
Ultra-high surface area mesoporous carbons for colossal pre combustion CO2 capture and storage as materials for hydrogen purification†
Michael
Cox
and
Robert
Mokaya
 *
*
School of Chemistry, University of Nottingham, University Park, NG7 2RD Nottingham, UK. E-mail: r.mokaya@nottingham.ac.uk
First published on 30th June 2017
Abstract
Carbon capture and storage (CCS) by solid adsorbents is currently attracting a great deal of attention. In this study, a new direction in the treatment of activatable carbon-containing precursors generated a family of mesoporous carbons that possess extremely high mesopore volume and hardly any microporosity. The mesoporous carbons, with up to 95% mesoporosity, have ultra-high surface area (2800–4000 m2 g−1) and pore volume (2.5–3.6 cm3 g−1). The porosity of the carbons, i.e., mesopores of size 25–50 Å and hardly any micropores, is favourable for CO2 uptake under conditions that are relevant to pre combustion CCS, i.e., 25 °C and pressure of 20 to 50 bar. The best performing carbons have near total absence of micropores; our findings suggest that the presence of microporosity is a limiting factor in the CO2 uptake capacity especially at high pressure (30–50 bar). The gravimetric (mmol g−1) CO2 uptake capacity of the mesoporous carbons is impressive; up to 28 (20 bar), 37 (30 bar), 46 (40 bar) and 55 (50 bar), which is equivalent to 2.42 g of CO2 per g of carbon. Furthermore, due to their packing density (0.25–0.4 g cm−3), the mesoporous carbons exhibit colossal volumetric CO2 uptake (in g l−1) of up to 480 (20 bar), 640 (30 bar), 780 (40 bar) and 930 (50 bar). The performance of the mesoporous carbons is such that, at 30 bar, they can hold more than 10 times the CO2 in a pressurized cylinder, and at 50 bar can store up to 470 cm3 cm−3. The all-round pre combustion CCS performance of the mesoporous carbons is significantly higher than that of the best carbons to date, and outperforms that of benchmark materials such as metal organic frameworks (MOFs). The carbons are highly suited, in terms of their CO2 adsorption capacity and CO2 selectivity over H2, as materials for hydrogen purification under syngas flow conditions.
Introduction
There are two main ways by which the increase in levels of atmospheric carbon dioxide can be tackled, namely, (i) reducing the use of carbon-based fossil fuels by moving over to low carbon emission energy sources or simply reducing consumption, and (ii) removal of carbon dioxide from the atmosphere via capture and storage.1–5 The later requires materials that can efficiently capture and store CO2;1,3,6 a significant proportion of the CO2 in the atmosphere comes from burning (combustion) of fossil fuels for energy production.3 A wide range of materials have been studied for application in carbon capture and storage (CCS) via physical adsorption and/or separation.3,6–10 The materials studied so far, and which have performed admirably, are dominated by porous carbons,11–14 zeolites15–17 and metal organic frameworks (MOFs).18,19 Due to their structural flexibility and stability (chemical and mechanical), light-weight porous carbons have emerged as promising adsorbents for CCS.11–14 Recent studies have shown that the control and optimisation of pore size and surface area of porous carbons can provide a very useful tool in generating materials tailored towards CO2 capture under either pre or post combustion (of fossil fuels) conditions.20–26It has been demonstrated by several studies that to maximise CO2 uptake at low pressure (i.e., post combustion conditions) porous carbons need to have a high surface area arising from small micropores of size 7–9 Å.8–14,27–35 On the other hand, the presence of small micropores can limit the level of pore volume achieved, which is inimical to material requirements for pre combustion CO2 capture, which normally occurs at high (>20 bar) pressure.36 With regard to high pressure CCS, which is akin to conditions for pre combustion CO2 storage, we have recently shown that carbons that possess both micropores and mesopores have improved storage of CO2 at high pressure; up to 29.5, 34.5 and 38.3 mmol g−1 at 25 °C and 30, 40 and 50 bar, respectively.37 Furthermore, such micro–mesoporous materials show enhanced CO2 working capacity storage for pressure swing adsorption (PSA) systems arising from a combination of greater CO2 capture at high pressure and diminished uptake at the lower regeneration pressures.37 Other researchers have also, recently, reported what are claimed to be record levels of high pressure CO2 storage in micro–mesoporous carbons derived from (i) polyaniline cross-linked polymer networks (storage capacity of 28.3, 32.5 and 34.9 mmol g−1 at 25 °C and 30, 40 and 50 bar, respectively),38 or (ii) asphalt (storage capacity of 35.2 mmol g−1 at 25 °C and 50 bar).39 A common feature of the previously explored materials, which currently constitute the benchmark for high pressure CO2 storage in porous carbons, is that they have very high surface area (3400–4200 m2 g−1).37–39 However, they also contain a significant amount of microporosity, which is not beneficial for high pressure (i.e., pre combustion) CO2 storage; we have shown that the introduction of mesoporosity, along with a decrease in the proportion of microporosity, improves high pressure CO2 storage.37
It would appear, therefore, that the optimum porosity for pre combustion high pressure CO2 uptake in carbons may tend towards high surface area materials that are devoid of microporosity or have minimal levels of microporosity. Such a class of materials does not currently exist. To test our hypothesis on the role of mesoporosity vis-a-vis microporosity, we have explored new routes for the preparation of porous carbons that go beyond currently accepted limits in an effort to generate materials with very low levels of microporosity and have assessed their high pressure CO2 uptake under conditions similar to those used for pre combustion CCS involving, for example, the purification of syngas in power stations. Syngas, typically derived from hydrocarbons and coals, mainly contains 71–75% H2, 15–20% CO2, and ca. 5% of other gases (CH4, CO, H2O). The syngas is used as fuel in integrated gasification and combined cycle (IGCC) systems that incorporate steps for the pre combustion removal and sequestration of CO2 in order to enrich the H2 content.40,41 Removal of CO2 improves the burning properties and also reduces the amount of CO2 released by combustion of the syngas.40,41 Purification of the syngas, i.e. removal of CO2, may be achieved via solvent washing systems although solid adsorbents may also be used.40–44 A key requirement of any adsorbent for syngas (or H2) purification is a high CO2 adsorption capacity and selectivity for CO2 over H2, and thus there is need to develop solid state absorbers with improved CO2 capacity and selectivity under pre combustion CCS conditions.
This study unravels interesting trends between the high pressure CO2 uptake and the proportion of mesoporosity and/or microporosity in high surface area carbons and illustrates how the porosity may be tailored toward exceptional and hitherto unreached levels of moderate to high pressure CO2 adsorption (both in terms of gravimetric and volumetric capacity), that are relevant to pre combustion CCS and hydrogen purification in general.
