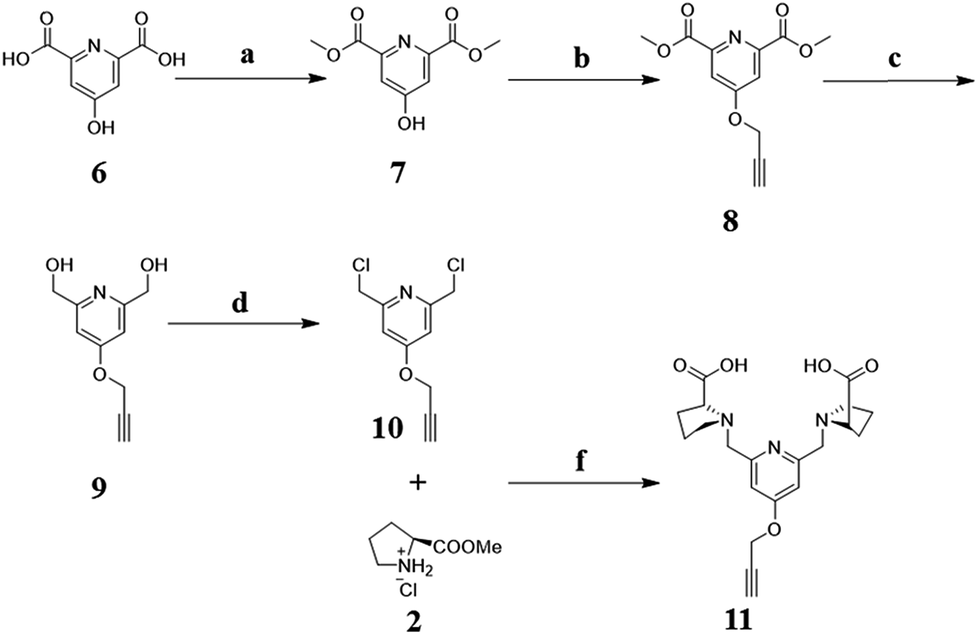
Herein, PEGylated Mn2+ complexes containing rigid rings were designed (Fig. 1). We first designed and synthesized a new rigid Mn2+ complex (MnL) with pyridine and pyrrolidine rings. Next, the complex was modified with alkynyl group and reacted with azido-terminated PEG. Finally, the relaxivity and rat contrast enhanced MRA of PEGylated MnL and free MnL were studied under clinical MRI scanners.
 |
| | Fig. 1 Scheme of PEGylated Mn2+ complex. Mn2+ was chelated by ligand with rigid pyridine and pyrrolidine rings, and linked PEG by rigid triazole ring. These rigid structures and PEG chain are quite important to hinder the local rotation of Mn2+ complexes (Prolong τR). | |
Results and discussion
Characterization of rigid Mn2+ complex (MnL)
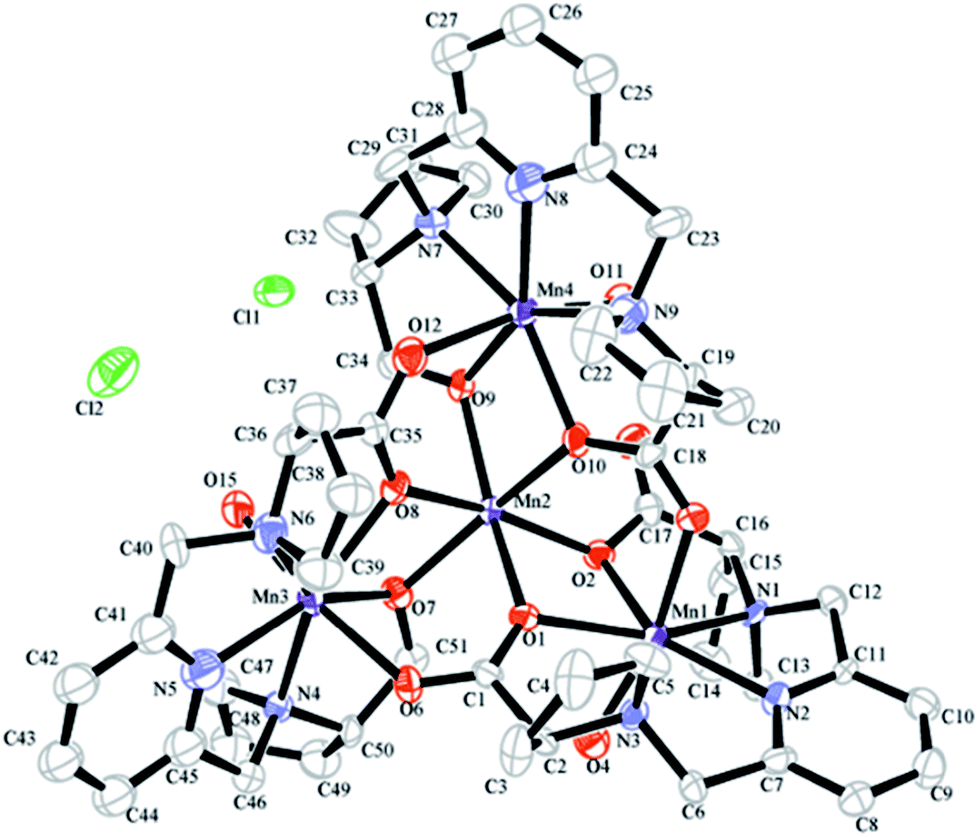
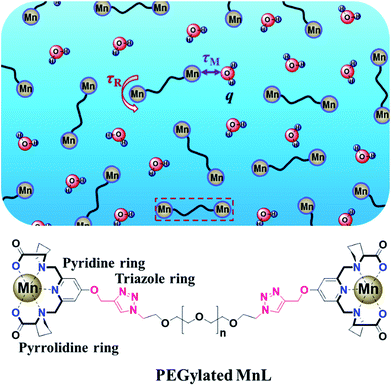
MnL complex was identified by mass spectrometry. MnL used to obtain single crystals were prepared in MilliQ water directly mixing of ligand and Mn2+ at alkaline condition. The product has not been further purified and was used to single crystal growth by slow vapor diffusion. Crystal structure of MnL is illustrated in Fig. 2. Two kinds of Mn2+ coordination number states were both found (coordination number 6 and 7) in one unit cell. Three Mn2+ ions are heptadentated at the cavity center of ligands in the same way. Five-coordinate atoms (a pyridine nitrogen atom, two tertiary amine nitrogen and carboxyl oxygen atoms) of one ligand and Mn2+ ion almost locate in the same plane, and one coordinate water molecule located on apical positions of this plane to form the pentagonal-bipyramidal coordination sphere. This coordination state is similar with previous reports.11 It's interesting that one Mn2+ is hexadentated with six carboxyl oxygen atoms of three ligands (two carboxyl oxygen atoms per ligand), forming a standard octahedral coordination sphere. This shows that there are free Mn2+ in our obtained manganese complexes. In the following experiments, MnL was further purified through Sephadex LH-20 column. But unfortunately, the corresponding single crystals were not obtained. So, we think free Mn2+ in MnL might be beneficial to the formation and growth of single crystal. Similar result was also discovered in our previous study.11 The crystals' structures informed us Mn2+ ions at the cavity center of ligands are heptadentated, and one coordinate water molecule was observed. In the following experiments, MnL was purified through Sephadex LH-20 column to ensure no free Mn2+.
