
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly(HPMA)-based copolymers with biodegradable side chains able to self assemble into nanoparticles†
Mattia Sponchioni a,
Lavinia Morosib,
Monica Lupib and
Umberto Capasso Palmiero*a
a,
Lavinia Morosib,
Monica Lupib and
Umberto Capasso Palmiero*a
aDepartment of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Via Mancinelli 7, 20131 Milano, Italy. E-mail: umberto.capasso@polimi.it
bDepartment of Oncology, IRCCS, Istituto di Ricerche Farmacologiche Mario Negri, Via La Masa 19, 20156 Milano, Italy
First published on 2nd November 2017
Abstract
N-(2-Hydroxypropyl)methacrylamide (HPMA) is a water soluble monomer used in the synthesis of biocompatible and non-immunogenic polymers. In particular, poly(HPMA) can be exploited to sterically stabilize nanoparticles (NPs) suitable for the delivery of lipophilic therapeutics without the concerns related to the use of the polyethylene glycol (PEG), such as allergic reactions and the accelerated blood clearance effect. In addition, the use of the ring opening polymerization (ROP) of a lactone in the presence of an initiator that bears a double bond and a hydroxyl group is a promising way (the so called “macromonomer method”) to produce oligoester-based monomers and, in turn, to obtain biodegradable NPs via free radical polymerization. However, HPMA cannot be used as initiator being a secondary alcohol and thus hampering the control over the polymer molecular weight (MW). For this reason, in this work, a novel class of amphiphilic block copolymers that consists of a poly(HPMA) backbone and several short oligo(ε-caprolactone) side chains were produced via the adoption of the reversible addition–fragmentation chain transfer (RAFT) polymerization and the “inversion” of the macromonomer method. The oligoester was first synthesized via the ROP of ε-caprolactone in the presence of a primary alcohol and then attached to HPMA using a succinic acid unit as spacer. The NPs obtained via the self-assembly of these novel block copolymers are designed to degrade into completely water soluble poly(HPMA) chains with a MW lower than the threshold value for the renal excretion. The cytotoxicity of these novel carriers and their ability to load trabectedin, a hydrophobic anticancer therapeutic, were assessed.
1. Introduction
Polymeric nanoparticles (NPs) are colloidal nano-assemblies widely adopted in medicine for the delivery of lipophilic therapeutics. They can be produced via different techniques, such as emulsion polymerization and nanoprecipitation.1 Among all the types of NPs produced in literature, those obtained via direct self-assembly in water of amphiphilic species, such as block copolymers, have the advantage to eliminate the need of toxic surfactants and to load the therapeutic directly during the NP formation. In this case the NP core is formed by the lipophilic part of the forming block copolymer and serves as a depot for hydrophobic compounds. For an effective delivery system, the lipophilic core forming chains should be able to load the highest amount possible of the therapeutic and to degrade in biocompatible, non-toxic and easily excretable compounds. Among the species adopted in the literature to fulfil these functions, poly(lactide) (PLA) and poly(ε-caprolactone) (PCL) chains have been chosen as “golden standards”.On the other hand, the hydrophilic part of the block copolymer forms the NP shell and provides colloidal stability and protects the loaded drug against protein adsorption and opsonisation. Among the hydrophilic species adopted for the stabilization of these carriers, poly(N-(2-hydroxypropyl)methacrylamide) (poly(HPMA)) is an attractive alternative to the polyethylene glycol (PEG) chains, by far the most adopted for biomedical applications. Besides its biocompatibility and its non-immunogenicity, an advantage of poly(HPMA) over PEG is its secondary alcohol functionality, which allows multiple targeting molecules to be conjugated to the same polymer chain. In addition, poly(HPMA) has been proved to not generate any accelerated blood clearance and allergic reaction compared to the PEG.2–4
The synthesis of well-defined amphiphilic block copolymers was made possible by the advent of living polymerization techniques, such as the reversible addition–fragmentation chain transfer (RAFT) polymerization that allows the fine tuning of the polymer architecture and, in turn, of the NP properties.5,6 Besides, the ring opening polymerization (ROP) has allowed to produce PCL and PLA based biodegradable macromonomers with different lengths exploiting 2-hydroxyethyl methacrylate (HEMA) as initiator, a monomer that possesses a primary alcohol functionality.7,8 Then, the combination of RAFT and ROP provides an effective technique to obtain well defined block copolymers able to self-assemble into NPs with different size. However, the so called “macromonomer method” for the production of oligoester-based macromonomers is not able to work properly in some conditions and the “inversion” of the method is sometimes necessary. It consists in the synthesis of the oligoester via ROP using a primary alcohol as initiator followed by the functionalization of the chain end with a double bond. As an example, in the work of Ferrari et al., where a macromonomer based on the low reactive decanolide and dodecanolide was produced, the polyester chain has been produced first and then conjugated with a monomer that bears a carboxylic acid via a N,N′-dicyclohexylcarbodiimide (DCC)-mediated esterification.9 In fact, these lactones require a strong base as catalyst that is also able to destroy the double bond in the direct method. The inversion of the macromonomer method is also a good strategy for the functionalization of the final side chain. As an example, when a fixed positive charge is wanted, it is not possible to directly use a quaternary ammonium salt that bears a hydroxyl group (i.e. choline chloride) as initiator in the ROP due to its insolubility in the lactone and in the common organic solvents. For this reason, in the paper of Rooney et al., a tertiary amine bearing an alcohol functionality was adopted as the initiator in the ROP of ε-caprolactone and the double bond was then added to the polyester chain by the reaction with methacryloyl chloride. In the end, the tertiary amine was transformed into the corresponding quaternary ammonium salt via reaction with methyl iodide.10,11 The macromonomer method fails also when HPMA is used as the initiator in the ROP of cyclic esters. This is due to its very low reactivity as a consequence of being a secondary alcohol. In the literature, a direct synthesis of a PLA based macromonomer exploiting the HPMA as the initiator was proposed.12 From a close inspection of the product obtained in that case, it is possible to note a low HPMA conversion that led to a poor control over the macromonomer structure, despite narrowly dispersed polymers could be obtained.
Here we report for the first time the synthesis of a well-defined HPMA-functionalized oligoester via the inversion of the macromonomer method. In particular, the macromonomer is obtained in three steps: (i) the ROP of caprolactone (CL) with benzyl alcohol (BA) as initiator; (ii) the acylation of the –OH bearing PCL chain with succinic anhydride and (iii) the further DCC-mediated esterification with HPMA. In particular, BA has been chosen as the co-catalyst in the ROP since it is a reactive primary alcohol with a high boiling point and because it is less toxic than the alkyl alcohols that are commonly adopted (e.g. dodecanol).13 The resulting macromonomer (hereinafter HPMA-CL5, where 5 is the number of caprolactone units added to the chain) was subsequently used to obtain biodegradable polymeric nanoparticles that are suitable for the drug delivery of poorly water soluble therapeutics. In order to produce these nanoparticles, a two-step sequential RAFT polymerization was adopted. In the first step, a poly(HPMA) water soluble block was obtained to provide steric stabilization to the NPs. Then, the obtained macro RAFT agent was chain extended with the novel lipophilic macromonomer containing HPMA. The obtained amphiphilic diblock copolymer is able to self-assemble in water via a simplified nanoprecipitation method with a rudimental apparatus, thus allowing the NP production shortly before the injection. Furthermore, the copolymer is designed to progressively degrade via the hydrolysis of the ester bonds in the PCL side chains. The mentioned NP degradation behaviour has been studied and resulted into the complete disappearance of the carriers leaving only the water soluble poly(HPMA) backbone, which is expected to be easily excreted by the kidneys due to its low molecular weight.1 The NP cytotoxicity and the ability to load trabectedin, a hydrophobic anticancer compound, have been finally assessed.
2. Materials and methods
2.1 Materials
Succinic anhydride (≥99%), 4-(dimethylamino)pyridine (DMAP, ≥99%), methacryloyl chloride (≥97%), DL-1-amino-2-propanol (≥98%), sodium carbonate (≥99%), N,N′-dicyclohexylcarbodiimide (DCC, 99%), tin(II)2-ethylhexanoate (Sn(Oct)2, 98%), ε-caprolactone (CL, 97%), benzyl alcohol (BA, ≥99%), anhydrous sodium sulphate (Na2SO4, ≥99%), 4,4′-azobis(4-cyanovaleric acid) (ACVA, ≥98%) and 4-cyano-4-(phenylcarbonothioylthio) pentanoic acid (CPA, ≥97%) were all purchased from Sigma Aldrich and used as received except when specifically noted. The phosphate buffered saline solution (PBS, Biowest) and the cell medium, that was composed of high glucose DMEM/F12 (Biowest, Nuaillé, France) and supplemented with 10% fetal bovine serum (Lonza, Basel, Switzerland) were used as received. All the solvents used were of analytical-grade purity and were purchased from Sigma Aldrich.2.2 HPMA synthesis
The HPMA was synthesized following a procedure reported in literature.14 Briefly, 25.22 g (238 mmol) of sodium carbonate were suspended in 56 mL of anhydrous dichloromethane (DCM). The temperature was kept at −10 °C using a brine\ice mixture and 16.25 g (216 mmol) of DL-1-amino-2-propanol were added. Finally, 22.60 g (216 mmol, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with respect to DL-1-amino-2-propanol) of freshly distilled methacryloyl chloride were dissolved in 22 mL of DCM and the solution was fed to the reaction flask kept at −10 °C in 45 minutes under vigorous stirring. The mixture was left to equilibrate to room temperature and allowed to react for further 30 minutes. Then, the product was recovered by filtration, dried twice with anhydrous Na2SO4, concentrated under vacuum and left to crystallize at −20 °C overnight. Finally it was washed with cold DCM and recrystallized from acetone. The purity of the product was assessed via proton Nuclear Magnetic Resonance (1H NMR) in deuterium oxide (D2O) using a Bruker 400 MHz spectrometer and the yield was up to 65%.
:1 with respect to DL-1-amino-2-propanol) of freshly distilled methacryloyl chloride were dissolved in 22 mL of DCM and the solution was fed to the reaction flask kept at −10 °C in 45 minutes under vigorous stirring. The mixture was left to equilibrate to room temperature and allowed to react for further 30 minutes. Then, the product was recovered by filtration, dried twice with anhydrous Na2SO4, concentrated under vacuum and left to crystallize at −20 °C overnight. Finally it was washed with cold DCM and recrystallized from acetone. The purity of the product was assessed via proton Nuclear Magnetic Resonance (1H NMR) in deuterium oxide (D2O) using a Bruker 400 MHz spectrometer and the yield was up to 65%.
2.3 HPMA RAFT polymerization
A hydrophilic poly(HPMA) with an average degree of polymerization (DP) equal to 70 was synthesized via RAFT polymerization in a mixture of acetic buffer/ethanol (Scheme 1). Specifically, 1.1 g of HPMA (7.69 mmol), 31 mg of CPA (0.11 mmol, HPMA/CPA mole ratio equal to 70), 10.3 mg of ACVA (0.04 mmol, CPA/ACVA mole ratio equal to 3) were dissolved in 3 g of ethanol and 7 g of acetic buffer and poured in a septa-sealed round bottom flask. The mixture was purged with nitrogen for 30 minutes and heated to 70 °C under stirring. The polymerization was left to proceed for 24 hours, the solvent evaporated under vacuum and the polymer precipitated three times in tetrahydrofuran (THF). Aliquots of the reactive mixture were taken at different times, dried under a nitrogen flow and analyzed via 1H NMR (in D2O) and gel permeation chromatography (GPC) to evaluate the monomer conversion and the MW distributions over time. GPC analysis was performed at 35 °C using a mixture of 20/80% acetonitrile (ACN)/0.05 M Na2SO4 as eluent and at a flow rate of 0.5 mL min−1. The instrument (Jasco apparatus) comprises a differential refractive index (RI) detector, three Suprema columns (Polymer Standards Service, particle size 10 μm, pore sizes of 100, 1000, and 3000 Å) and a pre-column. A universal calibration was applied based on polyethylene glycol (PEG) standards.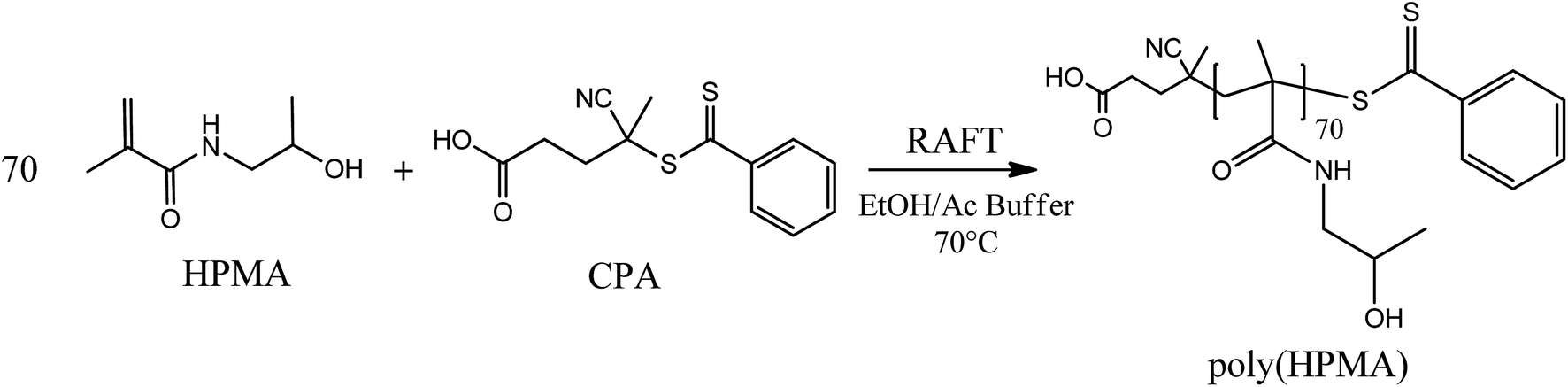 | ||
| Scheme 1 Synthesis of hydrophilic poly(HPMA) obtained via RAFT polymerization. | ||
2.4 Synthesis of HPMA-PCL5
As schematically reported in Scheme 2, the lipophilic HPMA-based macromonomer was synthesized by a three-step procedure: (i) ROP of CL with BA as the initiator and tin octoate as the catalyst, (ii) subsequent acylation of the product using succinic anhydride and (iii) DCC-mediated esterification of the obtained oligo(caprolactone) bearing a final carboxyl group with HPMA. | ||
| Scheme 2 Synthesis protocol to obtain the HPMA-based lipophilic macromonomer: (a) BA initiated ROP of the ε-caprolactone; (b) acylation of the product using succinic anhydride and (c) DCC-mediated esterification of the carboxylic acid terminated oligo(caprolactone) with the HPMA. | ||
In the first step (Scheme 2a), the ring opening polymerization was conducted with a CL/BA mole ratio and BA/Sn(Oct)2 ratio equal to 5 and 1/200, respectively. 10.55 g of CL (92.4 mmol) and 37 mg of Na2SO4, added to remove water from the system, were heated to 130 °C in a septa-sealed flask under stirring. 2 g (18.5 mmol) of BA were mixed with 37 mg (9.1 × 10−5 mol) of tin octoate and injected in the pre-heated CL containing flask. The polymerization was allowed to proceed for 2.5 hours.
The subsequent acylation (Scheme 2b) was obtained in bulk conditions by adding 1.2 mol equivalents of succinic anhydride to the obtained BA-CL5. Briefly, 2.22 g (22.2 mmol) of succinic anhydride were poured directly into the flask of the previously synthesized oligo(caprolactone) and the mixture was heated to 90 °C and left to react overnight. The final product was purified by dissolution in THF and further precipitation in water.
In the third step (Scheme 2c), HPMA-CL5 was obtained via DCC-mediated esterification between the previously synthesized BA-CL5Q and a 20% molar excess of HPMA, exploiting the DMAP as the catalyst. More in detail, 8.03 g (12 mmol) of BA-CL5Q and 2.09 g (15 mmol) of HPMA were dissolved in 40 mL of anhydrous DCM. The solution was poured in a 100 mL septa-sealed flask kept at 0 °C in a water/ice bath. Finally, 270 mg (2.2 mmol) of DMAP and 2.85 g (14 mmol) of DCC dissolved in 40 mL of anhydrous DCM were fed over a period of 1 h into flask. The reaction was left to equilibrate at room temperature and to react for additional 24 hours. Then the mixture was filtered to remove the white precipitate formed as the co-product of the esterification and the solvent evaporated under vacuum. The dry product was dissolved in THF and precipitated in a large excess of water. The obtained macromonomer was finally recovered as a white waxy solid.
For each intermediate step, an aliquot of the sample was withdrawn and analyzed via 1H NMR (in CDCl3), GPC and matrix-assisted laser desorption/ionization – time of flight (MALDI-TOF). In this case, GPC analysis was performed at 35 °C using THF as the eluent and a flow rate of 0.5 mL min−1. The instrument (Jasco apparatus) comprises a differential refractive index (RI) detector, three PL gel columns (Polymer laboratories Ltd., UK; two columns had pore sizes of the mixed-C type and one was an oligopore; 300 mm length and 7.5 mm ID) and a pre-column. A universal calibration was applied based on polystyrene (PS) standards. MALDI-TOF spectra were recorded using an Ultraflex II TOF Bruker spectrometer (Bremen, Germany) using 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene]-malononitrile (DCTB) as the matrix material. The samples that were co-crystallized with the matrix on the probe were ionized using a Smart Beam laser pulse (337 nm) and accelerated at 25 kV with a time-delayed extraction before entering the time-of-flight mass spectrometer. The matrix and sample were separately dissolved in DCM and mixed in a 10:1 matrix to sample ratio. To produce the specific adducts, sodium ions were added (1% sodium acetate in methanol). Approximately 1 mL of the mixture of matrix and sample was spread on the MALDI-TOF MS probe and air-dried. All the spectra were obtained in positive reflection mode, and the external calibration was performed by using the peptide calibration standard II (700–3200 Da) from Care (Bruker, Switzerland).
2.5 Synthesis of block copolymers
The amphiphilic block copolymers constituted by a uniform poly(HPMA) backbone were synthesized via the RAFT polymerization in ethanol of the HPMA-CL5 lipophilic macromonomer in the presence of the previously synthesized poly(HPMA), used as the macro-RAFT agent. Four different copolymers were synthesized with a final DP of the hydrophobic block equal to 2, 5, 10 and 20. The tuning of the DP was obtained by simply regulating the initial molar ratio between the HPMA-CL5 macromonomer and the macro-RAFT agent. The nomenclature adopted is composed by the hydrophilic block DP followed by the lipophilic block DP (i.e. 702 is applied to the polymer composed by 70 HPMA units and 2 HPMA-CL5 units). The synthesis procedure is represented in Scheme 3.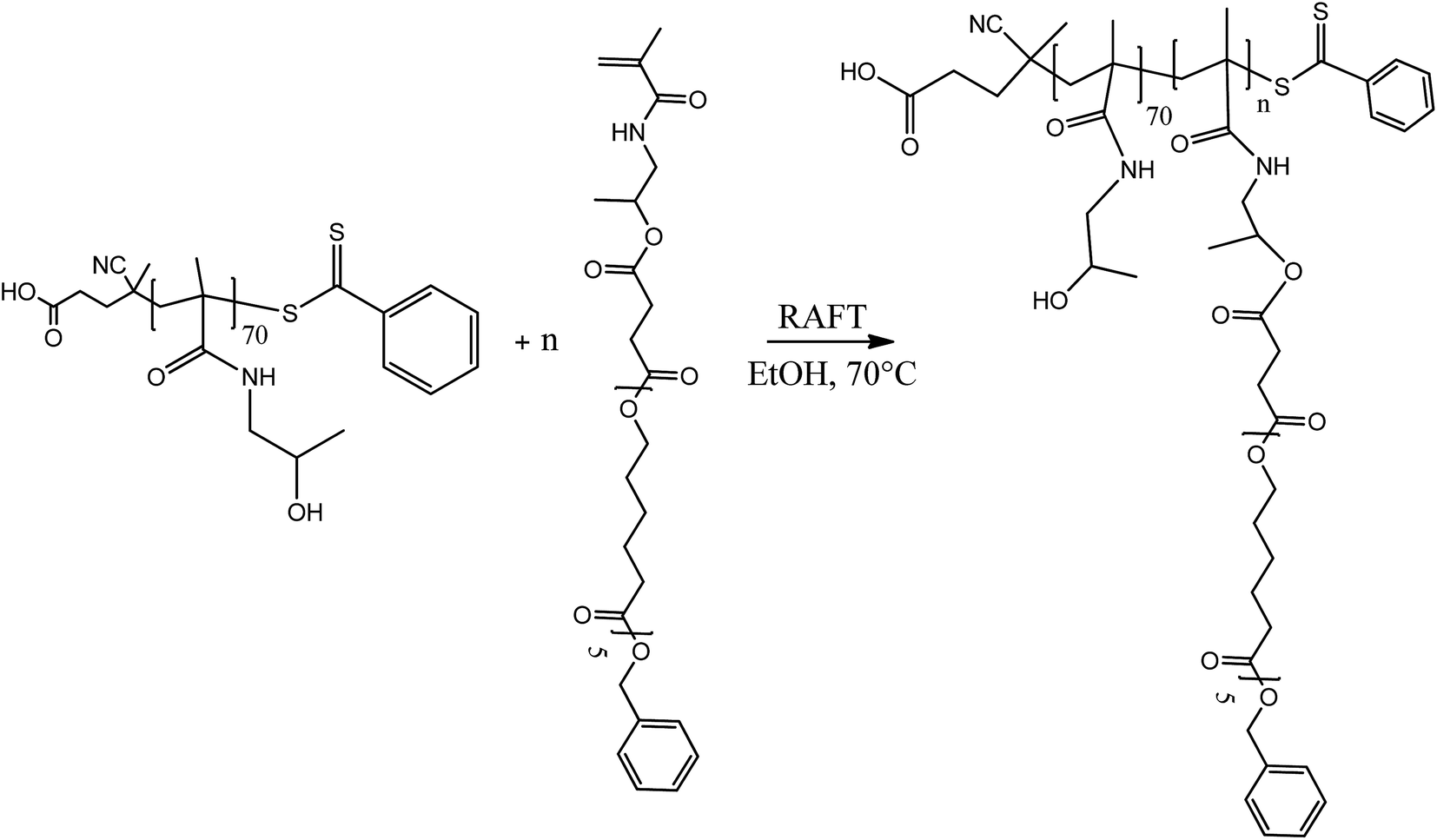 | ||
| Scheme 3 Synthesis of the HPMA-based amphiphilic block copolymers via RAFT polymerization. | ||
In a typical reaction, for the synthesis of 702 (70 units of HPMA and 2 units of HPMA-CL5), 0.647 g of poly(HPMA) and 0.141 g of HPMA-CL5 (HPMA-CL5/poly(HPMA) mole ratio equal to 2) were dissolved in 4 mL of ethanol. The solution was transferred in a septum-sealed flask and purged with nitrogen for 30 minutes. The solution was then heated to 70 °C and 10 mg of ACVA dissolved in 1 mL of ethanol was fed under stirring. The reaction was left to occur for 24 h, after which the same amount of ACVA was added and the reaction went on for additional 24 h. The solvent was then evaporated under vacuum and the polymer purified by precipitation in a large excess of diethyl ether. After drying under a flow of nitrogen, the polymer was collected as a pink powder and characterized via 1H NMR in deuterated dimethylsulfoxide (DMSO-d6).
2.6 NP formation via a simplified nanoprecipitation method
HPMA-based NPs have been obtained following a simple nanoprecipitation process without the use of any mixing devices that are commonly adopted to obtain strongly turbulent conditions between the organic and the aqueous phases. In this case, different amounts of the bulk block copolymers were dissolved in 0.3 g of DMSO. This organic solution was then added dropwise to 3 mL of PBS under vigorous agitation. In order to investigate the NP behaviour, different polymer concentrations in DMSO were tested. In particular, the amount of DMSO was kept constant (0.3 g) and different quantities of the block copolymer were used: 75 mg, 52 mg, 33 mg, 15 mg, 10 mg and 5 mg to achieve a final polymer concentration of 20%, 15%, 10%, 5%, 3.5% and 1.5% w/w respectively. These different concentrations were tested for all of the four block copolymers. The NP size was then analyzed via Dynamic Light Scattering (DLS, Malvern Zetasizer Nano ZS) at a scattering angle of 173°. The particle size distribution (PSD) is an average of three independent measurements. Transmission Electron Microscopy (TEM) images of the synthesized NPs were obtained using a FEI Morgagni 268 instrument, operated at 120 kV, equipped with an Orius SC1000 CCD camera.2.7 NP degradation
The NP degradability was investigated in PBS at 37 °C for several days. Briefly, the NP suspensions in PBS, obtained using a 10% w/w polymer concentration in the organic phase, were stored in a thermostated oven at 37 °C and periodically analyzed via DLS in order to evaluate the changes in their average size, polydispersity index (PDI) and intensity of the scattered light (count rate). DLS measurements were performed 1, 4, 7, 10, 14, 17, 22, 25 and 30 days after the synthesis.2.8 NP cytotoxicity
The 702 and 705 NP cytotoxicity was studied in vitro in the case of the triple negative breast (TNB) cancer MDA-MB-231 cell line (American Type Culture Collection, Manassas, VA). The cells were seeded in six-well plates (Costar) at the concentration of 20000 cells per mL. 24 h after the seeding, the cells were incubated with 0.9, 0.4 and 0.02 mg mL−1 of NPs. After 48 h of exposure, the cells were detached using 0.5 mL of trypsin, re-suspended in 2 mL of PBS and counted using a Coulter Counter ZM (Coulter Electronics, UK). The procedure was applied three times for each concentration and the average number of viable cells was considered and expressed as a percentage of the viable untreated cells (control) at the same time.
2.9 Trabectedin loading and release test
The loading of trabectedin was performed in the same step of the NP production. Briefly, 10 mg of the block copolymer (702 and 705) and trabectedin (75 μg) were dissolved in 100 mg of DMSO. After complete dissolution, the nanoprecipitation method was applied with 3 mL of PBS. The NP suspension was dialyzed against 200 mL of PBS for 24 h and the release was evaluated by withdrawing aliquots of the formulation in the dialysis cassette (Slide-A-Lyzer dialysis cassette, Thermo Scientific MWCO = 3.5 kDa) after 0, 0.25, 0.5, 1, 4, 24 h. The PBS was periodically changed in order to preserve the sink conditions. Each sample was then analyzed via liquid chromatography – tandem mass spectrometry (LC-MS/MS) as previously described.15,16 Briefly, 0.1 mL of the NP suspension were spiked with 100 ng of deuterated trabectedin as internal standard and 50 μL (NH4)2SO4:H2O = 1:3. Samples were extracted with 0.4 mL of CH3OH, HCl 0.1 M. After shaking for 10 s, samples were centrifuged at 13000 rpm for 10 min. The organic phase was separated and dried under nitrogen, and the residues were dissolved in 250 μL of mobile phase constituted by a 1:1 ammonium acetate buffer (10 mM pH 6.8): CH3OH solution. Two μL of the reconstitute samples were injected in the LC-MS/MS system. The apparatus comprises an Accucore C18 column. Control NPs without drug were used to prepare the calibration curve by the addition of trabectedin in the range 1–40 μg mL−1.
3. Results and discussion
3.1 Synthesis of poly(HPMA)-based macro RAFT agent
The HPMA monomer was synthesized according to the literature.12,14,17 To assess the purity of the produced monomer, 1H NMR analysis in D2O was performed and the obtained spectrum is reported in Fig. S1a.† The spectrum confirms the proper structure of the monomer and its purity after the workup protocol. The synthesized monomer was then polymerized via RAFT polymerization in order to obtain the hydrophilic macromolecular chain transfer agent (macro-CTA) employed in the synthesis of amphiphilic diblock copolymers and as steric stabilizer for the final NPs. The polymerization was carried out in an acetic buffer (pH = 5)/ethanol mixture with a low monomer concentration since it is well known that the propagation kinetic constant (kp) of hydrophilic monomers is higher in water due to its beneficial effect on the transition state of the propagation step and increases as the monomer concentration decreases.18,19 On the other hand, ethanol is necessary to dissolve the CPA. The DP of the polymer can be tuned by controlling the monomer/CPA mole ratio. In this case a DP equal to 70 was targeted. The 1H NMR spectrum of the poly(HPMA) is reported in Fig. S1b.† By evaluating the area of the vinyl signal and that of the signal E, it is possible to assess that a 97% monomer conversion is reached during the polymerization. Further from the NMR spectrum it is possible to evaluate the average DP according to eqn (1).
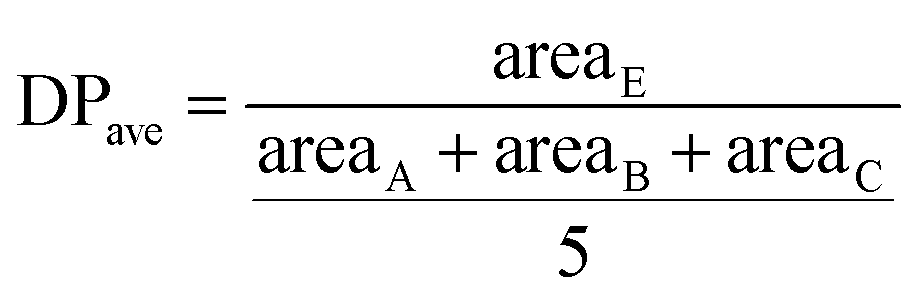 | (1) |
Being A, B and C the signals related to the benzyl chain-end group. From eqn (1) it is possible to demonstrate that the synthesized poly(HPMA) is constituted by an average DP of 75, that is in good accordance with the target (i.e. DP = 70). To demonstrate the living nature of the reaction, the kinetic of the process was evaluated by measuring the monomer conversion (via 1H NMR) and the molecular weight of the polymer over time. From Fig. S2a† it is possible to note that a logarithmic trend of the conversion versus time is obtained and a monomer conversion up to 80% is achieved within the first 24 hours of reaction. However, an initial inhibition time of 1.5 h is experienced. As reported in literature, this is a common feature of the RAFT polymerization, especially when the DP is low.20 Analyzing then the molecular weight provided by the GPC, a linear trend is obtained versus the conversion. This confirms the living character of the RAFT polymerization, for which a linear dependency of the molecular weight from the conversion is expected according to eqn (2).
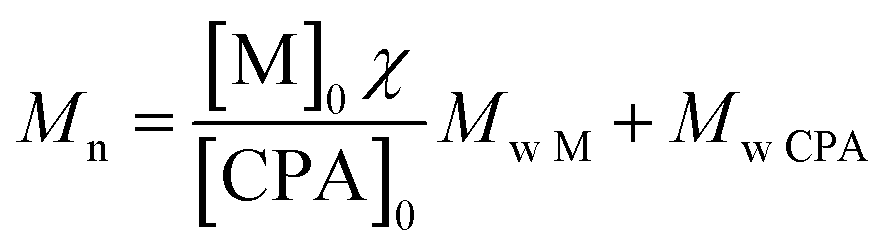 | (2) |
3.2 Synthesis of HPMA-CL5 hydrophobic macromonomer
The traditional way to synthesize oligoester-based lipophilic macromonomers is the direct ROP of a cyclic ester (i.e. ε-caprolactone, lactide, glycolide) exploiting a vinyl group bearing alcohol as the co-catalyst.7,21–26 Then, a direct synthesis exploiting HPMA as the co-catalyst in the ROP of the ε-caprolactone was tested using different HPMA/Sn(Oct)2 mole ratios and a reaction temperature of 130 °C. The conversion of both the HPMA and the caprolactone over time, evaluated via 1H NMR, is reported in Fig. 1.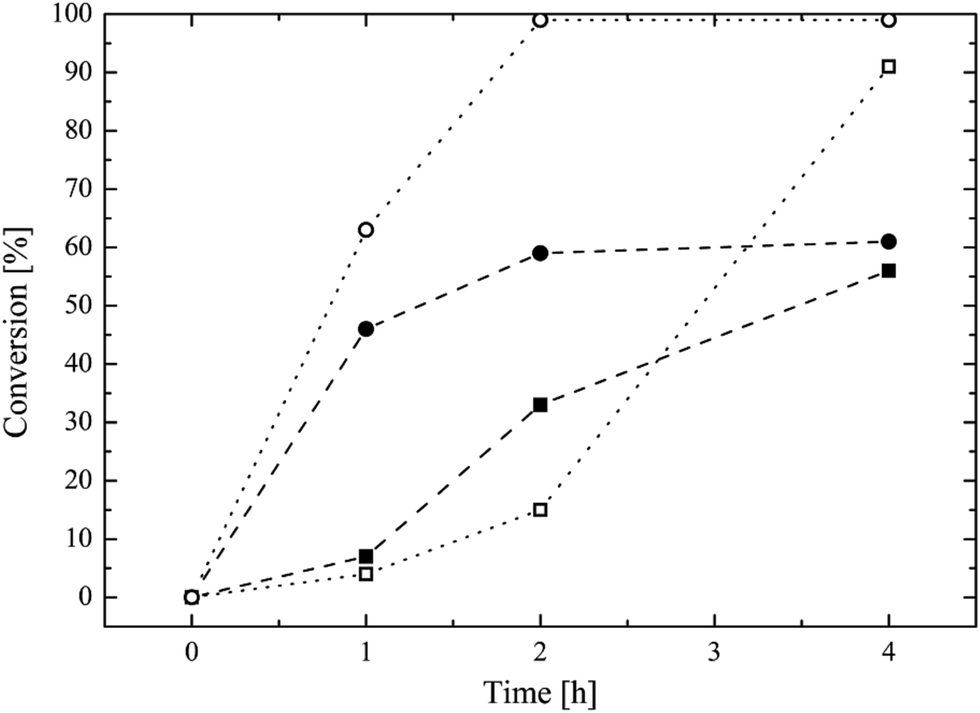 | ||
| Fig. 1 Direct ROP of ε-caprolactone using HPMA as the co-catalyst. Conversion over time for: HPMA (filled squares) and CL (open squares) using a HPMA/Sn(Oct)2 mole ratio of 100; HPMA (filled circle) and CL (open circle) in the case of a HPMA/Sn(Oct)2 mole ratio equal to 50. | ||
It is possible to observe that CL is very reactive during the ROP, and its conversion reaches values higher than 90% after 4 h for both the HPMA/Sn(Oct)2 mole ratios that were tested. However, the situation is different for the HPMA. In this case, the maximum conversion that can be obtained after 4 h is just of 60%. This very low HPMA conversion can be explained by considering the lower reactivity of a secondary alcohol compared to that of a primary alcohol like 2-hydroxyethyl methacrylate that is commonly used in the ROP of cyclic esters.7,21,23,26–30 This low HPMA conversion inevitably prevents the control of the molecular weight of the produced macromonomer. In fact, a DP (estimated via 1H NMR. Fig. S3 in the ESI section†) equal to 9 is obtained instead of the target value of 5. For this reason, in order to obtain a macromonomer with the desired molecular weight and number of CL units, the inversion of the macromonomer synthesis method is required and a three-step process is proposed in this work. The first step is the ROP of the CL exploiting the benzyl alcohol that is a very reactive primary alcohol, as the co-catalyst, in order to produce a well defined oligo (CL) with a target DP equal to 5. The 1H NMR spectrum of this first intermediate is reported in Fig. 2a.
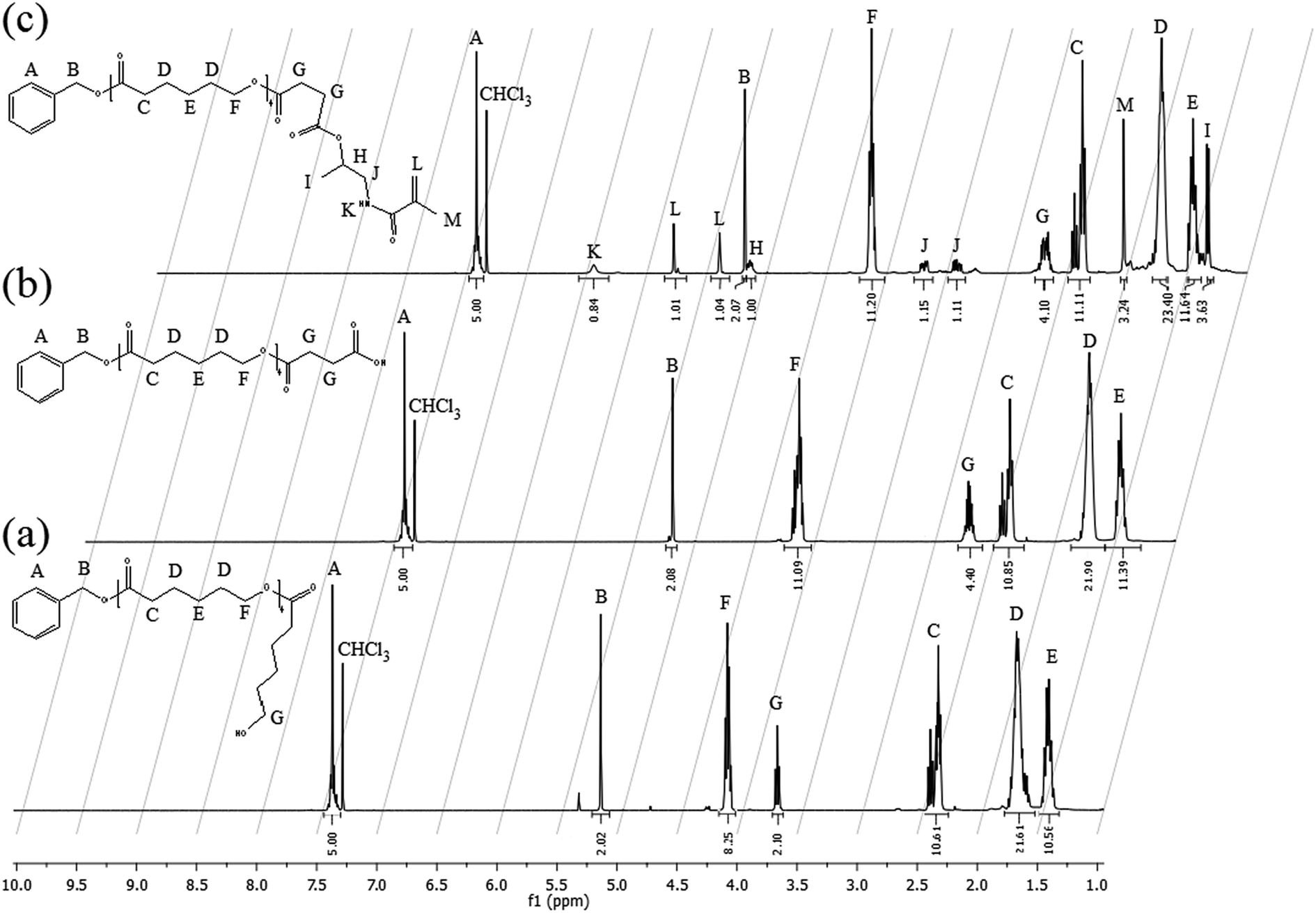 | ||
| Fig. 2 1H NMR spectra, recorded in CDCl3, for: (a) BACL5, (b) BACL5Q and (c) HPMA-CL5. | ||
In this case, the ROP reached a 94% conversion for the BA just after 3 h. The average number of CL units added to the oligomer can be evaluated according to eqn (3).
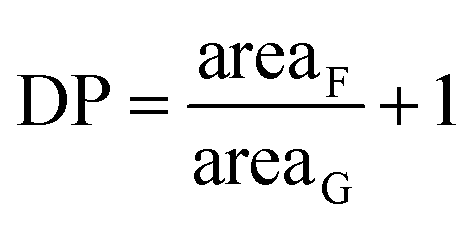 | (3) |
From Fig. 2a it is possible to calculate an average DP value of 4.9, which is close to the target. The second step in the synthesis of the HPMA-CL5 macromonomer is the acylation of the produced BACL5 using succinic anhydride in order to obtain a carboxylic acid-terminated molecule active in the esterification reaction. The acylation was conducted in bulk conditions at 90 °C. At this temperature, the succinic anhydride can be dissolved in the liquid BACL5. The 1H NMR spectrum of the produced BACL5Q is reported in Fig. 2b. From this spectrum, the disappearance of the peak at 3.7 ppm (G in Fig. 2a) confirms the complete functionalization of the BACL5. Further, no residual succinic anhydride is detected after the purification protocol. The synthesized BACL5Q was finally reacted with the HPMA in a DCC-mediated esterification reaction using DMAP as the catalyst. This led to the HPMA-CL5 macromonomer, whose 1H NMR spectrum is reported in Fig. 2c. In this case, the shift of the H peak to 5.15 ppm compared to the original chemical shift of 4 ppm in the HPMA NMR spectrum (peak D in Fig. S1a†) confirms the functionalization of the HPMA and the success of the esterification reaction. The molecular weight distribution of the final macromonomer was also studied via both MALDI-TOF and GPC analysis (Fig. S5 and S6 in the ESI section, respectively†) and an average value of 1179 g mol−1 with a polydispersity equal to 1.14 was obtained, in agreement with the theoretical values.
3.3 Synthesis of block copolymers
The poly(HPMA) macro-CTA was chain-extended with the synthesized HPMA-CL5 macromonomer via solution RAFT polymerization conducted in ethanol with a [macro-CTA]/[ACVA] mole ratio equal to 3. Different DPs for the lipophilic block were targeted (i.e. 2, 5, 10 and 20), in order to evaluate the impact of the HPMA-CL5 macromonomer over the self-assembly behaviour of the block copolymers, the NP size and their performance as drug delivery vectors. The monomer conversion during the process was evaluated via 1H NMR (Fig. S7 in the ESI section†) and summarized, for the four diblock copolymers, in Table 1.| Block copolymer | χ24 h [%] | χ48 h [%] | Mn [Da] |
|---|---|---|---|
| 702 | 46.23 | 88.02 | 11300 |
| 705 | 54.13 | 88.62 | 14400 |
| 7010 | 43.66 | 80.62 | 18400 |
| 7020 | 34.6 | 80.50 | 27600 |
In particular, the monomer conversion is quite low after the first 24 h at 70 °C, never reaching values higher than 54%. However, a considerable improvement is achieved by a successive initiator addition and further 24 h at 70 °C. In this case the monomer conversion significantly increases reaching up the 88% after an overall 48 h reaction. Besides, this further initiator addition has no detrimental effects on the polymer structure, as confirmed by the NMR spectrum reported in Fig. S7.†
3.4 Nanoparticle synthesis via a simplified nanoprecipitation method and degradation study
The synthesized block copolymers were assembled in monodisperse polymer NPs via a nanoprecipitation method that greatly simplifies the instrumental apparatus usually adopted for this purpose. The polymer is dissolved in few microliters of DMSO as the organic solvent and the nanoprecipitation occurs via the dropwise addition of the organic phase to PBS kept under vigorous agitation on a magnetic stirrer. This very simple method avoids the use of sonication, pumps and mixing devices that are commonly employed to obtain the efficient polymer self-assembly.30,31 In this way, the NP synthesis can be left to the end-user, thus avoiding intermediate steps like the lyophilization and dialysis. The polymer concentration in the organic phase can be in fact increased up to 20% w/w without a significant impact over the final PSD, so that a minimal amount of organic solvent is required to formulate the polymer. The PSD of the different block copolymers using this method and the influence of the polymer concentration in DMSO on the average NP size are reported in Fig. 3. | ||
Fig. 3 (a) PSD obtained from the DLS analysis for the 702 (green dotted line), 705 (blue dash-dotted line), 7010 (black solid line) and 7020 (red dashed line) diblock copolymers obtained following their nanoprecipitation in PBS using the simplified method with a polymer concentration in DMSO of 15% w/w. (b) TEM picture of the NPs obtained from the 702 diblock copolymer with a polymer concentration in DMSO of 10% w/w (scale bar 200 nm). (c) Influence of the polymer concentration in DMSO over the NP intensity-averaged size and (d) over the NP PDI for the 702 ( ), 705 ( ), 705 ( ), 7010 ( ), 7010 ( ) and 7020 ( ) and 7020 ( ) diblock copolymers. ) diblock copolymers. | ||
From Fig. 3a it is possible to observe that monodisperse polymer NPs can be obtained with the simplified nanoprecipitation method even at a high polymer concentration in the organic phase (i.e. 15% w/w). This is a very important feature since it allows using only small amount of DMSO, that is well tolerated and poorly cytotoxic at concentrations lower than 10% w/w in the infused solution.32–35 In addition, the formulation is obtained using only readily available instruments, without the necessity of any particular equipment, thus paving the way to the NP production directly by the end-user and few moments before their injection, thus solving important concerns that are usually associated to colloidal dispersions, such as storage and transport.36–39 The TEM picture reported in Fig. 3b for the 702 NPs as an example further confirms the data obtained by the DLS analysis, proving that monodisperse, round shaped NPs can be obtained.
A key point, when producing ready-to-use NPs is the minimization of the organic solvent used to dissolve the polymer. However, this inevitably leads to an increase in the viscosity of the organic phase, thus hardening the polymer self-assembly during the nanoprecipitation that is generally conducted under dilute conditions.40–42 Since high polymer concentration in the organic phase could have a detrimental effect on the quality of the final NPs, its effect over both the NP size and PDI has been investigated. In particular, from Fig. 3c it is possible to observe that by increasing the polymer concentration in DMSO the NP size initially increases for all the synthesized copolymers. However, for concentrations higher than 5% w/w, a sort of plateau is reached, with the size that only slightly fluctuates around an average value (i.e. 100 nm for the 702, 180 nm for the 705, 168 nm for the 7010 and 176 nm for the 7020). On the other hand, the PDI remains almost constant and around 0.2 over the whole concentration range, as it can be noticed from Fig. 3d. It was finally observed that with a concentration higher than 20% w/w, the polymer is only hardly soluble in DMSO.
In conclusion, a poly(HPMA70-b-HPMA-CL5n) biodegradable diblock copolymer constituted by a homogeneous HPMA backbone could be successfully assembled in monodisperse, round shaped NPs using a simplified nanoprecipitation method. This allows producing ready-to-use NPs due to the use of a very little amount of DMSO and rudimental equipment.
The NPs synthesized from the HPMA-based lipophilic macromonomer are composed of short oligo(caprolactone) side chains that can undergo hydrolytic degradation in water. The possibility for a nanovector specifically designed for intravenous administration to degrade leaving no traces is a main concern to avoid polymer accumulation into the bloodstream, as already stressed in the literature.1,8,43 Then, the degradation behaviour of the NPs obtained from the four different diblock copolymers has been studied at 37 °C using PBS (pH = 7.4) as the medium. This is a biologically relevant medium since it is commonly used for intravenous injections. The evolution of the NP size as obtained via DLS over time after the NP incubation at 37 °C is reported in Fig. 4a.
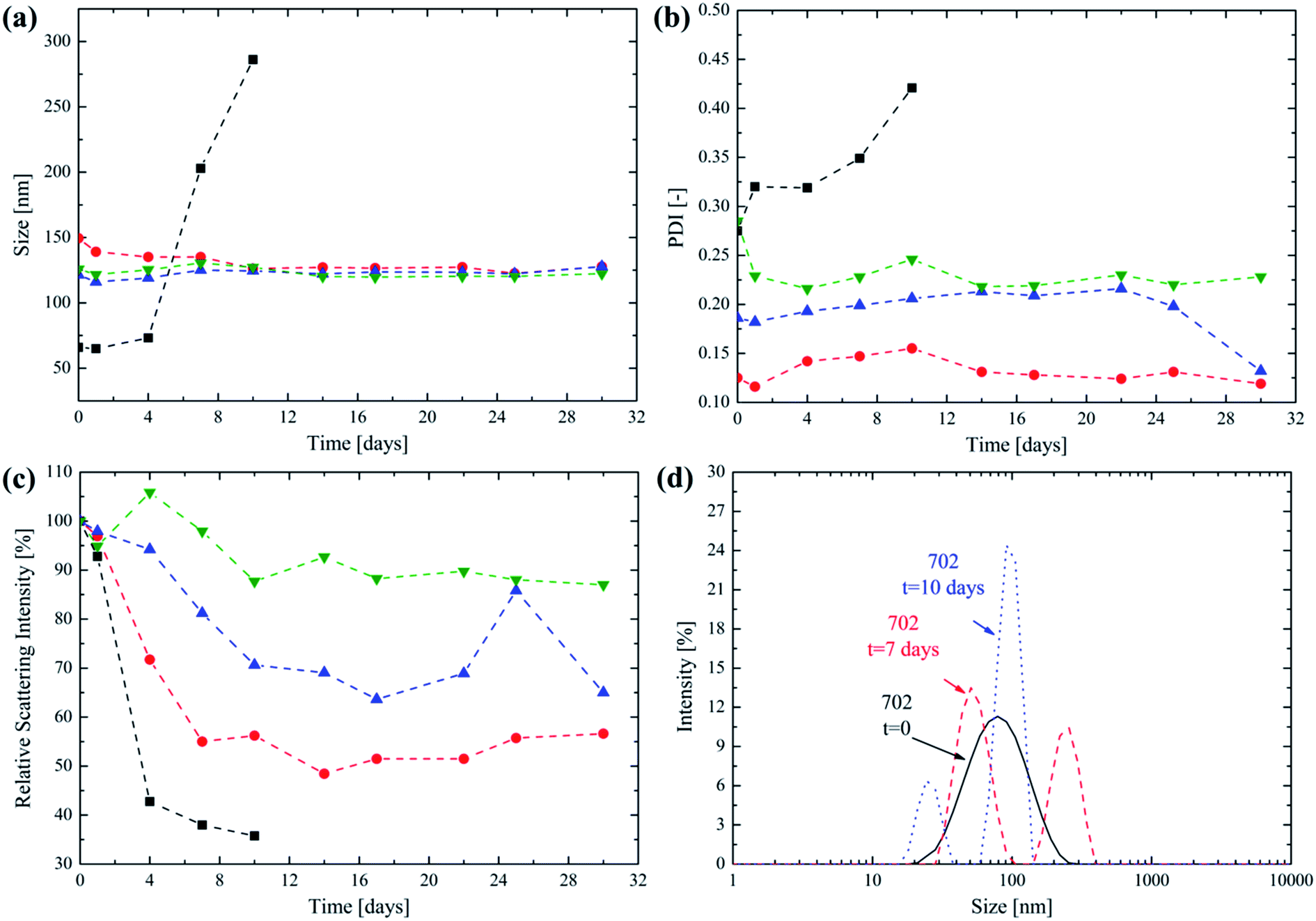 | ||
Fig. 4 Evolution of (a) intensity-averaged size, (b) PDI and (c) relative scattering intensity referred to the initial time t = 0 over time for the 702 ( ), 705 ( ), 705 ( ), 7010 ( ), 7010 ( ) and 7020 ( ) and 7020 ( ) NPs in PBS incubated at 37 °C over a month. (d) PSDs for the 702 NPs after the synthesis (black solid line), after 7 days (red dashed line) and after 10 days (blue dotted line) at 37 °C. ) NPs in PBS incubated at 37 °C over a month. (d) PSDs for the 702 NPs after the synthesis (black solid line), after 7 days (red dashed line) and after 10 days (blue dotted line) at 37 °C. | ||
From a close inspection, it is possible to observe that the size does not change significantly over 30 days for the copolymers 705, 710 and 7020. This suggests the ability of the poly(HPMA) block to provide colloidal stability to the produced NPs, avoiding their aggregation, over a significant period of time. An exception to this trend is represented by the 702 NPs. In this case their average size abruptly increases after 4 days. It is not surprising that this behaviour is recorded for the copolymer with the lowest molecular weight for its lipophilic block. The degradation of the few oligo(caprolactone) chains leads to an increase in the NP hydrophilicity that, in turn, causes them to significantly swell in water. After having reached a maximum size after 10 days the NPs cannot be detected any longer via DLS. In fact, the complete degradation of the lipophilic monomer leaves a highly hydrophilic poly(HPMA) backbone that is molecularly dissolved in PBS. From Fig. 4b it is clear that the PDI follows a trend that is similar to the NP size, being almost constant for the 705, 7010 and 7020 NPs thus providing a further proof of the colloidal stability provided by the poly(HPMA) block. Again, the PDI significantly increases for the 702 NPs starting from the fourth day, in correspondence with the size increase. An important parameter to be considered when studying NP degradation is the relative scattering intensity, which is directly related to the NP size and concentration.44,45 The trend of this parameter over time is reported in Fig. 4c. For the 702 NPs, the relative scattering intensity rapidly decreases, reaching the 35% of its original value after 10 days. Since the NP size is increasing during this period, this significant reduction in the scattering intensity suggests that the NPs are dissolving and that the degradation is almost complete after 10 days. The degradation is much slower for the 705, 7010 and 7020 NPs, for which the relative scattering intensity reaches the 56%, 65% and 88% of the original value after 30 days, respectively. This progressive reduction in the degradation rate as the lipophilic block DP increases is not surprising. In fact, the higher the lipophilic block DP, the lower the hydration that is expected for the core forming block. This reduces the local water concentration into the core and then slows down the kinetic of the hydrolysis reactions, as already proposed in the literature.23,46–48 Finally, the evolution of the PSD for the 702 NPs is reported in Fig. 4d. Here it is possible to observe that shortly upon the synthesis, a unimodal PSD is obtained. However, after 7 days, a second NP population centered at 260 nm is formed, thus suggesting the NP swelling as a consequence of the degradation and the progressively increasing hydrophilic character of the diblock copolymer. The presence of a second peak can be explained considering that the degradation mechanism is not homogeneous.8 Finally, after 10 days the presence of residual, low size micelles is detected by the DLS before the complete dissolution of the copolymer. This degradation mechanism was already observed and described in the case of NPs obtained via emulsion polymerization from PCL-based macromonomers.8,23,49 However, in this work, the synthesis of the lipophilic part of the block copolymer via a combination of ROP and RAFT polymerization allows to finely tune the NP degradation time in order to obtain drug carriers with an optimized half-life. The progressive release of the oligo(caprolactone) side chains is designed to leave the water soluble, low molecular weight poly(HPMA). The combination of complete solubility and low MW makes the degradation residue easily excreted by the kidneys, which is a key factor in avoiding the risk of polymer accumulation.1
3.5 Cytotoxicity and trabectedin release
To further prove the suitability of the produced NPs as nanovectors aimed at parenteral administration, their in vitro cytotoxicity was evaluated in the case of the TNB cancer MDA-MB-231 cells after exposing them to different concentrations of NP suspension for 48 h. The cell viability expressed as a percentage of the control is reported in Fig. 5a in the case of 702 and 705 NPs. | ||
Fig. 5 (a) Cell viability, expressed as the percentage of viable cells with respect to the control, after 48 h exposure with 702 (grey bars) and 705 (white bars) NPs at different NP concentration in the medium. (b) Evolution of the amount of the released trabectedin from 702 ( ) and 705 ( ) and 705 ( ) NPs. ) NPs. | ||
It can be noticed that both 702 and 705 proved to be highly biocompatible, even at the highest concentration in the medium. This is not surprising considered that poly(HPMA) is known to be non cytotoxic and that the biocompatibility of polyesters like PLA and PCL has been already demonstrated elsewhere.50–53 However, a slightly decrease in the cell viability (i.e. 88%) is recorded for cells exposed at 0.9 mg mL−1 of 705 NP suspension. This could be due to the sensitivity of the polymer towards hydrolysis of the dithioester of the RAFT agent.43,54
Finally, the ability of the produced amphiphilic NPs to encapsulate and mediate the release of a hydrophobic, anti-tumour drug was studied in the case of trabectedin. The loading efficiency, expressed as the amount of drug entrapped into the NP core compared to that loaded in the process, was equal to 75% and 90% for the 702 and the 705 NPs, respectively (i.e. 5.6 μg and 6.8 μg of trabectedin loaded per mg of polymer). These very high values can be explained considering the simultaneous NP formation and drug loading obtained with the proposed nanoprecipitation method, which leads to a higher loading efficiency compared to a post-synthesis loading process.49,55 Further, the higher lipophilic block MW in the case of the 705 NPs is responsible for a higher loading efficiency compared to the 702 NPs. Once the trabectedin was loaded into the NP core, its release was studied via LC-MS/MS and the results are reported in Fig. 5b. It can be observed that after a 30% initial burst release accomplished within 2 h, the drug was sustainedly released for over 24 h. The trabectedin amount released after this time was the 87% for the 702 NPs and the 75% for the 705 NPs referred to the loaded amount. Again, the higher MW of the lipophilic portion in the case of the 705 NPs accounts for a more efficient drug retention.
Finally, this study proved the ability of the proposed formulation to entrap and sustainedly release a hydrophobic drug over a time scale that is comparable to the NP residence time into the body.56,57 Further, it elucidates the dependence of both drug loading and drug release from the copolymer composition.
4. Conclusion
N-(2-Hydroxypropyl)methacrylamide, known as non toxic, non immunogenic and capable to be functionalized in mild conditions, has been exploited to both synthesize a novel lipophilic and biodegradable HPMA-CL5 macromonomer and to provide steric stability to the NPs produced starting from this macromonomer. In particular, to obtain a well defined and tunable structure for the lipophilic macromonomer, the adoption of an inverse macromonomer method was necessary due to the low reactivity of the HPMA as the co-catalyst in the ROP of cyclic esters. This inverse macromonomer approach comprises three steps: (i) the ROP of the ε-caprolactone in the presence of a reactive primary alcohol (i.e. benzyl alcohol), (ii) the acylation of the product with succinic anhydride and (iii) the DCC-mediated esterification with the HPMA. On the other hand, the direct ROP of the ε-caprolactone, using HPMA as the co-catalyst, proved to lead to a poor HPMA conversion and hence to a poor control over the final macromonomer MW. Then, to produce amphiphilic diblock copolymers able to self-assemble in an aqueous environment forming stable NPs, a poly(HPMA)70 macro-CTA was firstly synthesized via solution RAFT polymerization in ethanol/acetic buffer mixture. This was further chain extended with the HPMA-CL5 macromonomer to obtain a comb-like polymer composed of a homogeneous poly(HPMA) backbone and degradable oligo(caprolactone) pendants. A simplified nanoprecipitation strategy to assemble the final block copolymer in monodisperse core–shell NPs was also introduced. Since it was demonstrated that a high polymer concentration (up to 20% w/w) in the organic phase does not have a detrimental effect on the NP size and PDI, this strategy is promising in the production of ready-to-use nanovectors, avoiding intermediate steps like lyophilization and dialysis. The degradation kinetics of the produced NPs, as well as their ability to encapsulate and mediate the release of a hydrophobic drug was finally studied and related to the copolymer composition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from AIRC Special Program Molecular Clinical Oncology “5 per mille”. The authors acknowledge Viola Valenti for her help in the experimental part.References
- D. Moscatelli and M. Sponchioni, in Bioresorbable Polymers for Biomedical Applications: From Fundamentals to Translational Medicine, ed. G. Perale and J. Hilborn, Woodhead Publ Ltd, Cambridge, 2017, vol. 120, pp. 265–283 Search PubMed.
- R. Y. Liu, Y. Li, Z. Z. Zhang and X. Zhang, Regener. Biomater., 2015, 2, 125–133 CrossRef CAS PubMed.
- N. J. Ganson, T. J. Povsic, B. A. Sullenger, J. H. Alexander, S. L. Zelenkofske, J. M. Sailstad, C. P. Rusconi and M. S. Hershfield, J. Allergy Clin. Immunol., 2016, 137, 1610–1613 CrossRef CAS PubMed.
- P. H. Kierstead, H. Okochi, V. J. Venditto, T. C. Chuong, S. Kivimae, J. M. J. Frechet and F. C. Szoka, J. Controlled Release, 2015, 213, 1–9 CrossRef CAS PubMed.
- U. C. Palmiero, A. Agostini, S. Gatti, M. Sponchioni, V. Valenti, L. Brunel and D. Moscatelli, Macromolecules, 2016, 49, 8387–8396 CrossRef CAS.
- U. Capasso Palmiero, A. Agostini, E. Lattuada, S. Gatti, J. Singh, C. T. Canova, S. Buzzaccaro and D. Moscatelli, Soft Matter, 2017, 13, 6439–6449 RSC.
- R. Ferrari, Y. C. Yu, M. Morbidelli, R. A. Hutchinson and D. Moscatelli, Macromolecules, 2011, 44, 9205–9212 CrossRef CAS.
- C. Colombo, L. Dragoni, S. Gatti, R. M. Pesce, T. R. Rooney, E. Mavroudakis, R. Ferrari and D. Moscatelli, Ind. Eng. Chem. Res., 2014, 53, 9128–9135 CrossRef CAS.
- R. Ferrari, A. Agostini, L. Brunel, L. Morosi and D. Moscatelli, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3788–3797 CrossRef CAS.
- T. R. Rooney, S. P. Gumfekar, J. B. P. Soares and R. A. Hutchinson, Macromol. Mater. Eng., 2016, 301, 1248–1254 CrossRef CAS.
- T. R. Rooney, O. Monyatsi and R. A. Hutchinson, Macromolecules, 2017, 50, 784–795 CrossRef CAS.
- R. Ferrari, M. Callari and D. Moscatelli, RSC Adv., 2015, 5, 67475–67484 RSC.
- F. Brühne and E. Wright, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- M. Sponchioni, U. C. Palmiero and D. Moscatelli, Macromol. Chem. Phys., 2017, 1700380 CrossRef.
- L. Ceriani, M. Ferrari, M. Zangarini, S. A. Licandro, E. Bello, R. Frapolli, F. Falcetta, M. D'Incalci, R. Libener, F. Grosso, P. Aviles and M. Zucchetti, Bioanalysis, 2015, 7, 1831–1842 CrossRef CAS.
- M. Zangarini, L. Ceriani, F. Sala, E. Marangon, R. Bagnati, M. D'Incalci, F. Grosso and M. Zucchetti, J. Pharm. Biomed. Anal., 2014, 95, 107–112 CrossRef CAS PubMed.
- M. F. Ebbesen, D. H. Schaffert, M. L. Crowley, D. Oupicky and K. A. Howard, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5091–5099 CrossRef CAS.
- I. Lacík, S. Beuermann and M. Buback, Macromol. Chem. Phys., 2004, 205, 1080–1087 CrossRef.
- I. Lacík, A. Chovancová, L. Uhelská, C. Preusser, R. A. Hutchinson and M. Buback, Macromolecules, 2016, 49, 3244–3253 CrossRef.
- U. C. Palmiero, A. Chovancová, D. Cuccato, G. Storti, I. Lacík and D. Moscatelli, Polymer, 2016, 98, 156–164 CrossRef.
- R. Ferrari, C. Colombo, M. Dossi and D. Moscatelli, Macromol. Mater. Eng., 2013, 298, 730–739 CrossRef CAS.
- R. Ferrari, C. M. Pecoraro, G. Storti and D. Moscatelli, RSC Adv., 2014, 4, 12795–12804 RSC.
- Y. C. Yu, R. Ferrari, M. Lattuada, G. Storti, M. Morbidelli and D. Moscatelli, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 5191–5200 CrossRef CAS.
- A. Gallardo, A. Marcos-Fernandez, S. Egri, R. Lebron and E. Piskin, Int. J. Polym. Anal. Charact., 2008, 13, 83–94 CrossRef CAS.
- J. Kiehl, C. Delaite, S. Bistac, A. S. Schuller and H. Farge, Polymer, 2012, 53, 694–700 CrossRef CAS.
- Y. Xiao, M. Takwa, K. Hult, C. E. Koning, A. Heise and M. Martinelle, Macromol. Biosci., 2009, 9, 713–720 CrossRef CAS PubMed.
- X. M. Tong, X. W. Zhang, L. Ye, A. Y. Zhang and Z. G. Feng, Soft Matter, 2009, 5, 1848–1855 RSC.
- J. B. Xu, J. Z. Song, S. Pispas and G. Z. Zhang, Polym. Chem., 2014, 5, 4726–4733 RSC.
- R. Ferrari, Y. C. Yu, M. Lattuada, G. Storti, M. Morbidelli and D. Moscatelli, Macromol. Chem. Phys., 2012, 213, 2012–2018 CrossRef CAS.
- M. Sponchioni, R. Ferrari, L. Morosi and D. Moscatelli, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2919–2931 CrossRef CAS.
- F. Lince, S. Bolognesi, B. Stella, D. L. Marchisio and F. Dosio, Chem. Eng. Res. Des., 2011, 89, 2410–2419 CrossRef CAS.
- J. Hebling, L. Bianchi, F. G. Basso, D. L. Scheffel, D. G. Soares, M. R. O. Carrilho, D. H. Pashley, L. Tjaderhane and C. A. D. Costa, Dent. Mater., 2015, 31, 399–405 CrossRef CAS PubMed.
- J. GruberAllickson, B. Lenes, J. Bash and S. Smith, Transfusion, 1997, 37, S324 Search PubMed.
- E. S. Oz, E. Aydemir and K. Fiskin, Oncol. Lett., 2012, 3, 927–929 CAS.
- G. Da Violante, N. Zerrouk, I. Richard, G. Provot, J. C. Chaumeil and P. Arnaud, Biol. Pharm. Bull., 2002, 25, 1600–1603 CAS.
- W. Abdelwahed, G. Degobert, S. Stainmesse and H. Fessi, Adv. Drug Delivery Rev., 2006, 58, 1688–1713 CrossRef CAS PubMed.
- E. B. Souto, S. A. Wissing, C. M. Barbosa and R. H. Muller, Int. J. Pharm., 2004, 278, 71–77 CrossRef CAS PubMed.
- M. Chacon, J. Molpeceres, L. Berges, M. Guzman and M. R. Aberturas, Eur. J. Pharm. Sci., 1999, 8, 99–107 CrossRef CAS PubMed.
- F. Franks, Eur. J. Pharm. Biopharm., 1998, 45, 221–229 CrossRef CAS PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- P. Legrand, S. Lesieur, A. Bochot, R. Gref, W. Raatjes, G. Barratt and C. Vauthier, Int. J. Pharm., 2007, 344, 33–43 CrossRef CAS PubMed.
- S. Galindo-Rodriguez, E. Allemann, H. Fessi and E. Doelker, Pharm. Res., 2004, 21, 1428–1439 CrossRef CAS.
- W. Q. Shen, Y. L. Chang, G. Y. Liu, H. F. Wang, A. N. Cao and Z. S. An, Macromolecules, 2011, 44, 2524–2530 CrossRef CAS.
- V. Toncheva, E. Schacht, S. Y. Ng, J. Barr and J. Heller, J. Drug Targeting, 2003, 11, 345–353 CrossRef CAS PubMed.
- G. Gaucher, M. H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J. C. Leroux, J. Controlled Release, 2005, 109, 169–188 CrossRef CAS PubMed.
- A. S. Sawhney, C. P. Pathak and J. A. Hubbell, Macromolecules, 1993, 26, 581–587 CrossRef CAS.
- F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37–53 CrossRef CAS PubMed.
- R. Verber, A. Blanazs and S. P. Armes, Soft Matter, 2012, 8, 9915–9922 RSC.
- C. Colombo, L. Morosi, E. Bello, R. Ferrari, S. A. Licandro, M. Lupi, P. Ubezio, M. Morbidelli, M. Zucchetti, M. D'Incalci, D. Moscatelli and R. Frapolli, Mol. Pharm., 2016, 13, 40–46 CrossRef CAS PubMed.
- K. S. Soppimath, T. M. Aminabhavi, A. R. Kulkarni and W. E. Rudzinski, J. Controlled Release, 2001, 70, 1–20 CrossRef CAS PubMed.
- D. Lemoine, C. Francois, F. Kedzierewicz, W. Preat, M. Hoffman and P. Maincent, Biomaterials, 1996, 17, 2191–2197 CrossRef CAS PubMed.
- M. D. Coffin and J. W. McGinity, Pharm. Res., 1992, 9, 200–205 CrossRef CAS.
- A. Belbella, C. Vauthier, H. Fessi, J. P. Devissaguet and F. Puisieux, Int. J. Pharm., 1996, 129, 95–102 CrossRef CAS.
- W. Q. Shen, Q. A. Qiu, Y. Wang, M. A. Miao, B. S. Li, T. S. Zhang, A. N. Cao and Z. S. An, Macromol. Rapid Commun., 2010, 31, 1444–1448 CrossRef CAS PubMed.
- P. A. Grabnar and J. Kristl, J. Microencapsulation, 2011, 28, 323–335 CrossRef CAS PubMed.
- H. J. Lee and Y. Bae, Pharm. Res., 2013, 30, 2077–2086 CrossRef CAS PubMed.
- R. Ferrari, L. Talamini, M. B. Violatto, P. Giangregorio, M. Sponchioni, M. Morbidelli, M. Salmona, P. Bigini and D. Moscatelli, Mol. Pharm., 2017, 14, 124–134 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of the produced macromonomer and 1H NMR of the diblock copolymers. See DOI: 10.1039/c7ra11179g |
| This journal is © The Royal Society of Chemistry 2017 |