
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sequential catalysis: exploiting a single rhodium(I) catalyst to promote an alkyne hydroacylation–aryl boronic acid conjugate addition sequence†
Maitane
Fernández
,
Matthias
Castaing
and
Michael C.
Willis
 *
*
Department of Chemistry, University of Oxford, Chemical Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: michael.willis@chem.ox.ac.uk
First published on 9th September 2016
Abstract
We demonstrate that a single Rh(I) complex can promote two mechanistically distinct C–C bond-forming reactions – alkyne hydroacylation and aryl boronic acid conjugate addition – to deliver substituted ketone products from the controlled assembly of three readily available fragments. This is a rare example of a Rh(I)/Rh(III) cycle and a redox neutral Rh(I) cycle being promoted by a single catalyst. The process is broad in scope, allowing significant variation of all three reaction components. Incorporation of an enantiomerically pure bis-phosphine ligand renders the process enantioselective. Superior levels of enantioselectivity (up to >99% ee) can be achieved from using a two catalyst system, whereby two Rh(I) complexes, one incorporating an achiral bis-phosphine ligand and the second a chiral diene ligand, are introduced at the start of the reaction sequence.
Introduction
Since the initial report from Miyaura in 1997,1 the Rh(I)-catalysed addition of aryl boronic acids to activated alkenes has become established as a versatile method for the formation of C–C bonds (Scheme 1a).2 The variety of activating groups that can be employed on the alkene, the availability of a wide range of boronic acid derivatives and the predictable, often high levels of stereocontrol that can be achieved,3 have combined to make these transformations popular choices for synthetic chemists,4 including those working in industry.5 Although less developed than the conjugate addition chemistry, Rh(I)-catalysed hydroacylation processes are emerging as powerful methods for synthesis.6 Alkyne hydroacylation, combining aldehydes with alkynes, is dominated by the use of Rh(I)-catalysts,7 allowing the use of mild reaction conditions and low catalyst loadings and represents a potent method for the preparation of enones (Scheme 1b).8 The juxtaposition of Rh(I) catalysts in these two processes – alkyne hydroacylation delivering enones as products, and conjugate additions, consuming enones as substrates – although mechanistically distinct, suggested the possibility of merging these two transformations to provide a unique three-component route to substituted, stereodefined ketones (Scheme 1c). Although many examples of single catalysts controlling two bond forming events in a cascade sequence are known,9 examples in which two C–C bonds are forged in an intermolecular manner,10 using two mechanistically disparate processes, including control of enantioselectivity,11 are extremely rare: this contribution documents such a process. | ||
| Scheme 1 Rh(I)-catalysed boronic acid conjugate additions and alkyne hydroacylation reactions, together with a merged, sequential process. | ||
The Rh(I)-catalysed addition of aryl boronic acids to electron-poor alkenes is a redox neutral process which most commonly employs catalysts based on relatively large bite-angle bis-phosphine ligands such as BINAP.1,4,12 Conversely, Rh(I)-catalysed alkyne hydroacylation reactions involve a Rh(I)/Rh(III) cycle, and often employ complexes based on small bite-angle bis-phosphines.13 The key to developing the proposed sequential catalytic alkyne hydroacylation–boronic acid conjugate addition sequence would be to identify a rhodium complex capable of mediating both of these mechanistically distinct processes in an efficient manner.
Results and discussion
We began our study by exploring the combination of 2-aminobenzaldehyde 1a and 1-octyne, followed by the addition of phenyl boronic acid (Table 1). This sequence delivers β-phenyl substituted o-amino-ketone 2a as the product; o-amino-ketones such as this are useful synthetic units in their own right,14 and are also embedded in a variety of important heterocycles.15 We evaluated a range of bis-phosphine ligands in the proposed hydroacylation reaction and the results were comparable to our previous studies with amine-chelating aldehydes,16 with the smallest bite-angle dcpm and dppm bis-phosphines (entries 1 and 2), as well as dppe (entry 4), generating highly efficient catalysts. Increasing the bite angle further, as in the case of dppp, resulted in a poorly active hydroacylation catalyst (entry 5). As suggested from the literature,1 of the ligands successful in hydroacylation, only dppe, with a wider bite angle, was able to subsequently promote the conjugate addition, allowing for successful one-pot, two intermolecular C–C bond formation, to occur (entry 4).|
|
||||
|---|---|---|---|---|
| Entry | Ligand | HA convb. (%) | CA convb. (%) | Yield (%) |
| a Reaction conditions: 1a (1.0 equiv.), 1-octyne (1.3 equiv.), [Rh(nbd)2]BF4 (10 mol%), ligand (10 mol%), acetone, 55 °C, 30 min; then PhB(OH)2 (2.0 equiv.), K2CO3 (0.2 equiv.), acetone/water, 3 h. Isolated yield. b Determined by 1H NMR spectroscopy. DMB = 3,4-dimethoxybenzyl. | ||||
| 1 | dcpm | 100 | <5 | — |
| 2 | dppm | 100 | 10 | — |
| 3 | dcpe | 6 | — | — |
| 4 | dppe | 100 | 100 | 84 |
| 5 | dppp | 5 | — | — |

|
||||

We next explored the scope of the three-component transformation (Table 2), and for operational simplicity we used a pre-formed catalyst ([Rh(dppe)(C6H5F)]BArF).17 In general, the developed reaction was very broad in scope, allowing excellent variation of all three components. A wide range of aryl boronic acids could be employed successfully, including substitution at all three positions of the phenyl ring, and a variety of electronically varied functional groups (2a–n). The use of heterocyclic (2o–p), 1- and 2-naphthyl (2q–2r) and several alkenyl boronic acids (2s–2u) was also compatible with the process, delivering the final products in good yields. 2-Aminobenzaldehydes with various substituents on the amine could be employed, in all cases obtaining the final β-substituted ketones in very high yields (products 2v–y). Additionally, electronically varied substituents on the aromatic core of the aldehydes were also allowed (2z–2ad).


With respect to the alkyne, again, wide variation was possible, including the use of alkyl chains, carbocycles, acetals and aromatic groups (2ae–2as). Several examples in Table 2 show variation of more than one component from the standard reaction (2an–2as, 2aj), and give an indication of the structural range accessible using the developed chemistry. Ketones 2aj and 2an were prepared using both possible combinations of alkyne and boronic acid, demonstrating the flexibility of the approach to adapt to available feedstocks. Larger scale reactions were also possible; using 5 mol% of Rh, a 3 mmol scale experiment returned 1 gram of ketone 2w in a 97% yield.
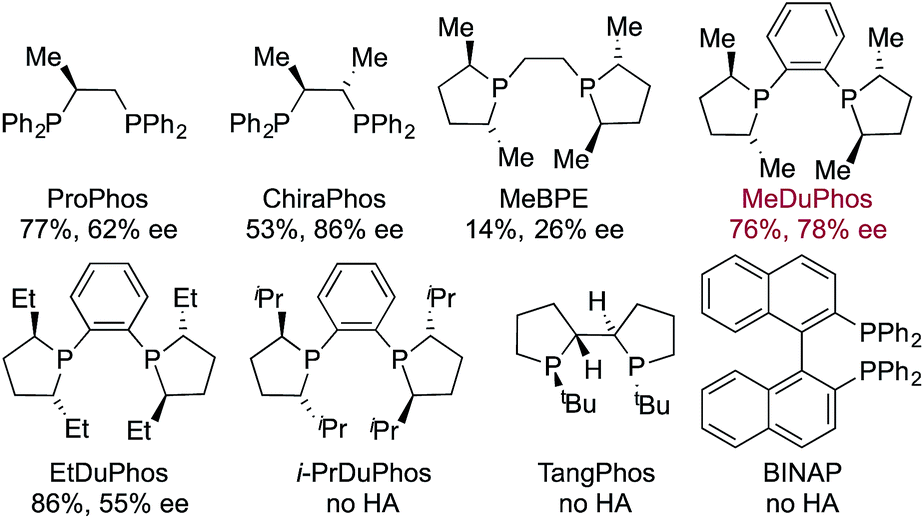
Having identified an achiral Rh-complex capable of delivering a hydroacylation-conjugate addition sequence of broad scope, our next task was to identify a chiral catalyst that would provide enantiomerically enriched products. We evaluated the performance of a series of chiral bis-phosphine ligands in our reaction (Table 3), mindful that the PCCP scaffold was the most efficient for the achiral reaction. Although the highest enantioselectivity was achieved with Chiraphos (86% ee), MeDuphos provided the best all round performance, delivering the ketone 2a in reasonable-good yield and ee (76% yield, 78% ee).
|
|
|---|
| a Reaction conditions: 1a (1.0 equiv.), 1-octyne (1.3 equiv.), [Rh(nbd)2]BF4 (10 mol%), ligand (10 mol%), acetone, 55 °C, 30 min; then PhB(OH)2 (2.0 equiv.), K2CO3 (0.2 equiv.), acetone/water, 3 h. Isolated yields. ees determined by chiral HPLC. |
|

Using a MeDuPhos-derived catalyst, we investigated if variation of the substrate would have an impact on enantioselectivity (Table 4). Overall, the reactions delivered the product ketones in high to excellent yields; however, the enantioselectives were broadly consistent with the trial system and remained in the 75–86% ee region. The exception was the use of aryl-substituted alkynes, which led to a significant reduction in ee (2ak, 2an).
|
|
|---|
| a Reaction conditions: 1 (0.20 mmol), alkyne (0.26 mmol), [Rh(R,R-MeDuPhos)(C6H5F)]BArF (10 mol%), acetone, 55 °C, 30 min; then boronic acid (0.40 mmol), K2CO3 (0.04 mmol), acetone/water, 3 h. Isolated yields. ees determined by chiral HPLC. |

|

The examples in Table 4 show that it is possible for a single Rh-complex to catalyze two distinct intermolecular C–C bond-forming reactions, delivering products with high, but not excellent, enantioselectivity. While we were confident that evaluating further chiral phosphines would deliver a more selective catalyst, we reasoned that a more expedient approach would be to explore the use of a two catalyst system, where one catalyst is optimized for the hydroacylation step, and the second is tailored to deliver an efficient and highly enantioselective conjugate addition. For practicality it would be ideal if both catalysts were present in the reaction vessel from the start. From Table 1, and earlier reports,16 we were confident that a dcpm-supported catalyst would be efficient for alkyne hydroacylation. For the conjugate addition step we turned our attention to the use of chiral diene ligands,18 and chose to evaluate three ligands in a model system involving the addition of phenyl boronic acid to enone 3a (Scheme 2). All three ligands provided efficient reactions. Although all three ligands also delivered levels of enantiocontrol that surpassed the results achieved using MeDuPhos, ligand L2, developed by Lam,19 was the stand-out choice, delivering ketone 2a in >99% ee. All three diene ligands generated inactive hydroacylation catalysts.
 | ||
| Scheme 2 The use of chiral diene ligands in the conjugate addition to enone 3a. | ||
We next explored the use of a two-catalyst system based on dcpm and chiral diene L2. For pragmatic reasons we used two preformed catalysts, [Rh(dcpm)(C6H5F)]BArF and [Rh(L2)(CH3CN)2]BArF, which allowed the addition of both complexes at the start of the reaction. Pleasingly, using this approach we were able to obtain the desired β-phenylketone 2a in 87% yield with an excellent 96% ee (Table 5). We explored the scope of this asymmetric process, and similar to the non-enantioselective variant, the reaction was broad in scope, allowing wide variation of the three components and providing the desired products in good yields and with excellent enantioselectivities (Table 5). A broad range of aryl boronic acids were successfully used, including those bearing substituents with different steric and electronic properties (products 2a–2n), as well as examples of heteroaromatic (2o–2p), naphthyl (2q–2r) and alkenyl boronic acids (2s). Aldehydes with different chelating groups, or substituents on the aromatic core were tolerated (products 2v–2ad), as were various alkyne reaction partners (2ae–2as). In particular, the use of ethynylbenzene derivatives offered very high levels of enantiocontrol, significantly improving the performance of several boronic acids that had shown only moderate selectivity when combined with 1-octyne (see 2ajvs.2s and 2apvs.2n). Finally, the ability to synthesize both enantiomers of the target ketones by simply reversing the combination of alkyne and boronic acid, for example ketone 2an, is a powerful feature of the developed sequence, significantly expanding the utility of the process.


Conclusions
We have shown that a dppe–Rh(I) complex can catalyze sequential alkyne hydroacylation and boronic acid conjugate additions to provide β-substituted ketones with high efficiency. This sequence is a rare example of a single catalyst mediating two distinct intermolecular C–C bond-forming reactions. Use of a MeDuPhos-derived catalyst renders the process enantioselective, however, the highest selectivities are obtained using a two-catalyst system involving a chiral diene ligand, delivering ketones with excellent enantioselectvities.Acknowledgements
This work was supported by the EPSRC and ERC Marie Curie Actions (DEGENHA 656493, to MF).Notes and references
- M. Sakai, H. Hayashi and N. Miyaura, Organometallics, 1997, 16, 4229–4231 CrossRef CAS.
- K. Yoshida and T. Hayashi, in Modern Rhodium-Catalyzed Organic Reactions, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 55–77 Search PubMed.
- (a) Y. Takaya, M. Ogasawara, T. Hayashi, M. Sakai and N. Miyaura, J. Am. Chem. Soc., 1998, 120, 5579–5580 CrossRef CAS; (b) P. Tian, H.-Q. Dong and G.-Q. Lin, ACS Catal., 2012, 2, 95–119 CrossRef CAS.
- (a) H. J. Edwards, J. D. Hargrave, S. D. Penrose and C. G. Frost, Chem. Soc. Rev., 2010, 39, 2093–2105 RSC; (b) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS PubMed.
- (a) S. Brock, D. R. J. Hose, J. D. Moseley, A. J. Parker, I. Patel and A. J. Williams, Org. Process Res. Dev., 2008, 12, 496–502 CrossRef CAS; (b) C. S. Burgey, D. V. Paone, A. W. Shaw, J. Z. Deng, D. N. Nguyen, C. M. Potteiger, S. L. Graham, J. P. Vacca and T. M. Williams, Org. Lett., 2008, 10, 3235–3238 CrossRef CAS PubMed.
- (a) A. Ghosh, K. F. Johnson, K. L. Vickerman, J. A. Walker and L. M. Stanley, Org. Chem. Front., 2016, 3, 639–644 RSC; (b) J. C. Leung and M. J. Krische, Chem. Sci., 2012, 3, 2202 RSC; (c) M. C. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef CAS PubMed.
- Non-Rh examples: (a) Q.-A. Chen, F. A. Cruz and V. M. Dong, J. Am. Chem. Soc., 2015, 137, 3157–3160 CrossRef CAS PubMed; (b) H. Miura, K. Wada, S. Hosokawa and M. Inoue, Chem.–Eur. J., 2013, 19, 861–864 CrossRef CAS PubMed; (c) V. M. Williams, J. C. Leung, R. L. Patman and M. J. Krische, Tetrahedron, 2009, 65, 5024–5029 CrossRef CAS PubMed; (d) S. Chen, X. Li, H. Zhao and B. Li, J. Org. Chem., 2014, 79, 4137–4141 CrossRef CAS PubMed; (e) S. Shi, T. Wang, V. Weingand, M. Rudolph and S. K. Hashmi, Angew. Chem., Int. Ed., 2014, 53, 1148–1151 CrossRef CAS PubMed; (f) F. Yang, T. Jin and Y. Yamamoto, Tetrahedron, 2012, 68, 5223–5228 CrossRef CAS; (g) S. Hatanaka, Y. Obora and Y. Ishii, Chem.–Eur. J., 2010, 16, 1883–1888 CrossRef CAS PubMed.
- Selected examples: (a) J. F. Hooper, S. Seo, F. R. Truscott, J. D. Neuhaus and M. C. Willis, J. Am. Chem. Soc., 2016, 138, 1630–1634 CrossRef CAS PubMed; (b) X.-W. Du and L. M. Stanley, Org. Lett., 2015, 17, 3276–3279 CrossRef CAS PubMed; (c) S. J. Poingdestre, J. D. Goodacre, A. S. Weller and M. C. Willis, Chem. Commun., 2012, 48, 6354–6356 RSC; (d) C. Gonzalez-Rodriguez, R. J. Pawley, A. B. Chaplin, A. L. Thompson, A. S. Weller and M. C. Willis, Angew. Chem., Int. Ed., 2011, 50, 5134–5138 CrossRef CAS PubMed.
- (a) Y. J. Jang, H. Yoon and M. Lautens, Org. Lett., 2015, 17, 3895–3897 CrossRef CAS PubMed; (b) R. Shintani, T. Yamagami and T. Hayashi, Org. Lett., 2006, 8, 4799–4801 CrossRef CAS PubMed; (c) B. M. Bocknack, L.-C. Wang and M. J. Krische, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5421–5424 CrossRef CAS PubMed.
- K. M. Gericke, D. I. Chai, N. Bieler and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 1447–1451 CrossRef CAS PubMed.
- (a) H. Clavier and H. Pellissier, Adv. Synth. Catal., 2012, 354, 3347–3403 CrossRef CAS; (b) H. Pellissier, Chem. Rev., 2013, 113, 442–524 CrossRef CAS PubMed.
- T. Hayashi, M. Takahashi, Y. Takaya and M. Ogasawara, J. Am. Chem. Soc., 2002, 124, 5052–5058 CrossRef CAS PubMed.
- (a) A. Prades, M. Fernández, S. D. Pike, M. C. Willis and A. S. Weller, Angew. Chem., Int. Ed., 2015, 54, 8520–8524 CrossRef CAS PubMed; (b) S. K. Murphy, A. Bruch and V. M. Dong, Chem. Sci., 2015, 6, 174–180 RSC; (c) A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 4885–4897 CrossRef CAS PubMed; (d) I. Pernik, J. F. Hooper, A. B. Chaplin, A. S. Weller and M. C. Willis, ACS Catal., 2012, 2, 2779–2786 CrossRef CAS.
- K. Hirai, T. Fujishita, T. Ishiba, H. Sugimoto, S. Matsutani, Y. Tsukinoki and K. Hirose, J. Med. Chem., 1982, 25, 1466–1473 CrossRef CAS PubMed.
- (a) H. Rapoport and K. G. Holden, J. Am. Chem. Soc., 1960, 82, 4395 CrossRef CAS; (b) H. Koga, A. Itoh, S. Murayama, S. Suzue and T. Irikura, J. Med. Chem., 1980, 23, 1358–1363 CrossRef CAS PubMed; (c) W. Zhang, K. F. Koehler, B. Harris, P. Skolnick and J. M. Cook, J. Med. Chem., 1994, 37, 745–757 CrossRef CAS PubMed; (d) J. X. Kelly, M. J. Smilkstein, R. Brun, S. Wittlin, R. A. Cooper, K. D. Lane, A. Janowsky, R. A. Johnson, R. A. Dodean, R. Winter, D. J. Hinrichs and M. K. Riscoe, Nature, 2009, 459, 270–273 CrossRef CAS PubMed.
- M. Castaing, S. L. Wason, B. Estepa, J. F. Hooper and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 13280–13283 CrossRef CAS PubMed.
- R. Dallanegra, A. P. Robertson, A. B. Chaplin, I. Manners and A. S. Weller, Chem. Commun., 2011, 47, 3763–3765 RSC.
- (a) C. Defieber, H. Grutzmacher and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4482–4502 CrossRef CAS PubMed; (b) T. Hayashi, K. Ueyama, N. Tokunaga and K. Yoshida, J. Am. Chem. Soc., 2003, 125, 11508–11509 CrossRef CAS PubMed; (c) C. Fischer, C. Defieber, T. Suzuki and E. M. Carreira, J. Am. Chem. Soc., 2004, 126, 1628–1629 CrossRef CAS PubMed; for an example of a chiral diene Rh complex used in sequential catalysis, see: (d) L. Zhang, Z. Qureshi, L. Sonaglia and M. Lautens, Angew. Chem., Int. Ed., 2014, 53, 13850–13853 CrossRef CAS PubMed.
- I. D. Roy, A. R. Burns, G. Pattison, B. Michel, A. J. Parker and H. W. Lam, Chem. Commun., 2014, 50, 2865–2868 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc03066a |
| This journal is © The Royal Society of Chemistry 2017 |