 |
| | Fig. 2 Molecular structure of MnL found in the structure of [Mn4L3(H2O)3Cl2]. All hydrogen atoms, noncoordinated solvent molecules have been removed for clarity. The thermal ellipsoids are drawn with 50% probability. | |
Synthesis and characterization of L-PEG-L
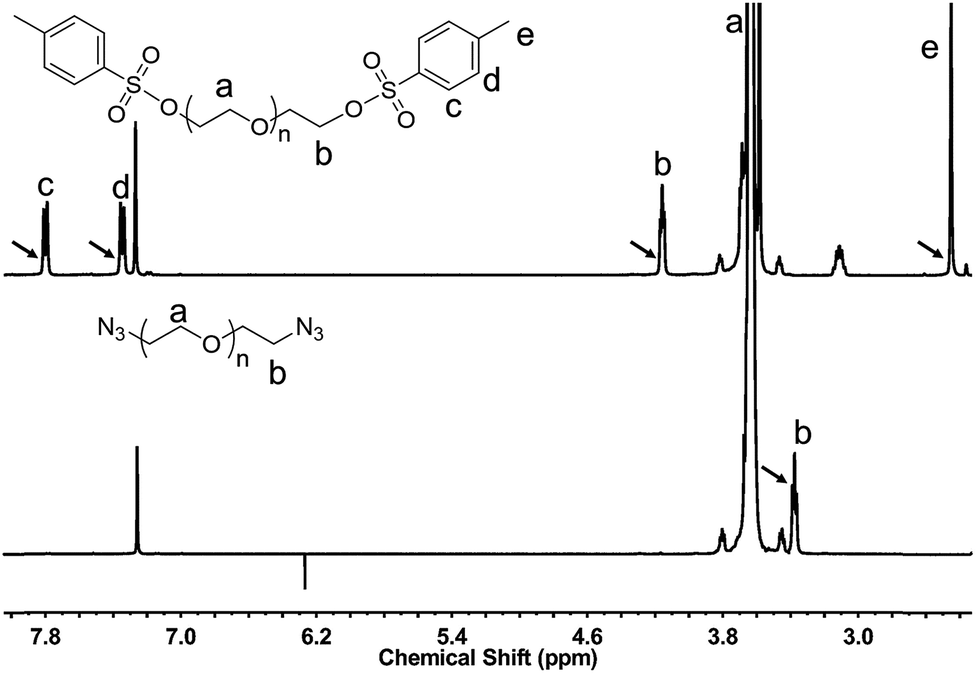
Synthesis of N3-PEG-N3. Hydroxyl-terminated PEG are commercially available in a variety of molecular weights with narrow dispersity (Mw/Mn ≤ 1.1). For covalent binding with other substrates, terminal hydroxyl group of PEG must be functionalized. In this work, azido-terminated PEG, N3-PEG-N3, was prepared using two steps modification.28 Hydroxyl-terminated PEG with different molecular weights (2 kDa and 4.6 kDa) were used. Tosylation of PEG diols is considered the most critical step because the conversion yield are involved in final replacement rate of azide. In order to achieve high conversion rate, p-toluenesulfonyl chloride with five equivalents to hydroxyl was added, and a long reaction time of 24 hours was kept. Results show that the degree of substitution were approximately 92% (PEG2k) and 91% (PEG4.6k) respectively, as calculated from the integral values of PEG (δ 3.9–3.4) and the methylene protons adjacent to the tosyl group (δ 4.15). Then, azido reaction carried out in DMF at 60 °C, and identified by disappearing of characteristic peaks of the tosyl group in 1H NMR spectrum (Fig. 3).
 |
| | Fig. 3 1H NMR spectrum of Ts-PEG-Ts and N3-PEG-N3 in CDCl3. | |
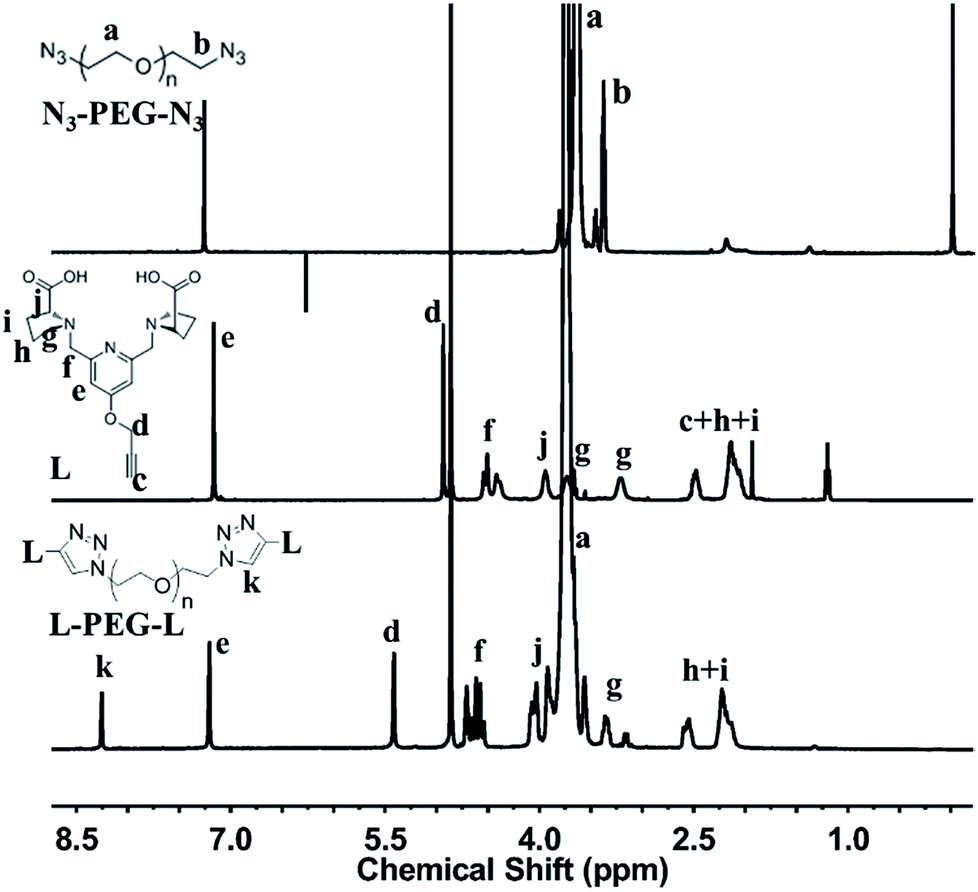
Synthesis of L-PEG-L. The click chemistry of L-Alkynyl and N3-PEG-N3 was carried out in water. The resulted solution was dialyzed 2 days against water (MWCO 1 kDa membrane) to remove free ligands and then lyophilized. Fig. 4 shows the 1H NMR spectra of N3-PEG-N3, L-Alkynyl and L-PEG-L, simultaneously. The characteristic peaks of PEG (a), ligand (d–j) and triazole ring linkage (k) appeared together in 1H NMR spectrum of L-PEG-L, and used to calculate grafting efficiency respectively (list in Table 1). The grafting ratio was calculated as the amount of ligand per PEG molecule. As a result, relatively higher ligand grafting rates are shown in NMR. However, lower molecular weight PEG2k has higher grafting rate (an average grafting ratio of 1.33) than that of PEG4.6k (an average grafting ratio of 1.10), depicting that the grafting efficiency was reduced with the hindering effect of longer PEG chains.
