
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of dietary oxidized tyrosine products on insulin secretion via the thyroid hormone T3-regulated TRβ1–Akt–mTOR pathway in the pancreas
Yin-Yi Ding ab,
Xue Tangab,
Xiang-Rong Cheng*ab,
Fang-Fang Wangac,
Zhu-Qing Liab,
Sha-Ji Wuab,
Xing-Ran Kouab,
Yonghui Shiab and
Guowei Le*ab
ab,
Xue Tangab,
Xiang-Rong Cheng*ab,
Fang-Fang Wangac,
Zhu-Qing Liab,
Sha-Ji Wuab,
Xing-Ran Kouab,
Yonghui Shiab and
Guowei Le*ab
aThe State Key Laboratory of Food Science and Technology, School of Food Science and Technology, Jiangnan University, Lihu Avenue 1800, Wuxi, 214122, China. E-mail: lgw@jiangnan.edu.cn; cheng-xiangrong@hotmail.com; Fax: +86 510 8591 7789; Tel: +86 510 8591 7789
bFood Nutrition and Functional Factors Research Center, School of Food Science and Technology, Jiangnan University, Wuxi, 214122, China
cSchool of Life Science, Linyi University, Linyi, 276000, China
First published on 29th November 2017
Abstract
Oxidized tyrosine products (OTPs) have been detected in commercial foods with high protein content. Dityrosine (Dityr) is a typical oxidized tyrosine product. The previous studies in our lab demonstrated that dityrosine administration impaired glucose tolerance and suppressed the bio-function of thyroid hormone T3 of mice. The T3-activated Akt–mTOR signaling pathway plays important roles in insulin synthesis in pancreatic β cells. Due to the structural homology between dityrosine and T3, the molecular binding domain for these two compounds in TRβ1 might be the same site. Therefore, the present study investigates the potential impact of dietary OTPs on the pancreatic function. Sprague Dawley (SD) rats were fed a diet containing OTPs for 12 weeks. In addition, a 10 week gavage experiment using C57BL/J mice was performed to explore whether dityrosine was responsible for the injury induced by OTPs. The blood glucose, plasma insulin levels, and plasma free thyroid hormones (THs) were then measured. After 12 week dietary OTPs or 10 week OTPs/dityrosine gavage, elevated fasting blood glucose and decreased plasma insulin levels were detected both in rats and mice in the presence of enhanced plasma free THs content, which indicated dysfunction of the pancreatic islets and that the regulation of T3 to insulin synthesis was suppressed by OTPs and dityrosine. A cell experiment using mouse MIN-6 cells was performed to explore the mechanism of the diminished T3 bio-function in pancreatic islets induced by dityrosine. Dityrosine incubation attenuated the T3-mediated insulin synthesis via an indirect way of regulating the mRNA expression of genes related to insulin synthesis and decreasing the protein level of TRβ1. In addition, dityrosine inhibited the Akt phosphorylation activated by T3 in MIN-6 cells. Dityrosine treatment altered the T3-activated translation factors involved in the Akt–mTOR signaling pathway. These findings indicate that decreased insulin secretion triggered by dietary OTPs may be mediated by suggested T3-stimulated protein synthesis in pancreatic β cells.
1. Introduction
Proteins in food systems are targets of various oxidants including radicals and non-radical oxidants. Food protein is vulnerable to oxidation during processing and storage.1,2 Tyrosine residues play a major role in several metabolic pathways, including insulin synthesis and insulin sensitivity.3,4 However, tyrosine is readily oxidized to dityrosine (Dityr)5 and 3-nitrotyrosine (3-NT).6 Dityrosine is a cross-linking oxidized protein product with a stable structure, which is resistant to acid hydrolysis and proteases.7,8 Previous studies in our lab illustrated that long-term exposure to oxidized tyrosine could impair redox status and induce fibrosis in the liver and kidneys.9,10 In addition, we also verified that dietary oxidized tyrosine products could induce dysfunction of insulin secretion in pancreas via oxidative stress-induced mitochondrial damage.11 OTPs and dityrosine exposure changed many systemic metabolic processes, including reduced choline bioavailability, led to fat accumulation in liver, induced hepatic injury, and renal dysfunction.12 However, the influence of dietary oxidized proteins on metabolic disturbance has still not been fully elucidated.It has been proved that accumulation of dityrosine in tissues is associated with many diseases, including Parkinson's disease, asthma, Alzheimer's diseases.13,14 It is suggested that dityrosine could induce oxidative damage to the brain, leading to novel object recognition deficits.15 Other researches also demonstrated the potential pathogenic role for dityrosine in age-induced bone loss.16,17 The previous studies in our lab also confirmed that dityrosine administration disrupted hypothalamus-pituitary-thyroid glands (HPT) axis and impaired glucose-stimulated insulin synthesis in pancreas.18 The studies of polychlorinated biphenyls (PCBs) and bisphenol A (BPA), a class of industrial compounds, have been shown to affect a number of endocrine targets.19,20 The endocrine disorder induced by PCBs and BPA is thought to be due to their toxicological effects, which may be related to the structural similarities shared by PCBs, BPA, and 3,5,3′-triiodothyronine (T3).21,22 One of the metabolism systems most frequently likely considered in connection with its disruption by dityrosine was the thyroid hormone T3 action due to the unexpected resemblance between the chemical structures of dityrosine and T3. According to the database of Chemical entities of biological interest (ChEBI, http://www.ebi.ac.uk/chebi/), the structure resemblance between dityrosine and T3 is 70% resemblance. It suggests an unexpected resemblance between the chemical structures of dityrosine and T3.
Given the findings of the aforementioned studies, examining the relationship between OTPs and T3-regulated insulin synthesis is a major research topic at present. Thyroid hormones (THs) are widely known for their ability to influence various cellular processes,23 energy expenditure, and metabolic rate.24,25 Thyroxine (T4) is the main secretory product of the thyroid gland, and the deiodination of this prohormone to the biologically active hormone, 3,5,3′-triiodothyronine (T3), is the first step in THs action.26 Evidence of protein–protein interactions between cytosolic thyroid hormone receptors (TRs) and phosphatidylinositol 3-kinase (PI3K), and activation of PI3K activity by T3 treatment have been reported.27,28 Previous observations demonstrated that the activation of Akt–mTOR signaling pathway is largely responsible for the T3-stimulated protein synthesis.29,30 TRβ1 seems to be an essential component in mediating T3 action on Akt pathway in pancreas.31 Moreover, T3-stimulated Akt–mTOR pathway activation has implicated in regulating insulin synthesis in pancreatic β cells.30,32
In the present study, we investigated the effects of OTPs on the ability of insulin secretion and bio-function of T3 in pancreas. As dityrosine accounts for 22% of OTPs,33 the potential role of dityrosine in pancreatic injury induced by OTPs was studied in a 10 week gavage experiment involving C57BL/6 mice administrated with OTPs or pure dityrosine. The current study also examined whether exposure to dityrosine resulted in suppressed T3-stimulated Akt–mTOR signaling pathway, subsequently attenuated insulin secretion, and ultimately triggered hyperglycemia.
2. Materials and methods
2.1. Oxidized tyrosine products preparation
OTPs was prepared by our laboratory according to the method of Kurahashi34 with a little modification as previously described.35 Briefly, L-tyrosine was dispersed in 0.05 M phosphate buffer (pH 7.4) with the concentration of 1 mg mL−1. The tyrosine solution was added with H2O2 (5 mM) and CuSO4 (0.05 mM) and mixed in sealed tubes. The mixtures were shaken at 60 rpm in a thermostatic bath at 45 ± 0.1 °C for 2 h. At the end of the reaction Cu2+ was removed using cation exchange resin. Freeze-drying was applied to remove the remaining hydrogen peroxide and dry the OTPs sample. The major constituents of OTPs were assessed in the previous study in our lab and it was found that OTPs sample mainly contains Dityr, the content of Dityr was 22% (w/w).332.2. Molecular docking simulations
Molecular docking simulations of the ligands (dityrosine and T3) into the TRβ1 binding sites were performed using AutoDock (version 4) in the present work,36 the crystal structures (1NAX, respectively) were extracted from Protein Data Bank (http://www.rscb.org/pdb). The grid-based docking program was employed to obtain the binding modes of the compounds. The interaction energy between ligand and TRβ1 was evaluated using atom affinity potentials calculated on a grid similar to that described by Goodford.37 During the molecular docking process, the molecule in the crystal structure was used as a standard docking model.For molecular docking, AutoGrid was used for the preparation of the grid map using a grid box with a docking box of 60 × 60 × 60 Å, the box spacing was set to 0.375 Å. And the grid center was designated at dimensions (xyz): 9.421 × 16.953 × 26.432. The docking possibilities were calculated by the genetic algorithm with local search (GALS). For each ligand, the simulation was composed of 100 docking runs using the standard AutoDock parameters.
2.3. In vitro transport studies in ussing chambers system
Mature male SD rats weighing 180–200 g fed a standard diet were used in this experiment. After a 12 h fast, rats were sacrificed and ≈3 cm long segments of jejunum were excised. The excised tissues were immediately rinsed with ice-cold Kreb-Ringer solution (118 mM NaCl, 4.75 mM KCl, 1.18 mM KH2PO4, 1.18 mM MgSO4, 2.5 mM CaCl2, 25 mM NaHCO3, and 11 mM glucose)38 to remove luminal content. Unstripped tissues were cut in 1 cm length pieces and opened along the mesenteric border. The fascia layer was then removed gently.Ussing chamber system (EM-CSYS-4, Physiological Instruments, Inc. San Diego, U.S.A.) was used in this experiment. Pieces of intestinal tissue were mounted as flat sheets between the two halves of acrylic chambers bathed on both mucosal and serosal sides by 5 mL Kreb-Ringer solution. The solution at mucosal side contained OTPs at concentration of 0, 0.5, 1 mg mL−1. The pH was 7.4 at 37 °C when bubbled with 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 O2/CO2 mixture used to circulate fluid in the chambers. 100 μL Kreb-Ringer solution was taken from the serosal side the indicated intervals (0, 30, 60, 90, 120 min) and complemented it with 100 μL of Kreb-Ringer solution. Dityrosine concentration in solution was measured by fluorescence spectrophotometer (FlourMax-4, HORIBA, Tokyo, Japan) with excitation wavelength of 320 nm and emission wavelength of 400 nm.
:5 O2/CO2 mixture used to circulate fluid in the chambers. 100 μL Kreb-Ringer solution was taken from the serosal side the indicated intervals (0, 30, 60, 90, 120 min) and complemented it with 100 μL of Kreb-Ringer solution. Dityrosine concentration in solution was measured by fluorescence spectrophotometer (FlourMax-4, HORIBA, Tokyo, Japan) with excitation wavelength of 320 nm and emission wavelength of 400 nm.
2.4. 12 week dietary OTPs experiment in SD rats
Male Sprague Dawley (SD) rats (weighting 300 ± 20 g) were purchased from Model Animal Research Center of Nanjing University. Rats were housed in individual cages in a temperature-controlled room at 24 ± 2 °C with a 12 h light–dark cycle (Research animal center of Jiangnan University).After 7 days of acclimatization, SD rats were divided into 2 groups (n = 8):
Control group: rats were received standard diet;
OTPs group: rats were received standard diet together with 8 g kg−1 diet OTPs.
Dityrosine level in feed were measured by fluorescence spectrophotometer (FlourMax-4, HORIBA, Tokyo, Japan) with excitation wavelength of 320 nm and emission wavelength of 400 nm.
The dose of OTPs we used in this experiment was consistent with the previous studies in our lab.33 And all animal treatments have been performed in accordance with the ethical standards laid down in the guidelines of National Institutes of Health Guide for the Care and Use of Laboratory Animals (China) and in the 1964 Declaration of Helsinki and its later amendments. All animal studies were approved by Laboratory Animals Ethics Committee of Jiangnan University.
2.5. 10 week gavage experiment using OTPs and dityrosine in C57BL/6 mice
The major constituent of OTPs is dityrosine, so we performed an additional dityrosine gavage experiment to explore whether dityrosine should be responsible for the injury induced by OTPs. Since the preparation of dityrosine is very costly and time-consuming, we chose to use mice (with body mass much lower than that of rats) for this gavage study. 4 week-old male C57BL/6 mice were purchased from Model Animal Research Center of Nanjing University, China. Mice were housed in a temperature-controlled room at 12 ± 2 °C with a 12 h light–dark cycle with free access to food and water. After 7 days of acclimatization, C57BL/J mice were randomly divided into 3 groups (n = 10):Control group: mice received with saline;
Dityr1 group: mice received with dityrosine (320 μg per kg per bw per day);
Dityr2 group: mice received with dityrosine (640 μg per kg per bw per day);
OTPs1 group: mice received with OTPs (1450 μg per kg per bw per day);
OTPs2 group: mice received with OTPs (2900 μg per kg per bw per day).
The dose of dityrosine we used in the animal experiment was consistent the precious studies in our lab.15 The mass percent of dityrosine in OTPs sample is 22%, the effect of 320 or 640 μg per kg per bw per day dityrosine should correspond to that of 1450 or 2900 μg per kg per bw per day OTPs. All animal studies have been performed in accordance with the ethical standards laid down in the guidelines of Laboratory Animals Care of Jiangsu Province (China) and in the 1964 Declaration of Helsinki and its later amendments.