Experimental section
Materials synthesis
The starting materials (precursors) used were polypyrrole and eucalyptus wood sawdust. The wood sawdust (designated as SD) was used after conversion to hydrochar,20,21 while the polypyrrole (designated as PPy), was prepared by adding 3 g of pyrrole to 200 ml of 0.5 M FeCl3 aqueous solution and magnetically stirring at room temperature for 2 h, following which the resulting solid product (PPy) was separated by filtration, washed with distilled water and dried in an oven at 120 °C. The PPy or SD were activated by mixing with KOH, in an agate mortar, at predetermined KOH/PPy or KOH/SD hydrochar ratios followed by heating at 700 to 900 °C. The KOH/precursor ratios used (3, 4 or 5) were those that were expected to offer high levels of activation. In a typical activation process the KOH/precursor mixture was heated under a flow of nitrogen in a tube furnace to the desired temperature (700, 800 or 900 °C) at a ramp rate of 3 °C min−1 and then held at the target temperature for 1 h. The resulting carbonaceous material was washed with 10 wt% HCl to remove any inorganic salts, and then washed several times with water (until the water was at pH 7), followed by drying in an oven at 120 °C. The carbons from PPy or SD were designated as PPY-xT or SD-xT, respectively, where x is the KOH/precursor ratio and T is the activation temperature. We also prepared samples of compactivated carbon20,21,45via a modified route that included compaction of the KOH/precursor mixture prior to thermal treatment. The KOH/PPy or KOH/SD hydrochar mixture, at a ratio of 4, was compacted for 10 min at a load of 10 tonnes in a 1.3 cm diameter die, equivalent to compaction pressure of 740 MPa. The resulting disk/pellet was placed in an alumina boat and heated in a horizontal furnace to 800 °C at heating ramp rate of 3 °C min−1 under a nitrogen gas flow and held at 800 °C for 1 h. The resulting compactivated carbon was washed and dried as described above except that no stirring was used during the washing steps. The compactivated carbons were designated as MPPY-4800 (from PPy) and MSD-4800 (from SD).We also prepared, for comparison purposes, a highly microporous carbon as follows; hydrochar was obtained by heating, at a ramp rate of 5 °C min−1, 6.4 g of cellulose acetate (CA) in 20 ml of water in a stainless steel autoclave to 250 °C for 2 h. The resulting hydrochar was obtained by filtration and dried overnight in an oven at 112 °C, and then activated as described above at KOH/CA hydrochar ratio of 4 and 700 °C. The resulting activated carbon (denoted as CA-4700) was washed as described above and dried at 112 °C.
Materials characterisation
Powder X-ray diffraction (XRD) patterns were recorded using a PANanalytical X'Pert PRO diffractometer with Cu-Kα radiation, operating at 40 kV and 40 mA with 0.02° step size and 30 s step time. Elemental (C, H, N and O) analysis was performed using a model CE-440 Elemental Analyzer (Exeter Analytical). Thermogravimetric analysis (TGA) was performed using alumina pans in a TA Instruments SDT Q600 analyzer. Samples for TGA were heated from room temperature to 1000 °C at 5 °C min−1 in air. Nitrogen sorption isotherms and textural properties of the activated and compactivated carbons were determined at −196 °C using nitrogen in a conventional volumetric technique by Micromeritics ASAP 2020 or 3FLEX sorptometers. Before their analysis, the carbons were dried in an oven at 120 °C and then degassed overnight at 200 °C under high vacuum. The surface area was calculated using the BET method based on adsorption data in the relative pressure (P/Po) range of 0.05 to 0.22, and the relative pressure range was selected to ensure a positive y-axis intercept from multipoint BET fitting (such that C > 0) and that Vads(1 − p/po) would increase with p/po.46 The total pore volume was determined from the amount of nitrogen adsorbed at a relative pressure of ∼0.99. The pore size distribution (PSD) was determined using a Non Local Density Functional Theory (NLDFT) model using nitrogen adsorption data.Gas uptake measurements
CO2 uptake measurements were performed at 25 °C or 0 °C over the pressure range of 0–50 bar with a Hiden XEMIS intelligent gravimetric analyzer or at 0–20 bar with a Hiden intelligent gravimetric analyzer (IGA-003). The carbon samples were outgassed under vacuum at 200 °C before determining the CO2 uptake isotherms. Buoyancy corrections were applied, and the measurements determined the excess CO2 uptake from which the total storage capacity could be determined.Hydrogen uptake capacity of the carbons was measured by gravimetric analysis with a Hiden XEMIS Intelligent Gravimetric Analyser using 99.9999% purity hydrogen additionally purified by a molecular sieve filter. Prior to analysis, the carbon samples were dried in an oven for 24 h at 80 °C and then placed in the analysis chamber and degassed at 200 °C and 10−10 bar for 4–6 h. The hydrogen uptake measurements were performed at room temperature (25 °C).
Results and discussion
The aim of this study was to explore the preparation of super porous carbons at very high levels of activation and to assess their CO2 uptake at high pressures that are relevant to pre combustion CCS. For this purpose we primarily used polypyrrole (PPy) as precursor as it is known to generate high surface area activated carbons that retain a high packing density.20,47 Following activation, we determined that the resulting PPy-derived activated carbons were fully carbonaceous by performing thermogravimetric analysis (TGA) in air. TGA curves of some representative samples (ESI Fig. S1†) indicated residual mass of 0–1.5 wt%, which is normal for KOH activated carbons. The TGA curves also offered information on the thermal stability of the carbons; in general samples activated at higher activation temperature show greater resistance to combustion (ESI Fig. S1†) perhaps due to higher levels of graphitisation induced during the thermal treatment. However, the overall level of graphitisation in the samples was generally low as indicated by powder XRD patterns (ESI Fig. S2†). Low levels of graphitisation are conducive for the generation of high levels of porosity, which is a target in this work. The elemental composition of the polypyrrole and the activated carbons is given in Table 1. As expected, the carbon content increases at higher levels of activation accompanied by a decrease in the N, H and O content. It is noteworthy that, due to their high level of activation, the N content of the activated carbons is very low (i.e., 0.1–0.7 wt%). This is consistent with the fact that the N content of activated carbons prepared from N containing precursors generally reduces at high levels of activation.37,47 We monitored the yield of activated carbon as a function of the amount of polypyrrole and despite the elevated activation conditions, the yields were 11–21 wt%. Sample PPY-4700 had the highest yield (21 wt%), which decreased at higher levels of activation to ∼11 wt% for sample PPY-4900 and PPY-5900. Such yields are typical for KOH activation and are higher than for activated carbons prepared via physical activation.20,47,48| Sample | C [%] | H [%] | N [%] | O [%] |
|---|---|---|---|---|
| PPY | 54.8 | 3.2 | 16.0 | 26.0 |
| PPY-4700 | 91.1 | 1.1 | 0.7 | 7.1 |
| PPY-4800 | 93.7 | 0.4 | 0.5 | 5.4 |
| MPPY-4800 | 94.0 | 0.3 | 0.4 | 5.3 |
| PPY-5800 | 86.8 | 0 | 0.4 | 12.8 |
| PPY-3900 | 92.1 | 0 | 0.4 | 7.7 |
| PPY-4900 | 94.1 | 0 | 0.2 | 5.7 |
| PPY-5900 | 88.7 | 0 | 0.1 | 11.2 |
The nitrogen sorption isotherms of the PPy-derived activated carbons are shown in Fig. 1A. All the samples exhibit type IV isotherms that are typical of mesoporous materials. All isotherms also exhibit significant adsorption at low relative pressure (P/Po < 0.1), which is an indication that the present activated carbons possess high surface area despite their mesoporous character. The total amount of nitrogen adsorbed increases for samples prepared at higher temperature and/or higher KOH/PPy ratio in the order PPY-4700 < PPY-4800 < PPY-3900 < PPY-5800–PPY-4900 < PPY-5900. As far as we know, polypyrrole-derived carbons have not previously been prepared via KOH activation at KOH/PPy ratio of 5 and/or 900 °C.20,47,48 The amount of nitrogen adsorbed is much higher than previously observed for activated carbons in general.20,47,48 The isotherms in Fig. 1A suggest that the main effect of greater levels of activation and compactivation (MPPY-4800 compared to PPY-4800) is an increase in the porosity without any significant change in the shape of the isotherms except for sample PPY-5900, which is prepared at the severest conditions.
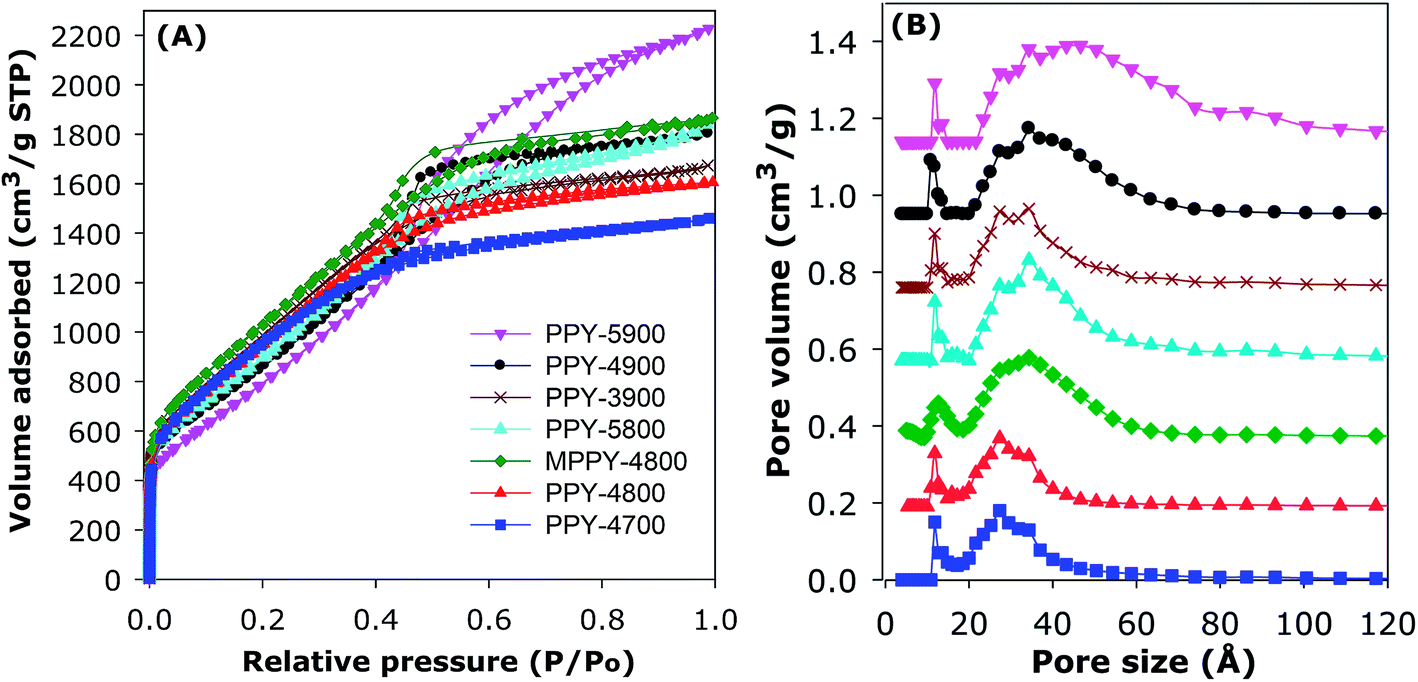 | ||
| Fig. 1 Nitrogen sorption isotherms (A) and corresponding pore size distribution (PSD) curves (B) of polypyrrole-derived (PPY-xT) activated carbons and compactivated (MPPY-4800) carbon. | ||
The textural parameters and packing density of the activated and compactivated carbons are summarised in Table 2. All the carbons possess high or ultra-high surface area, which ranges between 2800 and 3940 m2 g−1. In calculating the surface area, for all samples we confirmed that the data points used to determine the surface area gave a BET, or C, constant that is positive; this was evidenced by observing the Rouquerel plots and the value of the C constants (ESI Fig. S3†). Apart from their high to ultra-high surface area, the carbons also possess high to very high pore volume in the range 2.45–3.60 cm3 g−1. The highest surface area for an activated carbon is for PPY-4700 (3568 m2 g−1) and PPY-4800 (3589 m2 g−1), while samples PPY-5800 (2.85 cm3 g−1) and PPY-5900 (3.54 cm3 g−1) have the highest pore volume. Although all the activated carbons possess high porosity (i.e., surface area and pore volume), the balance between surface area and pore volume is such that the most highly activated carbons have the highest pore volume, but at the expense of a slight reduction in surface area (Table 2). All the carbons have very low levels of microporosity such that the proportion of volume arising from mesopores (referred to as mesoporosity) is above 90% and up to 95% (Table 2). This means that the present carbons are essentially mesoporous in character. The porosity of the carbons may be improved via compactivation as demonstrated by comparison between the analogously activated (PPY-4800) and compactivated (MPPY-4800) samples. The activated PPY-4800 sample has surface area and pore volume of 3589 m2 g−1 and 2.71 cm3 g−1, respectively, compared to 3934 m2 g−1 and 2.92 cm3 g−1 for the compactivated MPPY-4800 sample. The compactivated MPPY-4800 carbon appears to have an optimised mix of ultra-high surface area, pore volume and high (93%) mesoporosity.
| Sample | Surface area (m2 g−1) | Pore volumea (cm3 g−1) | Pore sizeb (Å) | Mesoporosityc (%) | Packing density (g cm−3) |
|---|---|---|---|---|---|
| a The values in the parenthesis are micropore volume. b Main pore size maxima obtained from NLDFT analysis. c Proportion of pore volume arising from mesopores. | |||||
| PPY-4700 | 3568 | 2.46 (0.28) | 27 | 89 | 0.34 |
| PPY-4800 | 3589 | 2.71 (0.25) | 28 | 91 | 0.32 |
| MPPY-4800 | 3934 | 2.92 (0.20) | 34 | 93 | 0.38 |
| PPY-5800 | 2974 | 2.85 (0.22) | 35 | 92 | 0.30 |
| PPY-3900 | 3285 | 2.60 (0.30) | 32 | 88 | 0.33 |
| PPY-4900 | 2842 | 2.77 (0.27) | 36 | 90 | 0.31 |
| PPY-5900 | 2798 | 3.54 (0.19) | 46 | 95 | 0.25 |
The mesoporous nature of the activated carbons and compactivated carbon is confirmed by the pore size distribution (PSD) curves in Fig. 1B. For all samples, the main pore size maxima is in the mesopore range of 27 to 46 Å. In general, the main pore size maxima increases with the severity of activation such that the sample prepared under the most severe conditions (PPY-5900) has the largest mesopores (46 Å). Table 2 gives the packing density of the carbons, which varies between 0.25 and 0.38 g cm−3. The packing density of the compactivated carbon (MPPY-4800) and some of the activated carbons was determined by pressing a known amount of sample in a 1.3 cm die at pressure of 74 MPa for 5 min. Such compaction has no effect on the porosity of the carbons.20 For activated carbons, the packing density (dcarbon) may also be estimated from skeletal density using the equation; dcarbon = (1/ρs + VT)−1, where ρs is skeletal density and VT is total pore volume. The skeletal density used (2.1 g cm−3) was obtained from helium sorption at 0 °C. There is an inverse relationship between the total pore volume and the packing density of the activated carbons. The compactivated carbon bucks this trend as it was already compacted prior to activation and therefore has a higher than expected packing density.20 Overall, therefore, with respect to porosity, we succeeded in generating mesoporous carbons with simultaneously high surface area and pore volume. In particular, the present carbons have very low levels of microporosity which is unusual for high surface area activated carbons.37–49 The near elimination of microporosity is achieved by choice of precursor and activation conditions.20,37–49
We assessed the CO2 uptake capacity of the activated carbons at room temperature (25 °C) and pressure range of 0 to 50 bar. The main focus was the CO2 uptake at moderate to high pressure (20–50 bar) under conditions that attempt to mimic pre combustion CCS. The total gravimetric CO2 uptake isotherms are shown in Fig. 2. Our measurements determined the excess CO2 uptake (ESI Fig. S4†) from which the total storage capacity was obtained by considering the density of the CO2 under the prevailing conditions and the carbon's pore volume using the equation; θT = θExc + dH2 × VT, where; θT is total CO2 uptake, θExc is excess CO2 uptake, dCO2 is density (g cm−3) of CO2 gas at the relevant temperature and pressure (the density of CO2 was obtained from the National Institute of Standards and Technology (NIST) website (http://www.nist.gov/)), and VT is pore volume (cm3 g−1) of the carbon. Table 3 summarizes the amount of CO2 adsorbed at various pressures. At 1 bar, the CO2 uptake is in the range 1.3–2.8 mmol g−1, which is low or moderate capacity compared to benchmark carbon materials.11–14,20–37 The storage capacity at 1 bar is consistent with the well-established fact that it is not the total surface area or pore volume, but the pore size that determines the CO2 uptake at low pressure.11–37 On the other hand, the CO2 uptake increases continuously at higher pressures to reach very high levels of storage at 50 bar. For all samples the CO2 uptake was fully reversible with no hysteresis, and no saturation was attained in the 0–50 bar pressure range except for sample PPY-4700 (Fig. 2 and ESI S4†). At 20 bar the PPy-derived carbons store between 23 and 28 mmol g−1, with excess uptake in the range 20.6–25.5 mmol g−1. This uptake is much higher than that previously reported for any carbon material.11–14,20–37 At 30 bar the CO2 storage capacity rises to between 29.6 and 37.7 mmol g−1, with excess uptake in the range 26.0–33.0 mmol g−1. This is higher than the current benchmark carbons, namely, polyaniline cross-linked polymer networks (SU-AC-400) that store 28.3 mmol g−1 under similar conditions (i.e., 25 °C and 30 bar).38 At 50 bar, which is the highest pressure used in this study, the carbons have exceptionally high CO2 storage capacity of 40 to 55 mmol g−1, with excess uptake in the range 33.0–45.4 mmol g−1. Total CO2 uptake of 55 mmol g−1 is exceptionally high and in any case is significantly greater (higher by ca. 60%) than that (34.9 mmol g−1) reported recently for polyaniline cross-linked polymer networks (SU-AC-400)38 or that (35.2 mmol g−1) for asphalt-derived carbon (UGil-900).39 The gravimetric CO2 uptake at high pressure (20–50 bar) closely matches the trend in pore volume such that the best performing samples (PPY-5900 and MPPY-4800) also have both the highest pore volume and highest levels of mesoporosity (93–95%). Comparison between PPY-4800 and MPPY-4800 illustrates how compactivation20 may be used to significantly enhance the porosity of activated carbons and consequently their CO2 uptake under conditions that are relevant to pre combustion CCS.
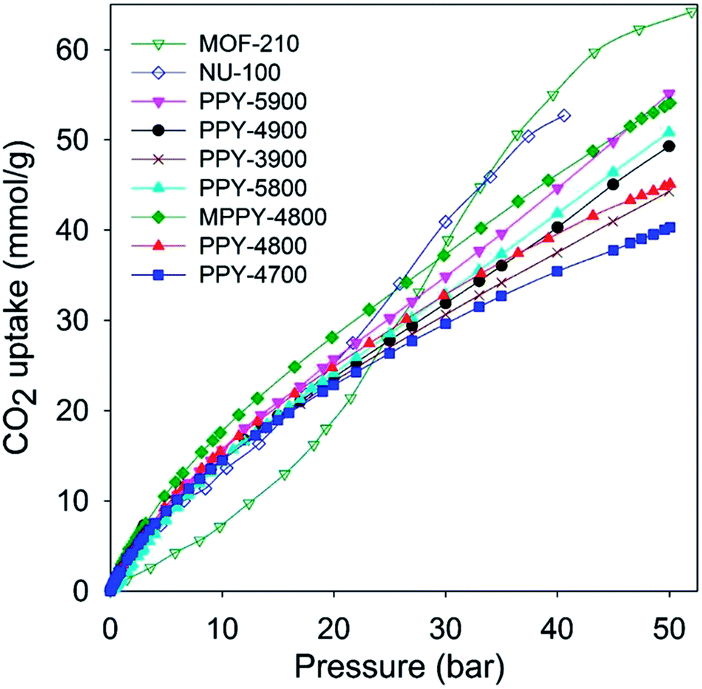 | ||
| Fig. 2 Total gravimetric CO2 uptake at 25 °C of polypyrrole-derived activated (PPY-xT) carbons, a compactivated (MPPY-4800) carbon and MOFs (MOF-210 and NU-100). | ||
| Sample | CO2 uptakea (mmol g−1) | ||||
|---|---|---|---|---|---|
| 1 bar | 20 bar | 30 bar | 40 bar | 50 bar | |
| a CO2 uptake at 25 °C and various pressures (i.e., 1, 20, 30, 40 and 50 bar). The values in the parenthesis are excess CO2 uptake. | |||||
| PPY-4700 | 2.3 | 22.8 (20.6) | 29.6 (26.0) | 35.4 (30.2) | 40.3 (32.9) |
| PPY-4800 | 2.5 | 25.0 (22.5) | 33.0 (29.0) | 39.5 (34.0) | 45.3 (37.2) |
| MPPY-4800 | 2.8 | 28.1 (25.5) | 37.2 (33.0) | 45.6 (39.5) | 54.1 (45.4) |
| PPY-5800 | 1.3 | 24.1 (21.5) | 32.8 (28.7) | 41.8 (35.8) | 50.8 (42.3) |
| PPY-3900 | 2.2 | 23.2 (20.9) | 30.7 (26.9) | 37.5 (32.0) | 44.5 (36.5) |
| PPY-4900 | 2.0 | 23.6 (21.2) | 31.9 (27.9) | 41.4 (34.4) | 49.3 (41.0) |
| PPY-5900 | 2.1 | 25.7 (22.5) | 34.9 (29.7) | 44.6 (37.1) | 55.1 (44.5) |
| MOF-210 | 0.9 | 19.2 (16.0) | 38.9 (33.7) | 55.0 (46.7) | 64.2 (52.5) |
| NU-100 | 2.7 | 24.6 (22.0) | 40.9 (36.3) | 52.7 (46.8) | |
The gravimetric CO2 uptake of the present PPy-derived carbons compares favourably (Fig. 2, ESI S4 and Tables S1 and S2†) with that of the best performing high surface area metal organic frameworks (MOFs), namely, Nu-100,50 and MOF-210,51 which are the current record holders with respect to gravimetric CO2 storage at ambient temperature and high pressure.52 It is noteworthy that the total gravimetric CO2 uptake of samples PPY-5900 and MPPY-4800 at 50 bar is similar to that of NU-100.50 This finding demonstrates the importance of the pore volume (and in particular the mesopore volume) in determining the high pressure CO2 uptake; for example although the surface area of NU-100 (6143 m2 g−1) is more than twice that of PPY-5900 (2798 m2 g−1), the two samples have similar high pressure CO2 uptake (Fig. 2) on account of their comparable mesopore volume (Tables 2 and 3 and ESI Tables S1 and S2†).
To further illustrate the importance of mesopore volume, we compared the CO2 uptake of the PPy-derived carbons to that of activated carbons that have similarly high surface area but lower levels of mesoporosity (Tables S1 and S2†). Comparison with a highly microporous carbon (designated as CA-4700) derived from activation of hydrochar of cellulose acetate (at 700 °C and KOH/hydrochar ratio of 4) is very revealing. Sample CA-4700 presents a type 1 nitrogen sorption isotherm (ESI Fig. S5†) and has surface area of 3771 m2 g−1 (with 3484 m2 g−1, i.e., 92% arising from micropores), pore volume of 1.75 cm3 g−1 and micropore volume of 1.54 cm3 g−1. Sample CA-4700, therefore, has mesopore volume of only 0.21 cm3 g−1 and a mesoporosity level of 12% (Table S1†). In line with its microporous nature, sample CA-4700 has high CO2 uptake (i.e., 3.7 mmol g−1) at 1 bar, which is higher than that of the mesoporous PPy-derived carbons (ESI Fig. S6 and S7, and Table S2†). However, at 20 bar, total CO2 uptake by the mesoporous PPy-derived samples (23–28 mmol g−1) far outperforms the microporous CA-4700 sample (19.5 mmol g−1). Furthermore, in the 20–50 bar pressure range, there is very little increase in the excess storage capacity of sample CA-4700; uptake increases from 18.0 to 20.5 mmol g−1, a rise of only 6% (Fig. S6 and Table S2†). On the other hand, the excess CO2 uptake of PPy-derived carbons nearly doubles from 21–26 mmol g−1 to a high of 45 mmol g−1 as pressure rises from 20 to 50 bar (Fig. S6 and Table S2†). A similar trend is observed in the total CO2 uptake within the 20–50 bar pressure range (Fig. S7 and Table S2†). We attribute this stark difference in CO2 uptake behaviour between CA-4700 and the PPy-derived carbons (ESI Fig. S6 and S7†) to differences in mesoporosity, and especially the balance between microporosity and mesoporosity. This conclusion is supported by the fact that activated carbon samples (AC-0 and AC-2M),37 with mesoporosity that is intermediate between the PPy-derived carbons and CA-4700 (Table S1†) also show intermediate CO2 uptake (ESI Fig. S8 and Table S2†) at high pressure (20–50 bar). This confirms that mesoporosity favours high pressure CO2 uptake while the presence of micropores can be a limiting factor.
A key measure of the efficiency of an adsorbent for pre combustion CCS is the working capacity, which is the difference in CO2 uptake between the adsorption and desorption (regeneration) pressure. For desorption at atmospheric pressure (1 bar), a microporous carbon such as CA-4700 has pressure swing adsorption (PSA) working capacity (in mmol g−1) of 16 (20 bar), 19 (30 bar), 20 (40 bar) and 22 (50 bar). For the mesoporous carbons, the working capacity vastly increases up to 25 (20 bar), 33 (30 bar), 43 (40 bar) and 53 (50 bar), wherein it is more than twice that of the microporous CA-4700 at 40 bar and above. Such working capacity is superior to that of all previously reported carbons.13,37–39,53–56
The PPy-derived carbons have very low N-content (Table 1), which means that their exceptional CO2 uptake may not be ascribed to N-doping. However, to ascertain that N-doping does not play a key role, and also to test the general applicability of the link between high mesoporosity and high pressure CO2 uptake, we prepared and analysed an activated carbon (designated as SD-4800) and compactivated carbon (designated as MDS-4800) derived from wood sawdust as precursor and prepared at a KOH/carbon ratio of 4 at 800 °C. The nitrogen sorption isotherms of both sawdust derived carbons (Fig. 3) are type IV with much higher nitrogen adsorption for the compactivated carbon (MSD-4800). The activated (SD-4800) and compactivated (MSD-4800) carbons have surface area of 2790 m2 g−1 and 3982 m2 g−1, respectively (Table 4). The pore volume is 1.77 cm3 g−1 (SD-4800) and 2.56 cm3 g−1 (MSD-4800) with micropore volume of 0.35 (SD-4800) and 0.28 cm3 g−1 (MSD-4800), and mesoporosity of 80% and 89%, respectively (Table 4). As shown in Fig. 4, both samples, and in particular MSD-4800, have high CO2 uptake at 25 °C, which is comparable to that of PPy-derived carbons. The sawdust-derived samples also show excellent storage of up to 33 mmol g−1 (SD-4800) and 37.5 mmol g−1 (MSD-4800) at 0 °C and 20 bar (ESI Fig. S9†). The CO2 uptake of these N-free samples indicates that it is primarily the optimised mesoporosity that is responsible for the high uptake rather than the presence or absence of N.
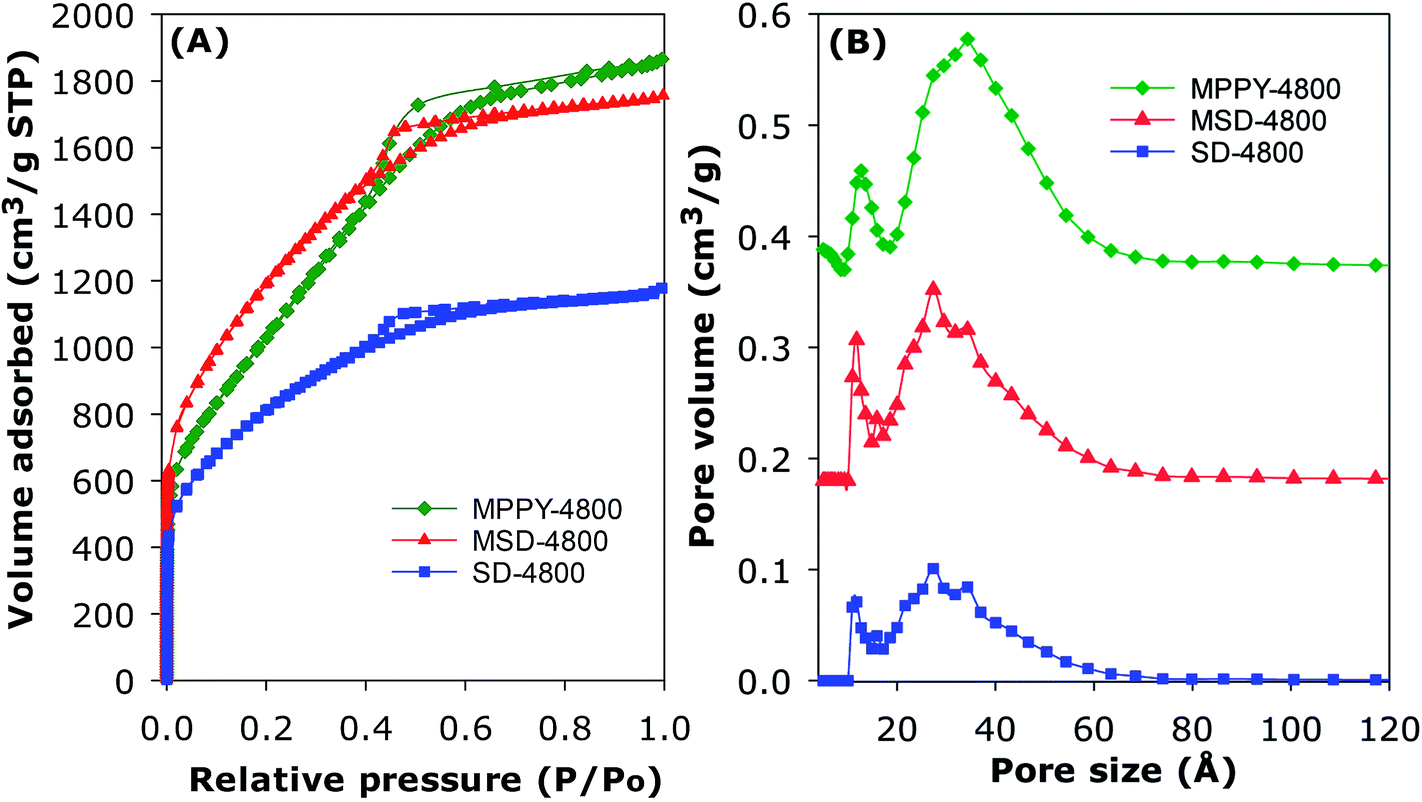 | ||
| Fig. 3 Nitrogen sorption isotherms (A) and corresponding pore size distribution (PSD) curves (B) of polypyrrole-derived compactivated carbon (MPPY-4800), and an activated (SD-4800) or compactivated (MSD-4800) carbon derived from wood sawdust. | ||
| Sample | Surface area (m2 g−1) | Pore volumea (cm3 g−1) | Pore sizeb,c (Å) | CO2 uptaked,e (mmol g−1) | ||||
|---|---|---|---|---|---|---|---|---|
| 1 bar | 20 bar | 30 bar | 40 bar | 50 bar | ||||
| a The values in the parenthesis refer to mesopore volume. b Main pore size distribution maxima obtained from NLDFT analysis. c The values in the parenthesis are % mesoporosity. d Total CO2 uptake at 25 °C and various pressures. e The values in the parenthesis are excess CO2 uptake. | ||||||||
| SD-4800 | 2790 | 1.77 (1.42) | 28 (80) | 2.9 | 22.8 (21.0) | 29.7 (27.1) | 35.4 (31.6) | 40.2 (34.8) |
| MSD-4800 | 3982 | 2.56 (2.28) | 31 (89) | 2.7 | 26.3 (24.0) | 34.8 (31.0) | 42.5 (37.5) | 50.4 (42.8) |
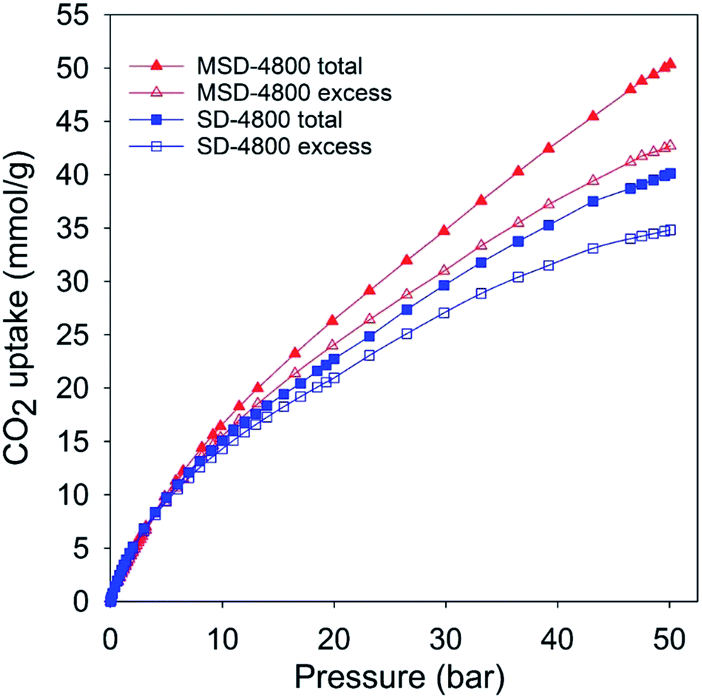 | ||
| Fig. 4 Total and excess CO2 uptake at 25 °C and 0–50 bar of activated (SD-4800) and compactivated (MSD-4800) carbon derived from wood sawdust. | ||
The foregoing discussion indicates that the present mesoporous carbons have exceptional gravimetric CO2 uptake. However, of equal or even greater importance is the volumetric CO2 uptake. This is because for pre combustion CCS applications the adsorbent is confined in restricted space such as a high pressure tank, and therefore the CO2 stored per given volume occupied by the adsorbent (i.e. volumetric uptake) is a key consideration. The volumetric gas uptake of porous materials is determined by their packing density, and gravimetric storage capacity. The PPy-derived carbons have packing density of 0.25–0.38 g cm−3 (Table 2). The volumetric CO2 uptake of the carbons is shown in Fig. 5 (and ESI S10†) and the capacity at various pressures is summarised in Table 5. At 20 bar the mesoporous carbons store an impressive 280 to 480 g l−1, with excess uptake of 240–440 g l−1. At 30 bar the capacity rises to between 380 and 640 g l−1, with excess uptake in the range 327–566 g l−1 while at 40 bar it is between 490 and 780 g l−1, with excess uptake in the range 408–680 g l−1. The volumetric uptake increases steadily at higher pressure and at 50 bar is exceptionally high; 600 to 940 g l−1, with excess uptake in the range 490–780 g l−1. Such high volumetric CO2 storage capacity is, to the best of our knowledge, the highest reported so far for any porous material. The volumetric uptake is much higher than that of high surface area metal organic framework materials, MOF210 and NU-100 (Fig. 5, Table 5 and Fig. S10†), which have uptake of, respectively; 90 and 165 g l−1 (20 bar), 190 and 270 g l−1 (30 bar), 270 and 350 g l−1 (40 bar), and 311 for MOF-210 at 50 bar. This means that the volumetric CO2 uptake of the mesoporous carbons is consistently 2 to 3 times higher than that of the benchmark MOFs. It is noteworthy that the mesoporous carbons still outperform MOFs even when crystal density (rather than packing density) is used to compute the volumetric CO2 uptake of MOF-210 and NU-100 (ESI Fig. S11†). The use of crystal density to estimate volumetric uptake is known to generate overestimated values and is not realistic for scenarios where the MOFs are packed into constrained space such as a cylinder.46,57–61
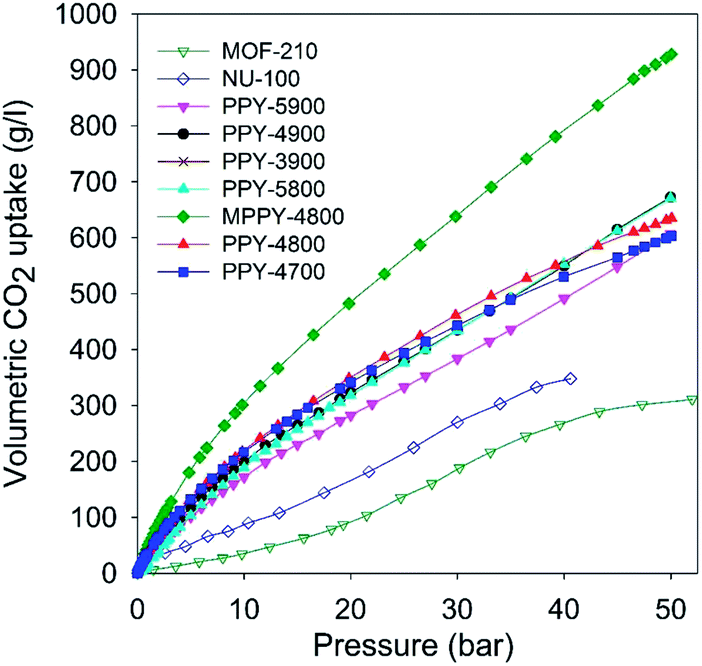 | ||
| Fig. 5 Total volumetric CO2 uptake at 25 °C of polypyrrole-derived activated (PPY-xT) carbons, compactivated (MPPY-4800) carbon and MOFs (MOF-210 and NU-100). | ||
| Sample | CO2 uptakea (g l−1) | ||||
|---|---|---|---|---|---|
| 1 bar | 20 bar | 30 bar | 40 bar | 50 bar | |
| a CO2 uptake at 25 °C and various pressures (i.e., 1, 20, 30, 40 and 50 bar). The values in the parenthesis are excess CO2 uptake. b Packing density values (g cm−3) used are: 0.15 (NU-100) and 0.12 (MOF-210).56 MOF data adapted from ref. 50 and 51. | |||||
| PPY-4700 | 34 | 341 (308) | 443 (389) | 530 (452) | 603 (492) |
| PPY-4800 | 35 | 352 (317) | 465 (408) | 556 (479) | 638 (524) |
| PPY-5800 | 17 | 318 (284) | 433 (379) | 552 (473) | 671 (558) |
| PPY-3900 | 33 | 338 (303) | 445 (390) | 545 (465) | 643 (530) |
| PPY-4900 | 27 | 322 (289) | 435 (381) | 565 (469) | 672 (559) |
| PPY-5900 | 23 | 282 (247) | 383 (327) | 491 (408) | 606 (490) |
| MPPY-4800 | 48 | 482 (438) | 638 (566) | 783 (678) | 928 (780) |
| MOF-210b | 4 | 93 (77) | 188 (163) | 266 (226) | 311 (254) |
| NU-100b | 18 | 163 (145) | 270 (240) | 348 (309) | |
The volumetric uptake may also be expressed per volume occupied by the adsorbent. This is especially useful for high pressure pre combustion CCS as it enables easy assessment of an adsorbent's uptake capacity in comparison with an empty cylinder of equal volume. The volumetric capacity of the mesoporous carbons (expressed as cm3 cm−3) as a function of pressure is shown in Fig. 6. The uptake is in the order pressurized CO2 < MOFs < activated (PPY-xT) carbons < compactivated (MPPY-4800) carbon. At 20 bar and 25 °C, pressurised CO2 equates to 20 cm3 cm−3 while MOFs store 47 and 83 cm3 cm−3 for MOF-210 and NU-100, respectively. On the other hand, PPY-xT activated carbons store 144 to 178 cm3 cm−3, and an impressive 246 for the compactivated carbon (MPPY-4800). At 30 bar the volumetric capacity of the mesoporous carbons rises to between 195 and 325 cm3 cm−3, compared to 95–137 cm3 cm−3 (MOFs) and 33 cm3 cm−3 for pressurized CO2. Thus at 30 bar, the present mesoporous carbons can enable CO2 storage at twice the capacity of MOFs and 10 times that of an empty cylinder. The volumetric uptake of the mesoporous carbons reaches, at 50 bar, exceptionally high capacity of between 310 and 470 cm3 cm−3, compared to 158 cm3 cm−3 for MOF-210 and 67 cm3 cm−3 for pressurized CO2.
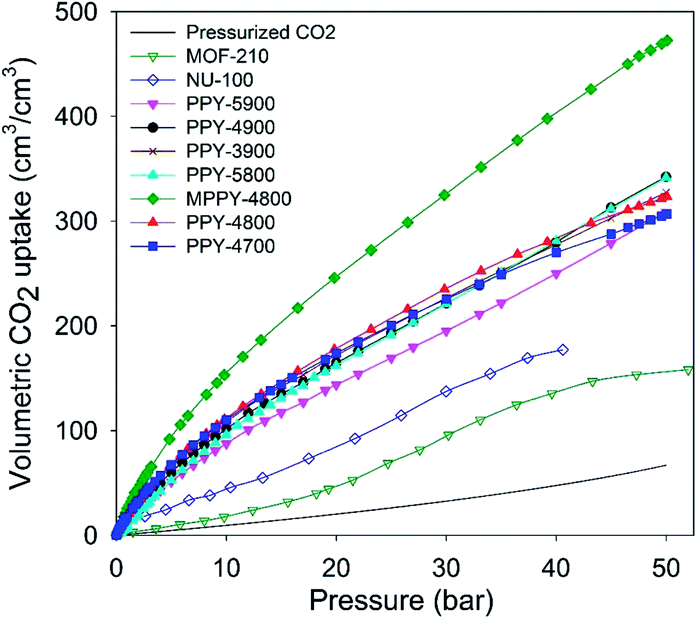 | ||
| Fig. 6 Total volumetric CO2 uptake at 25 °C of polypyrrole-derived activated (PPY-xT) carbons, compactivated (MPPY-4800) carbon and MOFs (MOF-210 and NU-100). | ||
The exceptional uptake of the present mesoporous carbons suggests that they may be suitable materials for the removal of CO2 from syngas during which the H2 content is enriched. Syngas, which is a fuel for energy generation from power stations typically contains 71–75% H2, 15–20% CO2, and ca. 5% of other gases (CH4, CO, H2O). We therefore compared the high pressure CO2 uptake with the H2 adsorption capacity for one of the best performing mesoporous carbons (MPPY-4800) so as to determine the selectivity for CO2. The selectivity for CO2 was also estimated for CO2/H2 mixtures with composition similar to that of syngas. We compared the performance of MPPY-4800 with that of a microporous carbon (CA-4700) of similar surface area. The excess CO2 and H2 uptake of mesoporous MPPY-4800 and microporous CA-4700 are shown in Fig. 7. For both samples, the CO2 uptake is much higher than the H2 uptake. There are, however, important differences in the CO2/H2 uptake ratio for the two samples that highlight the role played by mesoporosity in determining the relative adsorption of the two gases and thus the preference for CO2. Fig. 8A shows plots of the equilibrium CO2/H2 adsorption ratio for the two samples as a function of uptake pressure. At low pressure (1 bar), the microporous carbon (CA-4700) has a higher CO2/H2 ratio (10.2) compared to the mesoporous MPPY-4800 (7.8) due to a greater CO2 uptake arising from the presence of micropores in the former. However at higher pressures (>5 bar) that are more relevant to syngas purification, the CO2/H2 ratio for the mesoporous carbon increases significantly, while that of the microporous carbon drops drastically (Fig. 8A). Comparison of the relative excess uptake of CO2 and H2 at 50 bar indicates that CO2 uptake of MPPY-4800 (45.4 mmol g−1) is far higher than that of H2 (4.13 mmol g−1). Thus the equilibrium CO2/H2 adsorption ratio (at 50 bar) for MPPY-4800 is high at 11. For the microporous sample (CA-4700) the CO2 uptake at 50 bar is 20.5 mmol g−1 while for H2 it is 5.4 mmol g−1, giving an equilibrium CO2/H2 adsorption ratio of 3.8. The mesoporous carbon is therefore better suited for H2 purification under syngas flow conditions.