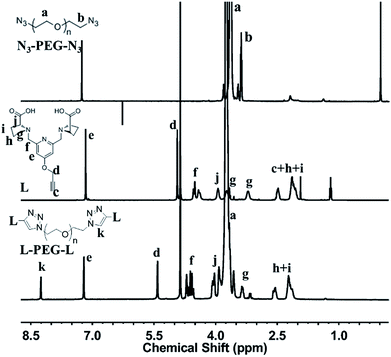 |
| | Fig. 4 1H NMR spectra and characteristic peaks assignment of N3-PEG-N3, L-Alkynyl and L-PEG-L. | |
Table 1 Grafting efficiency based on different characteristic peaks
| Characteristic peaks in 1H-NMR |
Grafting ratio (per PEG molecule) |
| PEG2k |
PEG4.6k |
| Peak a (PEG) |
Peak k |
1.29 |
0.96 |
| Peak d |
1.39 |
1.16 |
| Peak e |
1.27 |
1.06 |
| Peak f |
1.29 |
0.97 |
| Peak j |
1.34 |
1.23 |
| Peak h and i |
1.38 |
1.20 |
T1 relaxivity studies in vitro
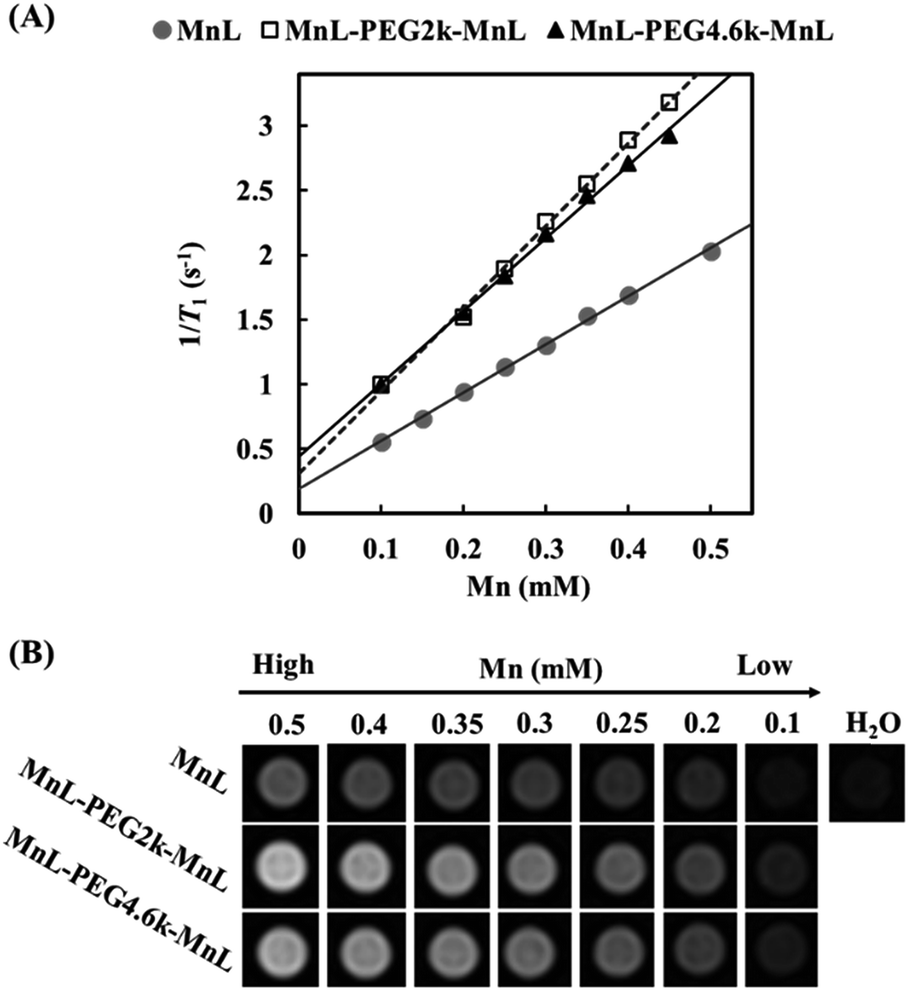
In order to study the relaxation property changes of manganese complexes before and after PEGylation, we simultaneously tested relaxivities of MnL, MnL-PEG2k-MnL and MnL-PEG4.6k-MnL under a 1.5 T clinical MRI scanner. Fig. 5A displays the T1 relaxation rates at different Mn concentrations. Three curve slopes represent the longitudinal relaxivities (r1) of MnL, MnL-PEG2k-MnL and MnL-PEG4.6k-MnL respectively. The relaxivity of free MnL complexes is 3.73 mmol−1 s−1. As expected, PEGylated manganese complexes show a higher relaxivities (MnL-PEG2k-MnL: r1 = 6.38 mmol−1 s−1; MnL-PEG4.6k-MnL: r1 = 5.63 mmol−1 s−1) than free MnL complexes. One contributing factor is the rigid rings and PEG chain, which usually results in a longer correlation time (τR).15 However, longer PEG chain with faster local motion does not lead to higher relaxivity, and it might be on account of the molecular structure of PEG, which is a flexible thread-like molecule.29 The similar result was also reported in the studies on PEGylated GdDOTA chelates.30 T1-weighted images of water at different concentration of MnL, MnL-PEG2k-MnL and MnL-PEG4.6k-MnL (from 0.5 to 0.1 mmol) were studied in Fig. 5B, water solutions with PEGylated MnL shown obviously higher signal than water solutions with free MnL contrast group in the same manganese concentrations.