2.6. Blood glucose, glucose tolerance test (GTT), plasma insulin content
For blood glucose tests, rats and mice were fasted overnight for 12 h, and blood samples were obtained from the tail vein. Animals were then given by gavage with 2 g kg−1 body weight of glucose, and blood samples were taken at the indicated intervals (0, 10, 30, 60, 90, 120 min after gavage). Blood glucose was tested using One Touch Sure Step test strip (Lifescan, Milpitas, California, U.S.A.). Plasma insulin concentration was measured using the ELISA kits (Huijia biotechnology, Xiamen, P. R. China) according to manufacturer's instruction.2.7. Measurements of plasma free T4 (FT4) and free T3 (FT3), dityrosine level in pancreas
Levels of FT4 and FT3 in plasma and dityrosine level in pancreas were measured using the ELISA kits (Huijia biotechnology, Xiamen, P. R. China) according to manufacturer's instruction.2.8. Cell experiment using mice MIN-6 cells
Cells were seeded in plates and permitted to adhere at 37 °C in a humidified atmosphere containing 95% O2 and 5% CO2 for 12 h. When the cells reached 40% confluence, they were stimulated with dityrosine in a final concentration of 0, 0.1, 1, 10 μM, and T3 in a final concentration of 0, 1 μM for 72 h.
2.9. Protein extracts and western blot analysis
Protein was extracted from MIN-6 cells using RIPA Lysis Buffer (P0013B) which was purchased from Beyotime Institute of Biotechnology (Shanghai, China). The lysis buffer mainly contains 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS and protease inhibitors including sodium orthovanadate, sodium fluoride, EDTA and leupeptin. According to the manufacturer's instructions, 1 mM (final concentration) phenylmethanesulfonyl fluoride (PMSF), phosphatase inhibitor (10 μL mL−1 lysis buffer), and protease inhibitor (5 μL mL−1 lysis buffer) were added to the lysis buffer before use. 200 mg of pancreas and cells was homogenized in 1.5 mL lysis buffer on ice. After complete lysis, homogenate was centrifuged for 5 min at 12000 g. Protein concentration in the supernatant was determined by the BCA assay kit from Nanjing Jiancheng Bioengineering Institute (Nanjing, China) according to the instructions of the manufacturer. All extracts were mixed with 5× loading buffer (50 mmol L−1 pH−1 6.8 Tris–HCl, 100 mm L−1 dithiothreitol, 2% SDS, 0.1% bromophenol blue, 10% glycerol and 5% β-mercaptoethanol) in the ratio of 4:1 and then were boiled at 95 °C for 10 min to denature the protein.
Protein extracts were separated on 12% SDS-PAGE gel using 10 μg protein per sample and then transferred onto NC membrane. The membrane was blocked in Tris-buffered saline (pH 7.4) with 5% BSA and 0.1% Tween 20. Afterwards, the blocked membrane was incubated with primary antibody including rabbit TRβ1, p-Akt (Ser473, Thr308), Akt, mTOR, eIF4E, S6K, p-4E-BP1 (Thr37/46), 4E-BP1 antibody (Cell Signaling Technology, Denvers, U.S.A.) and rabbit β actin antibody (Cell Signaling Technology, Denvers, U.S.A.) at dilution of 1:1000 at 4 °C for 10 hours. Blots were incubated with secondary antibody (Sigma-Aldrich, U.S.A.) at dilution of 1:4000 and fluorescence images were obtained with the Automatic chemiluminescence imaging analysis system (Tannon 5200, Shanghai, China).
2.10. Total mRNA isolation and quantitative RT-PCR (qRT-PCR)
