
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNeutral chiral cyclopentadienyl Ru(II)Cl catalysts enable enantioselective [2+2]-cycloadditions†
D.
Kossler
 and
N.
Cramer
*
and
N.
Cramer
*
Laboratory of Asymmetric Catalysis and Synthesis, Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne, EPFL SB ISIC LCSA, BCH 4305, CH-1015 Lausanne, Switzerland. E-mail: nicolai.cramer@epfl.ch
First published on 19th January 2017
Abstract
Cyclopentadienyl ruthenium(II) complexes with a large number of available coordination sites are frequently used catalysts for a broad range of transformations. To be able to render these transformations enantioselective, we have designed a chiral neutral CpxRu(II)Cl complex basing on an atropchiral cyclopentadienyl (Cpx) ligand which is accessed in a streamlined C–H functionalisation approach. The catalyst displays excellent levels of reactivity and enantioselectivity for enantioselective [2+2]-cycloadditions leading to strained chiral cyclobutenes, allowing for catalyst loadings as low as 1 mol%. A very strong counterion effect of a bound chloride anion transforms the corresponding unselective cationic complex into a highly enantioselective neutral version. Moreover, by adding norbornadiene at the end of the reaction the catalyst can be recovered and subsequently reused.
Introduction
Homogenous ruthenium complexes constitute a cornerstone of modern transition-metal catalysis.1 Besides metathesis catalysts2 and complexes employed for hydrogenations,3 cyclopentadienyl (Cp) or pentamethylcyclopentadienyl (Cp*) ruthenium(II) complexes, in particular CpRu(CH3CN)3PF6 and Cp*Ru(COD)Cl (COD = 1,5-cyclooctadiene), are highly valuable catalysts for atom-economic carbon–carbon and carbon-heteroatom bond formations.4 One reason is their high number of available and freely accessible coordination sites. However, this has rendered the design of chiral versions challenging.5 Recently, we introduced chiral Cp ligands (Cpx)6 with the cyclopentadienyl group being the only point of coordination. These developments provided cationic CpxRu(II) complexes able to mimic Trost's CpRu(CH3CN)3PF6 catalyst.7 Complementary to this positively charged CpRu+ fragment, neutral Ru(II) complexes catalyse different sets of transformations. In this domain, Cp*Ru(COD)Cl is the most versatile.8 Thus, a chiral version of this catalyst would be of great value. The bound halide ion is of critical importance and its role is going far beyond being only a bystander by occupying a coordination site, but altering the catalytic properties of the metal.9 Recently, the influence of the polarized [Ru–Cl] fragment has been subject to detailed investigations by Fürstner, showing the ability to pre-organize certain substrates and exert crucial influence on the reaction.10 Synthetically it is very attractive to form in situ neutral chiral Ru(II) complexes by reaction of the corresponding cationic CpxRu(II) complex 1 with a suitable coordinating anion (Fig. 1). One can use the same complex stock for a much broader set of transformations and conveniently interrogate possible counterion effects.9 This gives a complex with labile MeCN groups that easily dissociate. While isolated neutral Ru(II) complexes with tightly bound dienes are beneficial for the complex stability and storage, they reduce reactivity due to the required diene dissociation for the formation of catalytically active species. Ideally, the same chiral ligand could be used, increasing the overall generality and value of the established backbone design.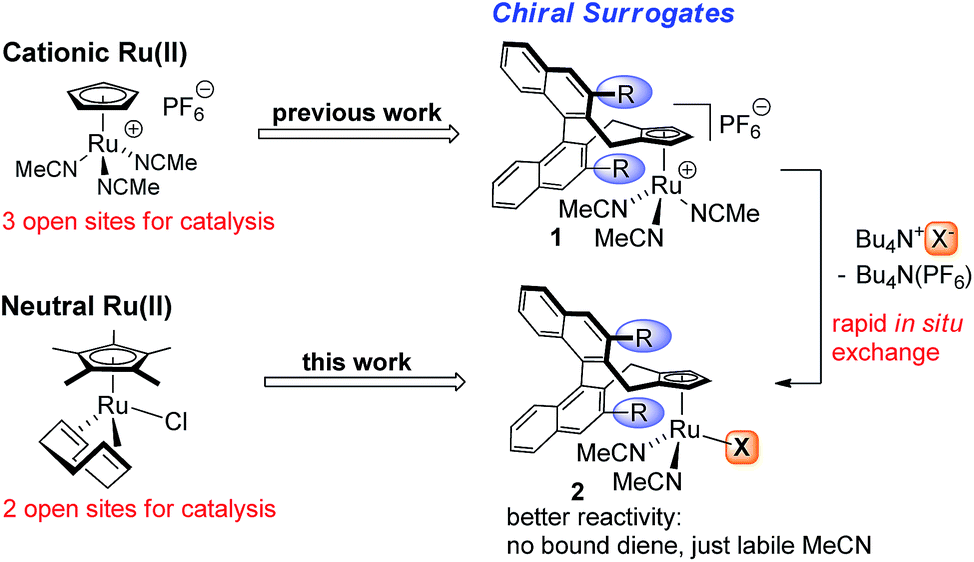 | ||