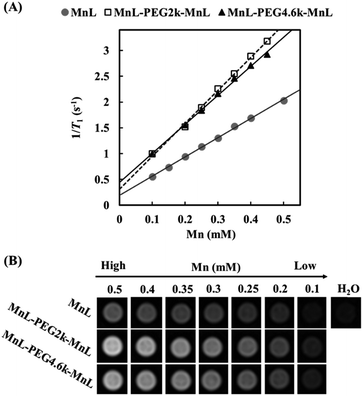 |
| | Fig. 5 (A) T1 relaxation rate as a function of Mn concentration. (B) T1-weighted images of water at different concentration of MnL, MnL-PEG2k-MnL and MnL-PEG4.6k-MnL (1.5 T, SE sequence, TE = 5.3 ms, TR = 70 ms, slice thickness = 3 mm, field of view = 292 × 146 mm, flip angle = 90°). | |
In vivo MRA studies
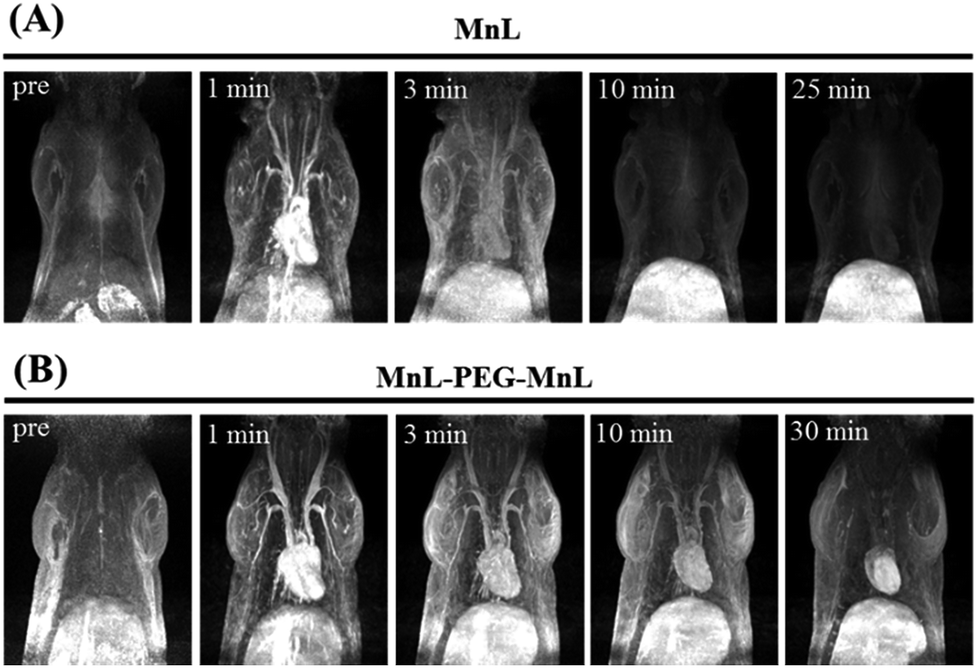
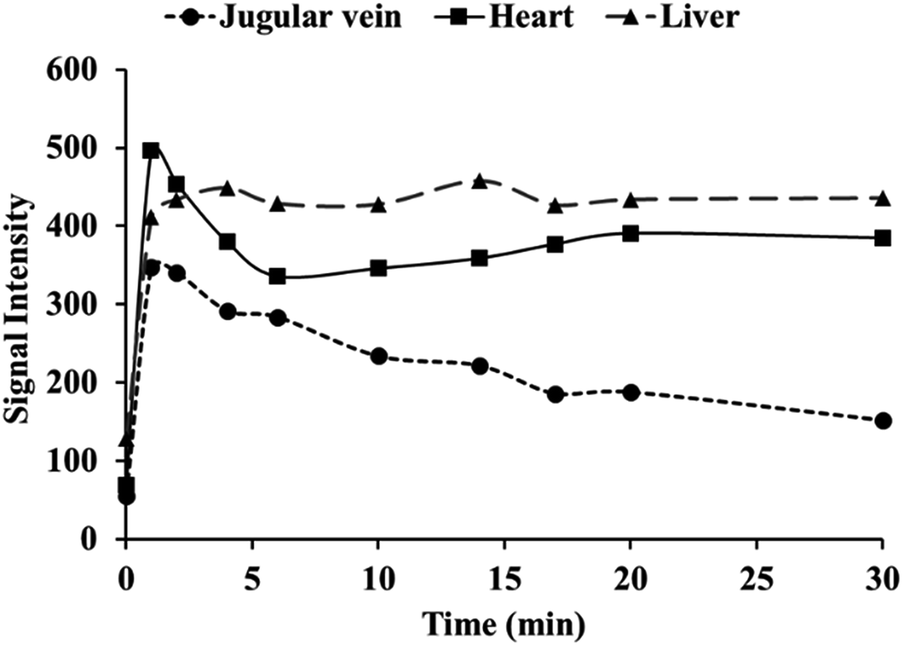
Contrast enhanced MRA is an important clinical tool to detect blood diseases, such as myocardial infarction, atherosclerotic plaque, and tumor angiogenesis.31,32 It is quite challenging to obtain sequential high-resolution blood vessel imaging in MRA using small molecule contrast agents due to low relaxivity and short plasma half-life. In this work, contrast enhanced MRA study of SD rats was carried out on a clinical 3.0 T MR scanner to evaluate vascular contrast enhancement effect of free MnL and PEGylated MnL. As shown in Fig. 6, blood vessel images of the chest and neck regions of rats were obtained after intravenous injection of free MnL and MnL-PEG4.6k-MnL at a dosage of 0.25 Mn mmol per kg body weight. The vascular imaging window of PEGylated MnL group reached 10 minutes, was obviously longer than that of the free MnL group (3 minutes of vascular imaging window). The results stated that PEGylation of small molecule agents are beneficial for prolonged plasma half-life, and probably due to lower kidney clearance rates. In Fig. 7, MRI signal changes in jugular vein, heart and liver at different time points (in 30 minutes) were quantitatively studied. After administration with MnL-PEG4.6k-MnL, signal intensity reached the peak at the same time (in 1 minute) for all organs, but presenting different variation tendency. The signal intensity in jugular vein is decreasing with time, while it is relatively constant in liver, and reduced to certain value and remaining constant for the heart. The results show that Mn concentration in blood vessel is gradually reducing, and there are certain uptake of agents in heart and liver. This phenomenon follows the metabolic pathway of manganese, uptake by mitochondria rich organs.2,33
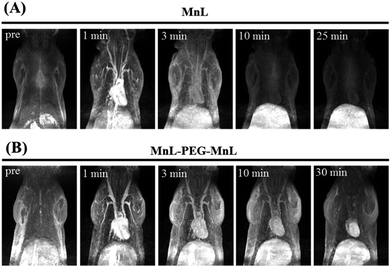 |
| | Fig. 6 Contrast enhancement MRA study of SD rat at a clinical 3.0 T scanner. Dosage: 0.25 Mn mmol kg−1 Bw of MnL (A) and MnL-PEG4.6k-MnL (B). | |
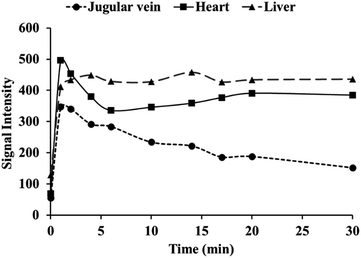 |
| | Fig. 7 Quantification of MRI signal changes in jugular vein, heart and liver at different time points after administration with MnL-PEG4.6k-MnL (dosage: 0.25 Mn mmol kg−1 Bw). | |