Total RNA of cells and tissues was extracted with Trizol reagent according to the manufacturer's protocol (Applied Biosystems, Foster City, CA, USA). The concentration of total RNA in each sample was quantified by NanoDrop Spectrophotometer (ND2000, Thermo, Waltham, MA, USA). An SYBR green based qRT-PCR kit was used according to the manufacturer's instructions in 7900HT instrument (Applied Biosystems, Forster, CA, USA). The specificity of the product was assessed from melting curve analysis. Gene expressions were determined using the 2−ΔΔCT method. The primer's sequences for the genes were shown in Tables 1 and 2.| Gene | Forward (5′ to 3′) | Reverse (5′ to 3′) |
|---|---|---|
| Pdx-1 | TCTGCCTCTGGGACTCTTTC | TCGCCTGGTGGCTGTTAT |
| MafA | TCCTCGCTCATTTGCTCTG | GGTAAGTTCCTCGGCTTCCT |
| Glut2 | TTTGGGTGTTCCTCTGGATG | CGTCTGGTGTCGTATGTGCT |
| Gck | TCACCTTCTCCTTCCCTGTG | ACGATGTTGTTCCCTTCTGC |
| Tpo | CCTTTCTACCGCTCCTCAGC | AGGTCAAGCCATTCATCTGC |
| Tg | TGGTGTGTGGATTCTGATGG | GGATTCGTTGCTTGTGTAGC |
| Nis | AGCCTCCTCTCTGGATGGTT | CTCTCTCTTTGGTGCGGGTA |
| Tshr | AACGCATTCCAGGGACTATG | GCATCCAGCTTTGTTCCATT |
| Creb | GGAGTTGTTATGGCGTCCTC | TCTTGCTGCTTCCCTGTTCT |
| Pax8 | CTCACTCCCTGTGTGGTCAA | TGGAACTCCTGTCACTCACG |
| FoxE | GGCGGCATCTACAAGTTCAT | CAGTCGTTGAGGGTGAGGTT |
| Nkx2-1 | CTCCTCTGGCTCGGACTATG | GGCGAATGGTGGTCTTTG |
| β actin | TGTCACCAACTGGGACGATA | GGGGTGTTGAAGGTCTCAAA |
| Gene | Forward (5′ to 3′) | Reverse (5′ to 3′) |
|---|---|---|
| Glut2 | ACCCCACTTACAGTCACACCA | CACAGACAGAGACCAGAGCATAG |
| Gck | AGGAGGCCAGTGTAAAGATGT | CTCCCAGGTCTAAGGAGAGAAA |
| Ins 2 | GCTTCTTCTACACACCCATGTC | AGCACTGATCTACAATGCCAC |
| Pdx1 | CTCACCTCCACCACCACCTTCC | CACCTCCTGCCCACTGGCCTTT |
| MafA | CTGGAGGATCTGTACTGGATGA | CGCACGGACATGGATACCA |
| mTOR | CAGTTCGCCAGTGGACTGAAG | GCTGGTCATAGAAGCGAGTAGAC |
| eIF4E | ACCCCTACCACTAATCCCCC | CAATCGAAGGTTTGCTTGCCA |
| 4E-BP1 | GGGGACTACAGCACCACTC | CTCATCGCTGGTAGGGCTA |
| S6K | ATGGATGAACCTATGGGAGAGG | CCAAATGATCCCTGCCCTAATAC |
| Creb | GTCCCAGGCTCTCTATCATCTC | ATAGGCATCAAGACGGCAGAA |
| Tpo | CCTTTCTACCGCTCCTCAGC | AGGTCAAGCCATTCATCTGC |
| Nis | AGCCTCCTCTCTGGATGGTT | CTCTCTCTTTGGTGCGGGTA |
| Tshr | AACGCATTCCAGGGACTATG | GCATCCAGCTTTGTTCCATT |
| Tg | TGGTGTGTGGATTCTGATGG | GGATTCGTTGCTTGTGTAGC |
| Nkx2-1 | CTCCTCTGGCTCGGACTATG | GGCGAATGGTGGTCTTTG |
| Pax8 | CTCACTCCCTGTGTGGTCAA | TGGAACTCCTGTCACTCACG |
| FoxE | GGACCCTGCTACAATCCCG | GCAGACACGAACCGATCCA |
| β actin | TGTCACCAACTGGGACGATA | GGGGTGTTGAAGGTCTCAAA |
2.11. Statistical analysis
All measurement values were expressed as mean ± S.E.M. Significant differences between groups were conducted using one-way or two-way ANOVA analysis of variance with the post hoc Duncan's test using SPSS 20.0 software (SPSS Inc. Chicago IL., U.S.A.) for Windows. A difference was considered significant at p < 0.05 level.3. Results
3.1. The binding modes of TRβ1-T3 and TRβ1-Dityr
The chemical structures of T3 and dityrosine were shown in Fig. 1A and B. The docking structures of TRβ1-T3 and TRβ1-dityrosine were superimposed based on main-chain atoms using http://cl.sdsc.edu/.40 The superposition result suggests that the two ligands are embedded into the active site with favorable poses. In addition, compound dityrosine in TRβ1 bearing a close resemblance to the pose of T3, indicating that the molecular docking domain for dityrosine and T3 might be the same sites (Fig. 1C). As models of 1NAX-DT, the combine pocket of the receptor is exposed in Fig. 1D, compound dityrosine is packaged in the pocket, which is composed by the residues Phe269, Phe272, Thr273, Ile275, Ile276, Ala279, Arg282, Met310, Met313, Ser314, Arg316, Arg317, Arg320, Leu341, Gly344, Gly345, Leu346, His435, Met442 and Phe455. In addition, the molecule is anchored in the receptor at 3 sites (Arg282, Arg320 and Thr329) with hydrogen bonds (Fig. 1E), further enhancing the ligand–receptor interactions. These data also suggest the effects of dityrosine on the TRβ1-T3 binding ability.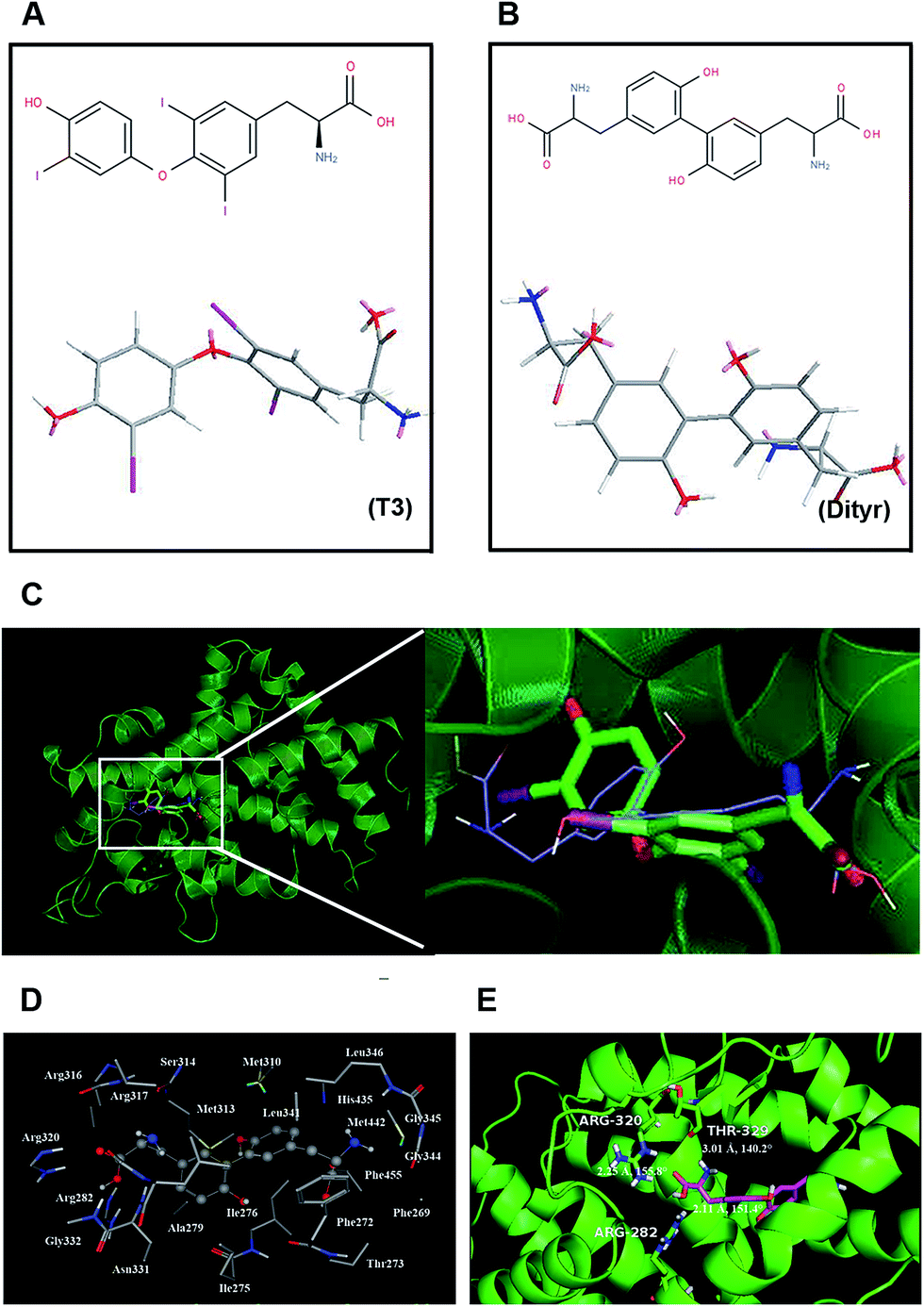 | ||
