
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flow synthesis of cyclobutanones via [2 + 2] cycloaddition of keteneiminium salts and ethylene gas†
C.
Battilocchio
 a,
G.
Iannucci
a,
S.
Wang
a,
E.
Godineau
b,
A.
Kolleth
b,
A.
De Mesmaeker
b and
S. V.
Ley
*a
a,
G.
Iannucci
a,
S.
Wang
a,
E.
Godineau
b,
A.
Kolleth
b,
A.
De Mesmaeker
b and
S. V.
Ley
*a
aInnovative Technology Centre, Department of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW, UK. E-mail: svl1000@cam.ac.uk
bSyngenta Crop Protection AG, Crop Protection Research, Schaffhauserstrasse 101, CH-4332, Switzerland
First published on 6th March 2017
Abstract
A flow chemistry process for the synthesis of 2-substituted cyclobutanones, via [2 + 2] cycloaddition of keteneiminium salts and ethylene gas, is reported. Our approach uses rapid and mild reaction conditions to access a diverse array of products with good to excellent yield, alongside a good level of functional group compatibility.
In recent years, there has been a resurgence in the importance of small rings in chemistry programmes.1 In particular, from a pharmaceutical and agrochemical perspective there is interest in developing new methods to access these molecular entities,2 with a focus towards developing more sustainable processes. In addition, the general demand to generate flexible building blocks, susceptible to further molecular elaboration, is driving the science in this area.3 Cyclobutanones represent an interesting class of small rings4 as they are associated with a reactive versatility,5 mainly due to their ring strain (ca. 25 kcal mol−1)4 (Fig. 1).
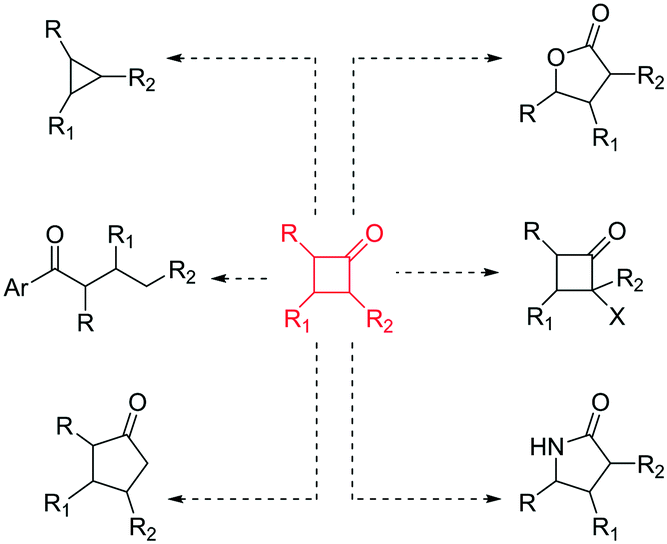 | ||
| Fig. 1 Chemical versatility of cyclobutanones.4,5 | ||
As part of an ongoing research programme, we became interested in the preparation of mono-substituted cyclobutanones as a generally underexplored class of small rings. We were interested in developing a reliable and robust procedure to the preparation of mono-substituted cyclobutanones, minimising risks and providing a potentially scalable process.