Experimental
Synthesis and characterization of rigid Mn2+ complexes (MnL)
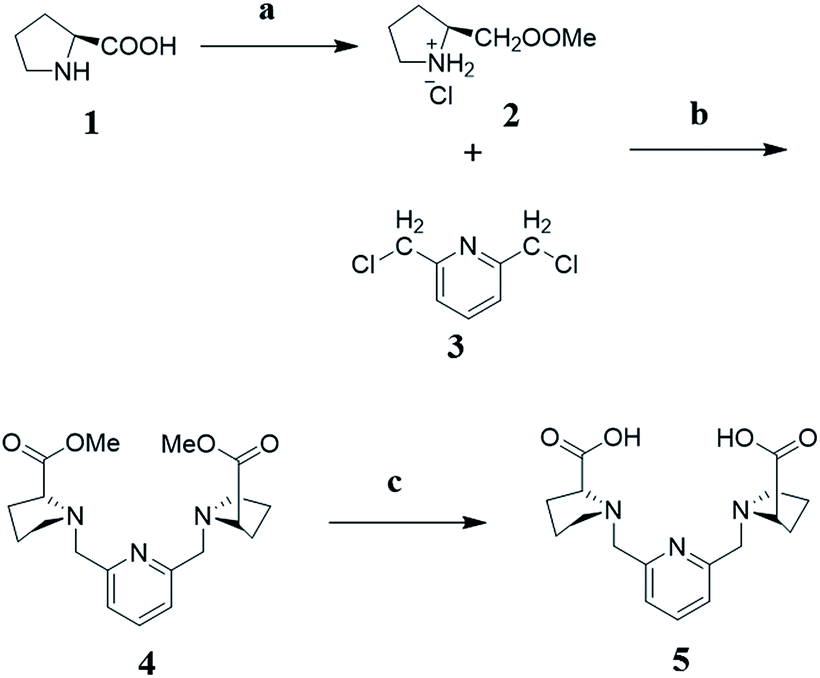
Synthesis of methyl pyrrolidine-2-carboxylate hydrochloride (2). L-Proline (1) (10 g, 90 mmol) was dissolved in 95 mL methanol. Then, thionyl chloride (8.5 mL) was dropped in at 0 °C in 5 min. The solution was heated to reflux for 1 h. The solvent was evaporated off to afford compound 2 without further purification. Yield: 14.8 g (100%). 1H NMR (400 MHz, D2O) δ 4.54 (s, 1H), 3.89 (s, 3H), 3.46 (s, 2H), 2.58–2.40 (m, 1H), 2.32–2.17 (m, 1H), 2.17–2.03 (m, 2H).
Synthesis of (2S, 2′S)-dimethyl-1,1′-(pyridine-2,6-diylbis(methylene))dipyrrolidine-2-carboxylate (4). Compound 2 (2.65 g, 16 mmol) and 2,6-bis (chloromethyl)pyridine (3) (1.4 g, 8 mmol) were dissolved in 30 mL acetonitrile, K2CO3 (11 g, 80 mmol) and KI (30 mg, 0.2 mmol) were added in. The mixture stirred at 45 °C for 12 h. Filtered inorganic salts from solution, acetonitrile was evaporated off. The product was purified by silica gel column chromatography (methylene dichloride/methanol = 20/1). Yield: 2.2 g (76%). 1H NMR (400 MHz, CDCl3) δ 7.62 (t, J = 7.7 Hz, 1H), 7.34 (d, J = 7.6 Hz, 2H), 4.02 (d, J = 13.7 Hz, 2H), 3.78 (d, J = 13.7 Hz, 2H), 3.66 (s, 6H), 3.43 (t, J = 13.5 Hz, 2H), 3.10 (d, J = 7.0 Hz, 2H), 2.62–2.45 (m, 2H), 2.22–2.07 (m, 2H), 2.03–1.87 (m, 4H), 1.87–1.74 (m, 2H). 13C NMR (400 MHz, CDCl3) δ 174.61 (s), 158.02 (s), 136.80 (s), 121.54 (s), 65.24 (s), 60.11 (s), 53.39 (s), 51.69 (s), 29.37 (s), 23.31 (s). HRMS (ESI): m/z = 362.1823 (M + H+), 384.1761 (M + Na+).
Synthesis of the ligand (5). Compound 4 (700 mg, 2.0 mmol) was dissolved in methanol (20 mL), NaOH (320 mg, 8 mmol) and 2 mL MilliQ water was added. The solution was heated to 45 °C and kept for 2 h. Evaporated off all the solvents to obtain light-yellow solid, which was dissolved in water and then acidified with concentrated HCl (37%, 1.0 mL). The water was evaporated off, ethanol added to dissolve the crude product. Filtered resulting the salts, ethanol was evaporated off to get the product. Yield: 390 mg (99%). 1H NMR (400 MHz, DMSO) δ 7.81 (t, J = 7.7 Hz, 1H), 7.40 (d, J = 7.7 Hz, 2H), 4.17 (d, J = 14.0 Hz, 2H), 3.95 (d, J = 14.0 Hz, 2H), 3.50 (dd, J = 9.0, 5.2 Hz, 2H), 3.23–3.12 (m, 2H), 2.74–2.60 (m, 2H), 2.11 (dq, J = 12.3, 8.5 Hz, 2H), 1.88 (tt, J = 12.8, 4.1 Hz, 2H), 1.83–1.65 (m, 4H). 13C NMR (400 MHz, DMSO) δ 171.84 (s), 155.24–153.24 (m), 137.93 (s), 122.94 (s), 66.26 (s), 58.15 (s), 53.75 (s), 28.50 (s), 22.84 (s). MS-ESI: m/z = 332.11 (M − H+), 368.09 (M + Cl−) (Fig. 8).
