
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultraviolet-responsive self-assembled metallomicelles for photocontrollable catalysis of asymmetric sulfoxidation in water†
Mengqiao Gao,
Rong Tan *,
Pengbo Hao,
Yaoyao Zhang,
Jiang Deng and
Donghong Yin
*,
Pengbo Hao,
Yaoyao Zhang,
Jiang Deng and
Donghong Yin
Key Laboratory of Chemical Biology and Traditional Chinese Medicine Research, Ministry of Education, National & Local Joint Engineering Laboratory for New Petro-chemical Materials and Fine Utilization of Resources, Hunan Normal University, Changsha 410081, P. R. China. E-mail: yiyangtanrong@126.com; Tel: +86-731-8872576
First published on 28th November 2017
Abstract
Self-assembled metallomicelles with ultraviolet (UV)-controlled morphologies were constructed from a synthesized azobenzene-containing amphiphilic chiral salen TiIV catalyst. The morphologies of the metallomicelles could be well adjusted by changing the UV irradiation time, and this was confirmed by TEM analyses. The UV-induced change in morphology potentially adjusted the catalyst concentration and/or accessibility in real-time, allowing photocontrollable catalysis of asymmetric sulfoxidation in water. UV-responsive catalytic activities with excellent selectivities were observed over the metallomicelles for a wide range of alkyl aryl sulfides. Moreover, a thermo-responsive NIPAAm block in the catalyst enables it to be easily recovered for steady reuse by thermo-controlled separation. This work constructed a photo-responsive metallomicellar system to carry out metallomicellar catalysis in a controllable way.
Introduction
Metallomicellar systems, which exhibit similar structural and kinetic properties to natural enzymes, have been extensively investigated as effective biomimetic systems for asymmetric catalysis in water.1 The ability to carry out metallomicellar catalysis in a controllable way has profound importance in chemical synthesis, and this area has garnered intense interest. The key to achieving this controllable catalysis was developing a metallomicellar system in which the concentration and/or accessibility of catalytic sites in reaction media could be adjusted in a controlled manner.2 Stimuli-responsive metallomicelles were thus developed to control catalytic reactions via external signals.3 The metallomicelles often undergo instantaneous morphological and/or architectural changes upon exposure to an external stimulus, consequently altering their catalytic properties.4 Lately, we have reported thermo-responsive metallomicelles for enantioselective sulfoxidation at room temperature in water by using a chiral salen TiIV complex as the catalytic site and NIPAAm as the thermo-trigger.3c The metallomicelles underwent reversible switching between self-assembly and precipitation in aqueous system in response to local temperature. At room temperature which was its lower critical solution temperature (LCST), catalysts were amphiphilic and self-assembled to form metallomicelles, accelerating the aqueous sulfoxidation by “concentration effect”. Upon heating above their LCST, they switched to hydrophobic compounds, and thus precipitated from aqueous system for facile recovery. The switchable behavior turned the enantioselective sulfoxidation on or off at will by adjusting local temperature. Nevertheless, in the metallomicellar catalysis, chemical processes were typically limited to a fixed rate and/or selectivity once the catalyst and reaction condition are chosen, control of catalytic activity as the reaction proceeds was difficult to achieve. Logically, if the morphology and/or architecture of the metallomicelles can be adjusted in real-time by external stimuli, controlled catalysis of asymmetric sulfoxidation should be realized.Light irradiation, a green and sustainable energy which could be precisely manipulated in a clean environment without pollution to reaction system, was considered as an ideal external stimulus for controllable catalysis. A variety of light-responsive moieties was introduced in catalysts to give the catalytic system response to light.2a,5 Among them, azobenzene has received much attention in recent years due to its well-known reversible photoisomerization from trans- to cis-configuration. Amphiphilic catalysts containing azobenzene group often undergo conformational and electronic changes upon light irradiation.6 It offered an attractive alternative to modulate morphology and/or architecture of metallomicelles in real-time. With these points in mind, we decided to incorporate azobenzene as the hydrophobic block of amphiphilic chiral salen TiIV catalyst to develop photo-controlled metallomicellar catalytic system of asymmetric sulfoxidation in water. We envisioned that in aqueous media, the azobenzene-containing amphiphilic catalyst should self-assemble through hydrophobic interaction and π–π staking of azobenzene group, forming micelle having hydrophobic azobenzene core. Catalytically active Ti(salen) was grafted on azobenzene segment to ensure the effective shielding of catalytic sites in the hydrophobic compartment, mimetic of enzyme. The self-assembled metallomicelles would behave as biomimetic nanoreactor, accelerating asymmetric sulfoxidation in water. In particular, the salient feature of azobenzene, reversible structural changes in the photoisomerization (trans/cis), should be beneficial for temporal control of the metallomicelles morphology using light. This unique property allowed one to control the metallomicellar catalysis of aqueous asymmetric sulfoxidation by external light irradiation. Thermo-responsive NIPAAm, which underwent hydrophilic-to-hydrophobic transformation at its LCST,7 was selected as the “smart” hydrophilic block to control self-assembly and precipitation of Ti(salen)-containing metallomicelle in the aqueous system.
Herein, well-defined diblock copolymer of poly(NIPAAm-co-azo) (NIPAAm, N-isopropylacrylamide; azo, azobenzene) was synthesized by copolymerizing NIPAAm with N-azo-acrylamide via atom transfer radical polymerization (ATRP).8 Covalently appending chiral salen TiIV complex on the azobenzene block provided an amphiphilic chiral salen TiIV catalyst responsive to temperature and UV irradiation. At the reaction temperature (25 °C), the catalyst was amphiphilic and self-assembled into metallomicelles in water, accelerating the aqueous asymmetric sulfoxidation by “concentration effect”. UV irradiation provoked the morphological change of metallomicelles, which thus controlled the metallomicellar catalysis of asymmetric sulfoxidation in water. After reaction, the catalyst became hydrophobic upon heating above its LCST, and precipitated from aqueous system for recovery. The activity switching was repeatable even after seven cycles.
Results and discussion
Preparation of catalysts
Stimuli-responsive metallomicellar catalysis, in which intrinsic activity could be finely modulated in real-time by external stimulus, was highly desirable in biomimetic catalysis.2,5e Light was attractive stimulus for this stimuli-responsive system, since it offered excellent temporal and spatial resolution, and could be precisely controlled with an appropriate source. The key to achieving the light-controlled catalysis was developing the metallomicelle whose morphology and/or architecture in reaction media could be instantaneously induced by light stimuli.2a,5 Azobenzene with reversible E/Z photoisomerization was an ideal photosensitive group for constructing the light-responsive metallomicelle.6 With these points in mind, we decided to incorporate hydrophobic azobenzene into amphiphilic chiral salen TiIV catalyst to fabricate a Ti(salen)-containing metallomicelle for photocontrollable catalysis of asymmetric sulfoxidation in water. PNIPAAm which underwent reversible switching between hydrophilicity and hydrophobicity at its LCST was incorporated as the “smart” hydrophilic block to ensure not only self-assembly of catalyst for metallomicellar catalysis, but also recovery of catalyst for efficient reuse in the aqueous system.The synthesis of thermo/photo-responsive chiral salen TiIV catalyst of PN427A10-C was illustrated in Scheme 1. First, well-defined diblock copolymer of poly(NIPAAm-co-azo) (denoted as PN427A10), featuring a terminated amino (–NH2) group on azobenzene block, was prepared via ATRP by using N,N-azobis(isobutyronitrile) (AIBN) as a radical initiator and 2-aminoethanethiol hydrochloride (AET) as the chain transfer agent. Successive N-alkylation of the terminal amino group (–NH2) with benzyl chloride group (–CH2Cl) at the 5-position of an reported asymmetric chiral salen ligand (CL) of (R,R)-N-(3,5-di-tert-butylsalicylidene)-N′-[3-tert-butyl-5-chloromethylsalicylidene]-1,2-cyclohexane-diamine9 resulted in covalently appending chiral salen ligand on the hydrophobic segment of PN427A10, as shown in Scheme 1. The strategy ensured shielding of the catalytic motif in hydrophobic domain (protected by the hydrophilic corona) upon self-assembly in water, which was a salient feature of enzyme catalysis. The resultant PN427A10-modified chiral salen ligand was coordinated with Ti(OiPr)4 and further treated with H2O, giving PN427A10-modified chiral salen TiIV complex. Excess Ti(OiPr) was hydrolyzed into TiO2 and then was removed by filtration, affording the thermo/photo-responsive chiral salen TiIV catalyst of PN427A10-C (where 427 and 10 represented the repeated units number of NIPAAm and azobenzene in copolymer, which were determined by 1H NMR spectrum, as shown in ESI†).