| Fig. 1 The binding modes of TRβ1-T3 and TRβ1-dityrosine. (A) The 2D chemical structure and the crystallographic structure of T3. (B) The 2D chemical structure and the crystallographic structure of dityrosine. (C) Structural superposition of TRβ1-T3, and TRβ1-Dityr. The projection highlights the structure of the active site with TRβ1 (green, helix), T3 (green, sticks), and dityrosine (blue, sticks). (D) The TRβ1 active site amino acid residues around dityrosine. (E) The enlargement for the ligand in the binding site after molecular docking, which is displayed in stick, hydrogen bonds are shown as dotted red lines, and the nonpolar hydrogens were removed for clarity. | ||
3.2. Transport studies in ussing chambers system
To confirm whether OTPs could be absorbed through intestinal wall, we performed a in vitro transport experiment using ussing chambers system. The dityrosine level was measured as an evaluation index of OTPs. As shown in Fig. 2, the fluorescence intensity of dityrosine in mucosal side solution increased in a time-dependent pattern. These results suggest that OTPs could be transported through the intestinal wall.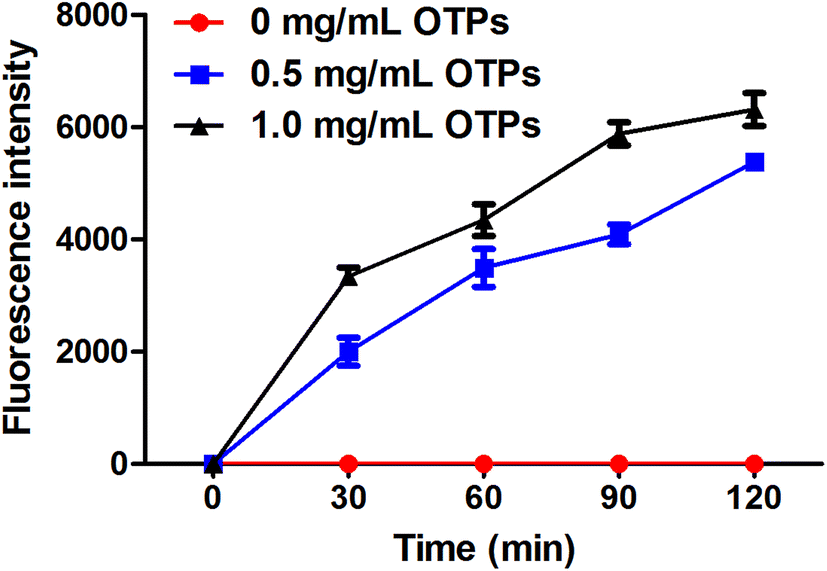 | ||
| Fig. 2 In vitro absorption pattern performed by ussing chamber experiment. | ||
3.3. 12 week dietary OTPs experiment in SD rats
| Group | Control | OTPs |
|---|---|---|
| Dityrosine content (mg kg−1) | 5.06 ± 0.63 | 1760.25 ± 186.32 |
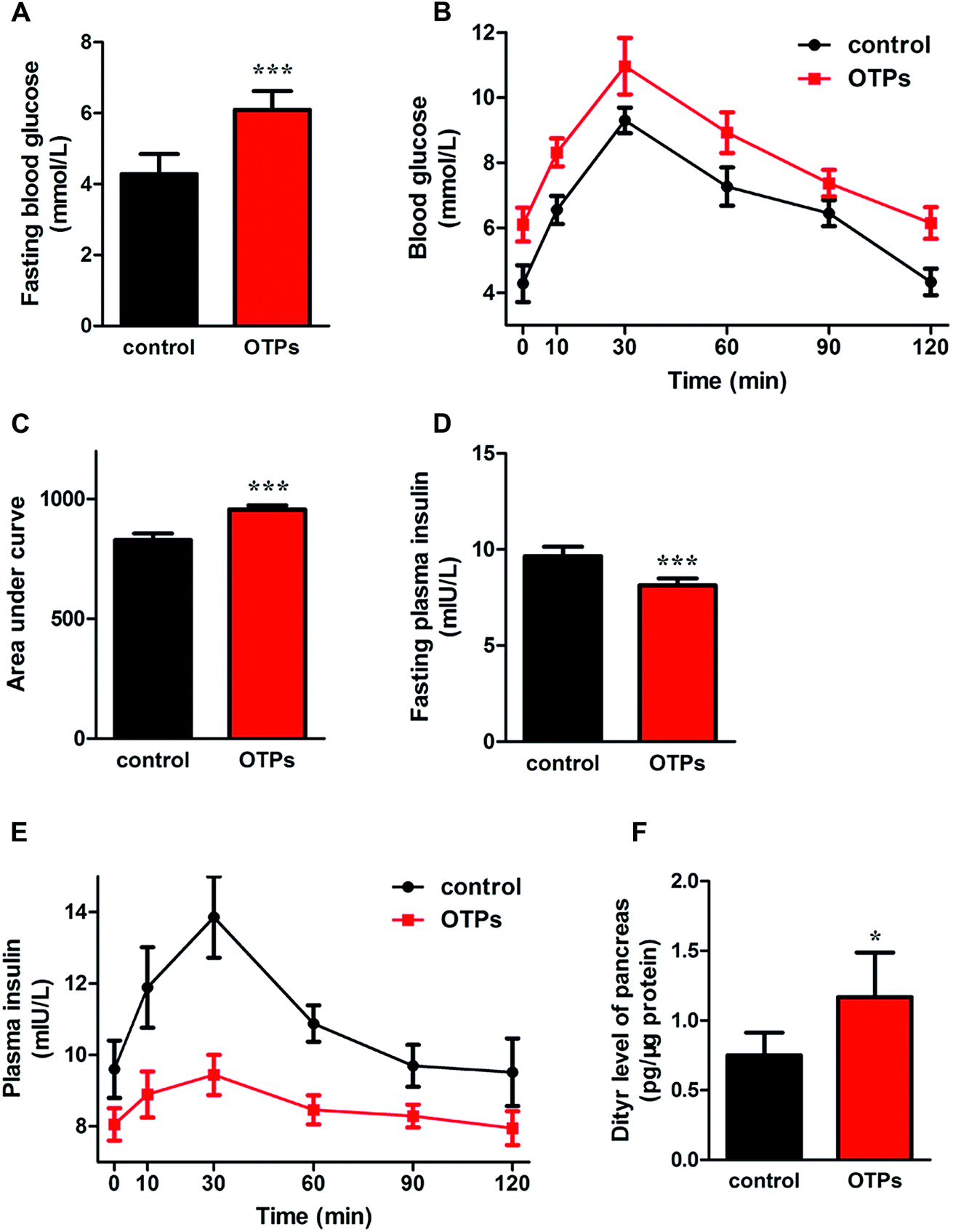 | ||
| Fig. 3 Effects of OTPs on glucose tolerance, insulin secretion and dityrosine accumulation in pancreas in 12 week dietary OTPs experiment. (A) OTPs elevated fasting blood glucose level. (B) Blood glucose curve in GTT. (C) Area under blood glucose curve in GTT. (D) OTPs declined fasting plasma insulin level. (E) Plasma insulin content curve in GTT. (F) OTPs induced Dityr accumulation in pancreas. *p < 0.05, ***p < 0.001 when compared with control group. | ||
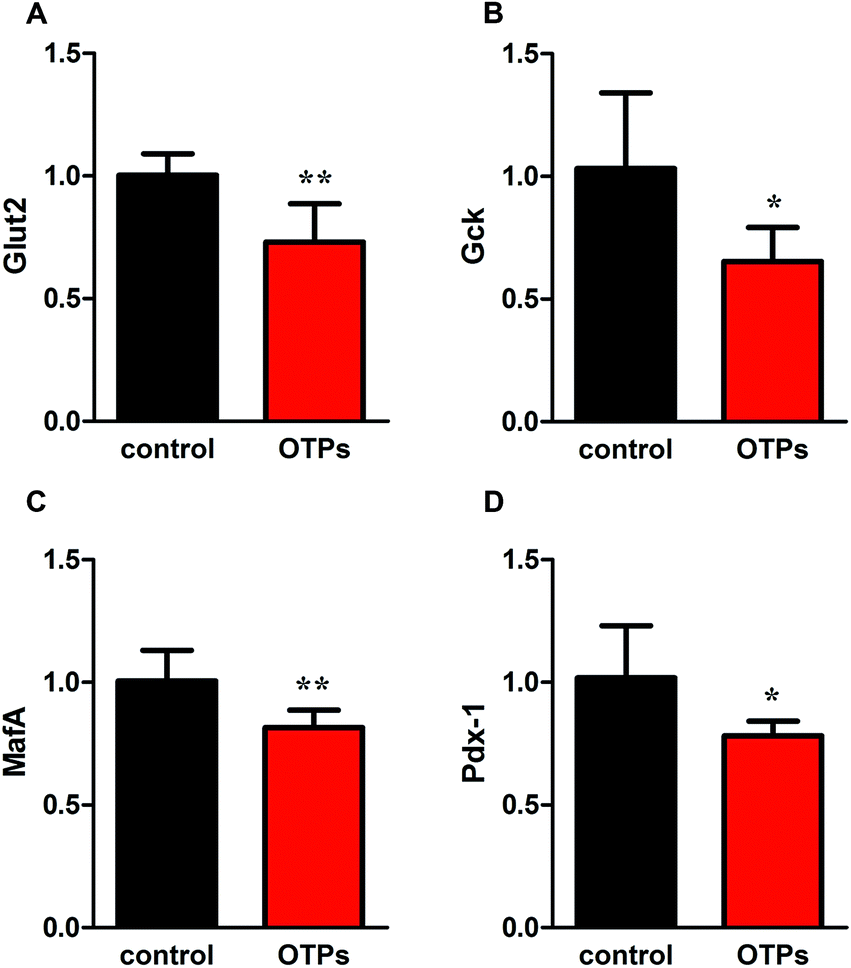 | ||
| Fig. 4 Effects of OTPs on the mRNA of insulin synthesis related genes in 12 week dietary OTPs experiment. (A) Glut2. (B) Gck. (C) MafA. (D) Pdx-1. *p < 0.05, **p < 0.01 when compared with control group. | ||
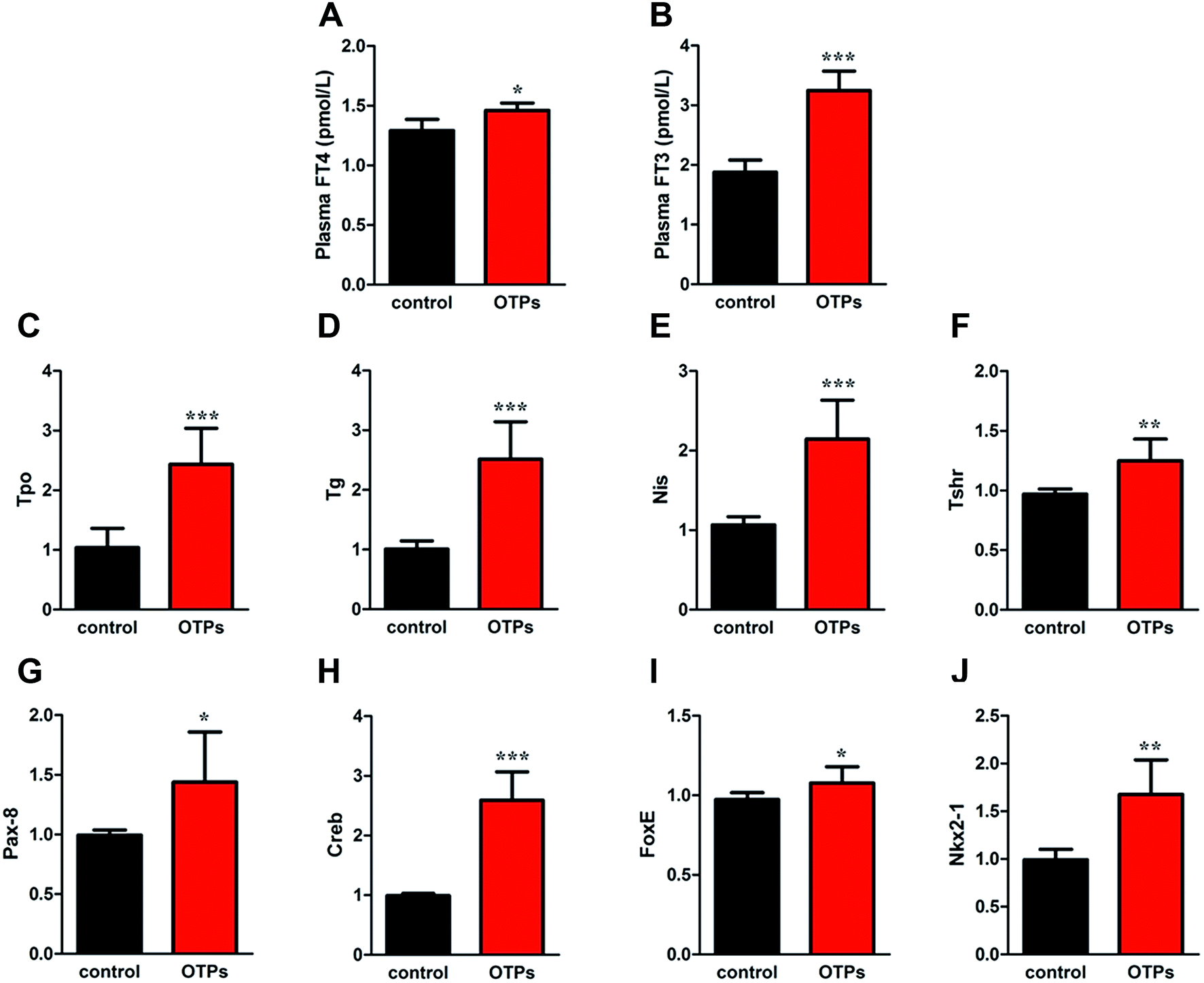 | ||
| Fig. 5 Effects of OTPs on plasma FT4 and FT3 in 12 week dietary OTPs experiment. (A) OTPs increased plasma FT4 level. (B) OTPs increased plasma FT3 level. (B) OTPs increased mRNA expression of THs synthesis related genes in thyroid gland. (C) Tpo, (D) Nis, (E) Tg, (F) Tshr. (G–J) mRNA expression of transcription regulators in thyroid gland (G) Nkx2-1, (H) Pax8, (I) FoxE1, (J) Creb **p < 0.01; ***p < 0.001 when compared with control group. | ||
3.4. 10 week gavage experiment with OTPs and dityrosine in C57BL/6mice
As shown in Fig. 6, the gavage of OTPs and dityrosine resulted in impaired glucose tolerance and decreased plasma insulin levels. OTPs and dityrosine also down-regulated the mRNA levels of genes related in GSIS pathway (Fig. 7). As might be expected, FT4 and FT3 in plasma increased in OTPs/dityrosine-treated mice. In addition, mRNA level of genes involved in THs synthesis in thyroid gland were increased attributed to dityrosine treatment (Tpo, Nis, Tg, Tshr, Nkx2-1, Pax8, FoxE, and Creb) (Fig. 8). All these changes, including increased blood glucose, decreased insulin concentration, and enhanced THs in plasma observed in the C57BL/6 mice after gavage were consistent with the effects induced by OTPs in the SD rats. The data revealed a dose effect. There was no significant difference between the OTPs and dityrosine treated mice. These data imply that dityrosine is responsible for the pancreatic damage caused by OTPs.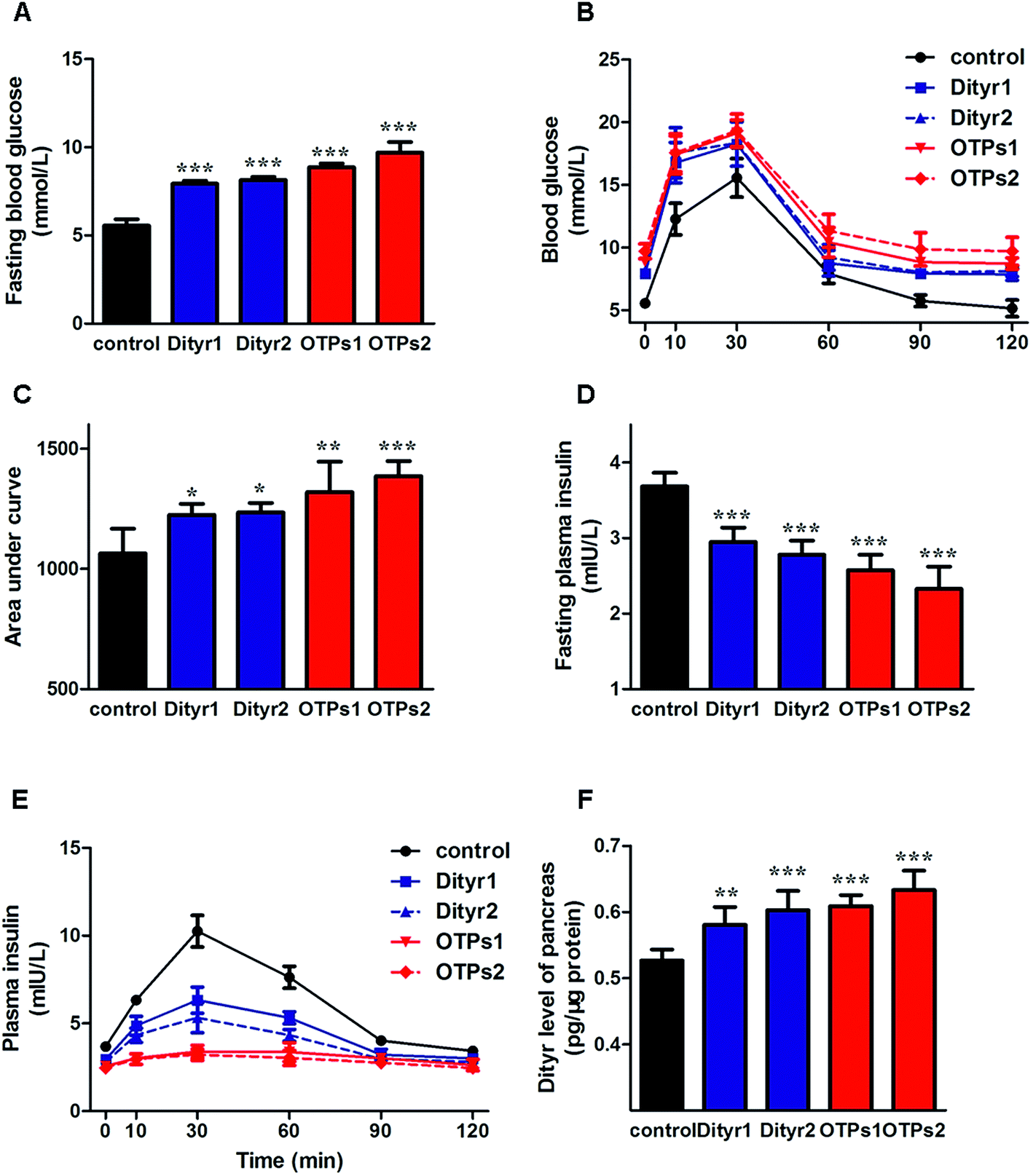 | ||