| Fig. 1 Powerful cationic and neutral CpRu(II) catalysts and their chiral Cpx surrogates for asymmetric catalysis. | ||
Among the many reactions catalysed by gold-standard complex Cp*Ru(COD)Cl – the formal [2+2]-cycloaddition between bicyclic alkenes and alkynes is an attractive benchmark transformation.11 The reaction provides a unique access to strained cyclobutenes12 which are very attractive intermediates for various valuable follow-up transformations.13 While its mechanism14 and the reactivity of different substrates15 have been investigated, no catalytic enantioselective Ru-catalysed [2+2] cycloaddition could be realised until now due to the lack of any suitable chiral CpRu(II) catalysts. Tam reported a diastereoselective version using chiral alkynes.16 Currently, an enantioselective transformation is only known for a Rh(I)-catalyst system with a significant substrate dependence of the enantioselectivity,17 and an Ir(I)-catalysed reaction which is restricted to benzo oxabicyclic alkenes and terminal alkynes.18
Results and discussion
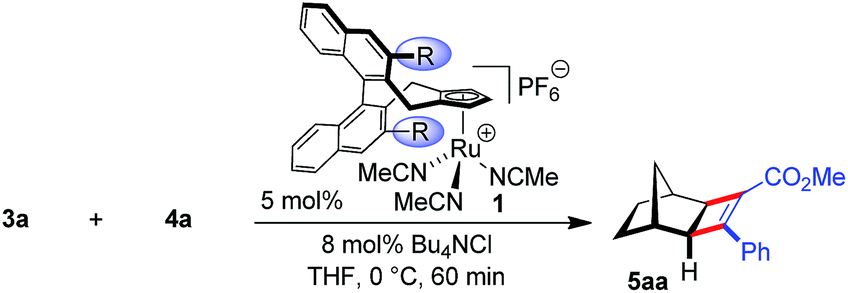
Initially, the envisioned transformation was investigated with norbornene 3a and alkyne 4a (Scheme 1). Exposure of the reactants to 5 mol% of the cationic CpxRu(II) complex 1a resulted in a rapid and clean conversion, giving cyclobutene 5aa in 98% yield after 60 min reaction time at 0 °C (entry 1). Despite the excellent reactivity of the cationic complex, no stereoinduction was observed, giving racemic cyclobutene 5aa. Notably, under these conditions the standard achiral catalyst, Cp*Ru(COD)Cl, displayed almost no reactivity at all, requiring 20 h for the reaction to go to completion at ambient temperature. This highlights the superior reactivity of this chiral CpxRu(II) catalyst. A dramatic improvement was observed when chloride anions were added to form in situ a neutral Ru(II) complex.19 The catalyst maintained its superb reactivity, now paired with excellent enantioselectivity, producing 5aa in 96.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.5 er.
:3.5 er.
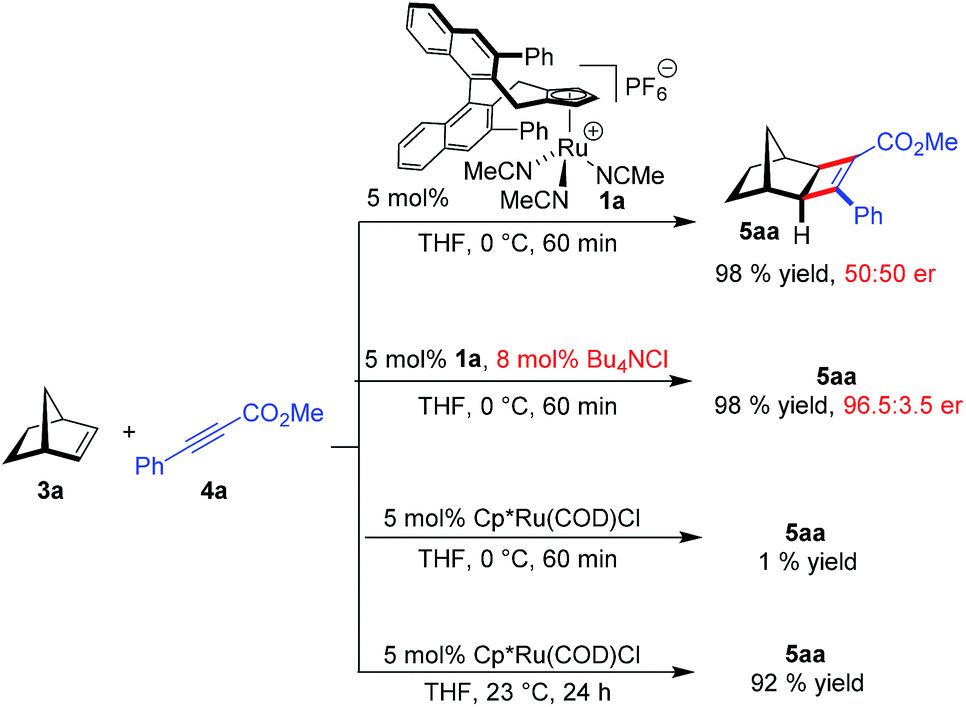 | ||
| Scheme 1 Very strong counteranion effect on the enantioselectivity of cyclobutene product 5aa. | ||
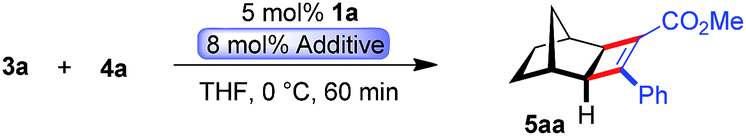
Intrigued by this dramatic selectivity enhancement, we then evaluated a range of additional anions in this transformation (Table 1).9 Bromide can replace chloride, providing largely comparable results (entry 3). However, an iodide diminished the reactivity, either due to its larger size or by altering the electronic properties of the ruthenium center (entry 4). Fluoride completely abolished the reactivity of the catalyst, as did the pseudohalides cyanide and azide (entries 5–7). Moreover, nitrate as well as acetate anions were not suitable (entries 8–9), likewise a neutral bound phosphine inhibited the reaction completely (entry 10). The reaction still proceeded with the corresponding CpxRu(II) carbonyl complex, however much slower and less selective (entries 11–12). These results demonstrate the unique match of the chloride anion for functionality tuning of the ruthenium catalyst.
|
|
||||
|---|---|---|---|---|
| Entry | Additive | % Conv.b | % yieldb | erc |
| a 37.5 μmol 3a, 25 μmol 4a, 2.0 μmol additive, 1.25 μmol 1a, 0.3 M in THF, 0 °C, 60 min. b Determined by 1H-NMR with an internal standard. c Determined by HPLC with a chiral stationary phase. d With preformed complex. | ||||
| 1 | None | 100 | 98 | 50:50 |
| 2 | (Bu4N)Cl | 100 | 98 | 96.5:3.5 |
| 3 | (Bu4N)Br | 100 | 98 | 96:4 |
| 4 | (Bu4N)I | 17 | 15 | 94:6 |
| 5 | (Bu4N)F | 2 | 1 | — |
| 6 | (Bu4N)CN | 10 | 0 | — |
| 7 | (Bu4N)N3 | 0 | 0 | — |
| 8 | (Bu4N)NO3 | 9 | 6 | 91.5:8.5 |
| 9 | (Bu4N)OAc | 8 | 0 | — |
| 10 | PPh3 | 0 | 0 | — |
| 11d | CO | 5 | 4 | 79:21 |
| 12d | CO | 92 (24 h, 20 °C) | 83 | 58:42 |
The influence of the ortho-substituents R of the ligand backbone were investigated next (Table 2). While their influence on the enantioselectivity was not very pronounced, the nature of the R groups had a large effect on the reaction rate. After 60 min reaction time, complexes 1b (3,5-xylyl) or 1d (4-methoxyphenyl), revealed a significant reduction in reactivity and yield (entries 1–4) compared to the parent complex 1a. Smaller ligands like 1e or 1f (entries 5–6) display lower reactivity which leads to the assumption that the phenyl groups help to pre-organise the substrates for enhanced reaction rate. The difference in electronic properties of the Ru center for different Cpx ligands is very small.20
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | R | % Conv.b | % yieldb | erc |
| a 37.5 μmol 3a, 25 μmol 4a, 2.0 μmol Bu4NCl, 1.25 μmol 1, 0.3 M in THF, 0 °C, 60 min. b Determined by 1H-NMR with an internal standard. c Determined by HPLC with a chiral stationary phase. | |||||
| 1 | 1a | Ph | 100 | 98 | 96.5:3.5 |
| 2 | 1b | 3,5-Me–C6H3 | 56 | 19 | 93:7 |
| 3 | 1c | C6F5 | 15 | 3 | 91:9 |
| 4 | 1d | 4-MeO–C6H4 | 39 | 26 | 96:4 |
| 5 | 1e | Me | 26 | 7 | 86:14 |
| 6 | 1f | OMe | 56 | 19 | 85.5:14.5 |
| 7 | 1g | OTIPS | 20 | 9 | 93:7 |
With Ph–Cpx being a uniquely efficient ligand in ruthenium catalysis, we aimed to shorten its synthesis (Scheme 2). Our previous discovery route was based on a rather lengthy diversity oriented strategy, giving several streamlining opportunities.6c,21 Bis-carboxylic acid 622 serves as directing group suited for Pd(II)-catalysed ortho-functionalisations,23 and coupling with iodobenzene would deliver 7. Biaryls are demanding substrates for such transformations, especially when a clean double functionalisation is required. Ligandless conditions24 were not very efficient, giving mixtures of starting material, mono- and diarylated products. Using Yu's mono-protected amino acid ligands (MPAA)25 improved the efficiency significantly. For instance, N-acetyl glycine yielded bis-arylated product 7 in 69% yield. Subsequent reduction and substitution of the resulting benzylic alcohol provided bis-bromide 8. Annulation of the Cp group and complexation yields air- and moisture-stable complex 10, from which 1a can be rapidly generated.7 With this approach, the ligand is synthesised in only four steps, cutting the previous step-count by half.
 | ||
| Scheme 2 Streamlined synthesis of Ph–Cpx ligand 9. | ||
The scope for the enantioselective Ru-catalysed cyclobutene formation was explored (Table 3). iPropyl or tbutyl propiolates increased the enantioselectivity to 98:2 er (entries 2–3). Even a free carboxylic acid (4e) was converted smoothly (entry 5). A ketone substrate reacted with somewhat lower enantioselectivity (entry 6). R1 can be a substituted arene or heteroarene (entry 7–11). The absolute configuration of 5ag was determined by X-ray crystallographic analysis after saponification to the corresponding free carboxylic acid. Besides arenes, vinyl or alkyl groups at the R1 position provide cyclobutenes (entries 12–14). Norbornadiene and benzo-fused derivatives 3 provided the cyclobutenes with similar enantioselectivity (entries 15–16). For 3e and 3f, the exo/endo orientation of the substituents R3 was important for the reactivity (entries 18–19). Moreover, a heteroatom was tolerated in the bridge position, leading to oxy-bridged compound 5gc (entry 20). However, less strained alkenes like cyclopentene or dihydrofuran were unreactive under these conditions.
|
|
||||
|---|---|---|---|---|
| Entry | 5 | % yieldb | erc | |
| a 0.15 mmol 3x, 0.10 mmol 4y, 8.0 μmol (Bu4N)Cl, 5.0 μmol 1a, 0.3 M in THF, 0 °C, 60 min. b Isolated yields. c Determined by HPLC with a chiral stationary phase. d At 23 °C. | ||||
| 1 | 5aa | R1 = Ph, R2 = CO2Me | 97 | 96.5:3.5 |
| 2 | 5ab | R1 = Ph, R2 = CO2iPr | 86 | 98:2 |
| 3 | 5ac | R1 = Ph, R2 = CO2tBu | 80 | 98:2 |
| 4 | 5ad | R1 = Ph, R2 = CO2Ph | 76 | 97:3 |
| 5 | 5ae | R1 = Ph, R2 = CO2H | 89 | 94:6 |
| 6 | 5af | R1 = Ph, R2 = C(O)Ph | 98 | 75.5:24.5 |
| 7 | 5ag | R1 = 3-Br–Ph, R2 = CO2Me | 85 | 97.5:2.5 |
| 8 | 5ah | R1 = 4-NO2–Ph, R2 = CO2Me | 40 | 89.5:10.5 |
| 9 | 5ai | R1 = 4-OMe–Ph, R2 = CO2Me | 91 | 98:2 |
| 10 | 5aj | R1 = 3-Me–Ph, R2 = CO2Me | 93 | 96.5:3.5 |
| 11 | 5ak |
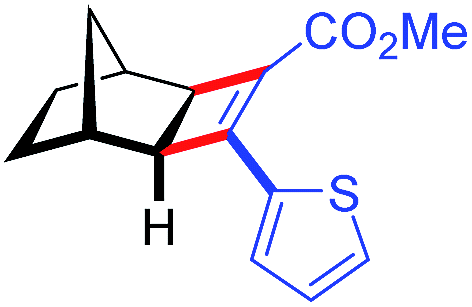
|
96 | 97.5:2.5 |
| 12d | 5al |
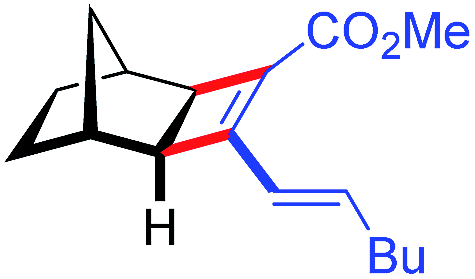
|
88 | 83:17 |
| 13d | 5am |
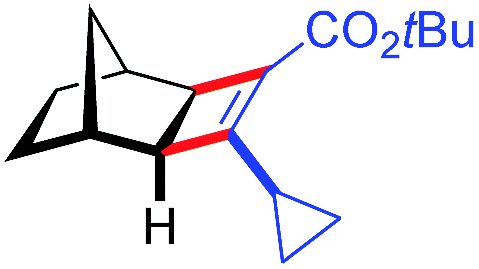
|
91 | 89:11 |
| 14d | 5an |
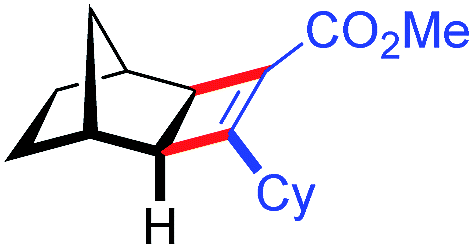
|
70 | 86:14 |
| 15d | 5bc |
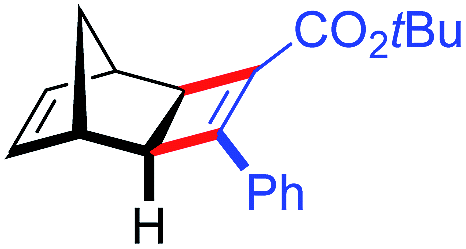
|
81 | 97:3 |
| 16 | 5cc |
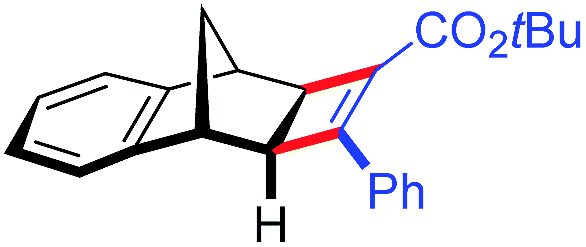
|
91 | 98:2 |
| 17 | 5dc |
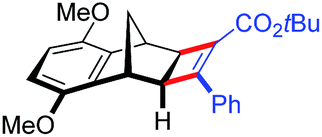
|
96 | 95:5 |
| 18 | 5ec |

|
97 | 99:1 |
| 19 | 5fc |
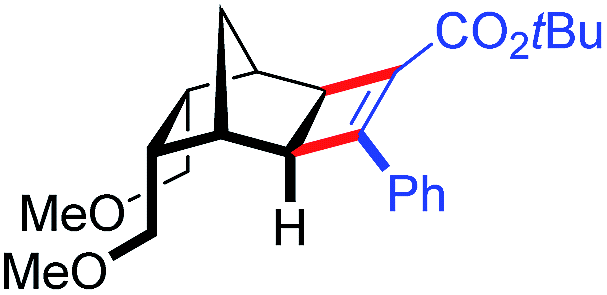
|
39 | 90.5:9.5 |
| 20 | 5gc |
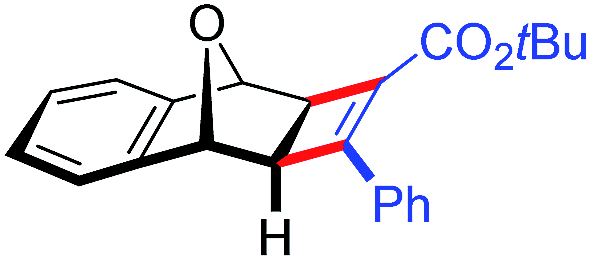
|
87 | 80:20 |

A larger scale allowed for a more concentrated reaction and the catalyst loading of 1a could be conveniently lowered to 1 mol% Ru (eqn (1)).
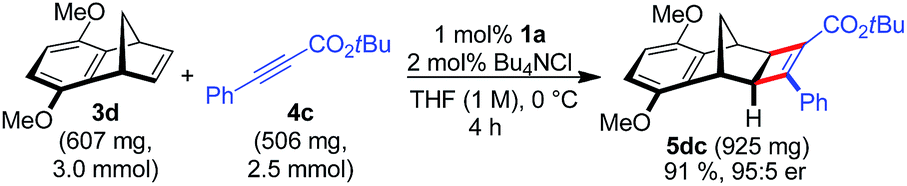 | (1) |
When using norbornadiene as substrate, ruthenium complex 11 (74% based on the employed amount of 1a) was isolated besides cyclobutene 5bc (Scheme 3a). The neutral complex 11 is remarkably stable and was isolated by silica gel chromatography and showed no sign of instability in air. The ruthenium center of 11 binds to one norbornadiene group and to a chloride atom. This well-defined complex allowed probing the chloride anion effect. Indeed, without any additional Bu4NCl, complex 11 provides 5bc in identical enantioselectivity (Scheme 3a). The dissociation of the tightly bound norbornadiene to free the required coordinating sites for catalysis slightly attenuates the reactivity (Scheme 3b).
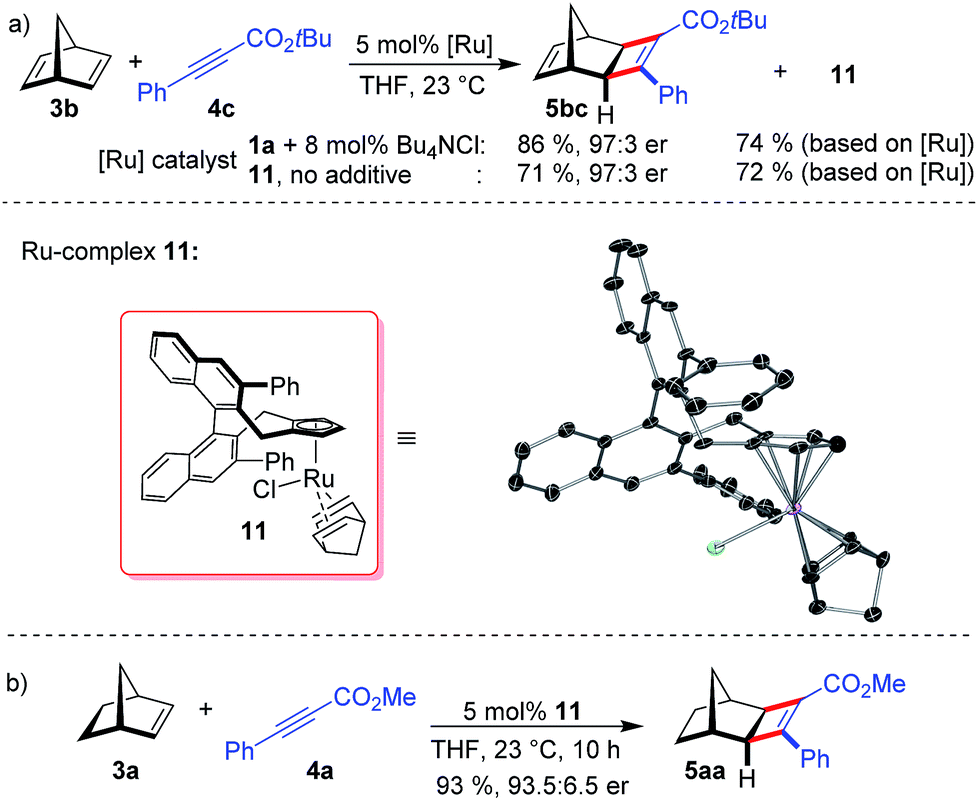 | ||
| Scheme 3 Isolation of the neutral CpxRuCl complex 12 and its recycling in catalysis. | ||
The X-ray structure of 11 provides important details on the binding pocket of the chloride ligated CpxRu catalysts (Scheme 4). Chloride occupies a coordination site at the ruthenium center allowing only for one single alkyne molecule to be coordinated in a productive cycle. In contrast, with the third vacant site of cationic complexes, a second alkyne molecule could coordinate causing an unselective participation leading to a racemic product as previously observed (Table 1, entry 1). Chloride, as smallest ligand, is oriented towards the naphthyl backbone. This differentiates the remaining two coordination sites. From the front view of 11, the left one is wide open allowing for the bulky bicyclic alkene to bind with its exocyclic face, due to orbital distortion of the double bond. In contrast, the right one is clearly restricted by the guiding phenyl group of the ligand. The alkyne with its rod-like geometry fits well. Within these boundaries, the enantioselection can be rationalised. Computational studies by Goddard with Cp*RuCl indicated a significantly lower transition state energy for the ruthenacyclopentene formation in which the ester points away from the chloride atom.14 Subsequent reductive elimination expels cyclobutene 5 and regenerates the initial complex. The complete picture fits well with the observations of higher selectivities of alkynes with bulkier esters, which point into the open space away from the complex.
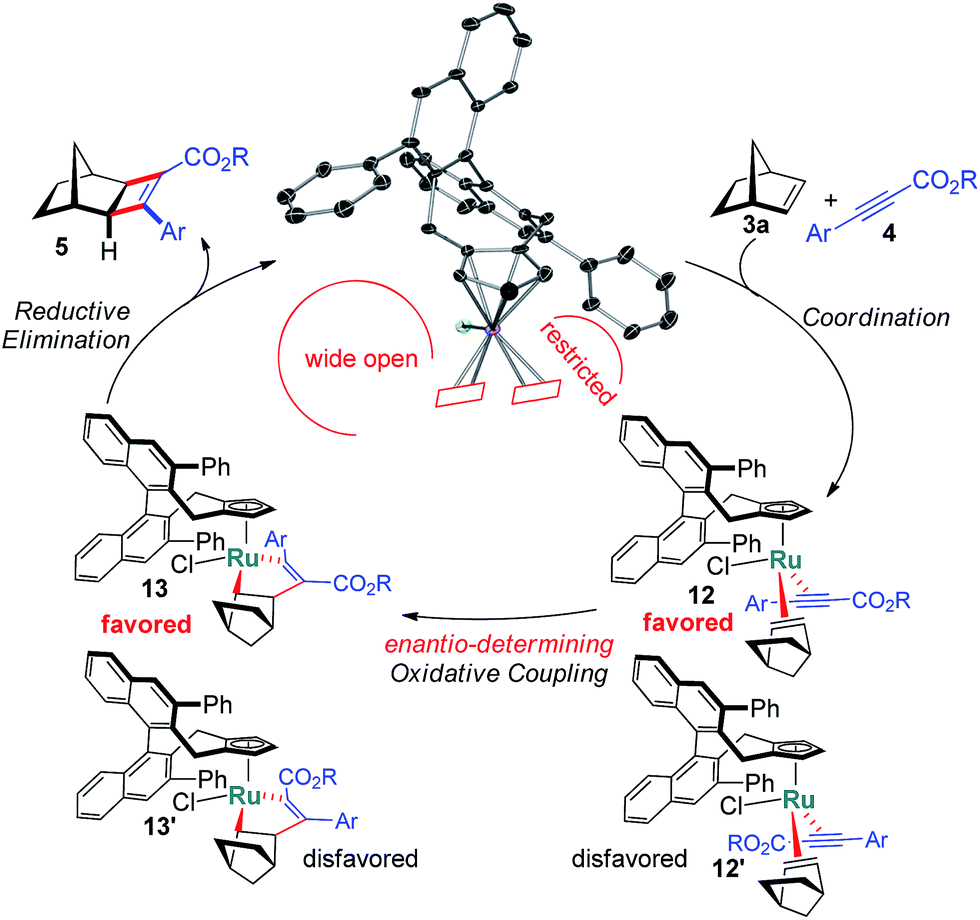 | ||
| Scheme 4 Selectivity model for the enantioselective cyclobutene formation. | ||
Conclusions
In summary, we report a chiral neutral CpxRu(II)Cl complex providing superior catalytic reactivity than the gold-standard Cp*Ru(COD)Cl complex. We have shown the excellent reactivity and selectivity of the catalyst for enantioselective [2+2]-cycloadditions leading to strained chiral cyclobutenes at catalyst loadings as low as 1 mol%. The bound chloride anion transforms the completely unselective cationic catalyst into the highly enantioselective neutral version. Moreover by adding norbornadiene at the end of the reaction, the catalyst can be recovered and subsequently reused. Finally, a streamlined C–H functionalisation approach allows an efficient preparation of the chiral cyclopentadienyl ligand.Acknowledgements
This work is supported by the Swiss National Science Foundation (No. 157741). We thank Dr R. Scopelliti and Dr F. F. Tirani for X-ray analysis of 5ag-CO2H and 11.Notes and references
- (a) S.-I. Murahashi, Ruthenium in Organic Synthesis, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) C. Bruneau and P. H. Dixneuf, Ruthenium in Catalysis, Springer, Berlin, 2014 Search PubMed.
- G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS PubMed.
- (a) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS; for C–C bond formation via transfer hydrogenation see: (b) F. Perez, S. Oda, L. M. Geary and M. J. Krische, Top. Curr. Chem., 2016, 374, 365–387 Search PubMed.
- (a) B. M. Trost, M. U. Frederiksen and M. T. Rudd, Angew. Chem., Int. Ed., 2005, 44, 6630–6666 CrossRef CAS PubMed; (b) B. M. Trost, Acc. Chem. Res., 2002, 35, 695–705 CrossRef CAS PubMed; (c) B. M. Trost, F. D. Toste and A. B. Pinkerton, Chem. Rev., 2001, 101, 2067–2096 CrossRef CAS PubMed.
- (a) B. M. Trost and M. C. Ryan, J. Am. Chem. Soc., 2016, 138, 2981–2984 CrossRef CAS PubMed; (b) B. M. Trost, M. C. Ryan and D. Maurer, Org. Lett., 2016, 18, 3166–3169 CrossRef CAS PubMed; (c) B. M. Trost, M. C. Ryan, M. Rao and T. Z. Markovic, J. Am. Chem. Soc., 2014, 136, 17422–17425 CrossRef CAS PubMed; (d) B. M. Trost, M. Rao and A. P. Dieskau, J. Am. Chem. Soc., 2013, 135, 18697–18704 CrossRef CAS PubMed; (e) S. Tanaka, T. Seki and M. Kitamura, Angew. Chem., Int. Ed., 2009, 48, 8948–8951 CrossRef CAS PubMed; (f) K. Fukamizu, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2008, 130, 10498–10499 CrossRef CAS PubMed; (g) N. Dodo, Y. Matsushima, M. Uno, K. Onitsuka and S. Takahashi, J. Chem. Soc., Dalton Trans., 2000, 35–41 RSC.
- (a) C. G. Newton, D. Kossler and N. Cramer, J. Am. Chem. Soc., 2016, 138, 3935–3941 CrossRef CAS PubMed; (b) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS PubMed; (c) B. Ye and N. Cramer, J. Am. Chem. Soc., 2013, 135, 636–639 CrossRef CAS PubMed; (d) B. Ye and N. Cramer, Science, 2012, 338, 504–506 CrossRef CAS PubMed.
- D. Kossler and N. Cramer, J. Am. Chem. Soc., 2015, 137, 12478–12481 CrossRef CAS PubMed.
- S. Dérien and P. H. Dixneuf, J. Organomet. Chem., 2004, 689, 1382–1392 CrossRef.
- K. Fagnou and M. Lautens, Angew. Chem., Int. Ed., 2002, 41, 26–47 CrossRef CAS.
- S. M. Rummelt, K. Radkowski, D.-A. Roşca and A. Fürstner, J. Am. Chem. Soc., 2015, 137, 5506–5519 CrossRef CAS PubMed.
- (a) T.-a. Mitsudo, H. Naruse, T. Kondo, Y. Ozaki and Y. Watanabe, Angew. Chem., Int. Ed., 1994, 33, 580–581 CrossRef; (b) M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49–92 CrossRef CAS PubMed; for a cobalt catalysed [2+2] reaction see: (c) T. G. Baik, A. L. Luis, L.-C. Wang and M. J. Krische, J. Am. Chem. Soc., 2001, 123, 6716–6717 CrossRef CAS PubMed; (d) L.-C. Wang, H.-Y. Jang, Y. Roh, V. Lynch, A. J. Schultz, X. Wang and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 9448–9453 CrossRef CAS PubMed.
- (a) M. Eisold, A. N. Baumann, G. M. Kiefl, S. T. Emmerling and D. Didier, Chem.–Eur. J., 2016 DOI:10.1002/chem.201604585; (b) A. Misale, S. Niyomchon and N. Maulide, Acc. Chem. Res., 2016, 49, 2444–2458 CrossRef CAS PubMed; (c) Y. Xu, M. L. Conner and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 11918–11928 CrossRef CAS PubMed.
- (a) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740–7752 CrossRef CAS PubMed; (b) E. Lee-Ruff and G. Mladenova, Chem. Rev., 2003, 103, 1449–1484 CrossRef CAS PubMed; (c) J. C. Namyslo and D. E. Kaufmann, Chem. Rev., 2003, 103, 1485–1538 CrossRef CAS PubMed.
- P. Liu, W. Tam and J. D. Goddard, Tetrahedron, 2007, 63, 7659–7666 CrossRef CAS.
- (a) W. Tam and N. Cockburn, Synlett, 2010, 1170–1189 CrossRef CAS; (b) R. R. Burton and W. Tam, J. Org. Chem., 2007, 72, 7333–7336 CrossRef CAS PubMed; (c) R. W. Jordan, K. Villeneuve and W. Tam, J. Org. Chem., 2006, 71, 5830–5833 CrossRef CAS PubMed; (d) R. W. Jordan and W. Tam, Tetrahedron Lett., 2002, 43, 6051–6054 CrossRef CAS; (e) R. W. Jordan and W. Tam, Org. Lett., 2001, 3, 2367–2370 CrossRef CAS PubMed.
- K. Villeneuve and W. Tam, Angew. Chem., Int. Ed., 2004, 43, 610–613 CrossRef CAS PubMed.
- T. Shibata, K. Takami and A. Kawachi, Org. Lett., 2006, 8, 1343–1345 CrossRef CAS PubMed.
- B.-M. Fan, X.-J. Li, F.-Z. Peng, H.-B. Zhang, A. S. C. Chan and Z.-H. Shao, Org. Lett., 2010, 12, 304–306 CrossRef CAS PubMed.
- (a) J. A. Varela, S. G. Rubín, C. González-Rodríguez, L. Castedo and C. Saá, J. Am. Chem. Soc., 2006, 128, 9262–9263 CrossRef CAS PubMed; for recent examples of counterion effects in Rh catalysis see: (b) X. Wu, Z. Chen, Y.-B. Bai and V. M. Dong, J. Am. Chem. Soc., 2016, 138, 12013–12016 CrossRef CAS PubMed; (c) S. K. Murphy, J.-W. Park, F. A. Cruz and V. M. Dong, Science, 2015, 347, 56–60 CrossRef CAS PubMed.
- [Cpx(Ph)Ru(CO)(MeCN)2]PF6:
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) (CO) = 1992 cm−1, [Cpx(OMe)Ru(CO)(MeCN)2]PF6: (CO) = 1989 cm−1.
(CO) = 1992 cm−1, [Cpx(OMe)Ru(CO)(MeCN)2]PF6: (CO) = 1989 cm−1. - T. Ooi, M. Kameda and K. Maruoka, J. Am. Chem. Soc., 2003, 125, 5139–5151 CrossRef CAS PubMed.
- (a) H. Egami, K. Sato, J. Asada, Y. Kawato and Y. Hamashima, Tetrahedron, 2015, 71, 6384–6388 CrossRef CAS; (b) T. Hoshi, E. Nozawa, M. Katano, T. Suzuki and H. Hagiwara, Tetrahedron Lett., 2004, 45, 3485–3487 CrossRef CAS.
- K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed.
- (a) D.-H. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 17676–17677 CrossRef CAS PubMed; (b) H. A. Chiong, Q.-N. Pham and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879–9884 CrossRef CAS PubMed.
- (a) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927–8955 CrossRef CAS PubMed; (b) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315–319 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of all new compounds. CCDC 1499170 and 1499171. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05092a |
| This journal is © The Royal Society of Chemistry 2017 |