 | ||
| Scheme 1 Synthesis of PN427A10-C. | ||
Unlike traditional chiral ((R,R)-salen)TiIV(Oipr)2 complex10 (denoted as neat complex, as shown in Chart 1) that was completely insoluble in water, the obtained PN427A10-C presented temperature-controlled water-solubility with the LCST of 26 °C. The LCST which was approximate to room temperature (25 °C) made PN427A10-C self-assemble at room temperature and separate also at mild temperature (slightly higher than room temperature) in the aqueous asymmetric sulfoxidation. Actually, to regulate the LCST close to room temperature, various rations of NIPAAm to azobenzene blocks have been modulated in the chiral salen TiIV-containing amphiphiles. The detail synthesis and identification of the controlled copolymers were available in the ESI.†
 | ||
| Chart 1 Structure of neat complex. | ||
Characterization of samples
 | ||
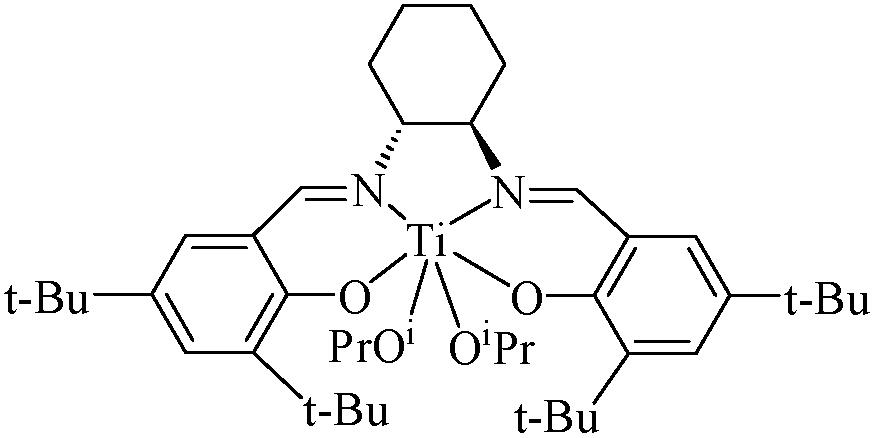
| Fig. 1 Transmittance curves of the PN427A10-C aqueous solutions (concentration: 0.8 mg mL−1, (A)), and optical transmittance at 440 nm of PN427A10-C solution observed upon several cycles under heating at 32 °C and then cooling to 25 °C (B). | ||
 | ||
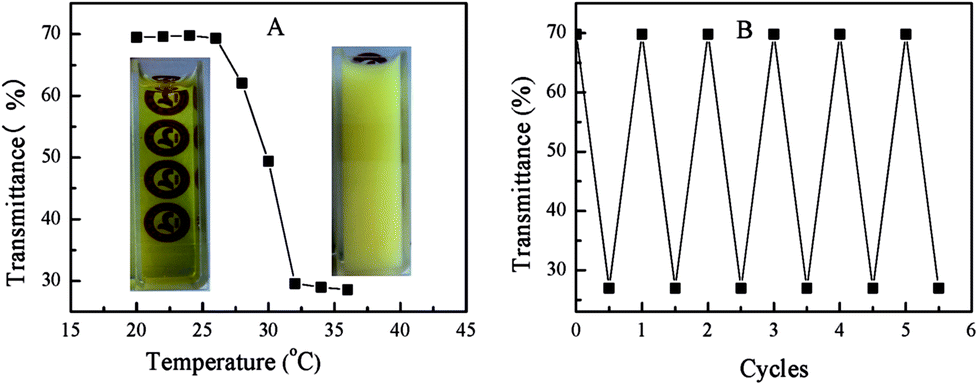
| Fig. 2 UV-vis spectra of PN427A10-C aqueous solution (concentration: 0.5 mg mL−1) upon UV irradiation (365 nm) for different times (A), and optical absorbance at 443 nm of PN427A10-C aqueous solution observed upon several cycles under irradiation at 365 nm for 140 s and keeping in dark for 72 h (B). | ||
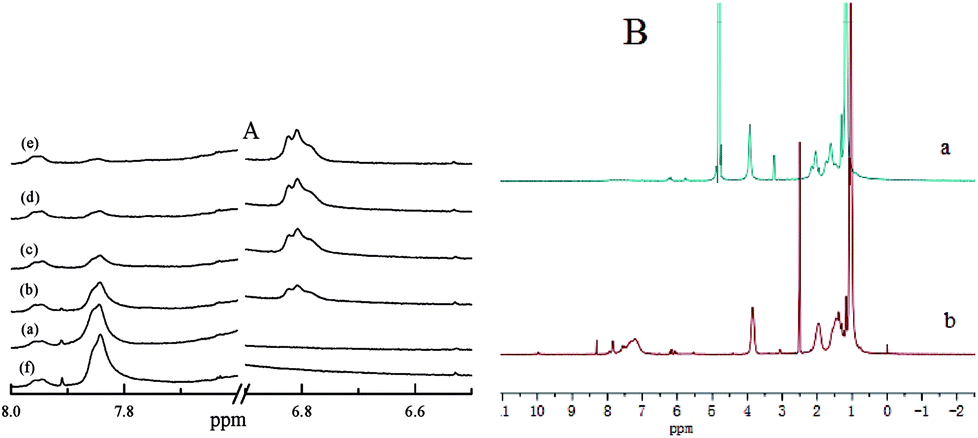 | ||
| Fig. 3 Changes in 1H NMR spectra of PN427A10-C upon photoisomerization in DMSO-d6 (A) (at dark-adapted state (a), after 1 min of UV irradiation (b), after 3 min of UV irradiation (c), after 5 min of UV irradiation (d), after 10 min of UV irradiation (e), and the UV-treated samples kept in dark for 72 h (f)); 1H NMR spectra of dark-adapted PN427A10-C (B) in D2O (a) and in DMSO-d6 (b). | ||
Interestingly, dark-adapted PN427A10-C in DMSO-d6 and D2O gave different 1H NMR spectra, as shown in Fig. 3B. 1H NMR spectrum of PN427A10-C in DMSO-d6 exhibited the distinct H signals of PNIPAAm moiety (δ = 3.84 ppm, O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CNHCH(CH3)2), azobenzene group (δ = 7.84 ppm, NN–Ph–H), and Ti(salen) unit (δ = 1.24, cyclohexyl-H). It indicated the presence of hydrophilic PNIPAAm block, hydrophobic azobenzene block, and catalytically active Ti(salen) unit in PN427A10-C. Whereas, the H signals associated with azobenzene and Ti(salen) moieties almost disappeared when 1H NMR spectrum was carried out in D2O. The difference gave an evidence for the effective shielding of azobenzene and Ti(salen) groups from the aqueous environment upon self-assembly of PN427A10-C in water.13d,15 Hydrophobic shielding of active site made the PN427A10-C an efficient enzyme-mimetic metallomicelle for asymmetric catalysis in water.
CNHCH(CH3)2), azobenzene group (δ = 7.84 ppm, NN–Ph–H), and Ti(salen) unit (δ = 1.24, cyclohexyl-H). It indicated the presence of hydrophilic PNIPAAm block, hydrophobic azobenzene block, and catalytically active Ti(salen) unit in PN427A10-C. Whereas, the H signals associated with azobenzene and Ti(salen) moieties almost disappeared when 1H NMR spectrum was carried out in D2O. The difference gave an evidence for the effective shielding of azobenzene and Ti(salen) groups from the aqueous environment upon self-assembly of PN427A10-C in water.13d,15 Hydrophobic shielding of active site made the PN427A10-C an efficient enzyme-mimetic metallomicelle for asymmetric catalysis in water.