| Fig. 6 Effects of OTPs/dityrosine on glucose tolerance, insulin synthesis and dityrosine accumulation in pancreas in 10 week gavage experiment. (A) OTPs/dityrosine elevated fasting blood glucose level. (B) Blood glucose curve in GTT. (C) Area under blood glucose curve in GTT. (D) OTPs/dityrosine decreased fasting plasma insulin level. (E) Plasma insulin content curve in GTT. (F) OTPs/dityrosine administration induced dityrosine accumulation in pancreas. *p < 0.05, **p < 0.01, ***p < 0.001 when compared with control group. | ||
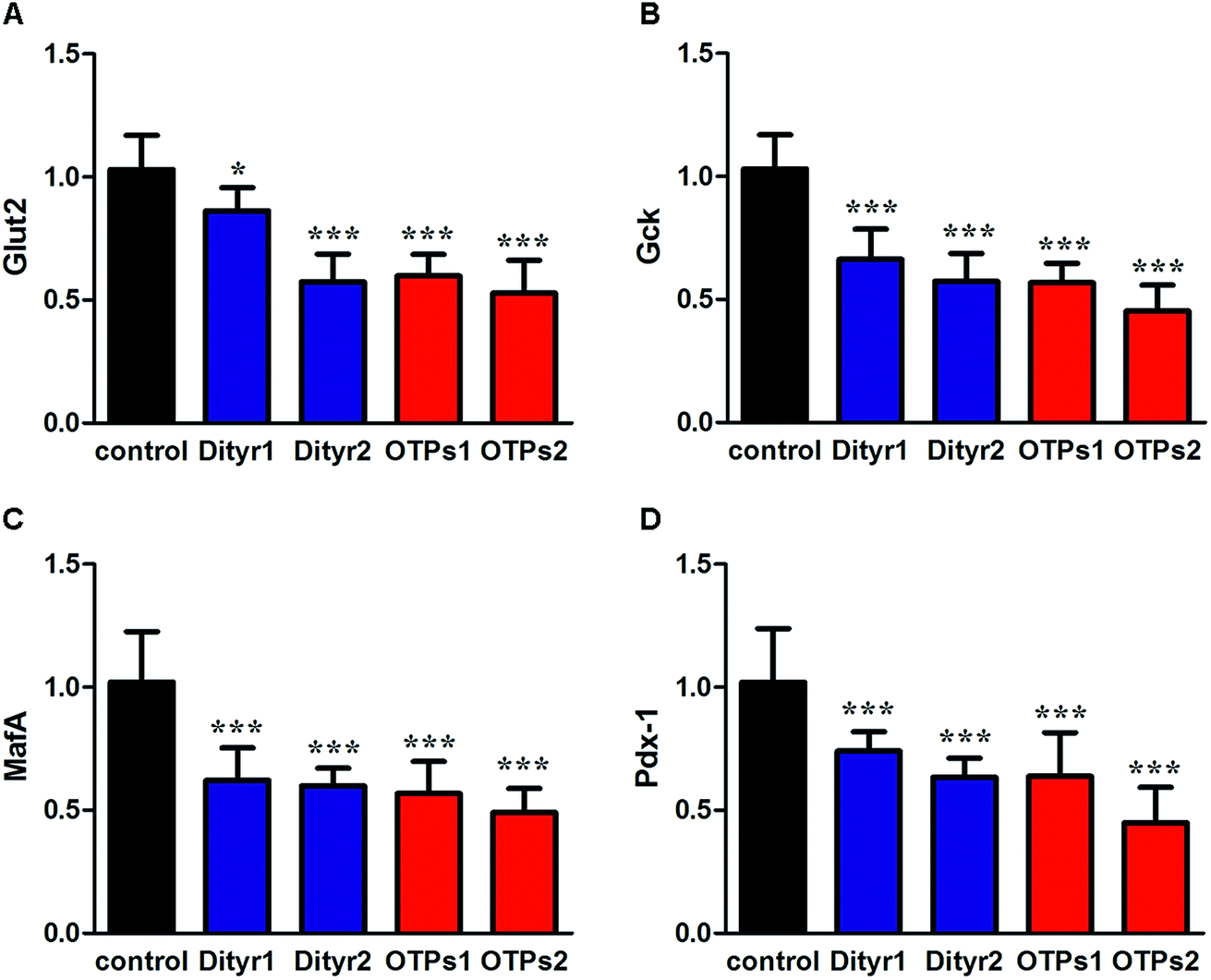 | ||
| Fig. 7 Effects of OTPs/dityrosine on the mRNA expression of insulin synthesis related genes of pancreas in 10 week gavage experiment. (A) Glut2. (B) Gck. (C) MafA. (D) Pdx-1. *p < 0.05, ***p < 0.001 when compared with control group. | ||
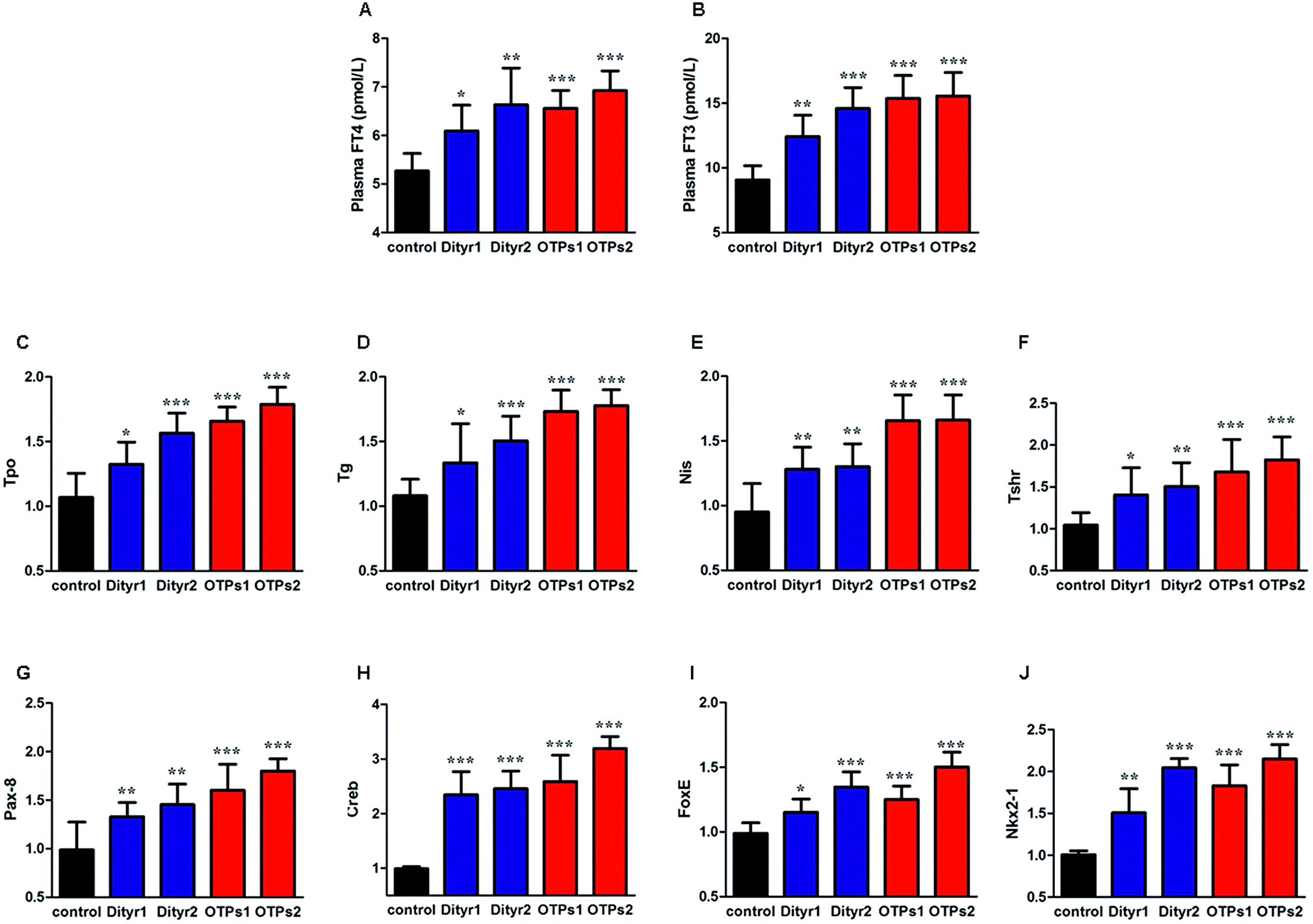 | ||
| Fig. 8 Effects of OTPs/dityrosine on plasma FT4 and FT3 in 10 week gavage experiment. (A) OTPs/dityrosine increased plasma FT4 level. (B) OTPs/dityrosine increased plasma FT3 level. (B) OTPs/dityrosine increased mRNA expression of THs synthesis related genes in thyroid gland. (C) Tpo, (D) Nis, (E) Tg, (F) Tshr. (G–J) mRNA expression of transcription regulators in thyroid gland (G) Nkx2-1, (H) Pax8, (I) FoxE1, (J) Creb. *p < 0.05, **p < 0.01, ***p < 0.001 when compared with control group. | ||
3.5. The effects of dityrosine on the T3-regulated GSIS in MIN-6 cells
We performed a cell experiment to verify the hypothesis that T3 action was disrupted by dityrosine using mice MIN-6 cell. The dityrosine content in OTPs-fed rats and OTPs/dityrosine-treated mice were approximately 1.2 pg μg−1 protein and 0.6 pg μg−1 protein, respectively. Thus, the concentration of dityrosine in the medium was 0.1 μM (0.036 mg L−1), 1 μM (0.36 mg L−1), 10 μM (3.6 mg L−1). As shown in Fig. 9, cells exposed to 1 μM T3 displayed a significant increase in basalt insulin release (2.5 mM glucose) when compared with untreated cells. The mRNA levels of genes involved in insulin synthesis significantly increased in T3-treated cells (Ins2, Pdx1 and MafA). Dityrosine-only treatment caused a markedly decrease of insulin release both in response to 2.5 mM glucose and 16.7 mM glucose, which was accompanied by decreased mRNA level of insulin synthesis-related genes. In the meanwhile, the insulin secretion of cells with 1 μM T3 treatment in the presence of dityrosine was notably declined when compared with cells exposed to T3 only. Furthermore, incubation with dityrosine attenuated the mRNA expression of genes related in insulin synthesis to certain concentration of T3.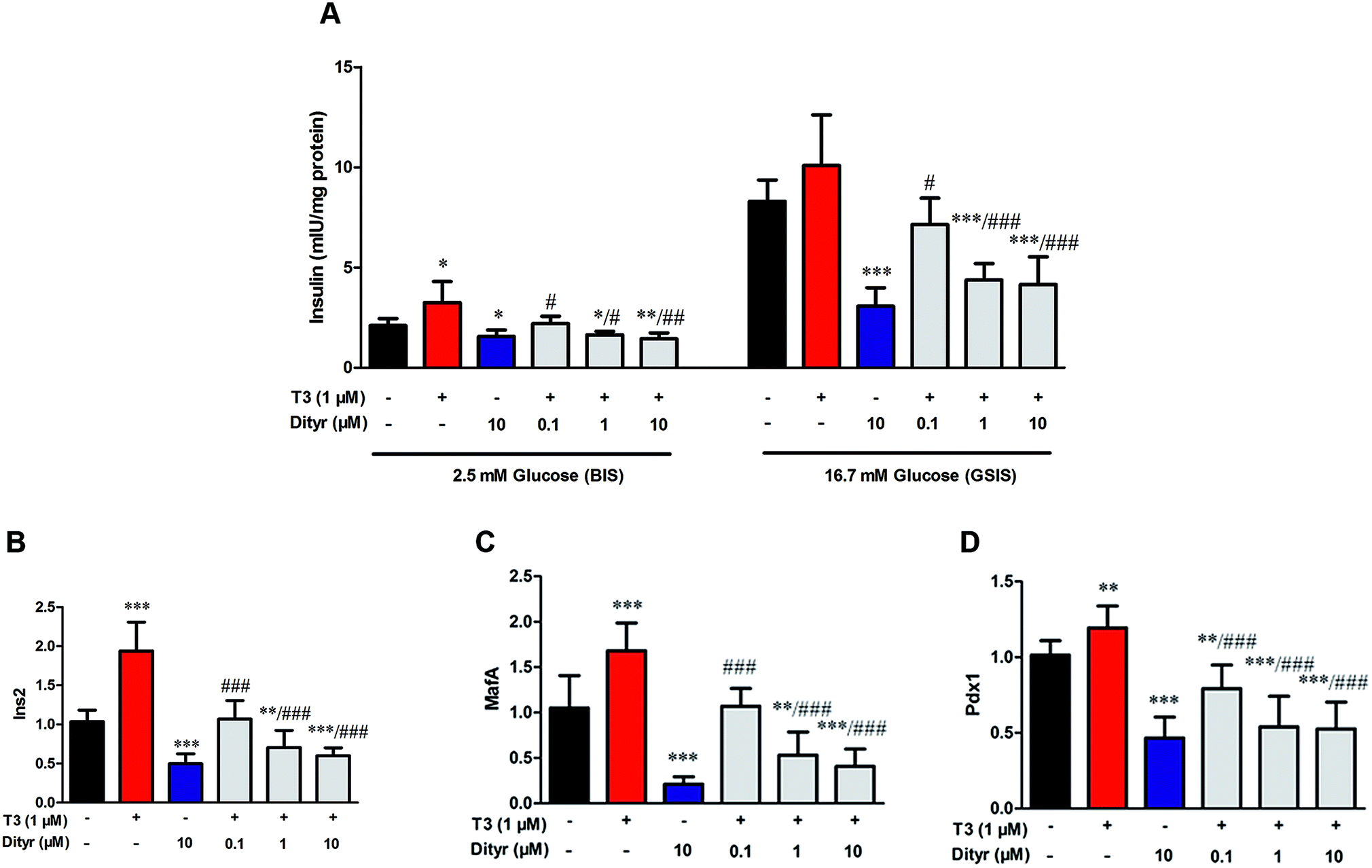 | ||
| Fig. 9 Effects of dityrosine or T3 on GSIS pathway in MIN-6 cells. (A) Insulin secretion by MIN-6 cells triggered by glucose after dityrosine and/or T3 exposure. (B–D) mRNA expression of GSIS-related genes in MIN-6 cells. (B) Ins2, (C) Pdx1, (D) MafA. *p < 0.05; **p < 0.01; ***p < 0.001 when compared with untreated cells. #p < 0.05 ##p < 0.01, ###p < 0.001 when compared with T3-only treated cells. | ||
3.6. The effects of dityrosine on the protein level of TRβ1 in MIN-6 cells
As shown in Fig. 10, T3 displayed a 27% increase of TRβ1 protein level in MIN-6 cells. However, the increase of TRβ1 protein level induced by T3 was significantly inhibited by dityrosine. These observations suggest that TRβ1 seems to be a component of the regulatory involved in the loss of T3 action caused by dityrosine.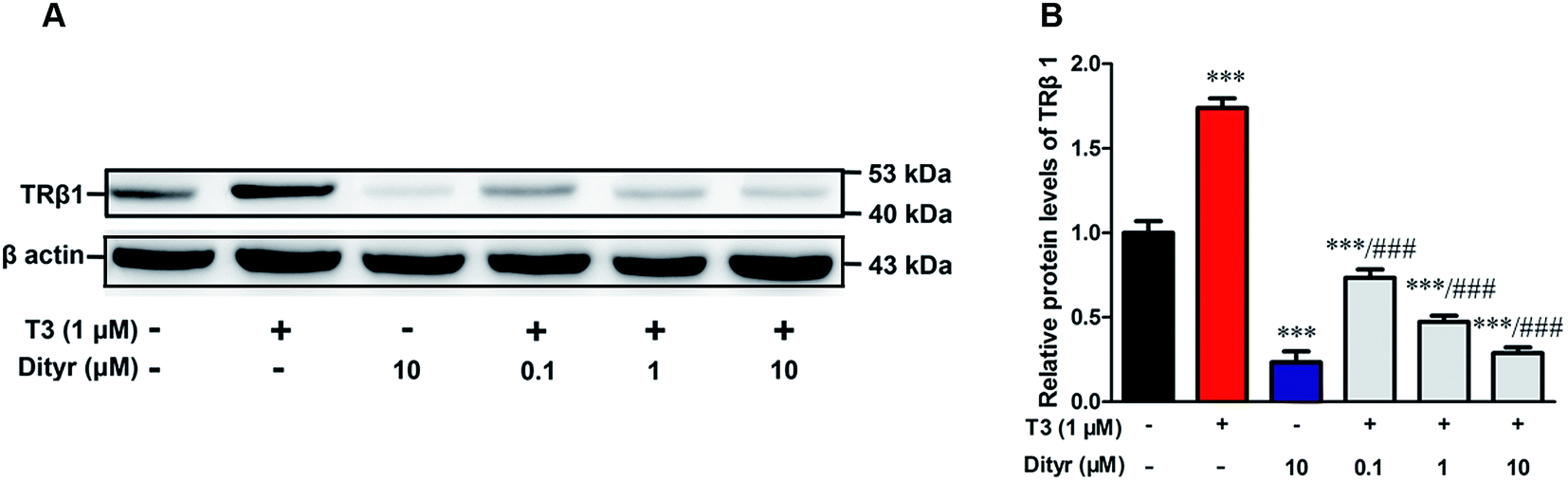 | ||