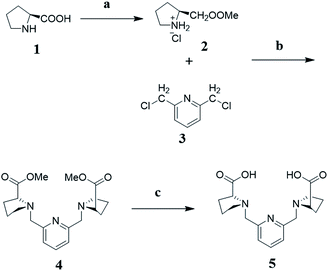 |
| | Fig. 8 Synthesis scheme of rigid ligand (L). (a) SOCl2, MeOH, reflux for 1 h; (b) KI, K2CO3, CH3CN, 45 °C, 12 h; (c) NaOH, H2O/MeOH, 45 °C, 2 h. | |
Preparation of Mn2+ complexes (MnL). Ligand (5) (180 mg, 0.55 mmol) was dissolved in MilliQ water (12 mL) and the pH of the solution was adjusted to 6 by adding NaOH solution (0.2 M). Manganese chloride tetrahydrate (100 mg, 0.5 mmol) was added under nitrogen protection. The resulting solution was stirred at room temperature for 4 h, and then its pH was readjusted to 7 by addition of NaOH (0.2 M aq.). The water was removed by rotary evaporation and the resulting solid was suspended in ethanol. The insoluble salts were removed by filtration and the filtrate was evaporated. The product was obtained as a colorless solid. HRMS-ESI: m/z = 385.0835 (M − H+), 421.0371 (M + Cl−). Single crystals of MnL suitable for X-ray diffraction analysis were obtained by slow vapor diffusion of acetone into water solution (MnL concentration: 10 mg mL−1). MnL complexes in the following experiments were further purified through Sephadex LH-20 column before use.
Synthesis of the ligand with alkynyl group
Synthesis of dimethyl 4-hydroxypyridine-2,6-dicarboxylate (7). Chelidamic acid 6 (10.0 g, 54.6 mmol) was suspended in 150 mL absolute methanol (MeOH). Then, 2,2-dimethoxypropane (DMP, 60 mL, 75 mmol) and concentrated HCl (7.5 mL, 75 mmol) were slowly added in with magnetic stirring. The result mixture was heated to reflux for 4 h under a CaCl2 drying tube. Following, cooling to room temperature and evaporating off the solvents. Anhydrous ether (100 mL) was added in to stir, the insoluble hydrochloride (compound 7) was filtered to be collected (11.9 g, yield 88%). 1H NMR (400 MHz, DMSO) δ 7.65 (s, 2H), 3.89 (s, 6H).
Synthesis of dimethyl 4-(prop-2-yn-1-yloxy)pyridine-2,6-dicarboxylate (8). Compound 7 (5.0 g, 20 mmol) was dissolved in 150 mL acetonitrile, anhydrous K2CO3 (27.6 g, 200 mmol) was added in. The solution was stirred for 30 min at room temperature. Then, 3-bromo-1-propyne solution in toluene (3.4 mL, 30 mmol) was added, and heated to reflux for 12 h. Filtered the insoluble from solution, acetonitrile was evaporated off to obtain crude product. 50 mL chloroform added to dissolve the product, washed three times with 20 mL water in a separatory funnel. Chloroform layer was dried with anhydrous magnesium sulfate. Filtered and evaporated off chloroform to get the product 8 (4.5 g, 90%). 1H NMR (400 MHz, CDCl3) δ 7.91 (s, 2H), 4.88 (d, J = 1.4 Hz, 2H), 4.02 (s, 6H), 2.63 (s, 1H).
Synthesis of (4-(prop-2-yn-1-yloxy)pyridine-2,6-diyl)dimethanol (9). Compound 8 (4.5 g, 18 mmol) was dissolved in 80 mL ethanol, NaBH4 (2.7 g, 72 mmol) was added in slowly at 0 °C. The reaction mixture was stirred at room temperature for 2 h and heated to reflux for another 5 h. The crude mixture was concentrated under vacuum and 100 mL saturated aqueous solution of potassium carbonate was added. After stirred at 60 °C for 2 h, products were extracted out with chloroform (100 mL, three times). The organic phase dried over sodium sulfate and concentrated. Compound 9 was obtained as a white solid; 2.8 g (80%). 1H NMR (400 MHz, DMSO) δ 6.92 (s, 2H), 5.41 (s, 2H), 4.90 (d, J = 2.4 Hz, 2H), 4.48 (s, 4H), 3.66 (t, J = 2.4 Hz, 1H).
Synthesis of 2,6-bis (chloromethyl)-4-(prop-2-yn-1-yloxy)pyridine (10). Compound 9 (2.5 g, 13 mmol) was slowly added in 20 mL of SOCl2 at 0 °C. The reaction mixture was stirred at room temperature for 1 h and heated to reflux for another 2 h. The crude mixture was removed solvent under vacuum and 20 mL of H2O was added. The solution was filtrated and saturated aqueous solution of sodium bicarbonate was added in drops into the filtrate. The precipitate was isolated by filtration to afford compound 10; 2.4 g, (80%). 1H NMR (400 MHz, CDCl3) δ 7.05 (s, 2H), 4.81 (d, J = 2.4 Hz, 2H), 4.64 (s, 4H), 2.61 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 166.49 (s), 157.20 (s), 109.51 (s), 77.59 (s), 77.23 (s), 56.34 (s), 44.87 (s). ESI-MS: m/z = 230.01 (M + H+).
Synthesis and characterization of ligand with alkynyl group (L-Alkynyl, 11). Compound 2 (2.65 g, 16 mmol) and compound 10 (2.1 g, 8 mmol) were dissolved in 30 mL dry acetonitrile, K2CO3 (11 g, 80 mmol) and KI (30 mg) were added. The mixture was stirred at 45 °C for 12 h. Filtered inorganic salt from solution, solvent was evaporated off. The product was purified by silica gel column chromatography (methylene dichloride/ethyl acetate = 2/1). 1H NMR (400 MHz, CDCl3) δ 6.98 (s, 2H), 4.75 (d, J = 1.6 Hz, 2H), 3.97 (t, J = 15.7 Hz, 2H), 3.76 (d, J = 14.2 Hz, 2H), 3.67 (s, 6H), 3.49–3.37 (m, 2H), 3.18–3.04 (m, 2H), 2.54 (q, J = 8.2 Hz, 3H), 2.25–1.70 (m, 8H). Then, the product (830 mg, 2 mmol) was dissolved in 20 mL methanol, NaOH (320 mg, 8 mmol) was dissolved in 2 mL MilliQ water and added in. Stirring at 40 °C for 5 h, methanol and water was evaporated off. The light-yellow solid was dissolved in 5 mL water, added concentrated HCl to adjust pH to 6.5, and water was evaporated off. Ethanol added to dissolve the product, filtered resulting salt. Solvent was evaporated off to get the ligand (765 mg, 99%). 1H NMR (400 MHz, D2O) δ 7.16 (s, 2H), 4.93 (s, 2H), 4.52 (d, J = 13.8 Hz, 2H), 4.40 (d, J = 12.4 Hz, 2H), 3.94 (m, 2H), 3.82–3.63 (m, 2H), 3.21 (m, 2H), 2.58–2.39 (m, 2H), 2.26–1.96 (m, 7H). 13C NMR (101 MHz, D2O) δ 165.21 (s), 141.91 (s), 129.68 (s), 110.63 (s), 77.70 (s), 76.77 (s), 69.07 (s), 58.89 (s), 57.38 (s), 56.13 (s), 28.96 (s), 22.61 (s). HRMS (ESI): m/z = 410.1647 (M + Na+) (Fig. 9).