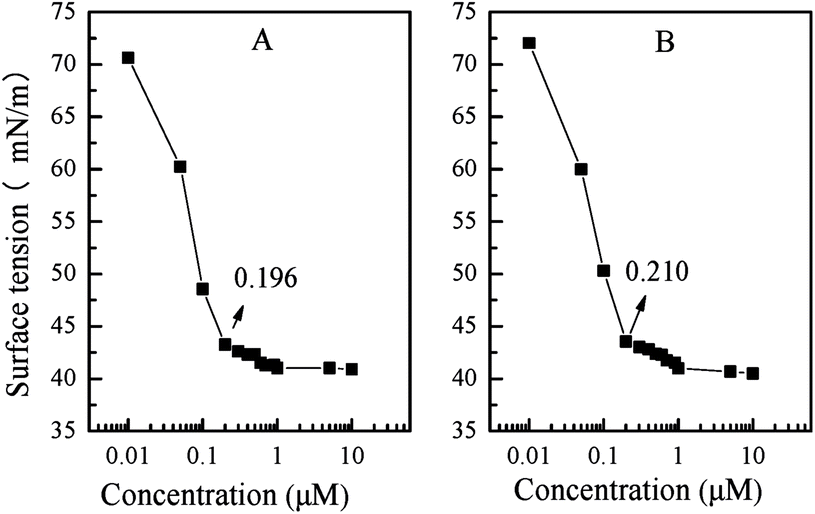 | ||
| Fig. 4 Plots of surface tension against logarithm of concentration for PN427A10-C in water at 25 °C (at dark-adapted state (A), after 5 min of UV irradiation (B)). | ||
 | ||
| Fig. 5 TEM images of PN427A10-C in a dark-adapted state (a) and after UV irradiation for 10 s (b), 30 s (c), 40 s (d), 60 s (e), 70 s (f), 90 s (g), and the UV-treated PN427A10-C kept in dark for 72 h (h). | ||
Interestingly, the adjacent vesicles underwent close contact and mergence to become the bigger vesicles, when the UV irradiation time was prolonged from 30 to 60 s (Fig. 5d and e). Larger vesicles with size greater than 20 nm were observed when the sample was exposed to UV light for 60 s. Increasing cis-isomer in vesicular membrane was the ultimate cause of this fusion process. As the cis-isomer increasing, a larger volume was needed for the cis-form to exist in stable form. It thus brought a loose stacking formation in the vesicular membrane, or even disrupted the bilayer of vesicles. In addition, the surface area of vesicles should expand as a result of the transition from tight to loose packing, leading to an increase in instantaneous surface free energy. Therefore, fusion was the most likely pathway for the vesicles to become stable and reduce their surface free energy. Furthermore, fusion proceeded through a highly bent stalk intermediate, which was formed when hydrophobic tails from two apposing membranes came into contact via a fluctuation and made a hydrophobic bridge between them.18 The formation of the stalk structure required overcoming the free energy barriers. In fact, the change from tight to loose packing could increase the entropy without adversely affecting the enthalpy. It thus lowered the free energy barriers, which benefited the fusion process.18 In other words, the defects in loose packing state with more structural flexibility and plasticity may facilitate the restructuring of the PN427A10-C bilayer in the fusion process. Notably, the regions of defects were not large enough to cause the rupture of individual vesicle, since the rupture would overcome the entanglement of polymer chains and increase the surface free energy. The disruption extent of membranes gradually increased with continuous irradiation. When the vesicles with large disruption areas appeared, rupture of vesicular membrane took place. Subsequently, the disintegrated fragments rearranged to a majority of small vesicles by virtue of the hydrophobic interaction. Indeed, after the sample was irradiated for 60 s, those larger vesicles with damages and defects severely ruptured until almost complete disintegration occurred, and then rearranged to form a large number of very small vesicles (Fig. 5g vs. f). Similar observations of the photo-induced process including fusion, disintegration and rearrangement have also been reported in the literature.19 Upon keeping the UV-treated PN427A10-C in dark for 72 h, spherical nanoparticular emerged again with a size close to 2.0 nm (Fig. 5h). Such morphological changes indicated the responsiveness of the metallomicelles towards UV light, which has the potential to adjust catalyst concentration and/or accessibility in real-time, allowing for fine control of the asymmetric sulfoxidation in water.
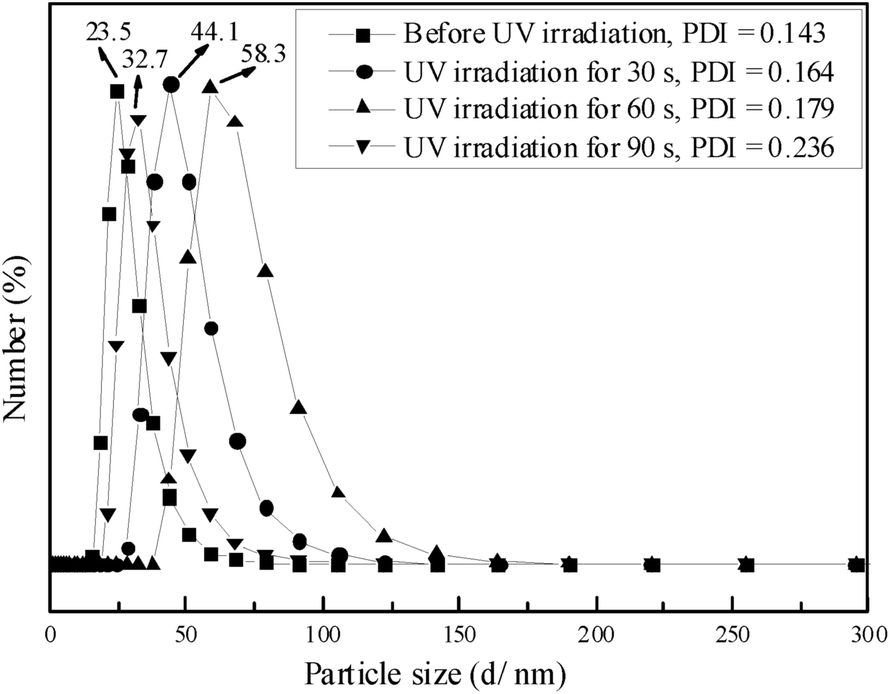 | ||
| Fig. 6 Size distribution of self-assembled PN427A10-C in water at a concentration of 0.5 mg mL−1 under different UV irradiation time. | ||
Catalytic performances
Encouraged by the photo-induced change in morphology of PN427A10-C, the self-assembled Ti(salen)-containing metallomicelles was employed as the photo-sensitive bio-nanoreactor for controlled catalysis of asymmetric sulfoxidation in water. Phenyl methyl sulfide was used as a substrate to investigate the photo-mediated catalytic performance of PN427A10-C. Before reaction, dark-adapted PN427A10-C in which all the azobenzene chromophores were in trans-state was exposed to UV light (λ = 365 nm) for a certain time, and then wrapped with aluminum foil to avoid the disturbance of natural light. The results are presented in Table 1. Traditional chiral salen TiIV complex (denoted as neat complex, Chart 1) was also employed for comparison.| Entry | Catalyst | Substrates | Products | UV-irradiation time (s) | T (min) | Conv.b/% | Sel.c/% | eed/% |
|---|---|---|---|---|---|---|---|---|
| a Catalyst (0.1 mol% of substrate, based on titanium content), substrate (1.0 mmol), H2O2 (30 wt%, 1.2 mmol, slowly added within 15 min), H2O (2.0 mL), 25 °C.b Determined by GC.c Chemoselectivity to sulfoxide (determined by GC).d Determined by HPLC (Daicel Chiralpak AD column). | ||||||||
| 1 | PN427A10-C |  |
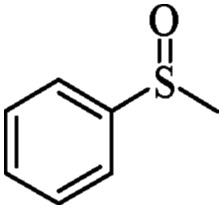 |
0 | 75 | 82 | 95 | 99 (R) |