| Fig. 10 Effects of dityrosine on T3-regulated TRβ1 level in MIN-6 cells. ***p < 0.001 when compared with untreated cells. ###p < 0.001 when compared with T3-only treated cells. | ||
3.7. The effects of dityrosine on T3-induced phosphorylation of Akt
We examined whether the observed dityrosine-downregulated TRβ1 level could suppress the T3-induced phosphorylation of Akt. As shown in Fig. 11, Ser473 and Thr308 phosphorylation of Akt increased when cell were treated by T3 only. Dityrosine exposure markedly declined Ser-473 and Thr-308 phosphorylation of Akt. Meanwhile, the phosphorylation of Akt induced by T3 was suppressed when cells were exposed with T3 and dityrosine. These data provide evidence that dityrosine affect T3-stimulated protein synthesis through inhibition of the Akt phosphorylation.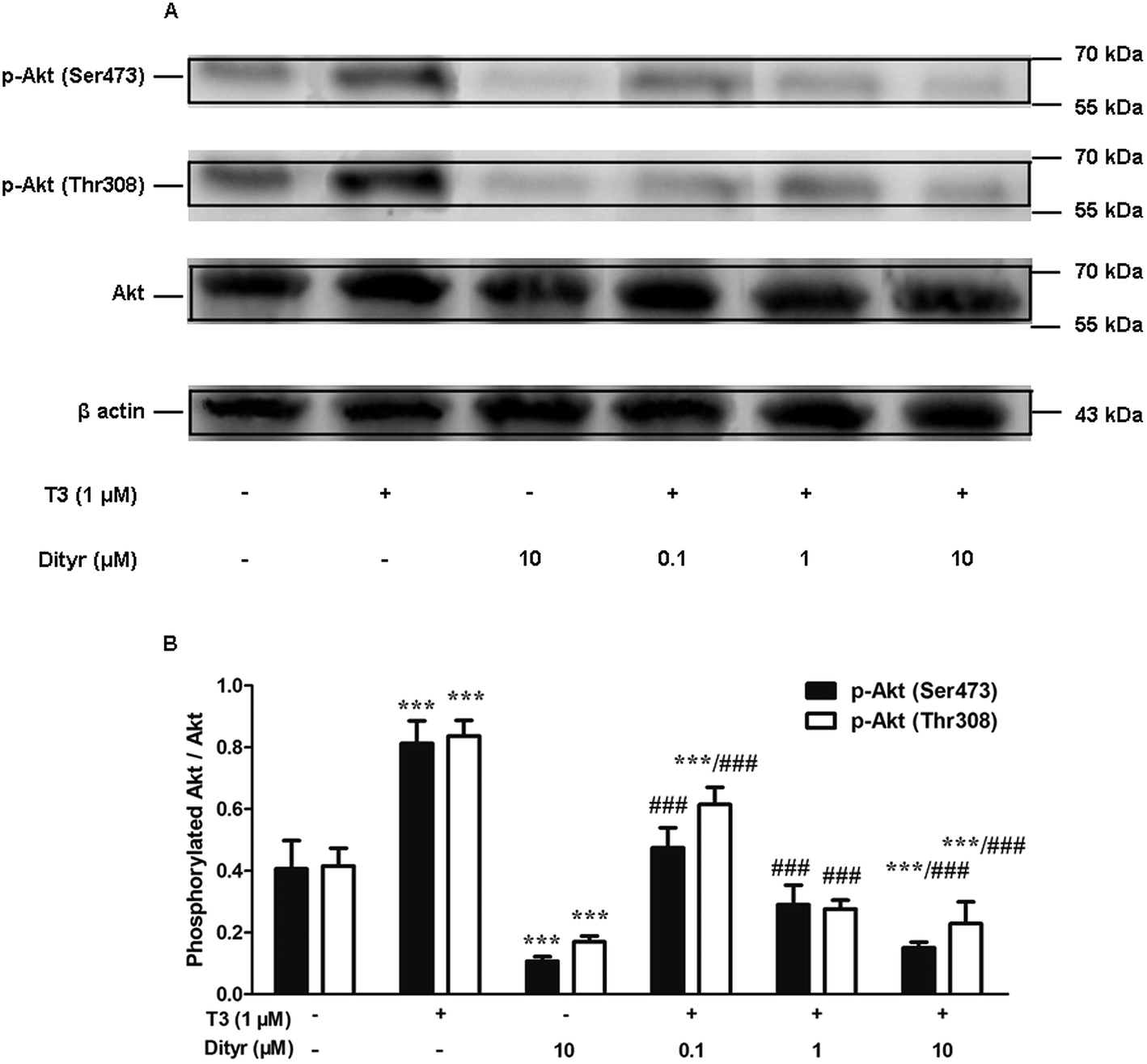 | ||
| Fig. 11 Effects of dityrosine on T3-induced phosphorylation of Akt in MIN-6 cells. (A) Protein level of p-Akt (Ser473), p-Akt (Thr308), and Akt in MIN-6 cells. (B) Ratio of phosphorylated Akt and Akt. *p < 0.05; ***p < 0.001 when compared with untreated cells. ##p < 0.01; ###p < 0.001 when compared with T3-only treated cells. | ||
3.8. The effects of dityrosine on the T3-activated translational factors involved in the downstream targets of Akt
We subsequently examined whether inhibited Akt phosphorylation could downregulate the mRNA expression and protein levels of translational factors involved in the protein translation. As expected, when cells were treated by T3 only, the mRNA levels of mTOR, S6K, and eIF4E, were upregulated and mRNA level of 4E-BP1 was downregulated. Dityrosine-only exposure markedly declined the mRNA levels of mTOR, S6K, and eIF4E, and enhanced the mRNA level of 4E-BP1. When cells were treated with T3 and dityrosine, mRNA level of mTOR, S6K, and eIF4E, significantly decreased and mRNA level of 4E-BP1 significantly increased compared with T3-only treated cells (Fig. 12). Fig. 13 shows the western-blot assay results of the protein expression of factors in Akt/mTOR signaling cascade. Dityrosine exposure significantly inhibited the changes of mTOR, S6K, eIF4E, and 4E-BP1 induced by T3.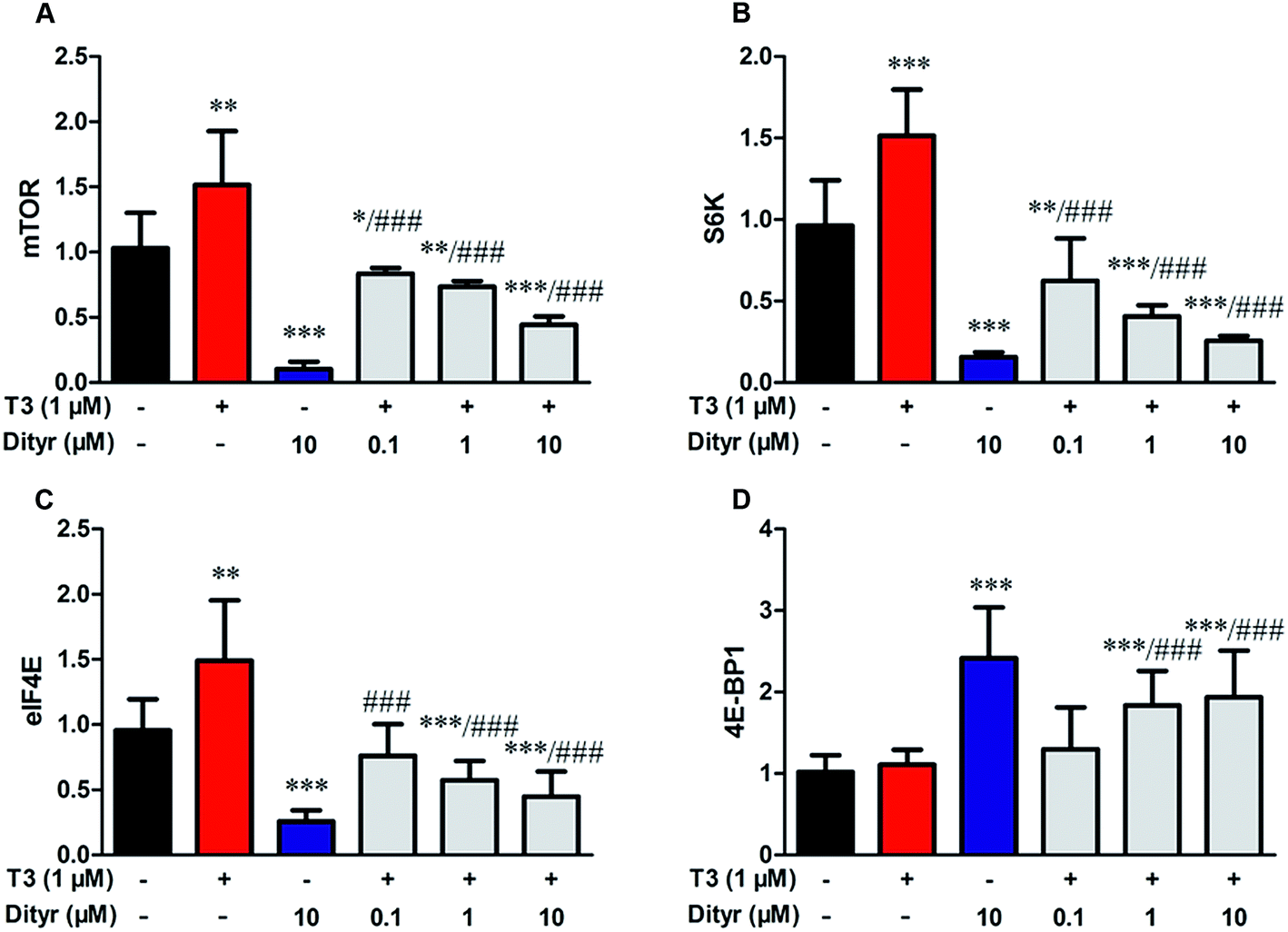 | ||
| Fig. 12 Effects of dityrosine on the mRNA level of T3-activated translational factors involved in the downstream targets of Akt in MIN-6 cells. (A) mTOR, (B) eIF4E, (C) 4E-BP1, (D) S6K. *p < 0.05; *p < 0.01; ***p < 0.001 when compared with untreated cells. #p < 0.05; ##p < 0.01; ###p < 0.001 when compared with T3-only treated cells. | ||
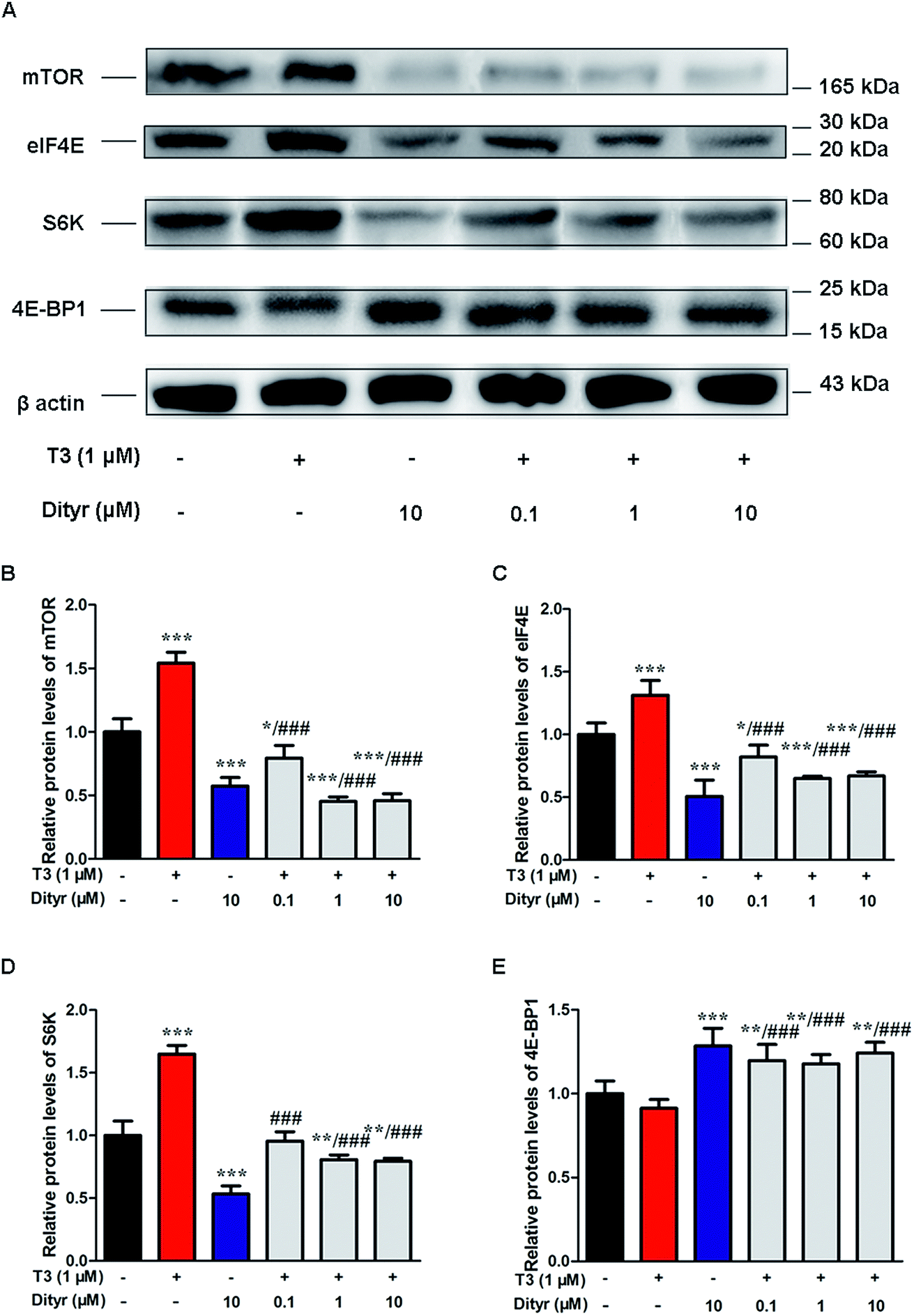 | ||
| Fig. 13 Effects of dityrosine on protein expression of T3-activated translational factors involved in the downstream targets of Akt in MIN-6 cells. *p < 0.05; *p < 0.01; ***p < 0.001 when compared with untreated cells. ###p < 0.001 when compared with T3-only treated cells. | ||
mTOR has been shown to increase protein translation by phosphorylating 4E-BP1, and thus prevent its association with the translation initiation factor eIF4E.41 We examined whether dityrosine-induced mTOR downregulation could inhibit the phosphorylation of 4E-BP1. As shown in Fig. 14, phosphorylating 4E-BP1 decreased in pancreas of mice treated with dityrosine. Furthermore, phosphorylating 4E-BP1 was evident with T3-only treatment, and this effect of T3 was significantly inhibited by supplement with dityrosine. The 4E-BP1 protein level also decreased after dityrosine treatment. These observations further demonstrate that the effect of T3 on the protein synthesis is inhibited by dityrosine.