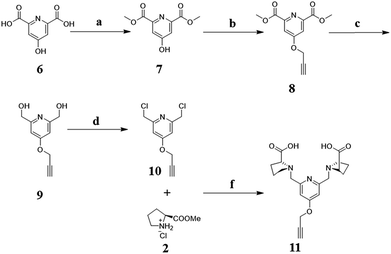 |
| | Fig. 9 Synthesis scheme of the ligand with alkynyl group. (a) HCl, DMP, MeOH, reflux for 4 h; (b) C3H3Br, K2CO3, CH3CN, reflux for 12 h; (c) NaBH4, EtOH, reflux for 5 h; (d) SOCl2, reflux for 2 h; (e) (i) KI, K2CO3, CH3CN, 45 °C, 12 h; (ii) NaOH, H2O, MeOH, 40 °C, 5 h. | |
Synthesis of PEGylated Mn2+ complex (MnL-PEG-MnL)
Synthesis of PEG with terminal azide group (N3-PEG-N3). PEG2k (Mw 2000 Da) or PEG4.6k (Mw 4600 Da) (1 mmol) and triethylamine (1 mL) were dissolved in 100 mL of methylene chloride. P-Toluenesulfonyl chloride (0.95 g, 5 mmol) dissolved in 20 mL methylene chloride was added in slowly at 0 °C. The solution was stirred at room temperature for 24 h. The mixture was washed three times with 50 mL water in a separatory funnel. Methylene chloride layer was dried with anhydrous magnesium sulfate and concentrated to 30 mL. The products were precipitated in cold diethyl ether, and dried in vacuo to give a white powder. Then, the product was dissolved in 50 mL DMF, NaN3 (325 mg) was added in slowly. The mixture was heat to 60 °C and stirred for 24 hours. DMF was evaporated off via vacuum distillation. The product was dissolved in 30 mL chloroform, and filtrated. Filtrate dropped into cold diethyl ether to obtain precipitate. Dried in vacuo to give a white powder product.
Synthesis of PEGylated ligand (L-PEG-L) by click chemistry reaction. L-Alkynyl (1.2 g, 3 mmol), N3-PEG2k-N3 or N3-PEG4.6k-N3 (1 mmol), anhydrous cupric sulfate and sodium ascorbate were added in 100 mL radius flask. 40 mL deoxygenated water was added to dissolve compounds. The mixture was stirred under inert gas protection and heated to 60 °C for 48 h. The solution was filtrated and dialyzed 3 days against water (MWCO 1000 Da membrane) and then lyophilized to obtain PEGylated ligand (L-PEG2k-L or L-PEG4.6k-L).
Preparation of PEGylated Mn2+ complexes solution. PEGylated ligand (L-PEG2k-L or L-PEG4.6k-L) and manganese chloride (5–10 times molar excess with regard to the L) were dissolved in 10 mL MilliQ water. After stirring for 1 hour, the solution was dialyzed 2 days against water (MWCO 1000 Da membrane) to ensure no free Mn2+. Mn concentration of PEGylated Mn2+ complexes solution was measured by elemental analyses using atomic absorption spectroscopy.
T1 relaxivity and MRI phantom studies
T1 relaxivities of free MnL, PEGylated Mn2+ complexes (MnL-PEG2k-MnL or MnL-PEG4.6k-MnL) were measured at 1.5 T on a clinical MR scanners (Siemens, Sonata) at 25 °C as previous reports.9 In brief, samples were diluted to different concentrations (0.1, 0.15, 0.2, 0.25, 0.3, 0.35, 0.4, and 0.5 mM of Mn). The T1-weighted MRI phantom were acquired with a conventional spin-echo (SE) acquisition with TR values ranging from 20 to 1000 ms (TE = 5.3 ms, slice thickness = 3 mm, 292 × 146 mm, flip angle = 90°). Signal intensities at different TR were collected and used to fitting signal decay curve by OriginPro 8, and therefore obtained T1 time parameters. Relaxivity values (r1) were calculated through the linear fitting of 1/T1 time (s−1) versus the Mn concentration (mM) in Microsoft excel 2013.
In vivo rat MRA studied
All studies involving animals were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Sichuan University and approved by the Animal Ethics Committee of Sichuan University. MR imaging was carried out on a clinical 3.0 T scanner (Achieva, Philips). Sprague-Dawley (SD) rats (180–220 g) were anaesthetized by pentobarbital sodium at the dose of 40 mg kg−1 body weight and placed within a 50 mm rat coil. After intravenous injection (via tail vein) of a solution of MnL or MnL-PEG4.6-MnL with the dosages of 0.25 Mn mmol kg−1 body weight, dynamic T1-weighted images of chest and cervical region were obtained by a 3DCEMRA sequence (T1FFE, TE = 3 ms, TR = 7 ms, slice thickness = 0.9 mm, field of view = 100 mm × 100 mm, flip angle 30°). MRI signal intensity values in jugular vein, heart and liver at different time points were read and collected by Philips DICOM Viewer R3.0.
Conclusions
In summary, a rigid Mn2+ complex (MnL) was designed and synthesized. Then, further PEG (PEG2k and 4.6k) modification of MnL complexes was achieved by click chemistry with high ligand grafting rates (above one ligand per PEG molecule). PEGylated MnL present higher T1 relaxivities (MnL-PEG2k-MnL: r1 = 6.38 mmol−1 s−1; MnL-PEG4.6k-MnL: r1 = 5.63 mmol−1 s−1) than free MnL complexes. In vivo SD rat MRA studies show that PEGylated MnL have a longer vascular imaging window (up to 10 minutes) than that of free MnL at the same manganese dosage (0.25 Mn mmol kg−1 Bw).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by grants from the Chinese Ministry of Sciences 973 Program (2013CB933903), National Key Technology R&D Program (2012BAI23B08), and National Natural Science Foundation of China (81621003, 20974065, 51173117, 50830107, 81671675 and 81601490). Science and Technology Project of Sichuan, China (2016JY0172), the Scientific Research Start-up Fund of North Sichuan Medical College (CBY15-QD02).