| 2 | PN427A10-C | 10 | 75 | 76 | 99 | 99 (R) | ||
| 3 | PN427A10-C | 20 | 75 | 88 | 99 | 99 (R) | ||
| 4 | PN427A10-C | 30 | 75 | 99 | 99 | 99 (R) | ||
| 5 | PN427A10-C | 40 | 75 | 76 | 99 | 99 (R) | ||
| 6 | PN427A10-C | 50 | 75 | 85 | 99 | 99 (R) | ||
| 7 | PN427A10-C | 60 | 75 | 99 | 99 | 99 (R) | ||
| 8 | PN427A10-C | 70 | 75 | 78 | 99 | 99 (R) | ||
| 9 | PN427A10-C | 80 | 75 | 86 | 99 | 99 (R) | ||
| 10 | PN427A10-C | 90 | 75 | 98 | 99 | 99 (R) | ||
| 11 | Neat complex | — | 75 | 8 | 86 | 83 (R) | ||
| 12 | PN427A10-C | 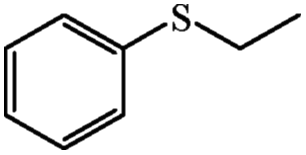 |
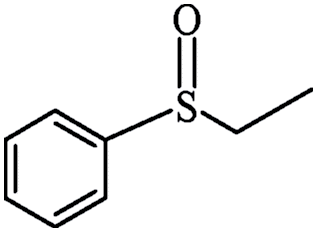 |
0 | 100 | 54 | 90 | 97 (R) |
| 13 | PN427A10-C | 60 | 100 | 90 | 91 | 97 (R) | ||
| 14 | Neat complex | — | 100 | 35 | 66 | 82 (R) | ||
| 15 | PN427A10-C | 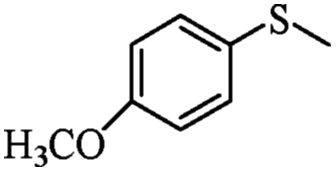 |
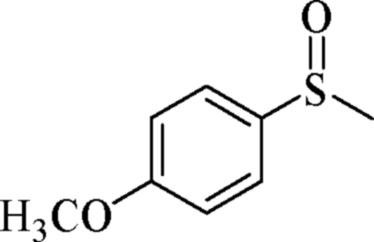 |
0 | 75 | 90 | 92 | 94 (R) |
| 16 | PN427A10-C | 60 | 75 | 94 | 92 | 97 (R) | ||
| 17 | Neat complex | — | 75 | 23 | 78 | 70 (R) | ||
| 18 | PN427A10-C | 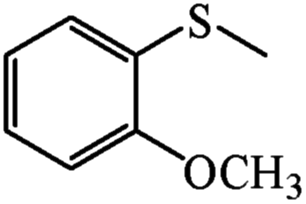 |
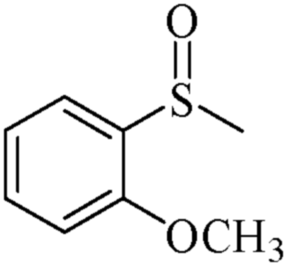 |
0 | 105 | 75 | 98 | 99 (R) |
| 19 | PN427A10-C | 60 | 105 | 91 | 99 | 99 (R) | ||
| 20 | Neat complex | — | 105 | 43 | 91 | 76 (R) | ||
As expected, PN427A10-C-based metallomicelles significantly accelerated the asymmetric oxidation of methyl aryl sulfides in water using H2O2 as oxygen source, giving high activity and remarkable selectivity (in terms of chemo- and enantioselectivity) (Table 1, entries 1–10). Only 0.1 mol% of PN427A10-C was sufficient for giving almost quantitative yield (>99%) of phenyl methyl sulfoxide with outstanding chiral induction (99%) when catalyst was treated with UV irradiation for 30, 60, or 90 s (Table 1, entries 4, 7, and 10). In contrast, extremely low conversion (8%) with disappointing chemoselectivity (86%) and ee value (83%) was obtained under identical conditions when the reaction was catalyzed by neat complex (Table 1, entry 11). The results demonstrated the advantages of metallomicellar catalytic approach over self-assembled PN427A10-C. The formed metallomicelles not only showed good water-solubility, but also possessed a hydrophobic compartmentalized structure which allowed effectively shielding active sites from aqueous environment, reminiscent of enzyme.21 Dense chiral salen TiIV complexes confined in the hydrophobic domain may enforce a cooperative reaction pathway favourable for the asymmetric sulfoxidation.3c,20,22 Substrates were also effectively sequestered by the hydrophobic compartment from the surrounding aqueous environment, creating a highly concentrated environment for efficient catalysis. Furthermore, synergistic effects between the metal-catalyzed center and the hydrophobic microenvironment in the metallomicelle led to high selectivity.23
Photo-responsive behavior of the metallomicelles made catalytic efficiency of PN427A10-C sensitive to UV irradiation. Indeed, the catalytic activity of PN427A10-C was adjusted by UV irradiation in the aqueous asymmetric sulfoxidation. Before UV treatment, dark-adapted PN427A10-C gave 82% conversion of phenyl methyl sulfide in water within 75 min (Table 1, entry 1). UV irradiation mediated the catalytic performance of PN427A10-C in the aqueous asymmetric sulfoxidation (Table 1, entries 1–10). In particular, almost quantitative yield (>99%) of (R)-phenyl methyl sulfoxide was obtained when the dark-adapted PN427A10-C was exposed to UV light for 30 s (Table 1, entry 4). Enhanced activity should be related to the micelle to vesicle transition behavior of PN427A10-C upon UV treatment, as shown in TEM images. Despite both confined catalysis, vesicles represented bubble-like structures in which a water-containing interior was enclosed by a double-layer membrane. The bilayer membrane was a more conducive structure for sequestering substrates from the surrounding aqueous environment, creating a highly concentrated environment favorable for efficient sulfoxidation.3c Varied UV irradiation time resulted in the variation of catalytic efficiency of PN427A10-C (Table 1, entries 5–10). Quantitative yield of (R)-phenyl methyl sulfoxide was also observed when PN427A10-C was treated by UV irradiation for 60 or 90 s. Various activities should be related to the photo-induced change in morphology of the metallomicelles. During the irradiation of UV light, the vesicles underwent photo-provoked disruption, fusion, disintegration and rearrangement in water. Relatively integrated double-layer membrane structure of the vesicles was observed when PN427A10-C was treated by UV light for 60 or 90 s, which was favourable for the metallomicellar catalysis. The UV-responsive catalytic efficiency of PN427A10-C made controlled catalysis of asymmetric sulfoxidation in water come true.