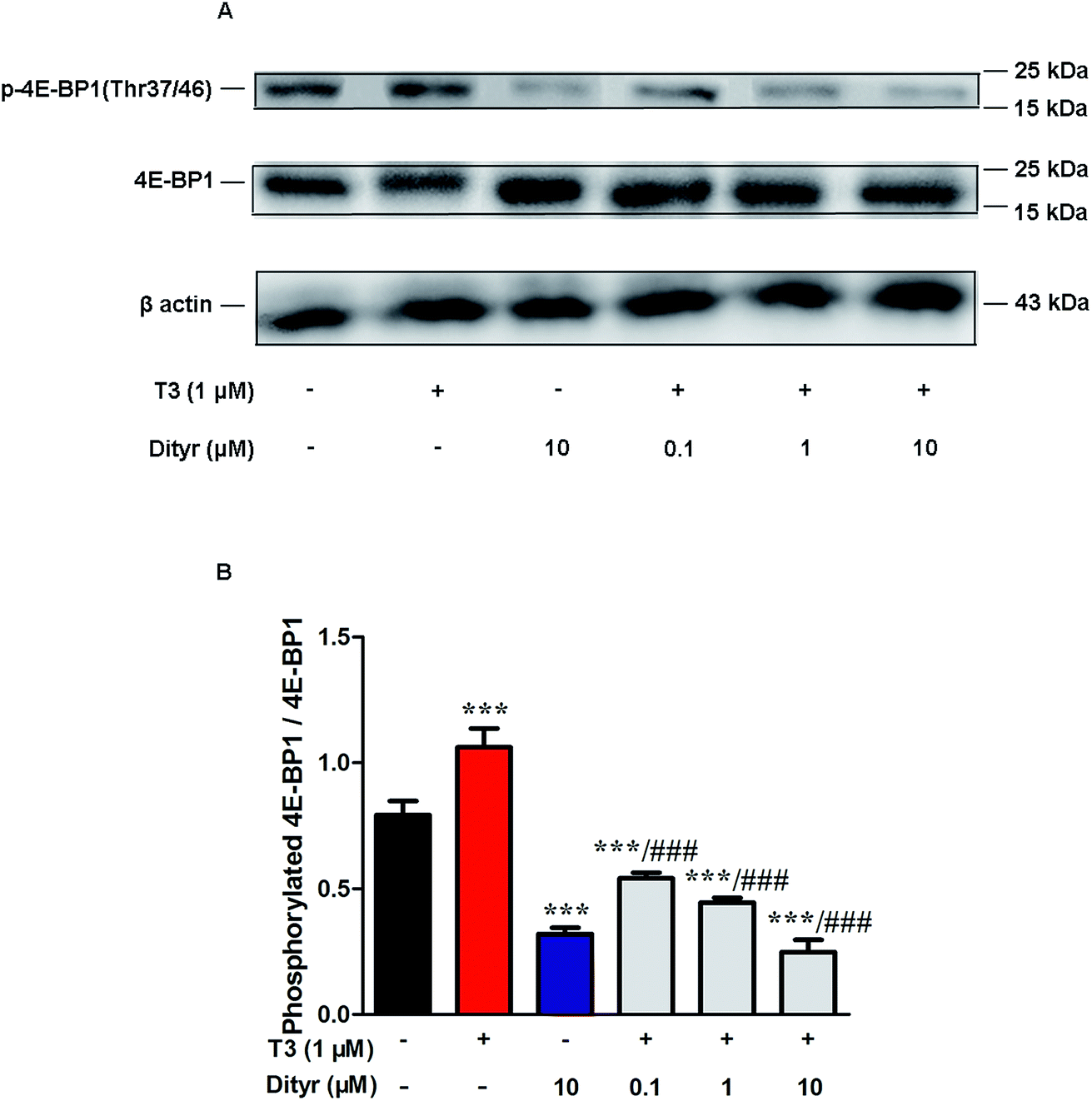 | ||
| Fig. 14 Effects of dityrosine on the T3-induced phosphorylation of 4E-BP1 in MIN-6 cells. (A) Protein level of p-4E-BP1 (Thr37/46) and 4E-BP1 in MIN-6 cells. (B) Ratio of phosphorylated 4E-BP1 and 4E-BP1. *p < 0.05; *p < 0.01; ***p < 0.001 when compared with untreated cells. #p < 0.05; ##p < 0.01; ###p < 0.001 when compared with T3-only treated cells. | ||
4. Discussion
Structure altering of proteins induced by oxidation result in loss of nutritional value and the functionality of food proteins. Oxygen radicals can oxidize apolipoproteins and other plasma proteins leading to the conversion of L-tyrosine to 3–3′-dityrosine.42 Tyrosine residues are susceptible to oxidation and nitration. The structure of oxidized tyrosine products (OTPs) is stable and not easy to be enzymatic hydrolysis, therefore, OTPs are widely detected in food system including meat and dairy products.43,44 Dityrosine is a main oxidative tyrosine product that is reported to be associated with diabetes.45,46 According to some clinical researches, serum dityrosine concentration is reported to be approximately 16.7 mM in individuals with diabetic mellitus, which is significantly higher than that in healthy subjects (0.26 mM).47 It has been suggested some of oxidized tyrosine products, such as dityrosine, can be transported to some important location by systemic circulation to produce harmful effects, including impaired protein synthesis and cytotoxicity.16,48,49 Indeed, we found that OTPs could be absorbed in the intestinal and caused a dityrosine accumulation in pancreas, which was associated with high dityrosine content in feed. However, previous study confirmed that dietary dityrosine would be excreted through the urine within 4–6 h.50 In the current research, the dityrosine in pancreas was quantified after the animal was sacrificed which was done 12 h after the last feeding or gavage. Further experiments are needed to clarify the relationship between dietary OTPs and protein oxidation in pancreas.The elevated fasting blood glucose and impaired glucose tolerance found in this study suggest that OTPs consumption would lead to dysfunction of pancreatic islets. The OTPs treatment apparently resulted in down-regulation of mRNA expressions of genes in the GSIS pathway (Glut2 and Gck) as well as down-regulation of transcription factors that bind to promoters of insulin genes (Pdx-1 and MafA). These findings confirmed that the consumption of OTPs impaired the GSIS pathway via the regulation of glucose uptake and glycolysis. In the former and current researches in our lab, we observed that both OTPs-fed rats and OTPs/dityrosine-treated mice appeared decreased insulin level in the present of enhanced free THs content, which suggested that OTPs might disrupt the T3 action in pancreatic islet,18 and dityrosine was responsible for the pancreatic damage caused by OTPs. T3 signaling in pancreatic β cells is important for normal islet bio-function and glucose homeostasis.51 The disruption of T3 physiological action is associated with hyperglycemia and hypoinsulinemia,52 this symptom is reversible after recovery of T3 function.53,54 A novel finding from the current study is that the molecular docking domain in TRβ1 for dityrosine and T3 might to be the same sites because of the unexpected structure resemblance between dityrosine and T3 according to the data from molecular docking simulations. Thus, we assumed that dityrosine might act as an antagonist to T3. The present study has provided insight into the molecular mechanisms by which dityrosine disrupt T3-induced protein synthesis in pancreatic β cells and potentially inhibit insulin secretion via the TRβ1–Akt–mTOR pathway.
One of the confounding factors which induce the disruption of T3 action may come from the deficiency of thyroid hormone receptors (TRs). The TRα gene encodes for the TRα1 and some structurally related proteins, such as TRα2, which do not bind T3. The thyroid hormone receptor β (TRβ) encodes for the receptor protein TRβ1 and TRβ2, TRβ is the predominant isoform in adult islets.55 MafA is a key transcription factor that binds to the promoter in the insulin gene and has been postulated to regulate insulin transcription in response to blood glucose levels.56 The TRs directly interact with two thyroid response elements (TREs) in the promoter of MafA gene.57 In the present study, dityrosine suppressed T3-regulated glucose-stimulated insulin synthesis might due to the diminished protein level of TRβ1, which in turn caused insufficient of mRNA level of insulin transcription factors.56,57 It has been evidenced the action of T3 on the PI3K/Akt pathway, showing that T3 can stimulate the phosphatidylinositol 3-kinase (PI3K) at the plasma membrane.58 This activation involves the binding of the TRβ1 to the submit p85 of the PI3K.59 The T3/TRβ1-stimulated PI3K activation leads to a series of events including the triggering of the Akt kinase and its downstream mTOR.60 In pancreas, T3 increase pancreatic β cell function potentially via the direct activation of Akt and by the specific action of TRβ1.61 Thus, dityrosine counteracted T3-mediated Akt phosphorylation observed in the current research might contribute to the absence of TRβ1. Mammalian target of rapamycin (mTOR) has been considered an important regulator in maintaining cell size through regulating protein translation, the regulation of mTOR is linked to PI3K/Akt.41,62 As might be expected, phosphorylation of Akt induced by T3 resulted in the upregulation of mTOR when cells were treated by T3 only. This effect was inhibited by dityrosine, which further supported the observed antagonistic effect of dityrosine on T3-mediated Akt activity.
Three downstream targets of mTOR, including eukaryotic translation initiation factor 4E (eIF4E), its binding proteins (4E-BP1, 2, and 3), and ribosomal protein S6 kinases (S6K), are important regulators of protein translation.63 mTOR controls cell growth and proliferation by modulating mRNA translation through regulation of the expressions of eIF4E, 4E-BPs, S6K.64,65 The unwound of mRNA before their coupling with the 43S ribosomal subunit requires specific translation protein, such as eIF4E. eIF4E is attached to specific cytoplasmic proteins, known as 4E-BPs. This attachment suppresses the activity of eIF4E and impairs protein synthesis initiation. 4E-BP1 plays a critical role in regulation of protein translation by binding to and inhibiting eIF4E when it is hypophosphorylated.41,62,66 Thus, the 4E-BPs phosphorylation has been recognized as a critical step in mRNA decoding, as its phosphorylation results in dissociation between eIF4E and 4E-BP, a process that initiates protein synthesis.67 The S6K is also an important regulator of protein translation as their ability to phosphorylate a 40S ribosomal subunit protein, S6, which enables the translation of mRNA containing a 5′-terminal oligopyrimidine track.68 The T3-stimulated mTOR activation is mediated by Akt signaling cascade and leading to the upregulation of S6K expression.60 Some previous observations suggested that T3 stimulated the phosphorylation of 4E-BPs and upregulated the expression of eIF4E and S6K, and T3-mediated phosphorylation and activation of Akt and mTOR were necessary for these responses.30,69 Other evidences also suggested that TRs are essential in regulating T3 action on Akt/mTOR pathway.70,71 The data in the present study indicate a clear antagonistic effect of OTPs and dityrosine on T3-mediated insulin synthesis. This led us to postulate that proteins from T3-regulated translational machinery may be targets for the regulatory action of OTPs on insulin synthesis. Indeed, we found that T3 increased the amount of phosphorylated 4E-BP1, suggesting the binding of eIF4E to 4E-BP1 was blocked. However, cells exposed to dityrosine inhibited the T3-induced 4E-BP1 phosphorylation. In addition, this study demonstrated that T3 upregulated the mRNA expressions of mTOR and S6K in MIN-6 cells, and these effects were repressed by dityrosine. The changes of mTOR and S6K expression induced by dityrosine or T3 reinforced the concept that OTPs acts on T3-regulated insulin production. This may further explain, in part, the decrease in insulin release observed in OTPs-fed rats which appeared symptoms of hyperthyroidism.
Food intake and dietary habits have straightforward impacts on health status. The intake of oxidized proteins and amino acids is silent threat to the health of people all over the world. However, the understanding of biology that governs the beneficial/harmful effects of food components is largely ignored by food scientists, nutritionists, and medical doctors. The current increase in the intake by over-processed food with high protein content and presumably high oxidation rates predicts rise in health disorders already associated to in vivo or dietary oxidative stress.72 As oxidation has been recognized as a potential cause of diseases, some researches have centered on the inhibition effect of antioxidants on protein oxidation during food processing and storage. The addition of vitamin C, E, and rosemary can significantly result in the decrease in polymerization of proteins to inhibit the oxidation of protein.73–77 The former published studies in our lab also demonstrated that lipoic acid could prevent or at least attenuate the metabolic disorders caused by dietary OTPs.11 The underlying mechanisms of the pathological effects of oxidized protein are not fully clear and require further investigation.
In summary, data in the present study lead us to conclude that high-OTPs diet could induce dysfunction of insulin secretion in the pancreas potentially resulted in the disruption of glucose metabolism. The antagonistic effect of OTPs on T3-mediated insulin production depends, at least in part, on alteration of protein synthesis. TRβ1 and translational factors involved in Akt–mTOR signaling pathway are modulated by OTPs. This study can improve the current understanding of the molecular mechanisms whereby OTPs affects glucose metabolism and establishes fundamental data for future experimental studies on dietary OTPs.
Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work is supported by the Chinese Nature Science Foundation (31571841), Natural Science Foundation of China (31700301), Postgraduate Scientific Research Innovation Project of Jiangsu Province (KYLX15_1139).References
- M. J. Davies, Biochim. Biophys. Acta, 2005, 1703, 93–109 CrossRef CAS PubMed.
- M. Estevez, Poult. Sci., 2015, 94, 1368–1378 CrossRef CAS PubMed.
- C. Hellmuth, F. F. Kirchberg, N. Lass, U. Harder, W. Peissner, B. Koletzko and T. Reinehr, J. Diabetes Res., 2016, 2016, 2108909 Search PubMed.
- B. Lu, P. Gu, Y. Xu, X. Ye, Y. Wang, H. Du and J. Shao, Minerva Endocrinol., 2016, 41, 1–9 Search PubMed.
- M. A. Malik, S. N. Basahel, A. Y. Obaid and Z. Khan, Colloids Surf., B, 2010, 76, 346–353 CrossRef CAS PubMed.
- E. R. Stadtman and R. L. Levine, Ann. N. Y. Acad. Sci., 2000, 899, 191 CrossRef CAS PubMed.
- Y. Fukuchi, Y. Miura, Y. Nabeno, Y. Kato, T. Osawa and M. Naito, J. Atheroscler. Thromb., 2008, 15, 185–192 CrossRef CAS PubMed.
- D. A. Malencik and S. R. Anderson, Amino Acids, 2003, 25, 233–247 CrossRef CAS PubMed.
- Z. L. Li, L. Mo, G. Le and Y. Shi, Food Chem. Toxicol., 2014, 64, 86–93 CrossRef CAS PubMed.