A classical approach to these systems involves cycloaddition reactions of ketene intermediates6 or, more effectively, keteneiminium salts species.7 In the latter case, Ghosez7a–d and co-workers have pioneered the use of keteneiminium salts and alkenes in [2 + 2] cycloadditions for the generation of substituted cyclobutanones (Scheme 1). Although these protocols provide an insightful approach towards the making of this scaffold, there are clear issues related to the original batch methods. Whilst the use of triflic anhydride on a small batch scale is acceptable, dosing at large scales can be quite problematic. Also, as the preparation of mono-substituted cyclobutanones requires the use of ethylene gas, there are clear safety issues associated with the use of flammable gaseous reagents.7b
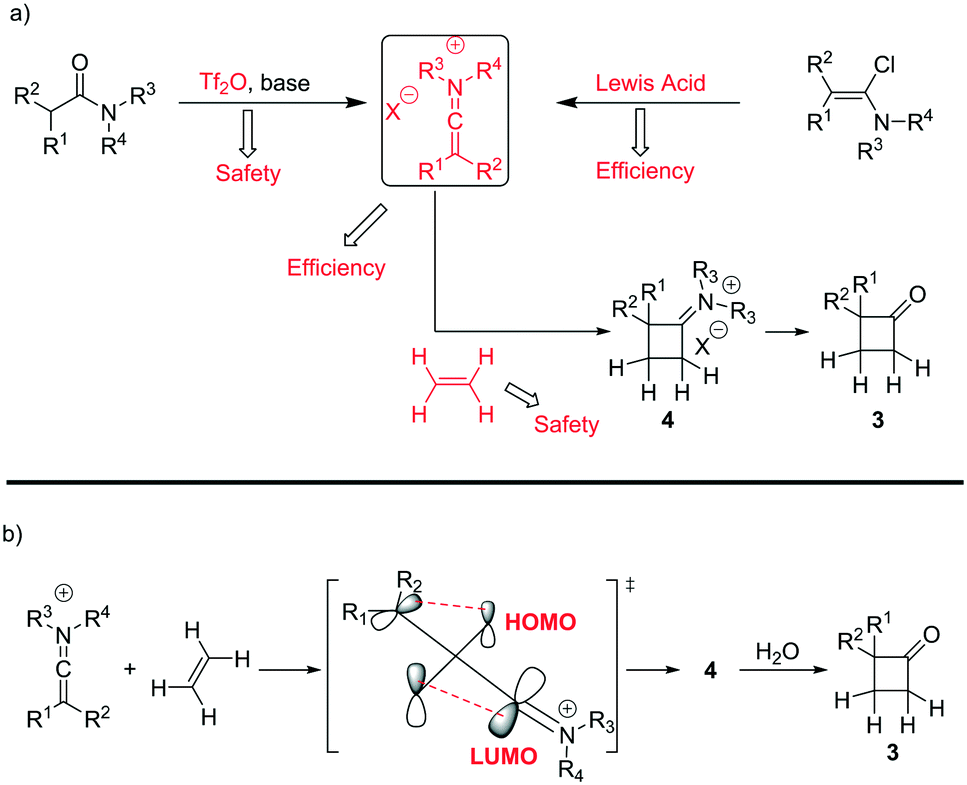 | ||
| Scheme 1 a) Synthesis strategies for the preparation of mono-substituted cyclobutanones and b) mechanistic overview for the cycloaddition of keteniminium salts and ethylene. | ||
On the other hand, the advent of new technologies and advanced synthesis equipment8,9 has opened improved safety windows.
Based on our expertise in the use of these enabling methods to solve chemical synthesis problems,8 we undertook a program to study the preparation of 2-mono-substituted cyclobutanones with the use of the tube-in-tube10 technology as an advantageous gas-feeding method for chemical reactions, which overcomes reactive gas handling issues.10a More precisely, the approach focused on the use of a standard flow system with in-line tube-in-tube reactor to control the introduction of ethylene gas (Scheme 2).
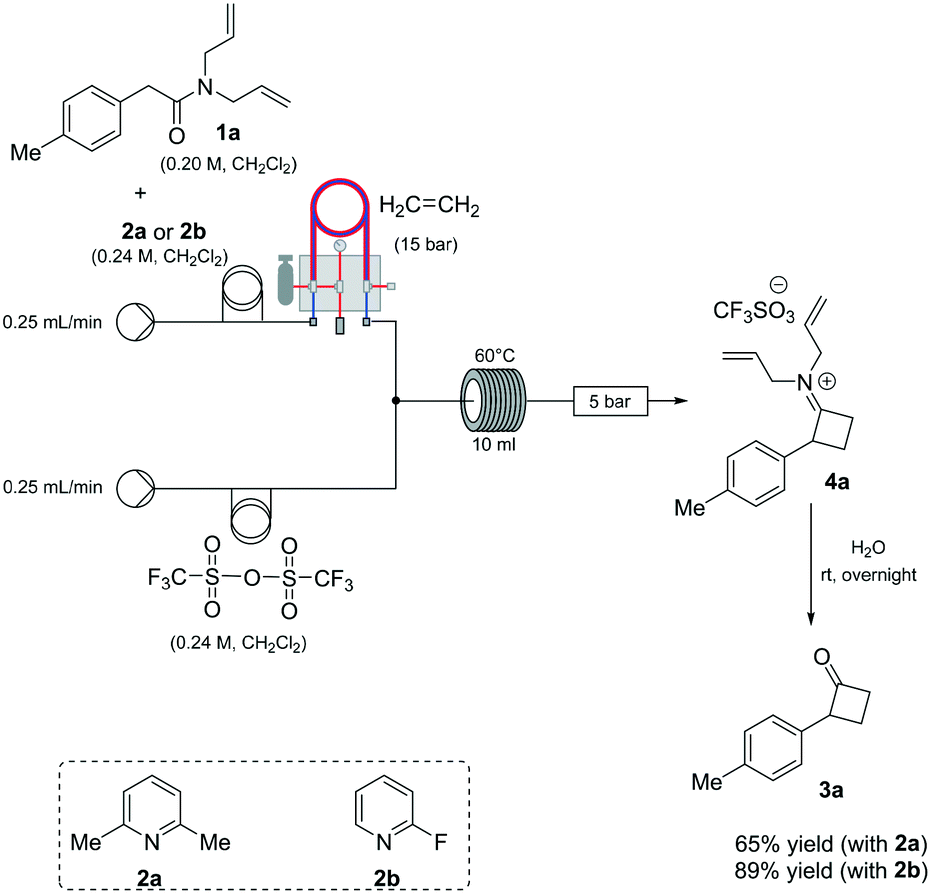 | ||
| Scheme 2 Machine-assisted reaction set up for screening. | ||
Previous investigations highlighted the importance of using bis-allyl amides, as precursors on these cycloaddition reactions, to afford a reasonably yielding process.7h For the first set of experiments, we explored the use of 2,6-lutidine as an additive base, particularly owing to its low cost.
Whilst ethylene gas was dosed using the tube-in-tube reactor, triflic anhydride and the reagents were loaded via 10 mL reaction loops. A first screening of conditions highlighted that temperature, residence time and pressure were all crucial parameters. The best reaction profile was obtained at 60 °C, whilst reducing or increasing the temperature gave no benefits to the overall reaction. We identified the optimum residence time to be 20 min, with a pressure differential of 10 bar (see Table S1 in the ESI†). However, as shown in Scheme 2, our attempts to optimise the reaction conditions beyond 65% for the preparation of 3a were not successful nor with other amides could this be improved.
An analysis of the reaction mechanism suggested the basicity of the 2,6-lutidine may be an obstacle to the reaction outcome, as noted in previous works.7d–h Consequently, switching to 2-fluoropyridine11 afforded a process that could be applied to a wide range of substituted amides, mostly in high isolated yields (Fig. 2). We then extended our investigation to study the reaction scope with respect to the aromatic acetic precursors (Fig. 2). Under these new conditions, a 0.2 M solution of amide 1a (containing 1.2 equiv. of 2-fluoropyridine) was pumped (0.25 mL/min) through the tube-in-tube system where ethylene gas was fed at a pressure differential of 10 bar and the solution directed to a T-piece where it combined with a solution of triflic anhydride (0.25 mL/min);‡ the reaction stream was then reacted in a 10 mL coil reactor at 60 °C for 20 min. The output of the reaction was directed to a quenching pot which contained distilled water. Under these conditions, compound 3a was obtained with an improved 89% yield.
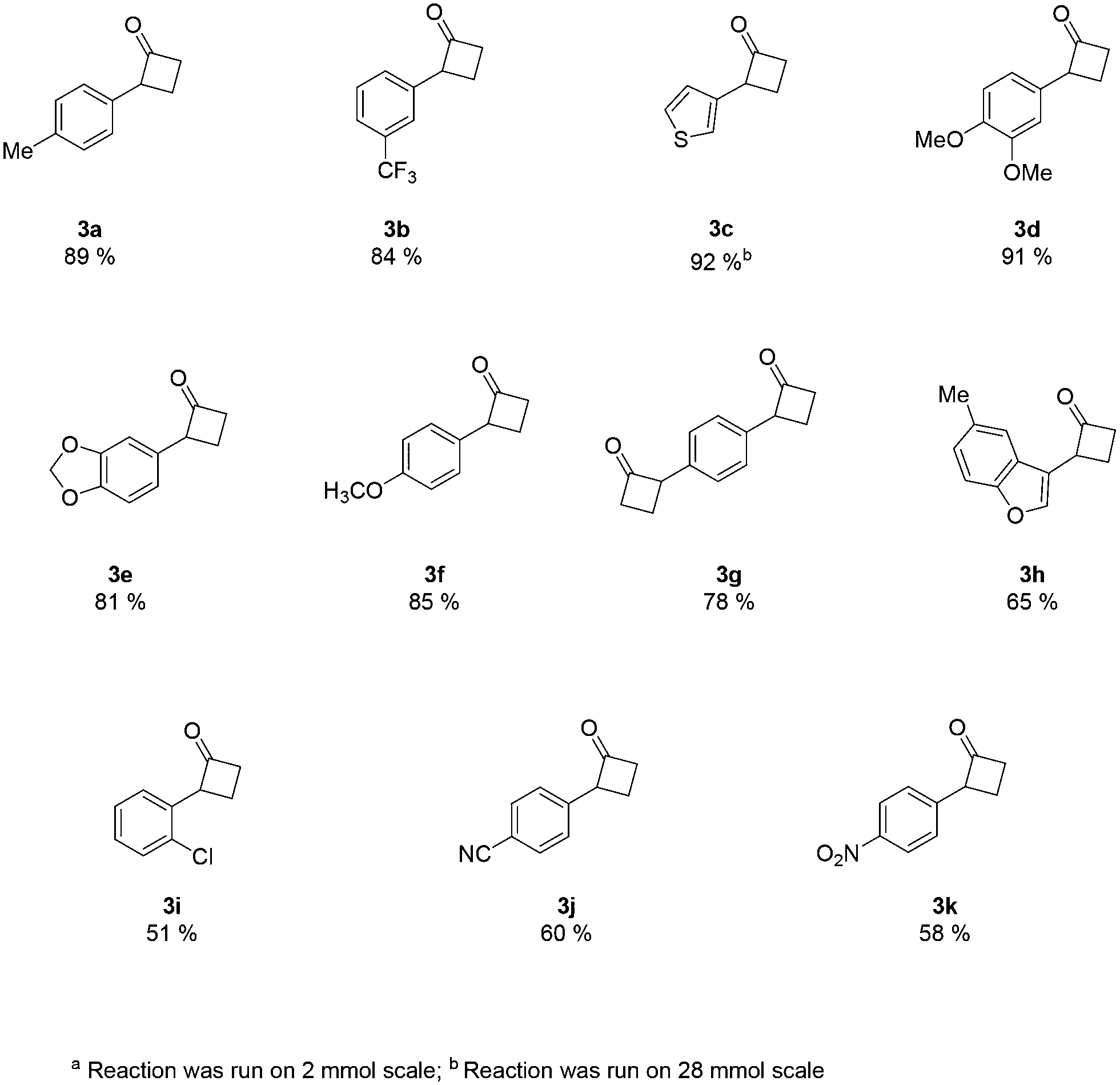 | ||
| Fig. 2 Scope of the reaction with respect to the use of aryl acetic amidesa. | ||
The presence of both-electron-rich and electron-poor substituents was tolerated, giving acceptable yields in all cases. In general, the presence of electron-rich aromatic rings was associated with slightly better outcomes. The protocol tolerated hetero-aromatic rings (3c and 3h, 92% and 65% yield respectively) and proved to be suitable for bis-derivatives (3g, 78% yield). The presence of ortho-substituents gave product 3i in slightly lower yield (51% isolated).
Next, we increased further the scope of the method using aliphatic derivatives (Fig. 3). Generally, these derivatives gave good yields with no major by-products being detected. The presence of ester and halogen groups was well tolerated, with compounds 3l and 3m being obtained with good yields (52% and 68%, respectively). An interesting example containing a terminal alkyne moiety, readily afforded 3p (47% yield). Moreover, 2-alkylcyclobutanones (2-ACBs), such as 2-hexylcyclobutanone (3n, 96% yield), represent important markers for the analysis of irradiated food; this protocol can easily apply to the synthesis of such compounds, where previous methods of synthesis failed to provide reasonable yields.12
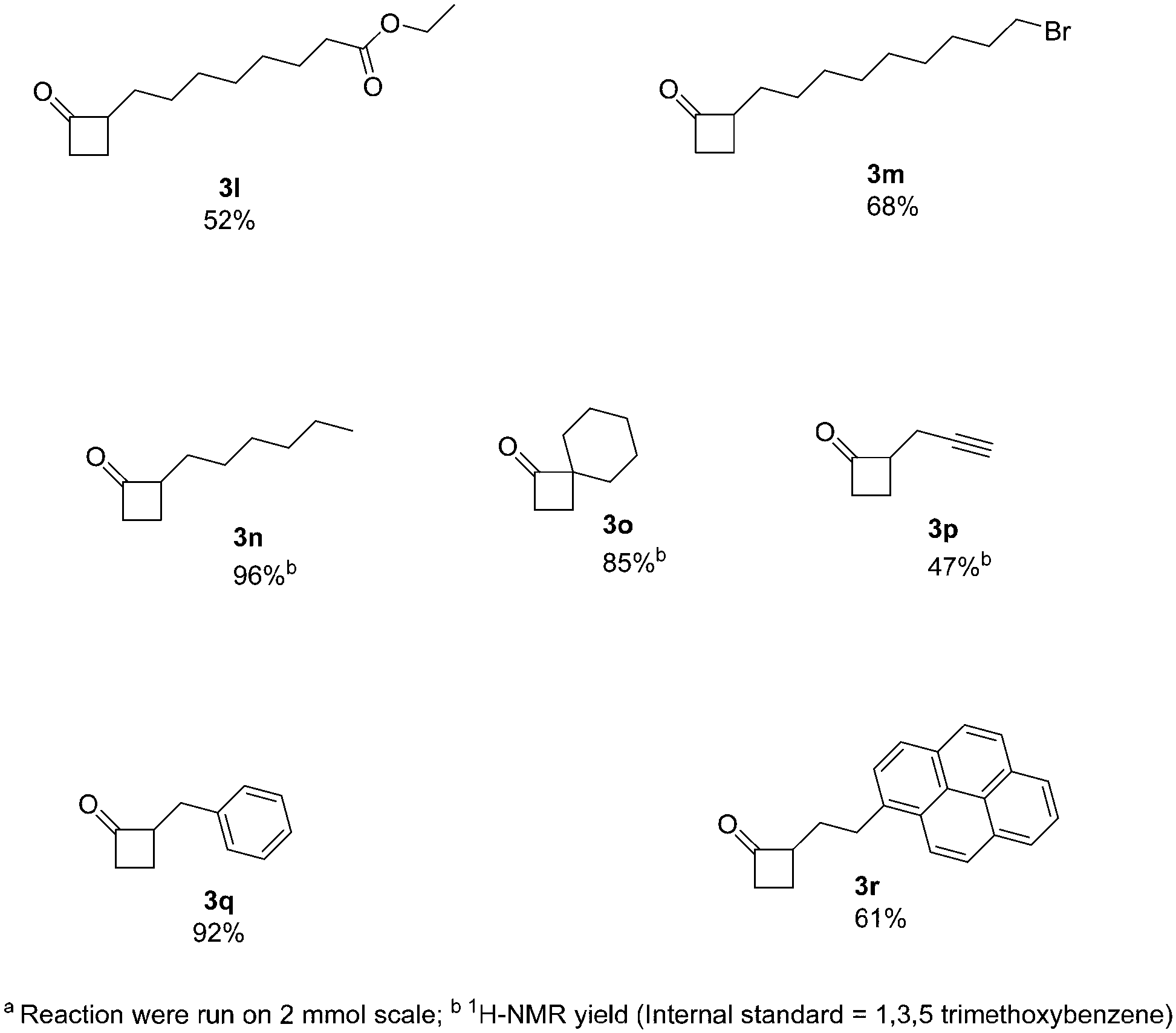 | ||
| Fig. 3 Expanding the scope of the reactiona. | ||
To prove the robustness of method, we examined the ability of the machine-assisted method to afford larger amounts of material, obtaining 3.92 g of 3c in a consistent 92% yield (Scheme 3). To make the system fully continuous, we collected the iminium intermediate 4c in a reservoir and then combined the organic solution with an aqueous stream (2.5 mL/min flow rate for each channel), reacting the biphasic mixture at 80 °C in a static-mixer coil13 (35 mL, 7 min residence time) (see ESI†), and used a membrane-based liquid–liquid separator to continuously collect the organic output.14
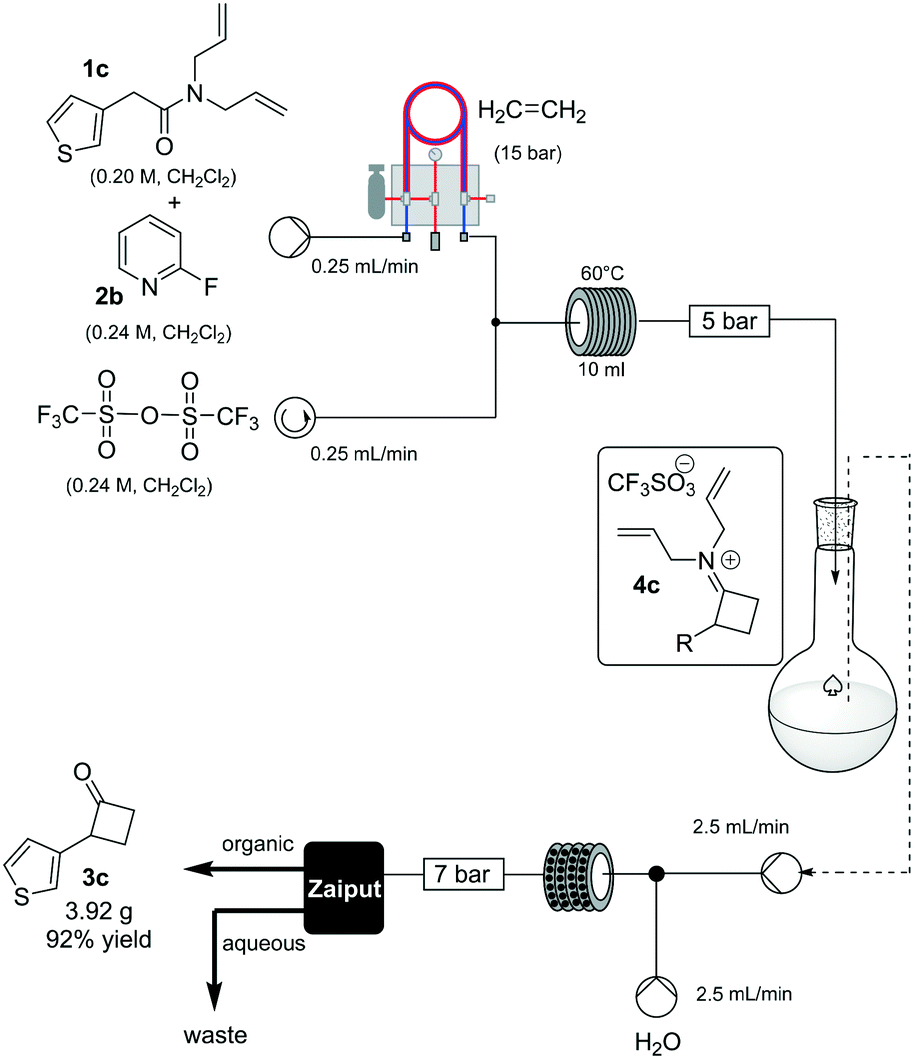 | ||
| Scheme 3 Scale up to afford compound 3c. | ||
In conclusion, we have developed a safe and reliable route for the generation of mono-substituted cyclobutanones, using ethylene as gaseous coupling partner. The use of flow chemistry and the combination of the tube-in-tube technology clearly offers advantages of safety, reproducibility and potential scalability.
The authors would like to thank Syngenta Crop Protection (CB) and the EPSRC (SVL, grants EP/K009494/1, EP/M004120/1 and EP/K039520/1) for financial support.
Notes and references
- (a) T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531–541 CrossRef CAS PubMed; (b) A. Figueras, R. Miralles-Llumà, R. Flores, A. Rustullet, F. Busqué, M. Figueredo, J. Font, R. Alibés and J.-D. Maréchal, ChemMedChem, 2012, 7, 1044–1056 CrossRef CAS PubMed.
- (a) L. Malet-Sanz and F. Susanne, J. Med. Chem., 2012, 55, 4062–4098 CrossRef CAS PubMed; (b) D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, ACS Cent. Sci., 2016, 2, 131–138 CrossRef CAS PubMed.
- (a) A. Guan, C. Liu, X. Yang and M. Dekeyser, Chem. Rev., 2014, 114, 7079 CrossRef CAS PubMed; (b) C. Lamberth, S. Jeanmart, T. Luksch and A. Plant, Science, 2013, 341, 742 CrossRef PubMed.
- (a) D. Bellus and B. Ernst, Angew. Chem., Int. Ed. Engl., 1988, 27, 797–827 CrossRef; (b) F. Secci, A. Frongia and P. P. Piras, Molecules, 2013, 18, 15541–15572 CrossRef PubMed.
- (a) M. H. Shaw and J. F. Bower, Chem. Commun., 2016, 52, 10817–10829 RSC; (b) H. M. Ko and G. Dong, Nat. Chem., 2014, 6, 739–744 CrossRef CAS PubMed; (c) M. Murakami, T. Itahashi and Y. Ito, J. Am. Chem. Soc., 2002, 124, 13976–13977 CrossRef CAS PubMed; (d) R. C. D. Brown, C. J. R. Bataille and J. D. Hinks, Tetrahedron Lett., 2001, 42, 473–475 CrossRef CAS; (e) B. Brown and L. S. Hegedus, J. Org. Chem., 2000, 65, 1865–1872 CrossRef CAS PubMed.
- M. D. Lawlor, T. W. Lee and R. L. Danheiser, J. Org. Chem., 2000, 65, 4375–4384 CrossRef CAS PubMed.
- (a) J. Marchand-Brynaert and L. Ghosez, J. Am. Chem. Soc., 1972, 94, 2870–2872 CrossRef CAS; (b) J.-B. Falmagne, J. Escudero, S. Taleb-Sahraoui and L. Ghosez, Angew. Chem., Int. Ed. Engl., 1981, 20, 879–880 CrossRef; (c) A. Sidani, J. Marchand-Brynaert and L. Ghosez, Angew. Chem., Int. Ed. Engl., 1974, 13, 267 CrossRef; (d) C. Genicot and L. Ghosez, Tetrahedron Lett., 1992, 33, 7357–7360 CrossRef CAS; (e) A. Lumbroso, S. Catak, S. Sulzer-Mossé and A. De Mesmaeker, Tetrahedron Lett., 2014, 55, 5147–5150 CrossRef CAS; (f) A. Lumbroso, S. Catak, S. Sulzer-Mossé and A. De Mesmaeker, Tetrahedron Lett., 2014, 55, 6721–6725 CrossRef CAS; (g) A. Lumbroso, S. Catak, S. Sulzer-Mossé and A. De Mesmaeker, Tetrahedron Lett., 2015, 56, 2397–2401 CrossRef CAS; (h) A. Kolleth, A. Lumbroso, G. Tanriver, S. Catak, S. Sulzer-Mossé and A. De Mesmaeker, Tetrahedron Lett., 2016, 57, 3510–3514 CrossRef CAS.
- For selected reviews regarding enabling synthesis technologies, see: (a) M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC; (b) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef CAS PubMed; (c) S. V. Ley, D. E. Fitzpatrick, R. M. Myers, C. Battilocchio and R. J. Ingham, Angew. Chem., Int. Ed., 2015, 54, 10122–10136 CrossRef CAS PubMed; (d) V. Hessel, D. Kralisch, N. Kockmann, T. Noël and Q. Wang, ChemSusChem, 2013, 6, 746–789 CrossRef CAS PubMed; (e) J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8801–8869 RSC; (f) D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680 RSC; (g) R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- For selected recent examples from our group, see: (a) C. Battilocchio, S.-H. Lau, J. M. Hawkins and S. V. Ley, Org. Synth., 2017, 94, 34–45 CrossRef CAS; (b) C. Battilocchio, F. Feist, A. Hafner, M. Simon, D. N. Tran, D. M. Allwood, D. C. Blakemore and S. V. Ley, Nat. Chem., 2016, 8, 360–367 CrossRef CAS PubMed; (c) T. Ouchi, R. J. Mutton, V. Rojas, D. E. Fitzpatrick, D. G. Cork, C. Battilocchio and S. V. Ley, ACS Sustainable Chem. Eng., 2016, 4, 1912–1916 CrossRef CAS; (d) S.-H. Lau, A. Galván, R. R. Merchant, C. Battilocchio, J. A. Souto, M. B. Berry and S. V. Ley, Org. Lett., 2015, 17, 3218–3221 CrossRef CAS PubMed; (e) T. Ouchi, C. Battilocchio, J. M. Hawkins and S. V. Ley, Org. Process Res. Dev., 2014, 18, 1560–1566 CrossRef CAS; (f) R. Chorgade, C. Battilocchio, J. M. Hawkins and S. V. Ley, Org. Lett., 2013, 15, 5698–5701 CrossRef PubMed; (g) C. Battilocchio, I. R. Baxendale, M. Biava, M. O. Kitching and S. V. Ley, Org. Process Res. Dev., 2012, 16, 798–810 CrossRef CAS.
- (a) M. Brzozowski, M. O'Brien, S. V. Ley and A. Polyzos, Acc. Chem. Res., 2015, 48, 349–362 CrossRef CAS PubMed; (b) C. Schotten, D. Plaza, S. Manzini, S. P. Nolan, S. V. Ley, D. L. Browne and A. Lapkin, ACS Sustainable Chem. Eng., 2015, 3, 1453–1459 CrossRef CAS PubMed; (c) J. C. Pastre, D. L. Browne, M. O'Brien and S. V. Ley, Org. Process Res. Dev., 2013, 17, 1183–1191 CrossRef CAS; (d) S. L. Bourne, M. O'Brien, S. Kasinathan, P. Koos, P. Tolstoy, D. X. Hu, R. W. Bates, B. Martin, B. Schenkel and S. V. Ley, ChemCatChem, 2013, 5, 159–172 CrossRef CAS.
- (a) B. Peng, D. Geerdink and N. Maulide, J. Am. Chem. Soc., 2013, 135, 14968–14971 CrossRef CAS PubMed; (b) B. Peng, D. Geerdink, C. Farès and N. Maulide, Angew. Chem., Int. Ed., 2014, 21, 5462–5466 CrossRef PubMed.
- (a) M. Miesch, B. Ndiaye, C. Hasselmann and E. Marchioni, Radiat. Phys. Chem., 1999, 55, 337–344 CrossRef CAS; (b) A. Hartwig, A. Pelzer, D. Burnouf, H. Titeeca, H. Delincee, K. Briviba, C. Soika, C. Hodapp, F. Raul, M. Miesch, D. Werner, P. Horvatovich and E. Marchioni, Food Chem. Toxicol., 2007, 45, 2581–2591 CrossRef CAS PubMed; (c) E. M. K. Leung, P. N. Y. Tang, Y. Ye and W. Chan, J. Agric. Food Chem., 2013, 61, 9950–9954 CrossRef CAS PubMed; (d) A. Breidbach and F. Ulberth, Food Chem., 2016, 201, 52–58 CrossRef CAS PubMed; (e) X. Meng and W. Chan, Food Chem., 2017, 217, 352–359 CrossRef CAS PubMed.
- The static mixer reactors are available from Vapourtec Ltd (https://www.vapourtec.com/).
- The membrane-based liquid-liquid separator was supplied by Zaiput Flow Technologies (http://www.zaiput.com/).
Footnotes |
| † Additional data is available from the University of Cambridge Data Repository website: https://doi.org/10.17863/CAM.7110. Electronic supplementary information (ESI) available: Reactions set up and compounds characterisation. See DOI: 10.1039/c7re00020k |
| ‡ The solution containing triflic anhydride was pumped with a peristaltic pump; the polymer tubing must tolerate this reagent. In our case, we used a SF-10 pump from Vapourtec (https://www.vapourtec.com/), with the red tubing. |
| This journal is © The Royal Society of Chemistry 2017 |