Notes and references
- D. Pan, S. D. Caruthers, A. Senpan, A. H. Schmieder, S. A. Wickline and G. M. Lanza, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 162–173 CrossRef CAS PubMed.
- E. Chabanova, V. B. Logager, J. M. Moller and H. S. Thomsen, Open Drug Saf. J., 2011, 2, 29–38 CrossRef CAS.
- D. Pan, A. H. Schmieder, S. A. Wickline and G. M. Lanza, Tetrahedron, 2011, 67, 8431–8444 CrossRef CAS PubMed.
- B. Drahos, J. Kotek, I. Cisarova, P. Hermann, L. Helm, I. Lukes and E. Toth, Inorg. Chem., 2011, 50, 12785–12801 CrossRef CAS PubMed.
- M. A. Sieber, T. Steger-Hartmann, P. Lengsfeld and H. Pietsch, J. Magn. Reson. Imag., 2009, 30, 1268–1276 CrossRef PubMed.
- T. S. Henrik, Radiol. Clin., 2009, 47, 827–831 CrossRef PubMed.
- R. Jin, B. Lin, D. Li and H. Ai, Curr. Opin. Pharmacol., 2014, 18, 18–27 CrossRef CAS PubMed.
- E. J. Werner, A. Datta, C. J. Jocher and K. N. Raymond, Angew. Chem., Int. Ed., 2008, 47, 8568–8580 CrossRef CAS PubMed.
- B. Drahos, J. Kotek, I. Cisarova, P. Hermann, L. Helm, I. Lukes and E. Toth, Inorg. Chem., 2011, 50, 12785–12801 CrossRef CAS PubMed.
- S. Wang and T. D. Westmoreland, Inorg. Chem., 2008, 48, 719–727 CrossRef PubMed.
- H. Su, C. Wu, J. Zhu, T. Miao, D. Wang, C. Xia, X. Zhao, Q. Gong, B. Song and H. Ai, Dalton Trans., 2012, 41, 14480–14483 RSC.
- A. Forgács, R. Pujales-Paradela, M. Regueiro-Figueroa, L. Valencia, D. Esteban-Gómez, M. Botta and C. Platas-Iglesias, Dalton Trans., 2017, 46, 1546–1558 RSC.
- A. Forgács, M. Regueiro-Figueroa, J. L. Barriada, D. Esteban-Gómez, A. de Blas, T. Rodríguez-Blas, M. Botta and C. Platas-Iglesias, Inorg. Chem., 2015, 54, 9576–9587 CrossRef PubMed.
- H. Ai, Adv. Drug Delivery Rev., 2011, 63, 772–788 CrossRef CAS PubMed.
- F. Kielar, L. Tei, E. Terreno and M. Botta, J. Am. Chem. Soc., 2010, 132, 7836–7837 CrossRef CAS PubMed.
- Y. Song, E. K. Kohlmeir and T. J. Meade, J. Am. Chem. Soc., 2008, 130, 6662–6663 CrossRef CAS PubMed.
- C. Wu, D. Li, L. Yang, B. Lin, H. Zhang, Y. Xu, Z. Cheng, C. Xia, Q. Gong, B. Song and H. Ai, J. Mater. Chem. B, 2015, 3, 1470–1473 RSC.
- L. O. Liepold, M. J. Abedin, E. D. Buckhouse, J. A. Frank, M. J. Young and T. Douglas, Nano Lett., 2009, 9, 4520–4526 CrossRef CAS PubMed.
- C. Wu, Y. Xu, L. Yang, J. Wu, W. Zhu, D. Li, Z. Cheng, C. Xia, Y. Guo, Q. Gong, B. Song and H. Ai, Adv. Funct. Mater., 2015, 25, 3581–3591 CrossRef CAS.
- Y. Xu, C. Wu, W. Zhu, C. Xia, D. Wang, H. Zhang, J. Wu, G. Lin, B. Wu, Q. Gong, B. Song and H. Ai, Biomaterials, 2015, 58, 63–71 CrossRef CAS PubMed.
- H. Y. Su, C. Q. Wu, D. Y. Li and H. Ai, Chin. Phys. B, 2015, 24, 127506 CrossRef.
- D. Wang, B. Lin, T. Shen, J. Wu, C. Xia, B. Song and H. Ai, Sci. Bull., 2016, 61, 1023–1030 CrossRef CAS.
- J. Xie, G. Liu, H. S. Eden, H. Ai and X. Chen, Acc. Chem. Res., 2011, 44, 883–892 CrossRef CAS PubMed.
- B. Podobnik, B. Helk, V. Smilović, Š. Škrajnar, K. Fidler, S. Jevševar, A. Godwin and P. Williams, Bioconjugate Chem., 2015, 26, 452–459 CrossRef CAS PubMed.
- C. Dhalluin, A. Ross, L.-A. Leuthold, S. Foser, B. Gsell, F. Müller and H. Senn, Bioconjugate Chem., 2005, 16, 504–517 CrossRef CAS PubMed.
- A. M. Mohs, X. Wang, K. C. Goodrich, Y. Zong, D. L. Parker and Z. R. Lu, Bioconjugate Chem., 2004, 15, 1424–1430 CrossRef CAS PubMed.
- A. A. Bogdanova, R. Weissleder, H. W. Frank, A. V. Bogdanova, N. Nossif, B. K. Schaffer, E. Tsai, M. I. Papisov and T. J. Brady, Radiology, 1993, 187, 701–706 CrossRef PubMed.
- S. Hiki and K. Kataoka, Bioconjugate Chem., 2007, 18, 2191–2196 CrossRef CAS PubMed.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512–523 RSC.
- A. Fontes, S. Karimi, L. Helm, P. M. Ferreira and J. P. André, Eur. J. Inorg. Chem., 2015, 2015, 4798–4809 CrossRef CAS.
- J. Svensson, Acta Radiol., 2015, 44, 1–30 CrossRef.
- R. Jain and S. Sawhney, Clin. Radiol., 2005, 60, 1171–1181 CrossRef CAS PubMed.
- V. Yousefi Babadi, L. Sadeghi, K. Shirani, A. A. Malekirad and M. Rezaei, J. Toxicol., 2014, 2014, 946372 Search PubMed.
Footnote |
| † Crystal data for [Mn4L3(H2O)3Cl2] complex. CCDC 1571263. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra09975d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Li Yanga,
Zhuzhong Chene,
Houbing Zhanga,
Danyang Lia,
Bingbing Lina,
Jiang Zhu*c,
Hua Ai*ad and
Xiaoming Zhangb
ab,
Li Yanga,
Zhuzhong Chene,
Houbing Zhanga,
Danyang Lia,
Bingbing Lina,
Jiang Zhu*c,
Hua Ai*ad and
Xiaoming Zhangb