- L. Zhuqing Leslie, Y. Shi, G. Le, Y. Ding and Q. Zhao, Oxid. Med. Cell. Longevity, 2016, 1, 1–12 Search PubMed.
- Y. Y. Ding, X. R. Cheng, Z. Q. Li, S. J. Wu, Y. Yang, Y. H. Shi and G. W. Le, RSC Adv., 2017, 7, 26809–26826 RSC.
- Y. Yang, H. Zhang, B. Yan, T. Zhang, Y. Gao, Y. Shi and G. Le, J. Agric. Food Chem., 2017, 65, 6957–6971 CrossRef CAS PubMed.
- M. Abdolvahab, K. Behnaz, B. Sima and M. Teimoorian, J. Global Infect. Dis., 2010, 2, 198–199 CrossRef PubMed.
- D. A. Butterfield, M. Perluigi and R. Sultana, Eur. J. Pharmacol., 2006, 545, 39–50 CrossRef CAS PubMed.
- Y. Ran, B. Yan, Z. Li, Y. Ding, Y. Shi and G. Le, Physiol. Behav., 2016, 164, 292–299 CrossRef CAS PubMed.
- M. El Refaey, C. P. Watkins, E. J. Kennedy, A. Chang, Q. Zhong, K. H. Ding, X. M. Shi, J. Xu, W. B. Bollag, W. D. Hill, M. Johnson, M. Hunter, M. W. Hamrick and C. M. Isales, Mol. Cell. Endocrinol., 2015, 410, 87–96 CrossRef CAS PubMed.
- M. El Refaey, Q. Zhong, W. D. Hill, X. M. Shi, M. W. Hamrick, L. Bailey, M. Johnson, J. Xu, W. B. Bollag, N. Chutkan and C. M. Isales, PloS one, 2014, 9, e91108 Search PubMed.
- Y. Y. Ding, Z. Q. Li, X. R. Cheng, Y. M. Ran, S. J. Wu, Y. Shi and G. Le, Amino Acids, 2017, 49, 1401–1414 CrossRef CAS PubMed.
- X. Terrien, J. B. Fini, B. A. Demeneix, K. W. Schramm and P. Prunet, Aquat. Toxicol., 2011, 105, 13–20 CrossRef CAS PubMed.
- J. D. McKinney and C. L. Waller, Environ. Health Perspect., 1994, 102, 290–297 CrossRef CAS PubMed.
- K. Moriyama, T. Tagami, T. Akamizu, T. Usui, M. Saijo, N. Kanamoto, Y. Hataya, A. Shimatsu, H. Kuzuya and K. Nakao, J. Clin. Endocrinol. Metab., 2002, 87, 5185–5190 CrossRef CAS PubMed.
- U. Rickenbacher, J. D. McKinney, S. J. Oatley and C. C. Blake, J. Med. Chem., 1986, 29, 641–648 CrossRef CAS PubMed.
- A. Oetting and P. M. Yen, Best Pract. Res., Clin. Endocrinol. Metab., 2007, 21, 193–208 CrossRef CAS PubMed.
- C. S. Mitchell, D. B. Savage, S. Dufour, N. Schoenmakers, P. Murgatroyd, D. Befroy, D. Halsall, S. Northcott, P. Raymond-Barker, S. Curran, E. Henning, J. Keogh, P. Owen, J. Lazarus, D. L. Rothman, I. S. Farooqi, G. I. Shulman, K. Chatterjee and K. F. Petersen, J. Clin. Invest., 2010, 120, 1345–1354 CAS.
- E. Danforth Jr and A. Burger, Clin. Endocrinol. Metab., 1984, 13, 581–595 CrossRef CAS.
- G. Roy, M. Nethaji and G. Mugesh, J. Am. Chem. Soc., 2004, 126, 2712–2713 CrossRef CAS PubMed.
- X. Cao, F. Kambe, L. C. Moeller, S. Refetoff and H. Seo, Mol. Endocrinol., 2005, 19, 102–112 CrossRef CAS PubMed.
- T. Simoncini, A. Hafezi-Moghadam, D. P. Brazil, K. Ley, W. W. Chin and J. K. Liao, Nature, 2000, 407, 538–541 CrossRef CAS PubMed.
- J. A. Kuzman, A. M. Gerdes, S. Kobayashi and Q. R. Liang, J. Mol. Cell. Cardiol., 2005, 39, 841–844 CrossRef CAS PubMed.
- A. Kenessey and K. Ojamaa, J. Biol. Chem., 2006, 281, 20666–20672 CrossRef CAS PubMed.
- V. F. Cecilia, P. Valentina, B. Barbara, M. Claudia, M. Simona, M. Giulia, S. Antonio, B. Ercole, T. Vincenzo and M. Silvia, J. Cell. Biochem., 2009, 106, 835–848 CrossRef PubMed.
- C. T. Bussiere, J. R. Lakey, A. M. Shapiro and G. S. Korbutt, Diabetologia, 2006, 49, 2341–2349 CrossRef CAS PubMed.
- Z. L. Li, Y. Shi, Y. Ding, Y. Ran and G. Le, Amino Acids, 2016, 49(2), 241 CrossRef PubMed.
- T. Kurahashi, A. Miyazaki, S. Suwan and M. Isobe, J. Am. Chem. Soc., 2001, 123, 9268–9278 CrossRef CAS PubMed.
- Z. L. Li, Y. Shi, G. Le, Y. Ding and Q. Zhao, Oxid. Med. Cell. Longevity, 2016, 2016, 3123294 Search PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- P. J. Goodford, J. Med. Chem., 1985, 28, 849–857 CrossRef CAS PubMed.
- Y. Gotoh, N. Kamada and D. Momose, J. Biomol. Screening, 2005, 10, 517–523 CrossRef CAS PubMed.
- M. Cao, Y. Long, Y. Tong, J. Wan and N. Tong, Mol. Cell. Biochem., 2012, 366, 183–189 CrossRef CAS PubMed.
- I. N. Shindyalov and P. E. Bourne, Protein Eng., 1998, 11, 739–747 CrossRef CAS PubMed.
- A. R. Tee and J. Blenis, Semin. Cell Dev. Biol., 2005, 16, 29–37 CrossRef CAS PubMed.
- C. Leeuwenburgh, J. E. Rasmussen, F. F. Hsu, D. M. Mueller, S. Pennathur and J. W. Heinecke, J. Biol. Chem., 1997, 272, 3520–3526 CrossRef CAS PubMed.
- T. K. Dalsgaard, J. H. Nielsen, B. E. Brown, N. Stadler and M. J. Davies, J. Agric. Food Chem., 2011, 59, 7939–7947 CrossRef CAS PubMed.
- E. R. Stadtman, Annu. Rev. Biochem., 1993, 62, 797–821 CrossRef CAS PubMed.
- J. W. Baynes, Diabetes, 1991, 40, 405–412 CrossRef CAS PubMed.
- B. Lipinski, J. Diabetes Complications., 2001, 15, 203–210 CrossRef CAS PubMed.
- N. Rabbani, M. V. Chittari, C. W. Bodmer, D. Zehnder, A. Ceriello and P. J. Thornalley, Diabetes, 2010, 59, 1038–1045 CrossRef CAS PubMed.
- H. Gurer-Orhan, N. Ercal, S. Mare, S. Pennathur, H. Orhan and J. W. Heinecke, Biochem. J., 2006, 395, 277–284 CrossRef CAS PubMed.
- S. W. Chan, R. A. Dunlop, A. Rowe, K. L. Double and K. J. Rodgers, Exp. Neurol., 2012, 238, 29–37 CrossRef CAS PubMed.
- S. Bhattacharjee, S. Pennathur, J. Byun, J. Crowley, D. Mueller, J. Gischler, R. S. Hotchkiss and J. W. Heinecke, Arch. Biochem. Biophys., 2001, 395, 69–77 CrossRef CAS PubMed.
- M. C. Medina, J. Molina, Y. Gadea, A. Fachado, M. Murillo, G. Simovic, A. Pileggi, A. Hernandez, H. Edlund and A. C. Bianco, Endocrinology, 2011, 152, 3717–3727 CrossRef CAS PubMed.
- Y. Taguchi, Y. Tasaki, K. Terakado, K. Kobayashi, T. Machida and T. Kobayashi, J. Endocrinol., 2010, 206, 195–204 CrossRef CAS PubMed.
- C. Rochon, I. Tauveron, C. Dejax, P. Benoit, P. Capitan, A. Fabricio, C. Berry, C. Champredon, P. Thieblot and J. Grizard, Clin. Sci., 2003, 104, 7–15 CrossRef CAS PubMed.
- S. Stanicka, K. Vondra, T. Pelikanova, P. Vlcek, M. Hill and V. Zamrazil, Clin. Chem. Lab. Med., 2005, 43, 715–720 CrossRef CAS PubMed.
- X. G. Zhu, J. A. Hanover, G. L. Hager and S. Y. Cheng, J. Biol. Chem., 1998, 273, 27058–27063 CrossRef CAS PubMed.
- H. M. Docherty, C. W. Hay, L. A. Ferguson, J. Barrow, E. Durward and K. Docherty, Biochem. J., 2005, 389, 813–820 CrossRef CAS PubMed.
- C. Aguayo-Mazzucato, A. Koh, I. El Khattabi, W. C. Li, E. Toschi, A. Jermendy, K. Juhl, K. Mao, G. C. Weir, A. Sharma and S. Bonner-Weir, Diabetologia, 2011, 54, 583–593 CrossRef CAS PubMed.
- F. C. Verga, L. Panacchia, B. Bucci, A. Stigliano, M. G. Cavallo, E. Brunetti, V. Toscano and S. Misiti, J. Cell. Physiol., 2010, 206, 309–321 CrossRef PubMed.
- F. Furuya, J. A. Hanover and S.-y. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1780–1785 CrossRef CAS PubMed.
- X. Cao, F. Kambe, L. C. Moeller, S. Refetoff and H. Seo, Mol. Endocrinol., 2005, 19, 102 CrossRef CAS PubMed.
- C. Verga Falzacappa, V. Patriarca, B. Bucci, C. Mangialardo, S. Michienzi, G. Moriggi, A. Stigliano, E. Brunetti, V. Toscano and S. Misiti, J. Cell. Biochem., 2009, 106, 835–848 CrossRef PubMed.
- T. Schmelzle and M. N. Hall, Cell, 2000, 103, 253–262 CrossRef CAS PubMed.
- C. J. Richardson, S. S. Schalm and J. Blenis, Semin. Cell Dev. Biol., 2004, 15, 147 CrossRef CAS PubMed.
- D. A. Guertin and D. M. Sabatini, Cancer Cell, 2007, 12, 9–22 CrossRef CAS PubMed.
- R. J. O. Dowling, I. Topisirovic, T. Alain, M. Bidinosti, B. D. Fonseca, E. Petroulakis, X. S. Wang, O. Larsson, A. Selvaraj, Y. Liu, S. C. Kozma, G. Thomas and N. Sonenberg, Science, 2010, 328, 1172–1176 CrossRef CAS PubMed.
- C. J. Richardson, S. S. Schalm and J. Blenis, Semin. Cell Dev. Biol., 2004, 15, 147–159 CrossRef CAS PubMed.
- R. J. O. Dowling, I. Topisirovic, T. Alain, M. Bidinosti, B. D. Fonseca, E. Petroulakis, X. Wang, O. Larsson, A. Selvaraj and Y. Liu, Science, 2010, 328, 1172–1176 CrossRef CAS PubMed.
- H. B. Jefferies, S. Fumagalli, P. B. Dennis, C. Reinhard, R. B. Pearson and G. Thomas, EMBO J., 1997, 16, 3693–3704 CrossRef CAS PubMed.
- F. Goulart-Silva, S. Teixeira Sda, A. D. Luchessi, L. R. Dos Santos, E. Rebelato, A. R. Carpinelli and M. T. Nunes, Thyroid, 2012, 22, 637–642 CrossRef CAS PubMed.
- C. Pantos, I. Mourouzis, G. Galanopoulos, M. Gavra, P. Perimenis, D. Spanou and D. V. Cokkinos, Horm. Metab. Res., 2010, 42, 718–724 CrossRef CAS PubMed.
- S. Westerholz, A. D. de Lima and T. Voigt, Front. Cell. Neurosci., 2013, 7, 121 Search PubMed.
- M. Estevez and C. Luna, Crit. Rev. Food Sci. Nutr., 2017, 57, 3781–3793 CrossRef CAS PubMed.
- A. Sanchez-Escalante, D. Djenane, G. Torrescano, J. A. Beltran and P. Roncales, Meat Sci., 2001, 58, 421–429 CrossRef CAS PubMed.
- D. Djenane, A. Sanchez-Escalante, J. A. Beltran and P. Roncales, Food Chem., 2002, 76, 407–415 CrossRef CAS.
- S. Lauzurica, J. de la Fuente, M. T. Diaz, I. Alvarez, C. Perez and V. Caneque, Meat Sci., 2005, 70, 639–646 CrossRef CAS PubMed.
- S. Ventanas, M. Estevez, J. F. Tejeda and J. Ruiz, Meat Sci., 2006, 72, 647–655 CrossRef CAS PubMed.
- M. N. Lund, M. S. Hviid and L. H. Skibsted, Meat Sci., 2007, 76, 226–233 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |