
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRectification of current responds to incorporation of fullerenes into mixed-monolayers of alkanethiolates in tunneling junctions†
Li
Qiu
,
Yanxi
Zhang
,
Theodorus L.
Krijger
,
Xinkai
Qiu
,
Patrick van't
Hof
,
Jan C.
Hummelen
and
Ryan C.
Chiechi
 *
*
Stratingh Institute for Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: r.c.chiechi@rug.nl
First published on 20th December 2016
Abstract
This paper describes the rectification of current through molecular junctions comprising self-assembled monolayers of decanethiolate through the incorporation of C60 fullerene moieties bearing undecanethiol groups in junctions using eutectic Ga–In (EGaIn) and Au conducting probe AFM (CP-AFM) top-contacts. The degree of rectification increases with increasing exposure of the decanethiolate monolayers to the fullerene moieties, going through a maximum after 24 h. We ascribe this observation to the resulting mixed-monolayer achieving an optimal packing density of fullerene cages sitting above the alkane monolayer. Thus, the degree of rectification is controlled by the amount of fullerene present in the mixed-monolayer. The voltage dependence of R varies with the composition of the top-contact and the force applied to the junction and the energy of the lowest unoccupied π-state determined from photoelectron spectroscopy is consistent with the direction of rectification. The maximum value of rectification R = |J(+)/J(−)| = 940 at ±1 V or 617 at ±0.95 V is in agreement with previous studies on pure monolayers relating the degree of rectification to the volume of the head-group on which the frontier orbitals are localized.
1 Introduction
Molecular rectification is the asymmetric current response to an external voltage bias of equal magnitude but opposite sign mediated by the electronic structure of individual molecules. In contrast to comparisons of current (I) or current-density (J), rectification has the advantage that it is self-referencing, which eliminates the resistance of the contacts in studies comparing the electronic structure of molecular tunneling junctions. Molecular diodes are, therefore, interesting both for functionality and fundamental, phenomenological study. Rectification can be controlled to some degree by tailoring molecules and/or altering the contacts.1–23 While the details of the mechanism can vary between experimental platforms, Nijhuis and co-workers have elucidated it unambiguously for AgTS/S(CH2)11Fc//Ga2O3/EGaIn junctions, where EGaIn is eutectic GaIn alloy24,25 and Fc is either ferrocene or biferrocene incorporated into a self-assembled monolayer (SAM) supported by template-stripped Ag (AgTS) and ‘/’ and ‘//’ denote covalent and non-covalent interfaces, respectively.26–32 Depending on the sign of the applied bias, the difference in energy between the Fermi level (Ef) and the highest-occupied π-state (HOPS) of the Fc increases or decreases, increasing or decreasing the relative rate of charge-transfer.33,34Yoon and Whitesides have shown rectification that is mediated instead by the lowest-unoccupied π-state (LUPS) of bipyridine (Bp) and naphthoquinone (Nq) moieties in AgTS/S(CH2)11Bp//Ga2O3/EGaIn junctions, though the degree of rectification was significantly lower in the latter.29,35 In these junctions, the sign of the applied bias at which the current is higher is reversed compared to Fc because Ef lies closer to the LUPS than the HOPS. In both cases, the magnitude of rectification, defined as R = |J(+V)/J(−V)| (measured at ±1 V) depends both on the structure of the SAM and on the volume of the π-system (i.e., Fc or Bp), thus it can be tuned by altering the substrate36 or through dilution with disulfides (to introduce defects)37 or non-rectifying alkanethiols38 and, in principle, by increasing the size of the π-system.27,39 These studies demonstrated a decrease in R either through the introduction of defects into the substrate or by introducing non-rectifying (or defect-inducing) compounds into the solution from which the SAMs were formed, establishing the sensitivity of R to the packing of the π-systems that mitigates rectification.
Fullerenes, due to their spherical geometry and unique optical and electronic properties, are widely studied for potential applications ranging from sensors and photovoltaic cells to nanostructured devices.40–43 Their high affinity for noble metals and large surface area available for contact also make them attractive candidates for applications in Molecular Electronics (ME), both in single-molecule (e.g., break-junctions) and large-area (e.g., EGaIn) platforms. Fullerene-based anchoring groups have been demonstrated in single-molecule junctions44,45 and in large-area junctions46 and several studies on functionalized fullerene-based SAMs have shown that the electronic structure of [60]fullerene (C60) remains intact even when the carbon cages are confined to a surface.46–48 Thus, the low-lying lowest-unoccupied molecular orbital (LUMO) of C60 (−4.5 eV)49 should translate into an accessible LUPS in SAM-based junctions and, therefore, rectification via, the mechanism described above. The large volume and spherical symmetry of C60 should lead to large magnitudes of log|R| and decreased sensitivity to packing/ordering compared to Bp.
We functionalized C60 with 11-unedecanethiol (SC11) to afford FSC11 (structure and synthetic detail shown in Fig. S1 in ESI† and corresponding thiolate shown in Fig. 2A) and compared the J/V characteristics of AgTS/FSC11//Ga2O3/EGaIn junctions to those reported for junctions comprising SAMs of Fc and Bp functionalized with SC11. We prepared the SAMs of FSC11 by incubating SAMs of 10-dodecanethiol (SC10) with FSC11, observing an increase in R with exposure time. This method of preparing mixed-monolayers prevents phase-segregation between the two dissimilar compounds and preserves the packing of the SAM. Thus, the magnitude of R corresponds to the degree of incorporation of FSC11 into the non-rectifying SAM of SC10 and not a change in packing or an increase in disorder. We ascribe the rectification of current to the LUPS-mediated mechanism described above, which is summarized in Fig. 1.