Kinetics was used to further investigate the advantages of the metallomicellar catalysis approach in aqueous reaction, as well as the photo-controlled catalytic performance of PN427A10-C-based metallomicelle. Corresponding kinetic curves and rate curves of asymmetric sulfoxidation of methyl phenyl sulfide over PN427A10-C in water (after 60 s of UV irradiation, after 40 s of UV irradiation, and at dark-adapted state) were shown in Fig. 7. Obviously, gradient increase in conversion of sulfide was observed over PN427A10-C either at dark-adapted state or upon UV treatment, when asymmetric sulfoxidations were performed in water (Fig. 7A(a–c)). The corresponding rate constant (kobs) initially rose rapidly due to the increasing concentration of substrate in hydrophobic microenvironment, went through a maximum, and then drastically decreased due to a dilution effect (Fig. 7B(a–c)).24 The featured kinetics demonstrated confined catalysis of asymmetric sulfoxidation in water over PN427A10-C. To evaluate the advantages of metallomicelle, we carried out the reaction with dark-adapted PN427A10-C in ethanol, a solvent in which PN427A10-C, sulfide and H2O2 could be well dissolved. As expected, at the same catalyst concentration, dark-adapted PN427A10-C in ethanol was less efficient than that in water, although the ethanol-based system was homogeneous (Fig. 7d vs. c). Logically, in ethanol, PN427A10-C may gather together just via π–π stacking of azobenzene species from neighbouring catalyst, rather than packing to form the confined hydrophobic pocket. It thus couldn't sequester organic substrate from surrounding environment through hydrophobic effect. Low local concentration of substrate in the ethanol system resulted in the unsatisfied activity. Interestingly, despite also homogeneous system, traditional neat complex in ethanol was far less efficient than PN427A10-C under identical conditions (Fig. 7e vs. d). Extremely low kobs value was observed over the traditional neat complex (Fig. 7B(e)). It was reasonable that traditional neat complex without azobenzene moiety could not gather together in ethanol system to achieve concentrated catalytic sites and organic substrate. Low local concentration of catalytic species and sulfides were insufficient for the efficient sulfoxidation. The results were consistent with our hypothesis that the high local concentrations of catalytic species and substrate were crucial for this system to be active, and this was only achieved in the metallomicellar system in water. Notably, despite all metallomicellar catalysis, the kobs over PN427A10-C varied with the UV irradiation time, and highest efficiency was observed when PN427A10-C was treated with UV light for 60 s (Fig. 7a). The observations did agree with the ability to photo-modulate the activity of PN427A10-C in asymmetric sulfoxidation in water.
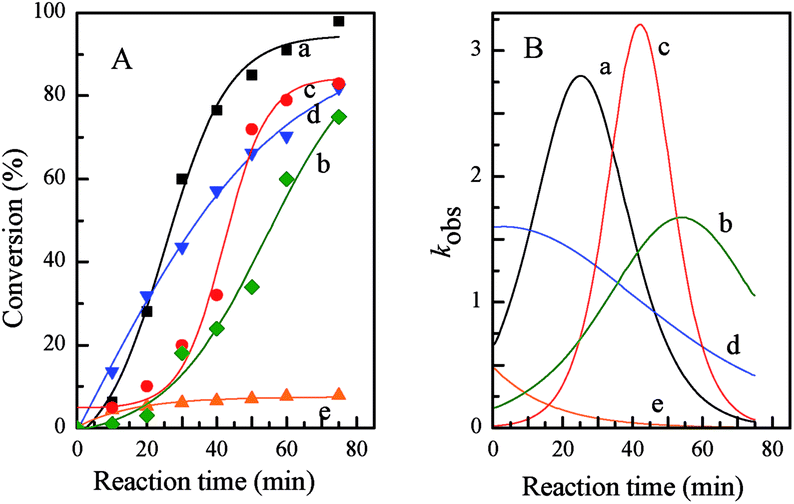 | ||
| Fig. 7 Kinetic curves (A) and rate curves (B) of asymmetric sulfoxidation of methyl phenyl sulfide over PN427D10-C in water (after 60 s of UV irradiation (a), after 40 s of UV irradiation (b), and at dark-adapted state (c)), over dark-adapted PN427D10-C (d) and over neat complex (e) in ethanol. | ||
The UV-responsive catalytic efficiency over PN427A10-C was also noticeable for other aryl methyl sulfides, such as ethyl phenyl sulfide, methyl p-methoxyphenyl sulfide, and methyl o-methoxyphenyl sulfide. PN427A10-C after 60 s of UV irradiation always gave high activity than the dark-adapted PN427A10-C in corresponding sulfoxidations (Table 1, entry 13 vs. 12, entry 16 vs. 15, entry 19 vs. 18). All the aryl methyl sulfides get quantitatively oxidized to the corresponding sulfoxides with excellent chemo- (91–99%) and enantioselectivity (97–99%) within 105 min in water with the employment of UV-treated PN427A10-C (Table 1, entries 13, 16, and 19). Especially, almost quantitative chiral induction to (R)-sulfide (>99%) was observed for the methyl o-methoxyphenyl sulfide over the PN427A10-C after 60 s of UV irradiation (Table 1, entry 16). The results suggested the universality of PN427A10-C in green asymmetric sulfoxidation. Traditional neat complex was also inactive in the aqueous sulfoxidations systems due to water-incompatibility (Table 1, entries 14, 17, and 20).
Apart from photo-responsive catalytic activity, PN427A10-C also exhibited the reversibility of thermal-driven water-solubility switching, as shown in Fig. 1. The salient features allowed for the facile recovery of PN427A10-C from the aqueous phase by thermo-controlled separation. Especially, the LCST close to room temperature made the recovery of catalyst for reuse under mild local temperature (at 32 °C). Fig. 8 showed the reusability of PN427A10-C after 60 s of UV treatment or in dark-adapted state in asymmetric oxidation of methyl phenyl sulfide in water.
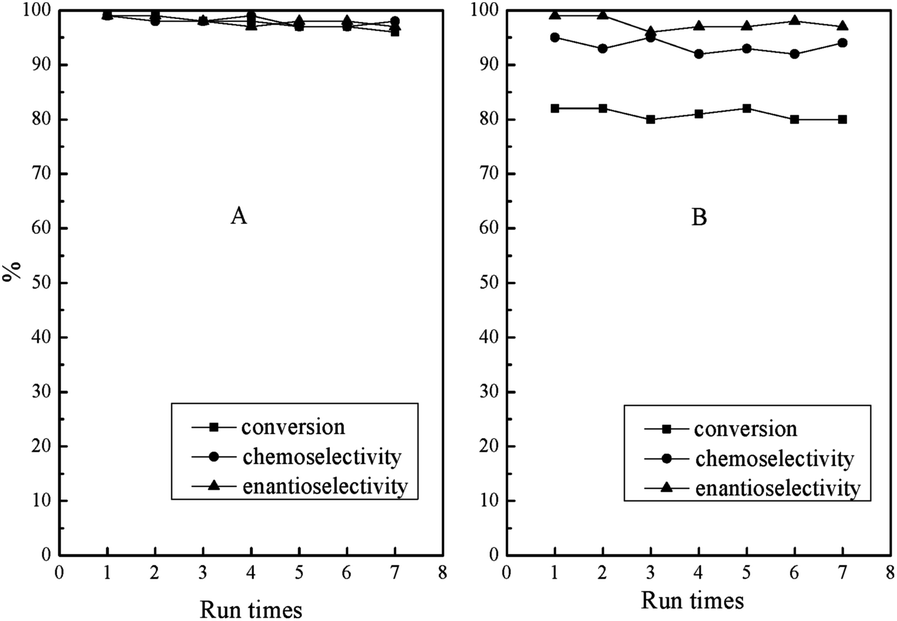 | ||
| Fig. 8 Reuse of PN427A10-C after 60 s of UV treatment (A) or in dark-adapted state (B) in asymmetric sulfoxidation of methyl phenyl sulfide in water using aq. H2O2 as an oxidant at 25 °C. | ||
To our delight, the catalyst either at dark-adapted state or upon UV treatment could be reused up to seven times without significant loss in activity and selectivity. Leaching test of catalyst revealed negligible leaching loss of titanium species (less than 0.1 ppm by ICP) to reaction medium during the oxidation. Chemical analysis of the recovered PN427A10-C gave the titanium content (0.0172 mmol g−1) almost identical to that of the fresh one (0.018 mmol g−1). Furthermore, oxidative decomposition of PN427A10-C, a main reason for the deactivation of chiral salen TiIV catalyst in H2O2-based oxidation,25 also did not occur during the reaction. The excellent stability of catalyst should arise from the shielding of the chiral salen TiIV complex in a hydrophobic pocket, which protected the catalytic site from oxidative decomposition by excluding excess H2O2 from the hydrophobic compartment.
Conclusions
In conclusion, controllable metallomicellar catalysis of aqueous asymmetric sulfoxidation has been realized by using photo-responsive self-assembled metallomicelle which contained azobenzene and chiral salen TiIV complexes in the hydrophobic compartment. Catalytic activity of the metallomicelle could be precisely controlled by UV irradiation time, due to the UV-induced change in morphology which adjusted catalyst concentration and/or accessibility in real-time. Furthermore, the catalysts could be facilely recovered from the aqueous system for efficient reuse by adjusting the local temperature. The photo-controlled catalytic efficiency, as well as outstanding reusability, made the metallomicelle highly promising for controllable catalysis of various organic reactions in water. And also, the responsive self-assembly approach inspired researchers to develop other controllable metallomicellar catalysis system for a much wider range of application.Experimental section
Materials and reagents
Tetra-isopropyl titanate, aminoazobenzene and methyl aryl sulfides were obtained by J&K. N,N-Azobis(isobutyronitrile) (AIBN), NIPAAm, 2-aminoethanethiol hydrochloride, and L(+)-tartaric acid were purchased from Acros. 2-tert-Butyl phenol was purchased from Alfa Aesar. Other commercially available chemicals were laboratory grade reagents from local suppliers. All solvents were purified by standard procedures. NIPAAm was purified by recrystallization from n-hexane and dried in vacuo before use.Methods
UV-vis spectroscopy was carried out on a UV-vis Agilent 8453 spectrophotometer. Molecular weight of the synthesized copolymers were obtained by gel permeation chromatography (GPC) on an Alltech Instrument (Alltech, America) equipped with an Alltech ELSD 800 detector. NMR spectrum of samples was recorded on a BRUKER AVANCE-500 spectrometer with TMS as an internal standard. The measurements were repeated for at least three times to ensure good reproducibility. The critical micellization concentration (CMC) was investigated using the surface tension method. The surface tension of an aqueous solution of catalyst was measured as a function of catalyst concentration (10 × 10−3 to 1.0 mmol L−1) on a Krüss K12 tensiometer using the Wilhelmy plate method at 25 °C. The surface tension vs. catalyst concentration plot gave information on the CMC. Morphologies of the self-assembled aggregates were observed by TEM on a Microscope JEM-2100F at an accelerating voltage of 200 kV. Samples were prepared by depositing aqueous solution (0.5 mg mL−1) onto a carbon-coated copper grid, followed by removal of excess solution by blotting the grid with filter paper. The samples were dried for 72 h at room temperature in a desiccator containing dried silica gel. After that, the samples were negatively stained by phosphotungstic acid and dried for another 72 h before examination. Dynamic light scattering (DLS) was performed using a MS2000 Laser Particle Size Analyzer (Malvern, UK). The sample solutions for measurements were prepared for concentrations of 0.5 mg mL−1 followed by filtering through a 0.45 lm disposable polyamide (PA) membrane to free it from dust particles. Light transmittance was fixed at 633 nm with the scattering angle of 90°. The mean diameters of self-assemblies were obtained from the number distribution curves produced by the particle analyzer. The titanium content in samples was determined by inductively coupled plasma mass spectrometry (ICP-MS) on a NexION 300X analyzer (Perkin-Elmer Corp.). Optical rotation of samples was measured in dichloromethane on a WZZ-2A Automatic Polarimeter. The dark-adapted PN427A10-C was obtained by the irradiation of visible light (λ = 410 nm) for 30 min, and then being kept in the dark for 72 h to ensure that all the azobenzene chromophores were in trans-state.Preparation of PN427A10-C
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at room temperature. Acryloyl chloride (4.5 mmol, 0.410 g) in acetonitrile (5 mL) was then dropwise added into the solution. The mixture was stirred at 55 °C for 4 h, and the amidation progress was monitored by thin-layer chromatography. After cooling to room temperature, the reaction mixture was filtered through a 0.2 μm PTFE filter. The orange residue was washed with water and purified by recrystallization in ethanol to get N-azo-acrylamide. FT-IR (KBr): γmax/cm−1 3280, 3199, 3134, 3068, 1670, 1600, 1560. 1H NMR (CDCl3, 500 MHz) δ (ppm): 7.94–7.89 (s, 4H, NN–Ph–H), 7.76 (s, 2H, OC–NH–Ph–H), 7.52–7.44 (s, 3H, aromatic), 6.50–5.81 (s, 3H, OC–CHCH2).N–Ph–H), 7.57–7.56 (m, 20H, OC–NH–Ph–H), 7.52 (m, 30H, NN–Ph–H), 7.30–7.19 (m, 437H, OC–NH–CH and OC–NH–Ph), 3.83 (m, 427H, CH3–CH–CH3 in NIPAAm), 2.93 (m, 2H, S–CH2–CH2–NH2), 2.63 (m, 2H, S–CH2–CH2–NH2), 2.36 (m, 2H, S–CH2–CH2–NH2), 1.96 (m, 437H, CH2–CH– in NIPAAm and N-azo-acrylamide), 1.44 (m, 874H, –CH2–CH– in NIPAAm and N-azo-acrylamide), 1.03 (s, 2562H, CH3–CH–CH3). The Mn of PN427A10 is about 30825 g mol−1 based on GPC.N–), 7.90 (s, 2H, Ph–H in Ti(salen)), 7.84 (m, 42H, –NN–Ph–H), 7.60–7.56 (m, 20H, OC–NH–Ph–H), 7.52 (m, 30H, NN–Ph–H), 7.29–7.20 (m, 437H, OC–NH–CH and OC–NH–Ph), 3.83 (m, 427H, CH3–CH–CH3 in NIPAAm), 3.17–3.16 (m, 2H, cyclohexyl-H), 2.92–2.90 (m, 2H, NH–CH2–Ti(salen)), 2.63 (m, 2H, S–CH2–CH2–NH), 2.36 (m, 2H, S–CH2–CH2–NH), 2.17 (m, 2H, CH3–CH–CH3 of iPrO- in Ti(salen)), 1.95 (m, 437H, CH2–CH– in NIPAAm and N-azo-acrylamide), 1.44 (s, 874H, –CH2–CH– in NIPAAm and N-azo-acrylamide), 1.22 (m, 8H, cyclohexyl-H), 1.17 (m, 39H, –CH3 of t-Bu and iPrO- in Ti(salen)), 1.03 (s, 2562H, CH3–CH–CH3). [α]20D = −16.0 (C = 0.005 g mL−1 in CH2Cl2). Titanium content: 0.018 mmol g−1. The LCST of PN427A10-C is 26 °C based on transmittance measurement.
:1, v/v) at room temperature. Acryloyl chloride (4.5 mmol, 0.410 g) in acetonitrile (5 mL) was then dropwise added into the solution. The mixture was stirred at 55 °C for 4 h, and the amidation progress was monitored by thin-layer chromatography. After cooling to room temperature, the reaction mixture was filtered through a 0.2 μm PTFE filter. The orange residue was washed with water and purified by recrystallization in ethanol to get N-azo-acrylamide. FT-IR (KBr): γmax/cm−1 3280, 3199, 3134, 3068, 1670, 1600, 1560. 1H NMR (CDCl3, 500 MHz) δ (ppm): 7.94–7.89 (s, 4H, NN–Ph–H), 7.76 (s, 2H, OC–NH–Ph–H), 7.52–7.44 (s, 3H, aromatic), 6.50–5.81 (s, 3H, OC–CHCH2).N–Ph–H), 7.57–7.56 (m, 20H, OC–NH–Ph–H), 7.52 (m, 30H, NN–Ph–H), 7.30–7.19 (m, 437H, OC–NH–CH and OC–NH–Ph), 3.83 (m, 427H, CH3–CH–CH3 in NIPAAm), 2.93 (m, 2H, S–CH2–CH2–NH2), 2.63 (m, 2H, S–CH2–CH2–NH2), 2.36 (m, 2H, S–CH2–CH2–NH2), 1.96 (m, 437H, CH2–CH– in NIPAAm and N-azo-acrylamide), 1.44 (m, 874H, –CH2–CH– in NIPAAm and N-azo-acrylamide), 1.03 (s, 2562H, CH3–CH–CH3). The Mn of PN427A10 is about 30825 g mol−1 based on GPC.N–), 7.90 (s, 2H, Ph–H in Ti(salen)), 7.84 (m, 42H, –NN–Ph–H), 7.60–7.56 (m, 20H, OC–NH–Ph–H), 7.52 (m, 30H, NN–Ph–H), 7.29–7.20 (m, 437H, OC–NH–CH and OC–NH–Ph), 3.83 (m, 427H, CH3–CH–CH3 in NIPAAm), 3.17–3.16 (m, 2H, cyclohexyl-H), 2.92–2.90 (m, 2H, NH–CH2–Ti(salen)), 2.63 (m, 2H, S–CH2–CH2–NH), 2.36 (m, 2H, S–CH2–CH2–NH), 2.17 (m, 2H, CH3–CH–CH3 of iPrO- in Ti(salen)), 1.95 (m, 437H, CH2–CH– in NIPAAm and N-azo-acrylamide), 1.44 (s, 874H, –CH2–CH– in NIPAAm and N-azo-acrylamide), 1.22 (m, 8H, cyclohexyl-H), 1.17 (m, 39H, –CH3 of t-Bu and iPrO- in Ti(salen)), 1.03 (s, 2562H, CH3–CH–CH3). [α]20D = −16.0 (C = 0.005 g mL−1 in CH2Cl2). Titanium content: 0.018 mmol g−1. The LCST of PN427A10-C is 26 °C based on transmittance measurement.Catalyst testing
Before reaction, PN427A10-C was irradiated by visible light (λ = 410 nm) for 30 min, and then kept in the dark for 72 h to ensure that all the azobenzene chromophores were in trans-state. The dark-adapted PN427A10-C (0.1 mol% substrate, based on the titanium content in catalyst) and methyl aryl sulfides (1.0 mmol) were added to H2O (2 mL) under stirring at 25 °C. The mixture was exposed to UV light (λ = 365 nm) for a certain time, and then wrapped with aluminum foil. H2O2 (30 wt%, 1.2 mmol) was dropwise added into the solution within 15 min. The resulting mixture was stirred at room temperature until the reaction was judged to be complete based on GC analysis. Then, the reaction mixture was heated to 32 °C. Catalyst was precipitated out from the reaction system completely, washed with diethyl ether (3 × 5 mL), dried in a vacuum, and finally recharged with fresh substrate and oxidant for the next catalytic cycle. The supernatants separated from reaction system were extracted with dichloromethane thrice. Combined organic phase was concentrated in vacuum. Further purification of the residue by chromatography on silica gel (petroleum ether/ethyl acetate, 1.5/1) afforded pure chiral sulfoxides. The products have been identified by 1H NMR and 13C NMR spectra. The conversion and chemoselectivity of chiral sulfoxides were measured by a 6890N gas chromatograph (Agilent Co.) equipped with a capillary column (HP19091G-B213, 30 m × 0.32 mm × 0.25 μm) and a FID detector. Ee values of corresponding chiral sulfoxides were determined by HPLC analysis using the Daicel Chiralpak AD columns. Detailed NMR spectra and HPLC analysis for the chiral sulfoxides were available in ESI.†:5 (v/v)); flow rate = 1.0 mL min−1; 25 °C; λ = 254 nm; major enantiomer tR = 4.4 min, minor enantiomer tS = 5.2 min; 1H NMR (CDCl3, 500 MHz) δ (ppm): 2.49 (s, 3H, Me), 7.27–7.29 (m, 3H, ArH), 7.42–7.44 (m, 2H, ArH); 13C NMR (CDCl3, 125 MHz) δ (ppm): 43.1 (SCH3), 122.8, 128.6, 130.3, 144.9 (ArC).:8 (v/v)); flow rate = 1.0 mL min−1; 25 °C; λ = 254 nm; major enantiomer tR = 6.4 min and minor enantiomer tS = 8.2 min; 1H NMR (CDCl3, 500 MHz) δ (ppm): 7.59–7.53 (m, 2H, ArH), 7.52–7.47 (m, 3H, ArH), 2.92–2.72 (m, 2H, –CH2–), 1.19–1.16 (m, 3H, Me); 13C NMR (CDCl3, 125 MHz) δ (ppm): 5.91 (CH3), 50.24 (SCH2), 124.13, 129.09, 130.88, 143.25 (ArC).:6 (v/v)); flow rate = 1.0 mL min−1; 25 °C; λ = 254 nm; major enantiomer tR = 5.5 min and minor enantiomer tS = 6.6 min; 1H NMR (CDCl3, 500 MHz) δ (ppm): 7.60–7.57 (d, 2H, ArH), 7.03–7.01 (d, 2H, ArH), 3.84 (s, 3H, OCH3), 2.69 (s, 3H, SCH3); 13C NMR (CDCl3, 125 MHz) δ (ppm): 43.9 (SCH3), 55.5 (OCH3), 114.8, 125.4, 136.6, 162.0 (ArC).:5 (v/v)); flow rate = 1.0 mL min−1; 25 °C; λ = 254 nm; major enantiomer tR = 4.8 min and minor enantiomer tS = 5.7 min; 1H NMR (CDCl3, 500 MHz) δ (ppm): 7.82–7.80 (m, 1H, ArH), 7.46–7.42 (m, 1H, ArH), 7.19–7.16 (m, 1H, ArH), 6.92–6.90 (m, 1H, ArH), 3.87 (s, 3H, OCH3), 2.76 (s, 3H, SCH3); 13C NMR (CDCl3, 125 MHz) δ (ppm): 41.16 (SCH3), 55.64 (OCH3), 110.53, 121.65, 124.58, 131.89, 133.07, 154.76 (ArC).Sulfoxidation reaction for kinetic measurement
Chiral salen TiIV catalyst (0.1 mol% of substrate, based on titanium ion content) was stirred with methyl phenyl sulfides (1.0 mmol) in solvent (2 mL) at 25 °C. H2O2 (30 wt%, 1.2 mmol) was then added into the stirred solution in one portion. To determine the rate of sulfoxidation, aliquots at an interval of 10 min were drawn from the reaction mixture, filtrated through silica gel with ethyl acetate as an eluent and analyzed by GC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21676078, 21476069), the Natural Science Foundation of Hunan Province for Distinguished Young Scholar (2016JJ1013), the Program for Excellent Talents in Hunan Normal University (ET14103), and Hunan Provincial Innovation Foundation For Postgraduate (CX2016B168).References
- (a) L. Marchetti and M. Levine, ACS Catal., 2011, 1, 1090 CrossRef CAS; (b) S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991 CrossRef CAS PubMed; (c) Y. Zhao, F. Sakai, L. Su, Y. Liu, K. Wei, G. Chen and M. Jiang, Adv. Mater., 2013, 25, 5215 CrossRef CAS PubMed; (d) Y. Liu, X. Ma, J. Xie, P. Liu, B. Dai and R. He, Appl. Organomet. Chem., 2013, 27, 494 CrossRef CAS; (e) A. Doring, W. Birnbaum and D. Kuckling, Chem. Soc. Rev., 2013, 42, 7391 RSC; (f) Y. Liu, T. Pauloehrl, S. I. Presolski, L. Albertazzi, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 13096 CrossRef CAS PubMed; (g) A. F. Cardozo, C. Julcour, L. Barthe, J. F. Blanco, S. Chen, F. Gayet and F. D'agosto, J. Catal., 2015, 324, 1 CrossRef CAS.
- (a) B. M. Neilson and C. W. Bielawski, ACS Catal., 2013, 3, 1874 CrossRef CAS; (b) X. Mao, W. Tian, J. Wu, G. C. Rutledge and T. A. Hatton, J. Am. Chem. Soc., 2015, 137, 1348 CrossRef CAS PubMed.
- (a) X. He, M. Aizenberg, O. Kuksenok, L. D. Zarzar, A. Shastri, A. C. Balazs and J. Aizenberg, Nature, 2012, 487, 214 CrossRef CAS PubMed; (b) W. Li, L. Huang, X. Ying, Y. Jian, Y. Hong, F. Hu and Y. Du, Angew. Chem., Int. Ed., 2015, 54, 3126 CrossRef CAS PubMed; (c) Y. Zhang, R. Tan, G. Zhao, X. Luo, C. Xing and D. Yin, J. Catal., 2016, 335, 62 CrossRef CAS; (d) W. C. de Vries, D. Grill, M. Tesch, A. Ricker, H. Nüsse, J. Klingauf, A. Studer, V. Gerke and B. J. Ravoo, Angew. Chem., Int. Ed., 2017, 56, 9603 CrossRef CAS PubMed; (e) Y. Zhou, Y. Chen, P. Zhu, W. Si, J. Hou and Y. Liu, Chem. Commun., 2017, 53, 3681 RSC.
- (a) S. Yagai, T. Karatsu and A. Kitamura, Chem.–Eur. J., 2005, 11, 4054 CrossRef CAS PubMed; (b) Y. Wang, H. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849 CrossRef CAS; (c) G. Shen, G. Xue, J. Cai, G. Zou, Y. Li and Q. Zhang, Soft Matter, 2013, 9, 2512 RSC.
- (a) T. Imahori, R. Yamaguchi and S. Kurihara, Chem.–Eur. J., 2012, 18, 10802 CrossRef CAS PubMed; (b) A. Parrot, G. Izzet, L. Chamoreau, A. Proust, O. Oms, A. Dolbecq, K. Hakouk, H. E. Bekkachi, P. Deniard, R. Dessapt and P. Mialane, Inorg. Chem., 2013, 52, 11156 CrossRef CAS PubMed; (c) Y. Shiraishi, K. Tanaka and T. Hirai, ACS Appl. Mater. Interfaces, 2013, 5, 3456 CrossRef CAS PubMed; (d) Z. Chen, L. Zhou, W. Bing, Z. Zhang, Z. Li, J. Ren and X. Qu, J. Am. Chem. Soc., 2014, 136, 7498 CrossRef CAS PubMed; (e) A. Wu, F. Lu, P. Sun, X. Gao, L. Shi and L. Zheng, Langmuir, 2016, 32, 8163 CrossRef CAS PubMed; (f) M. Karimi, P. S. Zangabad, S. Baghaee-Ravari, M. Ghazadeh, H. Mirshekari and M. R. Hamblin, J. Am. Chem. Soc., 2017, 139, 4584 CrossRef CAS PubMed.
- (a) L. Zhu, H. Yan, C. Y. Ang, K. T. Nguyen, M. Li and Y. Zhao, Chem.–Eur. J., 2012, 18, 13979 CrossRef CAS PubMed; (b) T. Tian, Y. Song, J. Wang, B. Fu, Z. He, X. Xu, A. Li, X. Zhou, S. Wang and X. Zhou, J. Am. Chem. Soc., 2016, 138, 955 CrossRef CAS PubMed; (c) Y. Guo, Y. Gong, Y. Gao, J. Xiao, T. Wang and L. Yu, Langmuir, 2016, 32, 9293 CrossRef CAS PubMed; (d) A. H. Gelebart, D. J. Mulder, M. Varga, A. Konya, G. Vantomme, E. W. Meijer, R. L. B. Selinger and D. J. Broer, Nature, 2017, 546, 632 CrossRef CAS PubMed; (e) Y. Sun, J. Ma, F. Zhang, F. Zhu, Y. Mei, L. Liu, D. Tian and H. Li, Nat. Commun., 2017, 8, 260 CrossRef PubMed; (f) H. Zhou, C. Xue, P. Weis, Y. Suzuki, S. Huang, K. Koynov, G. K. Auernhammer, R. Berger, H. Butt and S. Wu, Nat. Chem., 2017, 9, 145 CrossRef CAS PubMed.
- T. Sun, G. Qing, B. Su and L. Jiang, Chem. Soc. Rev., 2011, 40, 2909 RSC.
- J. E. Chung, M. Yokoyama, T. Aoyagi, Y. Sakurai and T. Okano, J. Controlled Release, 1998, 53, 119 CrossRef CAS PubMed.
- M. Holbach, X. Zheng, C. Burd, C. W. Jones and M. Weck, J. Org. Chem., 2006, 71, 2903 CrossRef CAS PubMed.
- C. Lv, D. Xu, S. Wang, C. Miao, C. Xia and W. Sun, Catal. Commun., 2011, 12, 1242 CrossRef CAS.
- T. Sun and G. Qing, Adv. Mater., 2011, 23, 57 CrossRef PubMed.
- K. Han, W. Su, M. Zhong, Q. Yan, Y. Luo, Q. Zhang and Y. Li, Macromol. Rapid Commun., 2008, 29, 1866 CrossRef CAS.
- (a) B. Yu, X. Jiang, R. Wang and J. Yin, Macromolecules, 2010, 43, 10457 CrossRef CAS; (b) J. Hu, H. Yu, L. H. Gan and X. Hu, Soft Matter, 2011, 7, 11345 RSC; (c) M. Baroncini, S. d'Agostino, G. Bergamini, P. Ceroni, A. Comotti, P. Sozzani, I. Bassanetti, F. Grepioni, T. M. Hernandez, S. Silvi, M. Venturi and A. Credi, Nat. Chem., 2015, 7, 234 CrossRef PubMed; (d) S. R. Deka, S. Yadav, M. Mahato and A. K. Sharma, Colloids Surf., B, 2015, 135, 150 CrossRef CAS PubMed.
- G. Markiewicz, D. Pakulski, A. Galanti, V. Patroniak, A. Ciesielski, A. R. Stefankiewicz and P. Samorì, Chem. Commun., 2017, 53, 7178 RSC.
- J. Rao and A. Khan, J. Am. Chem. Soc., 2013, 135, 14056 CrossRef CAS PubMed.
- M. T. Garcia, I. Ribosa, L. Perez, A. Manresa and F. Comelles, Colloids Surf., B, 2014, 123, 318 CrossRef CAS PubMed.
- H. Shaikh, M. Hassan and N. M. Ahmad, Phot. Nano. Fund. Appl, 2014, 12, 34 CrossRef.
- (a) J. Y. Lee and M. Schic, Biophys. J., 1994, 5, 1699 Search PubMed; (b) M. Müller, K. Katsov and M. Schick, Biophys. J., 2003, 85, 1611 CrossRef.
- L. Lin, Z. Yan, J. Gu, Y. Zhang, Z. Feng and Y. Yu, Macromol. Rapid Commun., 2009, 30, 1089 CrossRef CAS PubMed.
- (a) Y. N. Belokon, M. North, V. I. Maleev, N. V. Voskoboev, M. A. Moskalenko, A. S. Peregudov, A. V. Dmitriev, N. S. Ikonnikov and H. B. Kagan, Angew. Chem., Int. Ed., 2004, 43, 4085 CrossRef CAS PubMed; (b) M. Li, S. Qi, Y. Jin, W. Yao, S. Zhang and J. Zhao, Colloids Surf., B, 2014, 123, 852 CrossRef CAS PubMed.
- A. Lu and R. K. O'Reilly, Curr. Opin. Biotechnol., 2013, 24, 639 CrossRef CAS PubMed.
- Y. Zhang, R. Tan, M. Gao, P. Hao and D. Yin, Green Chem., 2017, 19, 1182 RSC.
- J. Li, Y. Tang, Q. Wang, X. Li, L. Cun, X. Zhang, J. Zhu, L. Li and J. Deng, J. Am. Chem. Soc., 2012, 134, 18522 CrossRef CAS PubMed.
- T. Dwars, E. Paetzold and G. Oehme, Angew. Chem., Int. Ed., 2005, 44, 7174 CrossRef CAS PubMed.
- C. Baleizão and H. Garcia, Chem. Rev., 2006, 106, 3987 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation and identity of the catalysts, NMR and HPLC analysis for the chiral sulfoxides. See DOI: 10.1039/c7ra11022g |
| This journal is © The Royal Society of Chemistry 2017 |