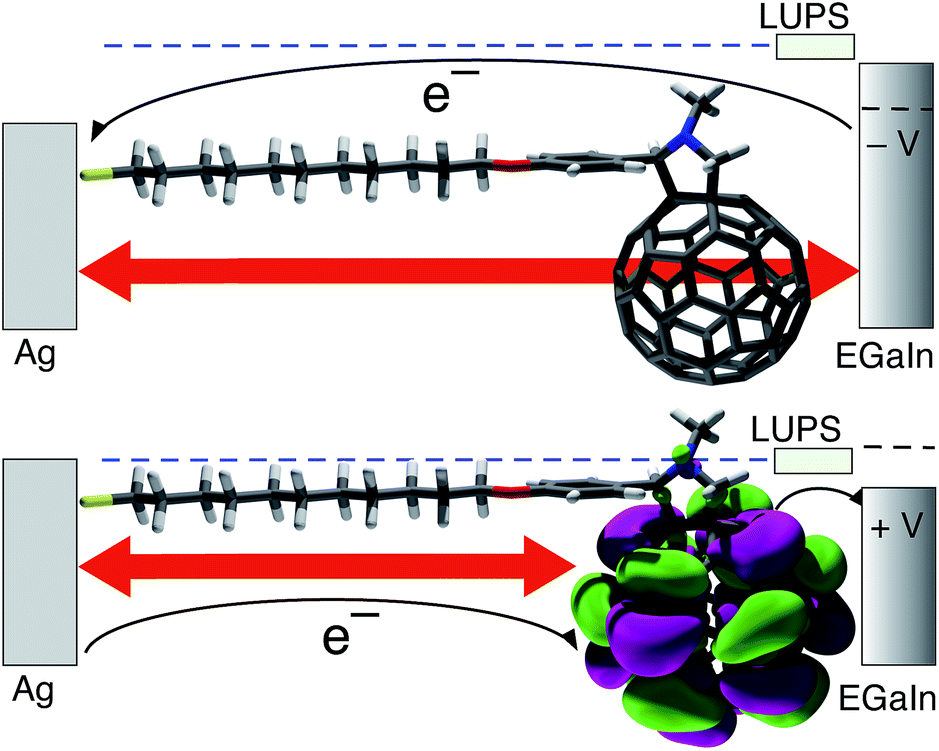 | ||
| Fig. 1 A schematic showing the mechanism of rectification. At negative bias (top) the lowest-unoccupied π-state (LUPS; indicated by a light green rectangle) is pushed out of resonance with the Ag electrode as is depicted by the blue dashed line. The width of the tunneling barrier (indicated with a double-headed red arrow) is therefore defined by the entire end-to-end length of the molecule and electrons must tunnel through the entire physical width of the junction. At positive bias (bottom) the LUMO (visualized as purple and green isosurfaces) is brought into resonance with the Ag electrode and electrons can tunnel to the LUPS, which is localized entirely on the C60 cage, and then hop to EGaIn. The width of the tunneling barrier is therefore defined only by the aliphatic portion of FSC11, thus electrons must tunnel through a distance approximately equal to SC10. | ||
2 Results and discussion
The preparation of SAMs of fullerene derivatives functionalized with thiols on metal surfaces has been extensively studied.50–56 Contrary to simple systems such as alkanethiols, the self-assembly of fullerene derivatives on gold is complicated by the formation of multilayers and head-to-tail assemblies due to competition from the strong fullerene–fullerene and fullerene–gold interactions.54,57–60 (The same complexities are expected on Ag.) These problems can be mitigated by pre-passivating the substrate with a SAM of decanethiol (SC10) and then forming a mixed-monolayer by incubating this SAM in a solution containing the fullerene derivatives (see Experimental).47,61 Throughout this manuscript we refer to mixed-monolayers of FSC11 and SC10 prepared by this method simply as SAMs of FSC11 unless specified otherwise.We measured the J/V characteristics of AgTS/FSC11//Ga2O3/EGaIn and AuTS/FSC11//Ga2O3/EGaIn junctions (Fig. 2A) by acquiring 1016 (650 on Ag and 366 on Au) sweeps between ±0.5 V for 61 (37 on Ag and 24 on Au) junctions across 7 (4 on Ag and 3 on Au) substrates (Table 1). The frequency of shorts increased dramatically above 0.5 V precluding the extraction of meaningful statistics above 0.5 V. We also measured AgTS/SAM//Ga2O3/EGaIn junctions based on SAMs of pure FSC11 and SC10 for comparison. We calculated R by dividing each value of J at positive bias into the corresponding value at negative bias for each value of |V| and then fitting a Gaussian to the resulting histogram of log|R| and expressing the error as the confidence interval of the fit. Histograms comparing log|R| for SAMs of pure FSC11 and mixed-monolayers of FSC11 SAMs on AgTS are shown in the ESI.† Although both monolayers show comparable peak values of log|R|, the histogram of the pure SAM is broad and possibly multi-modal, while that of the mixed-monolayer is a single, log-normal distribution. We ascribe this difference to the strong fullerene–metal interactions competing with thiol–metal interactions to form mixed phases of upright (thiol-down) and upside-down (fullerene-down) molecules. Despite these differences, the yields of working junctions for the pure SAMs of FSC11versus the mixed-monolayers are comparable (Table 1), which underscores the utility of using statistics and observables such as log|R| to characterize tunneling junctions comprising SAMs.
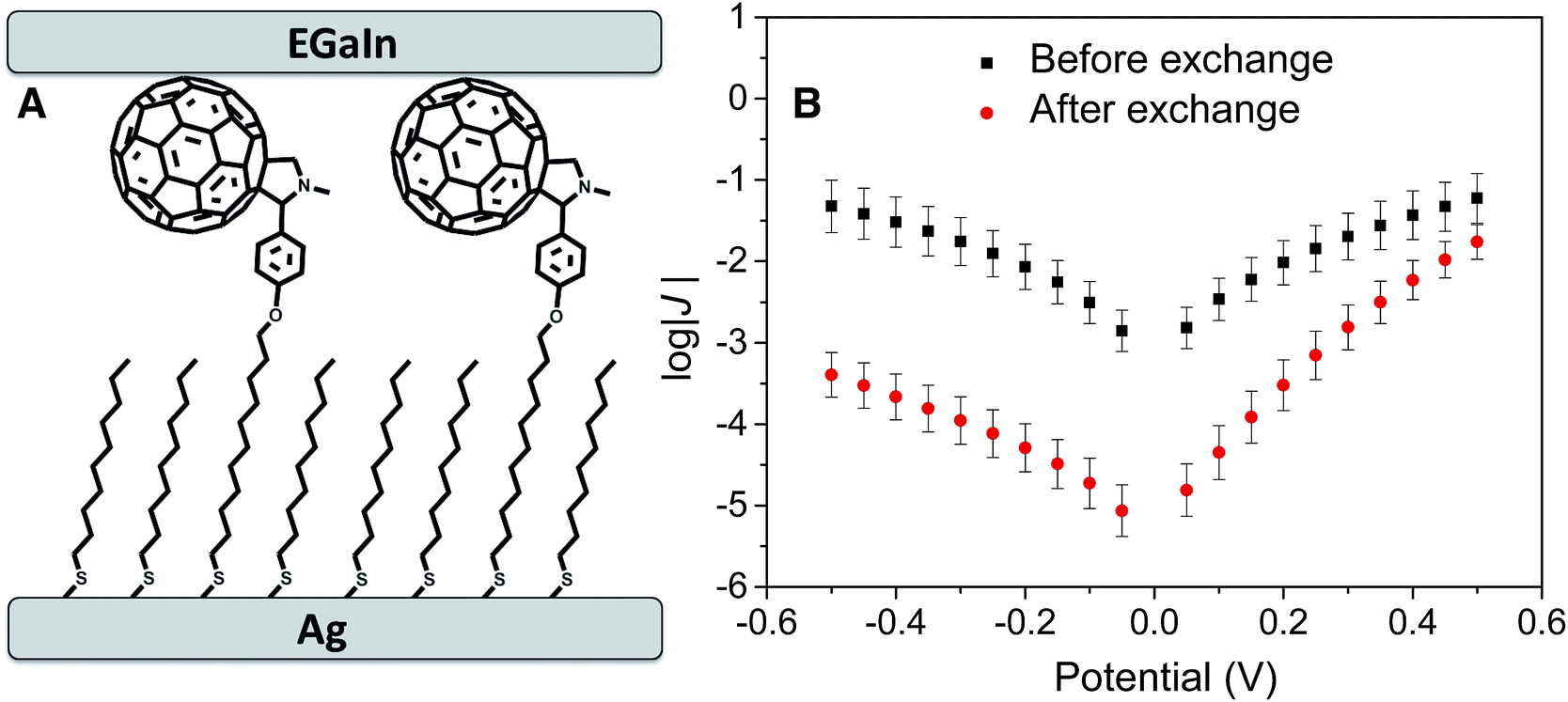 | ||
| Fig. 2 (A) Schematic diagram of a AgTS/FSC11//Ga2O3/EGaIn junction prepared by incubating a SAM of SC10 on AgTS in a solution of FSC11. (B) Plots of log|J| (the units of J are A cm−2) versus V for SAMs of SC10 on AgTS before and after incubation with FSC11. Each datapoint is the mean from a Gaussian fit to a histogram of log|J| for that value of V (see ESI† for details) and the error bars are the 95% confidence intervals of the fit. The J/V of SC10 is symmetric, with a maximum value of log|J| of approximately −1. After exchanging FSC11 into this SAM, log|J| decreases by approximately 2 at negative bias, giving rise to rectification. The magnitude of J at 0.5 V is almost identical before and after exchange because the width of the tunneling barrier at positive bias is nearly equal (see Fig. 1). | ||
| SAM | Substrates | Junctions | Traces | Unstablea (%) | Yieldb (%) | R@±0.5 V |
|---|---|---|---|---|---|---|
| a Junctions with noisy J/V curves that shorted readily. b Non-shorting junctions that gave smooth J/V curves. c Measurement was conducted at ±1 V. d The low yield is due to the high bias. e The value was obtained for the highest one at ±0.95 V. f Junction comprising pure SAMs formed directly from FSC11. | ||||||
| AgTS/FSC11 | 4 | 42 | 650 | 5 (12) | 88 | 29 ± 1 |
| AgTS/SC10 | 1 | 8 | 166 | 0 | 100 | 1 |
| AuTS/FSC11 | 3 | 28 | 366 | 4 (14) | 86 | 8 ± 1 |
| AgTS/FSC11c | 3 | 25 | 25 | 24 (96)d | 4 | 617e |
| AgTS/FSC11f | 3 | 34 | 568 | 3 (9) | 91 | 43 ± 9 |
Fig. 2B shows the J/V curves of the junctions before and after exchanging FSC11 into the passivating SAM of SC10 as described above (see ESI† for details of the data acquisition and processing). The J/V curve of SC10 is almost perfectly symmetric (log|R| = 0.07), reaching a maximum of log|J| ≈ −1 at ±0.5 V (the units of J throughout this paper are A cm−2). After exchange, however, the maximum value of log|J| at −0.5 V drops below −3, while the value at +0.5 V remains almost equal to that of SC10. This asymmetry results in a value of log|R| = 1.46 ± 0.018 and is characterized by a suppression of leakage current (at negative bias) compared to the SAM of pure SC10 caused by the increase in tunneling distance imposed by the C60 cage (Fig. 1). At positive bias, the LUPS is sufficiently close to Ef of the Ag electrode that electron transport is mediated by the LUPS of the C60 cage and tunneling occurs only through the aliphatic portion of the SAM followed by hopping between C60 cage and EGaIn. Hence the magnitude of J is nearly equal for SAMs of SC10 and FSC11 at +0.5 V.
Ferrocene-based rectifiers exhibit a strong odd–even effect in the magnitude of log|R| caused by the angle of the ferrocenes differing for even- and odd-numbered alkyl spacers.30,62,63 The observation of this effect provides compelling evidence that rectification is indeed caused by interactions between the ferrocene and the EGaIn electrode. The spherical symmetry of C60 precludes such a study for FSC11, however, varying the length of the alkanethiolates into which FSC11 is exchanged should have no influence on tunneling transport if it is indeed mediated by the C60 group. Fig. S9† shows the J/V curves of mixed-monolayers of FSC11 exchanged into SAMs of SC8, SC10 and SC12; they are nearly identical, giving both the same magnitudes of log|J| and log|R| and supporting the hypothesis that tunneling currents are mediated entirely by FSC11 in mixed-monolayers.
To exclude the possibility that the increase in log|R| after exposure of SC10 to FSC11 is due to electrochemical reactions between the AgTS electrode and the C60 cage, we measured AuTS/FSC11//Ga2O3/EGaIn junctions where AuTS is template-stripped Au prepared identically to AgTS. The lower work function of AuTS compared to AgTS has two potential consequences on log|R|: (i) the rectification of current is due to redox reactions taking place at the AgTS electrode, which will be pushed out of the experimental bias window; (ii) the lower (more negative) Ef of AuTS will increase the bias required to move the LUPS of FSC11 close enough to resonance to induce rectification. The corresponding observables are log|R| ≈ 1 (i) and a decrease in log|R| at ±0.5V (ii). We observe the latter: log|R| decreases from 1.46 ± 0.018 on AgTS to 0.92 ± 0.017 on AuTS from which we conclude that the origin of rectification in mixed SAMs of FSC11/SC10 is indeed the onset of a hopping as the LUPS comes close to Ef and not electrochemistry at the AgTS.
To support this conclusion, we compared the LUPS energies of SAMs of FSC11 and Bp and Nq using a combination of optical and photoelectron spectroscopy. The LUPS of FSC11 is approximately 3.72 eV (see ESI†), which is nearly identical to Bp (3.7 eV)38 and Nq (3.9 eV)29 and close to the assumed Ef of Ga2O3/EGaIn electrode (approximately 4.3 eV). Thus, it is plausible that the mechanism of rectification for FSC11 is the same as Bp since the electrodes and substrates are identical. We also measured the I|V characteristics of SAMs of FSC11 using conductive probe atomic force microscopy (CP-AFM)64 which replaces a conformal, liquid-metal electrode (EGaIn) with a rigid Au electrode. Fig. 3 compares log|R| as a function of voltage for AgTS/FSC11//EGaIn and AgTS/FSC11//Au. (Note that the wiring is reversed, thus we use the convention of EGaIn junctions to express log|R| as we have done previously.65) Interestingly, log|R| rises much faster with EGaIn, crossing 1.0 only above ±1.25 V with CP-AFM. The sudden onset with CP-AFM is due to the low-current limit (our CP-AFM module uses fixed op-amps, unlike our EGaIn setup) clipping data at low bias. Nonetheless, there is a significant difference between the values of V at which the magnitude of log|R| is equal between CP-AFM and EGaIn. This result suggests that both electrodes—that is, Ef or their mechanical properties—play an important role in the magnitude of rectification. The weaker dependence on bias for CP-AFM could simply be due to weaker coupling at the SAM//Au interface, such that the offset between Ef and the LUPS is higher for a particular voltage with CP-AFM compared to EGaIn. Changing the loading force of the AFM tip does change the magnitude of log|R|, but the onset of rectification is approximately 1 V higher than it is for EGaIn top-contacts. This result suggests that the different behavior is due to the difference in work function between EGaIn and Au and/or that conformal EGaIn contacts couple much more effectively to the SAM. Whatever the origins of the difference, log|R| is greater than 1 in both cases, meaning that the origin of rectification is the electronic structure of the SAM and not AgTS or EGaIn.29
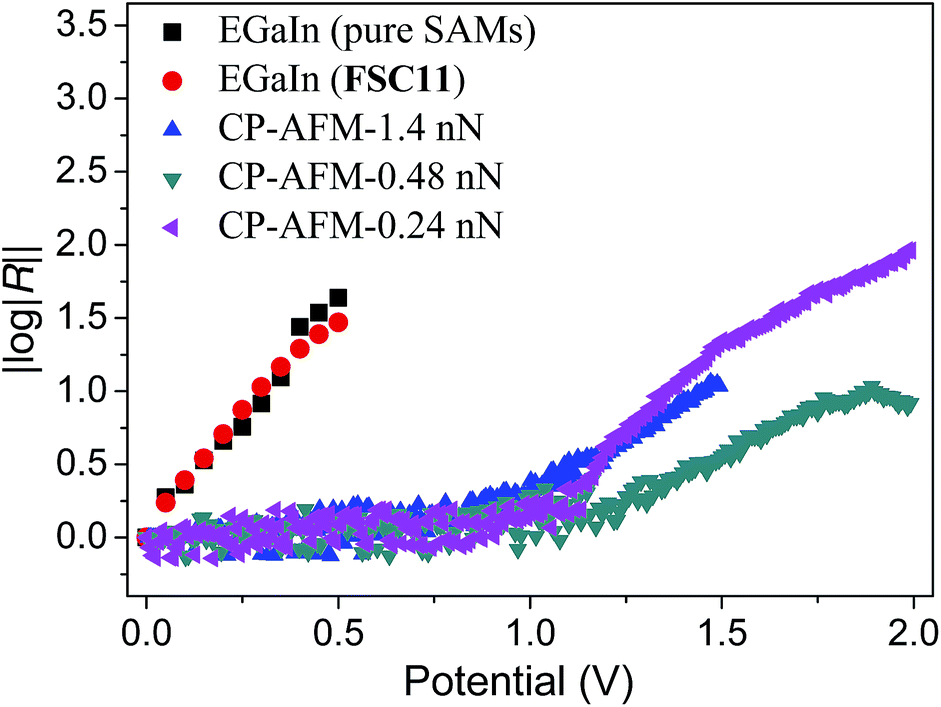 | ||
| Fig. 3 Comparison of log|R|–|V| plots of EGaIn (black squares and red dots) and CP-AFM (blue, green and magenta symbols) measurements. The Gaussian mean values for junctions based on pure and mixed FSC11 SAMs from EGaIn measurements show an identical trend, however, the variance (shown in the ESI†) is much higher for the pure SAMs. CP-AFM measurements gave values of log|R| > 1 at loading forces of 0.24 nN, 0.48 nN and 1.4 nN, but with opposite polarity from that of EGaIn due to the difference in wiring (as is described in ref. 65). The original curves with error bars are shown in ESI.† The onset of rectification occurs are at higher bias (2 V and 1.5 V) for CP-AFM than (0.5 V) for EGaIn, at least some of which is due to the low-current detection limit of CP-AFM at low bias. | ||
An interesting consequence of using log|R| as an observable is that the dynamics of the exchange between FSC11 and the passivating SAM of SC10 by observing the evolution of log|R| in time. (Assuming the magnitude of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) R corresponds directly to the amount of FSC11 incorporated into the SAM.) Fig. 4 shows log|R| versus exchange time (the amount of time a SAM of SC10 was exposed to a solution of FSC11). Following a relatively rapid increase, log|R| saturates after 24 h, implying that the mixed-monolayer reaches an equilibrium structure past which it becomes energetically unfavorable to incorporate any more FSC11. We interpret this saturation as the point at which the fullerene head-groups (which rise above the SAM of SC10) have reached maximum packing density as is depicted in Fig. 2A. We recently observed similar kinetics by following the on/off ratio of SAMs of spiropyran switches as a function of exposure time to hexanethiol.66 This timescale is also normal for place-exchange between adsorbed thiolates.67,68 Thus, EGaIn can be used to follow the dynamics of exchange in mixed-monolayers by observing the changing characteristics of the commensurate tunneling junctions.
R corresponds directly to the amount of FSC11 incorporated into the SAM.) Fig. 4 shows log|R| versus exchange time (the amount of time a SAM of SC10 was exposed to a solution of FSC11). Following a relatively rapid increase, log|R| saturates after 24 h, implying that the mixed-monolayer reaches an equilibrium structure past which it becomes energetically unfavorable to incorporate any more FSC11. We interpret this saturation as the point at which the fullerene head-groups (which rise above the SAM of SC10) have reached maximum packing density as is depicted in Fig. 2A. We recently observed similar kinetics by following the on/off ratio of SAMs of spiropyran switches as a function of exposure time to hexanethiol.66 This timescale is also normal for place-exchange between adsorbed thiolates.67,68 Thus, EGaIn can be used to follow the dynamics of exchange in mixed-monolayers by observing the changing characteristics of the commensurate tunneling junctions.
 | ||
| Fig. 4 A plot of log|R| versus the time that SAMs of SC10 were exposed to solutions of FSC11 (exchange time) using EGaIn top-contacts. The magnitude of log|R| increases gradually, saturating above 24 h, indicating the nearly complete FSC11 SAM was achieved after 24 h. The values of log|R| and the error bars are the mean and standard deviation from Gaussian fits to histograms of R for each value of V. | ||
If FSC11 does indeed rectify current via the mechanism described above, the maximum observed rectification should relate to the volume of the C60 cage because the LUPS is localized to the C60 π-system that is in contact with and (partially) pinned to the Ga2O3/EGaIn electrode. As is hypothesized for Bp, positive bias decreases Ef (relative to vacuum) at that electrode, which also decreases the LUPS and brings it into resonance with Ef at the AgTS electrode. At this point the LUPS becomes energetically accessible and charges tunnel from AgTS onto the C60 cage instead from AgTS to Ga2O3/EGaIn (or Au in the case of CP-AFM). Assuming a rectangular tunneling barrier, J = J0exp(−βd) where β is the tunneling decay coefficient, d is the barrier width and J0 is the extrapolated value of J when d = 0. Using this equation we can estimate log|R| by calculating J using the value of d corresponding to the end-to-end lengths of FSC11 and dCH2 corresponding that value with the volume of the LUPS removed (that is, the red arrows in Fig. 1). This methodology in turn allows a direct comparison to Bp.35 The results are summarized in Table 2.
| Assumed structure of T group | Molecular rectifiers | d RM (Å) | Rectification ratio (R) | |
|---|---|---|---|---|
| Calcdc (β ∼ 0.6 Å−1) | Obsd | |||
| a Calculations for FSC11 were based on the assumptions that either (i) the terminal group, which we treated as the molecular moiety excluding only the aliphatic spacer, is spherical in shape, and the width of tunneling barrier of terminal group is dRM = 2 × (3/4π)3; or (ii) that the terminal group is an extended trans structure. b Numbers for Bp and Fc were taken from ref. 35. c Values of |R|calcd were calculated with equation |R|calcd = exp(βRMdRM), assuming that the tunneling decay constants characteristic of attenuation through FSC11, Bp and Fc are equal to that of oligophenylenes (β ∼ 0.6). | ||||
| Spherical shape | FSC11 | 10.9 | 692 | 940 |
| Bpb | 6.8 | 59 | 85 | |
| Fc | 7.2 | 73 | 150 | |
| Extended trans structure | FSC11 | 11.2 | 829 | 940 |
| Bp | 7.2 | 75 | 85 | |
| Fc | 6.0 | 36 | 150 | |
We first calculated dRM (the width of the tunneling barrier for the rectifying moiety) based on two different approximations used in ref. 35 to be 10.9 Å for a spherical volume and 11.2 Å for the volume of an extended structure, respectively. Not surprisingly, the two different methods of calculating volume give very similar values for FSC11 because the C60 cage is nearly spherical. To compare observed values of rectification it is necessary to pick a value of |V| at which to compute |R|obsd. Thus far we have used ±0.5 V because the junctions were not sufficiently stable above this value to collect sufficient data for a robust statistical analysis. However, the standard value in ME is ±1.0 V. Fig. 5 shows a linear dependence of log|R| on |V| from which we extrapolated a value of R = 676 at ±0.95 V. We compared this value to R = 617 from a ‘hero junction’ that survived sweeping to ±0.95 V (which most junctions did not) and gave qualitatively similar curves to the more robust and reproducible curves acquired at ±0.5 V. This very close agreement and the fact that plots of log|R| versus |V| for SAMs of FSC11 at different exchange time (24, 36 and 60 h) gave the same slope (see ESI†) validates the extrapolated value of |R|obsd = 940 at ±1.0 V. Fig. 6 compares values of |R|obsd and |R|calcd to several other rectifiers, including Fc and Bp; FSC11 lies almost exactly on the diagonal, further validating the presumed mechanism.
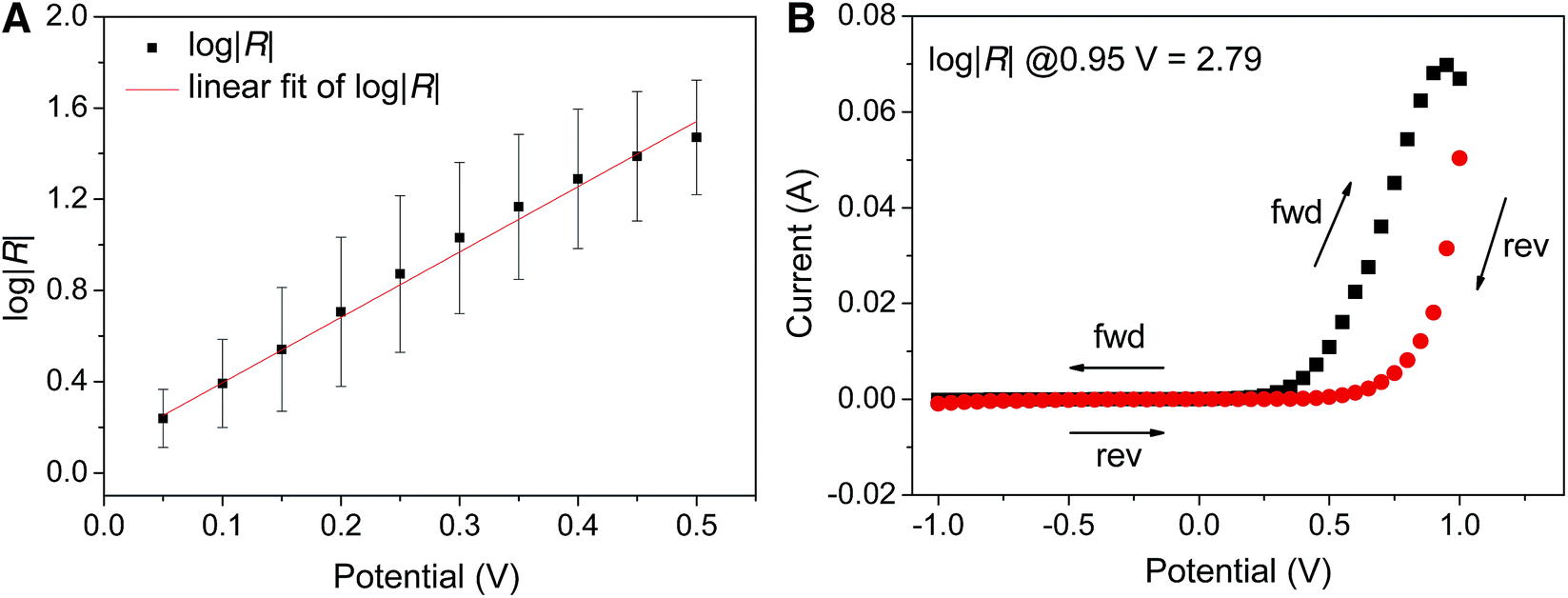 | ||
| Fig. 5 (A) Linear fit of log|R| vs. |V| of FSC11 with EGaIn as a top-contact. The data and error bars are the mean and standard deviations of Gaussian fits (adjusted R-square = 0.994) (B) I|V data for a representative ‘hero’ junction (which could survive sweeping to a voltage bias higher than ±0.5 V) that produce stable I|V curves above 0.5 V showing a typical degree of hysteresis and a maximum value of log|R| of 2.79. | ||
 | ||
| Fig. 6 A comparison of the observed and predicted magnitudes of rectification |R| at ±1 V based on the volume of the π-system confined to the head-group for FSC11 and other systems from the literatures following the methodology of ref. 35. The experimental value of |R| for FSC11 is the maximum value observed only in a few hero junctions. The two different methods of calculating volume give very similar values for FSC11 because of its spherical geometry. | ||
3 Conclusions
We have shown that FSC11 SAMs composed of decanethiolate (SC10) and functionalized C60 bearing undecanethiol groups (FSC11) reproducibly rectify current in AgTS/SAM//EGaIn junctions at ±0.5 V. The mechanism is identical to those of SAMs containing bipyridyl (Bp) and Nq since the LUMO of these compounds lie at nearly the same energy, translating into an accessible π-state in SAM-based junctions under positive bias. Further, we show unambiguously that rectification is the result of the electronic structure of C60 because it persists with AuTS bottom electrode and with Au top-contact. We circumvented the difficulties of growing SAMs from C60 derivatives by preparing mixed-monolayers via exchange into substrates pre-passivated with SAMs of SC10 such that the C60 cages are never exposed to bare metal surfaces.Among the molecular rectifiers included in Fig. 6, the fullerene head group of FSC11 is the second largest behind the copper phthalocyanine salt complex from ref. 19 measured by scanning tunneling microscopy (STM). Not only does FSC11 show the second-highest magnitude of rectification, it shows rectifying behavior with a large-area, conformal EGaIn top-contact and a nanoscopic, rigid Au top-contact. Moreover, the spherical symmetry of C60 and the use of mixed-monolayers mitigates the extreme sensitivity of molecular rectifiers to the details of packing and supramolecular structure. The magnitude of rectification for ferrocene moieties, for example, is sensitive to tilt angle30 and the purity of the thiol-precursor is also crucial; less than 5% of disulfide disrupts the packing and causes a drop in R and rectification vanishes completely at 15%.37 Similarly, forming mixed monolayers of Bp with n-alkanethiolates only decreases R from that of pure Bp and phase separation makes binary SAMs with relatively uniform composition difficult to achieve.38 Since we begin from non-rectifying SAMs of SC10 into which FSC11 is incorporated, R increases to a saturation point.
The magnitude of log|R| at ±0.5 V is 1.46 ± 0.018, which can reach as high as 940 at ±1 V in a few hero junctions (too few for statistical analysis). This value (940) is consistent with calculations assuming the proposed rectification mechanism, further supporting the proposed relationship between the volume and energy of the accessible π-state and the magnitude and direction of rectification. Future work will focus on stabilizing junctions containing C60 above ±1 V to utilize the large magnitude of rectification in the hero junctions in device platforms.
4 Experimental section
Materials
11-Bromoundec-1-ene, 4-hydroxybenzaldehyde, thioacetic acid, 1-decanethiol (SC10) were obtained from Sigma-Aldrich and used as received with the exception of SC10 which was purified by column chromatography (silica, hexane). The C60 used for the synthesis was of 99.5% purity (purchased from Solenne BV, Groningen, the Netherlands). All compounds were stored in nitrogen-flushed vials and in the dark. Their structures were verified by acquiring 1H-NMR and IR spectra immediately prior to use and comparing to the spectra acquired immediately after purification. FSC11 was prepared starting from 11-bromoundec-1-ene as described in the ESI.† All new compounds were all fully characterized by means of HRMS, NMR and IR. The AgTS and AuTS substrates used in this work were made by mechanic template stripping as described elsewhere;21 we deposited 200 nm of Ag and 100 nm of Au (99.99%), respectively, by thermal vacuum deposition onto a 3′′ silicon wafer (with no-adhesion layer). Using the UV-curable optical adhesive (OA) Norland 61, we glued 1 cm2 glass chips on the metal surfaces.SAM formation
SAMs of FSC11 were prepared through exchange of SC10 from its SAMs with FSC11 through two steps. Firstly, SAMs of SC10 were formed by incubating freshly cleaved 1 × 1 cm2 AgTS surfaces for 24 h in 2 mL of 2 mM solution of SC10 in degassed ethanol (100%; anhydrous) at room temperature. The substrates were then rinsed gently with 200 proof ethanol (3 × 1 mL) and residual solvent on surface was removed by gently blowing N2. SAMs of FSC11 were then prepared by incubation of the resulting SAMs of SC10 (bared AgTS surfaces used directly for the pure SAMs) in 0.5 mM solutions of FSC11 in degassed toluene at room temperature for 24 h. After incubation, they were then rinsed with toluene and dried as previously described and then used for the measurements.SAM characterization: contact angle measurement
The SAM of FSC11 was first evaluated with water contact angle measurements under ambient conditions on a SCA20 Data Physics instrument with software version 3.60.2. Equilibrium contact angles were obtained by applying 1 μL water droplets on SAMs using the sessile drop method. The contact angle was measured at three different locations on each surface and the results were averaged. The results showed an average contact angle of 68 ± 1°, which corresponds closely to values of C60-SAM reported by Tsukruk and co-workers.69 While, before the exchanging, the SAM of SC10 was determined to be more hydrophobic with a contact angle of 94 ± 1°, which also confirmed the formation of the fullerene SAM. See ESI† for a description of the EGaIn measurement setup.XPS thickness measurement
To measure the thickness of the SAM, XPS measurements were performed using a VG Microtech spectrometer with a hemispherical electron analyzer (Clam 100), and a MgKα (1253.6 eV) X-ray source. The Ag3p3/2 and Ag3d peaks were acquired with the sample rotated under 0, 10, 20, 30, 40, and 50 degrees with respect to the electron analyzer. A Gaussian fit with background was made to the peaks to obtain their intensities. To correct for slow fluctuations in the X-ray source intensity we acquired the spectra for each peak at 0° in between the measurements where the sample is rotated. These measurements are used to obtain a correction factor γI.The corrected peak intensities I* are given by I* = γII and can be used to determine the thickness of the layer. The values are given in ESI.† The measured electrons in the peaks are the electrons that make it from the silver through the layer without scattering. An expression for the intensity of the peaks for different lengths of the path that through the overlayer:
 | (1) |
Estimation of LUMO of FSC11
The UV-vis absorption spectrum of FSC11 enables the estimation of optical band gap (Eg) to be 1.73 eV from the onset wavelength of 718.13 nm. Ultraviolet photoelectron spectroscopy (UPS) analysis of the SAM-bound AgTS (AgTS/SC11P) for estimating the Fermi level of the silver, and the HOMO level of the FSC11 relative to the Fermi level. Binding energies are calculated with respect to the vacuum level. The vacuum level is found by summing the secondary electron cutoff and the photon energy (He I, hν = 21.2 eV). The valence band spectrum is shown in the ESI† as measured by UPS, showing the characteristic double peak of HOMO and HOMO-1 of C60. For this data a smooth background function has been subtracted. A multiple Gaussian peak fit is performed on the data and the width (σ) and center (μ) of the peaks are found from the fit. We take the value of μ + 2σ as the onset of the HOMO (−5.45 eV). Therefore estimated LUMO of the FSC11 could be derived to be −3.72 eV from the equation ELUMO = EHOMO + Eg (eV).AFM measurement
AFM and CP-AFM measurements were performed on a Bruker AFM Multimode MMAFM-2 equipped with a Peak Force TUNA Application Module (Bruker). Pure SAMs of SC10 before exchange and mixed-monolayers of FSC11 were characterized by AFM. While individual C60 cages could not be resolved Fig. S16† shows clear qualitative differences before and after exchange, but low roughnesses (Ra ≈ 1 nm) and no signs of aggregation or other irregularities. See ESI† for details.Acknowledgements
This is a publication by the FOM Focus Group ‘Next Generation Organic Photovoltaics’, participating in the Dutch Institute for Fundamental Energy Research (DIFFER). R. C. C. and Y. Z. also acknowledge the European Research Council for the ERC Starting Grant 335473 (MOLECSYNCON).References
- A. Aviram and M. A. Ratner, Chem. Phys. Lett., 1974, 29, 277–283 CrossRef CAS.
- H. Ashwell, J. Bonham and L. Lyons, Aust. J. Chem., 1980, 33, 1619–1623 CrossRef.
- A. Martin, J. Sambles and G. Ashwell, Phys. Rev. Lett., 1993, 70, 218 CrossRef CAS PubMed.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS PubMed.
- C. Kaes, A. Katz and M. W. Hosseini, Chem. Rev., 2000, 100, 3553–3590 CrossRef CAS PubMed.
- T. Xu, I. R. Peterson, M. Lakshmikantham and R. M. Metzger, Angew. Chem., Int. Ed., 2001, 40, 1749–1752 CrossRef CAS PubMed.
- M.-K. Ng and L. Yu, Angew. Chem., Int. Ed., 2002, 41, 3598–3601 CrossRef CAS PubMed.
- A. Troisi and M. A. Ratner, J. Am. Chem. Soc., 2002, 124, 14528–14529 CrossRef CAS PubMed.
- R. McCreery, J. Dieringer, A. O. Solak, B. Snyder, A. M. Nowak, W. R. McGovern and S. DuVall, J. Am. Chem. Soc., 2003, 125, 10748–10758 CrossRef CAS PubMed.
- R. M. Metzger, Chem. Rev., 2003, 103, 3803–3834 CrossRef CAS PubMed.
- A. Troisi and M. A. Ratner, Nano Lett., 2004, 4, 591–595 CrossRef CAS.
- G. J. Ashwell, W. D. Tyrrell and A. J. Whittam, J. Am. Chem. Soc., 2004, 126, 7102–7110 CrossRef CAS PubMed.
- A. Honciuc, A. Jaiswal, A. Gong, K. Ashworth, C. W. Spangler, I. R. Peterson, L. R. Dalton and R. M. Metzger, J. Phys. Chem. B, 2005, 109, 857–871 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- G. J. Ashwell and A. Mohib, J. Am. Chem. Soc., 2005, 127, 16238–16244 CrossRef CAS PubMed.
- R. L. McCreery, J. Wu and R. P. Kalakodimi, Phys. Chem. Chem. Phys., 2006, 8, 2572–2590 RSC.
- G. Ho, J. R. Heath, M. Kondratenko, D. F. Perepichka, K. Arseneault, M. Pézolet and M. R. Bryce, Chem.–Eur. J., 2005, 11, 2914–2922 CrossRef CAS PubMed.
- G. J. Ashwell and A. Chwialkowska, Chem. Commun., 2006, 1404–1406 RSC.
- G. J. Ashwell, B. Urasinska and W. D. Tyrrell, Phys. Chem. Chem. Phys., 2006, 8, 3314–3319 RSC.
- N. Armstrong, R. C. Hoft, A. McDonagh, M. B. Cortie and M. J. Ford, Nano Lett., 2007, 7, 3018–3022 CrossRef CAS PubMed.
- E. A. Weiss, G. K. Kaufman, J. K. Kriebel, Z. Li, R. Schalek and G. M. Whitesides, Langmuir, 2007, 23, 9686–9694 CrossRef CAS PubMed.
- X. Chen, S. Yeganeh, L. Qin, S. Li, C. Xue, A. B. Braunschweig, G. C. Schatz, M. A. Ratner and C. A. Mirkin, Nano Lett., 2009, 9, 3974–3979 CrossRef CAS PubMed.
- C. A. Nijhuis, W. F. Reus, J. R. Barber, M. D. Dickey and G. M. Whitesides, Nano Lett., 2010, 10, 3611–3619 CrossRef CAS PubMed.
- R. C. Chiechi, E. A. Weiss, M. D. Dickey and G. M. Whitesides, Angew. Chem., 2008, 120, 148–150 CrossRef.
- Y. Zhang, Z. Zhao, D. Fracasso and R. C. Chiechi, Isr. J. Chem., 2014, 54, 513–533 CrossRef CAS.
- C. A. Nijhuis, W. F. Reus and G. M. Whitesides, J. Am. Chem. Soc., 2009, 131, 17814–17827 CrossRef CAS PubMed.
- C. A. Nijhuis, W. F. Reus and G. M. Whitesides, J. Am. Chem. Soc., 2010, 132, 18386–18401 CrossRef CAS PubMed.
- K. S. Wimbush, W. F. Reus, W. G. van der Wiel, D. N. Reinhoudt, G. M. Whitesides, C. A. Nijhuis and A. H. Velders, Angew. Chem., Int. Ed., 2010, 49, 10176–10180 CrossRef CAS PubMed.
- W. F. Reus, M. M. Thuo, N. D. Shapiro, C. A. Nijhuis and G. M. Whitesides, ACS Nano, 2012, 6, 4806–4822 CrossRef CAS PubMed.
- N. Nerngchamnong, L. Yuan, D.-C. Qi, J. Li, D. Thompson and C. A. Nijhuis, Nat. Nanotechnol., 2013, 8, 113–118 CrossRef CAS PubMed.
- L. Yuan, N. Nerngchamnong, L. Cao, H. Hamoudi, E. del Barco, M. Roemer, R. K. Sriramula, D. Thompson and C. A. Nijhuis, Nat. Commun., 2015, 6, 6324 CrossRef CAS PubMed.
- A. R. Garrigues, L. Yuan, L. Wang, E. R. Mucciolo, D. Thompon, E. del Barco and C. A. Nijhuis, Sci. Rep., 2016, 6, 26517 CrossRef CAS PubMed.
- L. Cao, M. Yang, L. Yuan, N. Nerngchamnong, Y.-P. Feng, A. T. Wee, D.-C. Qi and C. A. Nijhuis, J. Phys.: Condens. Matter, 2016, 28, 094006 CrossRef PubMed.
- A. R. Garrigues, L. Wang, E. del Barco and C. A. Nijhuis, Nat. Commun., 2016, 7, 11595 CrossRef CAS PubMed.
- H. J. Yoon, K. C. Liao, M. R. Lockett, S. W. Kwok, M. Baghbanzadeh and G. M. Whitesides, J. Am. Chem. Soc., 2014, 136, 17155–17162 CrossRef CAS PubMed.
- L. Yuan, L. Jiang, D. Thompson and C. A. Nijhuis, J. Am. Chem. Soc., 2014, 136, 6554–6557 CrossRef CAS PubMed.
- L. Jiang, L. Yuan, L. Cao and C. A. Nijhuis, J. Am. Chem. Soc., 2014, 136, 1982–1991 CrossRef CAS PubMed.
- G. D. Kong, M. Kim, S. J. Cho and H. J. Yoon, Angew. Chem., Int. Ed., 2016, 55, 10307–10311 CrossRef CAS PubMed.
- L. Yuan, R. Breuer, L. Jiang, M. Schmittel and C. A. Nijhuis, Nano Lett., 2015, 15, 5506–5512 CrossRef CAS PubMed.
- S. Bosi, T. Da Ros, G. Spalluto and M. Prato, Eur. J. Med. Chem., 2003, 38, 913–923 CrossRef CAS PubMed.
- J.-F. Nierengarten, New J. Chem., 2004, 28, 1177–1191 RSC.
- Y. He and Y. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- L. Rincón-García, A. K. Ismael, C. Evangeli, I. Grace, G. Rubio-Bollinger, K. Porfyrakis, N. Agraït and C. J. Lambert, Nat. Mater., 2016, 15, 289–293 CrossRef PubMed.
- C. Zeng, H. Wang, B. Wang, J. Yang and J. Hou, Appl. Phys. Lett., 2000, 77, 3595 CrossRef CAS.
- C. A. Martin, D. Ding, J. K. Sørensen, T. Bjørnholm, J. M. van Ruitenbeek and H. S. van der Zant, J. Am. Chem. Soc., 2008, 130, 13198–13199 CrossRef CAS PubMed.
- D. Bonifazi, O. Enger and F. Diederich, Chem. Soc. Rev., 2007, 36, 390–414 RSC.
- K. Kelly, Y.-S. Shon, T. Lee and N. Halas, J. Phys. Chem. B, 1999, 103, 8639–8642 CrossRef CAS.
- P. Sudeep, B. I. Ipe, K. G. Thomas, M. George, S. Barazzouk, S. Hotchandani and P. V. Kamat, Nano Lett., 2002, 2, 29–35 CrossRef CAS.
- P. Peumans and S. Forrest, Appl. Phys. Lett., 2001, 79, 126–128 CrossRef CAS.
- W. B. Caldwell, K. Chen, C. A. Mirkin and S. J. Babinec, Langmuir, 1993, 9, 1945–1947 CrossRef CAS.
- X. Shi, W. B. Caldwell, K. Chen and C. A. Mirkin, J. Am. Chem. Soc., 1994, 116, 11598–11599 CrossRef CAS.
- R. R. Sahoo and A. Patnaik, J. Colloid Interface Sci., 2003, 268, 43–49 CrossRef CAS PubMed.
- L. Grüter, F. Cheng, T. T. Heikkilä, M. T. González, F. Diederich, C. Schönenberger and M. Calame, Nanotechnology, 2005, 16, 2143 CrossRef PubMed.
- Y. Shirai, L. Cheng, B. Chen and J. M. Tour, J. Am. Chem. Soc., 2006, 128, 13479–13489 CrossRef CAS PubMed.
- Y. Shirai, J. M. Guerrero, T. Sasaki, T. He, H. Ding, G. Vives, B.-C. Yu, L. Cheng, A. K. Flatt and P. G. Taylor, et al. , J. Org. Chem., 2009, 74, 7885–7897 CrossRef CAS PubMed.
- M. del Carmen Gimenez-Lopez, M. T. Räisänen, T. W. Chamberlain, U. Weber, M. Lebedeva, G. A. Rance, G. A. D. Briggs, D. Pettifor, V. Burlakov and M. Buck, et al. , Langmuir, 2011, 27, 10977–10985 CrossRef PubMed.
- S. Chase, W. Bacsa, M. Mitch, L. Pilione and J. Lannin, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 7873 CrossRef CAS.
- E. I. Altman and R. J. Colton, Surf. Sci., 1993, 295, 13–33 CrossRef CAS.
- M. Hunt, S. Modesti, P. Rudolf and R. Palmer, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 10039 CrossRef CAS.
- C.-T. Tzeng, W.-S. Lo, J.-Y. Yuh, R.-Y. Chu and K.-D. Tsuei, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 2263 CrossRef CAS.
- L. A. Bumm, J. J. Arnold, M. T. Cygan, T. D. Dunbar, T. P. Burgin, L. Jones, D. L. Allara, J. M. Tour and P. S. Weiss, Science, 1996, 271, 1705–1707 CAS.
- D. Thompson and C. A. Nijhuis, Acc. Chem. Res., 2016, 49, 2061–2069 CrossRef CAS PubMed.
- L. Yuan, D. Thompson, L. Cao, N. Nerngchangnong and C. A. Nijhuis, J. Phys. Chem. C, 2015, 119, 17910–17919 CAS.
- T. W. Kelley, E. Granstrom and C. D. Frisbie, Adv. Mater., 1999, 11, 261–264 CrossRef CAS.
- O. E. Castañeda Ocampo, P. Gordiichuk, S. Catarci, D. A. Gautier, A. Herrmann and R. C. Chiechi, J. Am. Chem. Soc., 2015, 137, 8419–8427 CrossRef PubMed.
- S. Kumar, J. T. van Herpt, R. Y. Gengler, B. L. Feringa, P. Rudolf and R. C. Chiechi, J. Am. Chem. Soc., 2016, 138, 12519–12526 CrossRef CAS PubMed.
- H. M. Saavedra, C. M. Barbu, A. A. Dameron, T. J. Mullen, V. H. Crespi and P. S. Weiss, J. Am. Chem. Soc., 2007, 129, 10741–10746 CrossRef CAS PubMed.
- S. Patole, C. Baddeley, D. O'Hagan and N. Richardson, J. Phys. Chem. C, 2008, 112, 13997–14000 CAS.
- V. V. Tsukruk, L. M. Lander and W. J. Brittain, Langmuir, 1994, 10, 996–999 CrossRef CAS.
- W. Li, J. A. Virtanen and R. M. Penner, J. Phys. Chem., 1994, 98, 11751–11755 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc04799h |
| This journal is © The Royal Society of Chemistry 2017 |