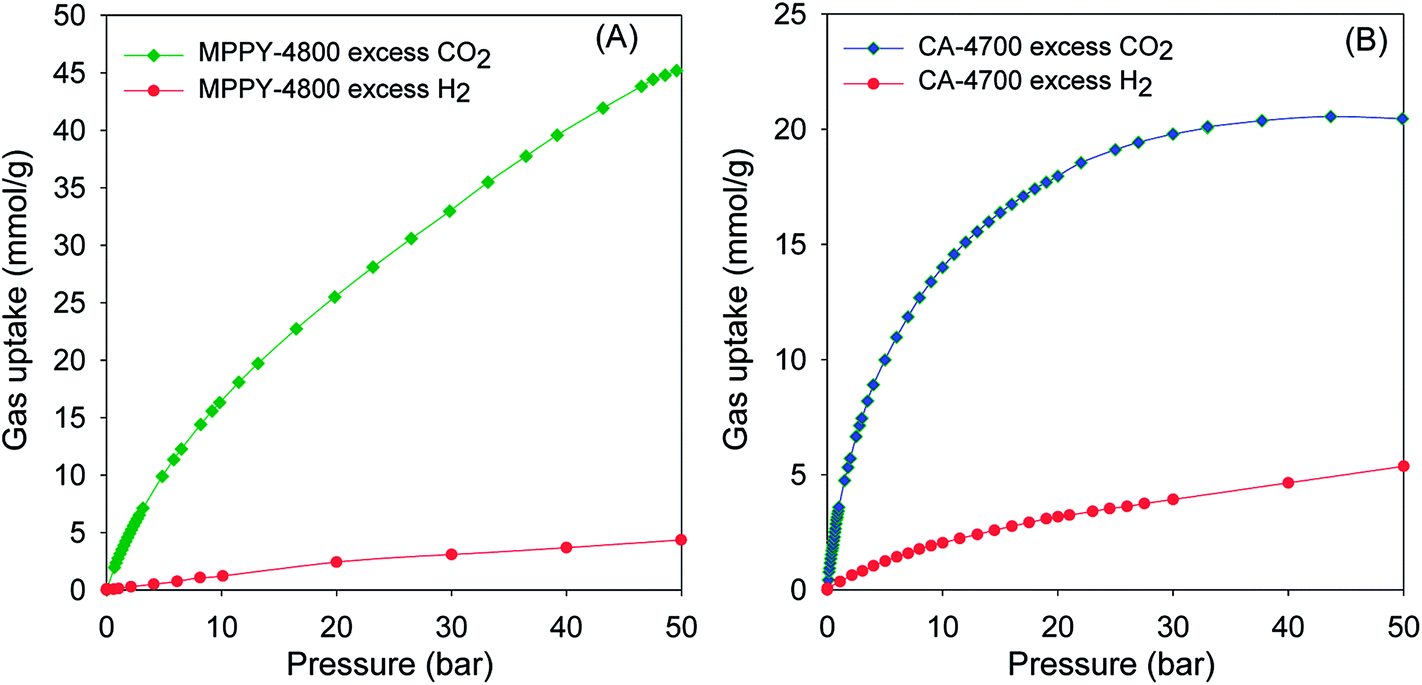 | ||
| Fig. 7 Comparison of excess CO2 and H2 uptake isotherms at room temperature for (A) mesoporous carbon (MPPY-4800) and (B) microporous carbon (CA-4700). | ||
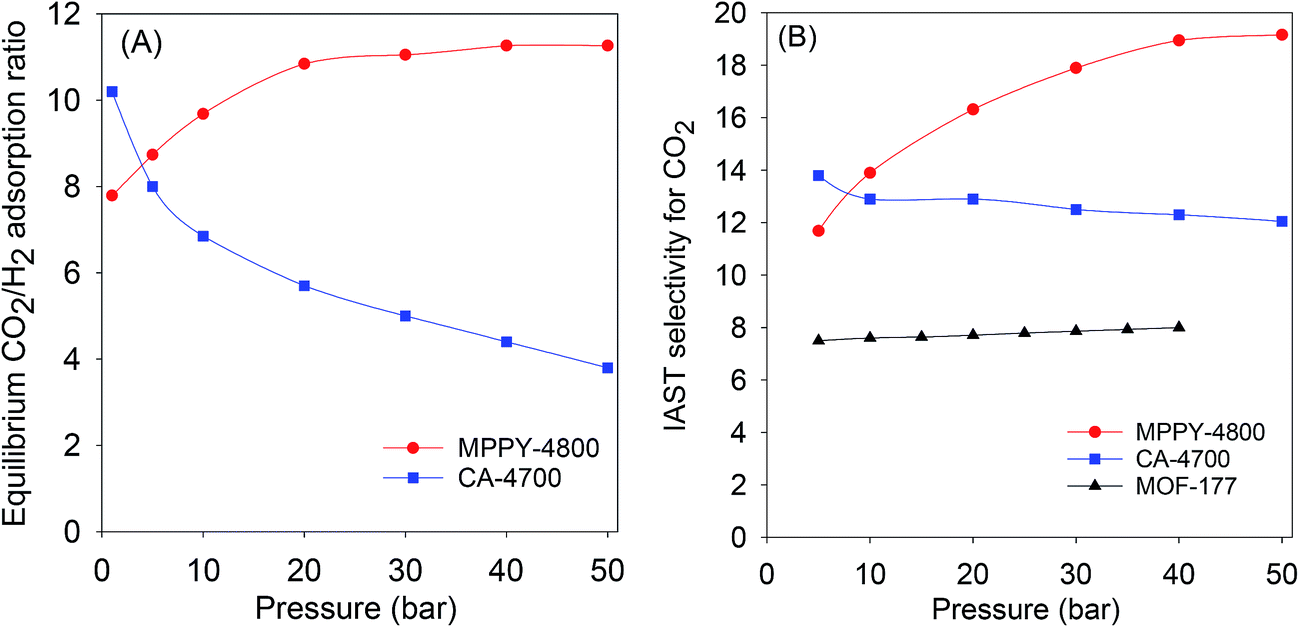 | ||
Fig. 8 (A) Equilibrium CO2/H2 adsorption ratio as a function of uptake pressure at 25 °C, and (B) IAST CO2/H2 selectivity for a 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 H2/CO2 mixture at 25 °C for mesoporous (MPPY-4800) carbon, microporous (CA-4700) carbon and a metal organic framework (MOF-177). Data for MOF-177 was obtained from ref. 61. :20 H2/CO2 mixture at 25 °C for mesoporous (MPPY-4800) carbon, microporous (CA-4700) carbon and a metal organic framework (MOF-177). Data for MOF-177 was obtained from ref. 61. | ||
The selectivity for CO2 adsorption may also be estimated from a simulated pre combustion syngas stream with a 80:20 H2/CO2 mixture. The simulation is based on the fact that syngas streams typically contain 71–75% H2 and 15–20% CO2 and thus comparison of the CO2 uptake from a 80:20 H2/CO2 mixture gives a realistic estimation of selectivity for CO2. The selectivity analysis was determined using the ideal adsorbed solution theory (IAST) model, which is commonly applied in calculating the relative uptake of adsorbents for any two gases in a binary gas mixture.61,62 According to the IAST model, the selectivity (S) for CO2 was derived from the following equation; S = n(CO2)p(H2)/n(H2)p(CO2), where n is uptake of CO2 and H2 in mmol g−1 at partial pressure of 0.2 and 0.8 bar, respectively, p(H2) is 0.8 and p(CO2) is 0.2. Fig. 8B shows plots of the IAST selectivity for CO2 as a function of uptake pressure. At 5 bar, the microporous carbon (CA-4700) has slightly higher IAST CO2 selectivity (13.8) compared to the mesoporous MPPY-4800 (11.7). However at higher pressures (>5 bar) that are more relevant to syngas purification, the IAST CO2 selectivity of the mesoporous carbon increases significantly to ≥18 at 40 bar and above. In contrast that of the microporous carbon drops to 12.0–12.5 at 20 to 50 bar (Fig. 8A). The IAST CO2 selectivity, therefore, demonstrates a clear advantage for the mesoporous carbons with respect to CO2 removal from syngas. Furthermore, as shown in Fig. 8B, the CO2 selectivity of the mesoporous carbon is higher than that of a metal organic framework (MOF-177) that has similar surface area.61 The mesoporous carbons, due to their large mesopore volume and high surface area, appear to have very high affinity for CO2 storage at pre combustion CCS conditions. The carbons fulfill the adsorbent requirements for syngas (or H2) purification, namely, high CO2 adsorption capacity and selectivity for CO2 over H2.
Conclusions
In summary, we have described a new family of ultra-high surface area mesoporous carbons that possess extremely high mesopore volume with hardly any micropores present. The mesoporous carbons have surface area and pore volume in the range of 2800–4000 m2 g−1 and 2.5–3.6 cm3 g−1, respectively. The porosity of the carbons can be readily tailored by choice of the activation temperature and the amount of activator used. The mesoporosity of the carbons is found to be ideal for CO2 uptake under conditions that closely mimic pre combustion carbon capture and storage, i.e., 25 °C and pressure of 20 to 50 bar. The best performing carbons are found to be those that have a near total absence of micropores; microporosity appears to limit high pressure CO2 uptake. The gravimetric (mmol g−1) CO2 uptake capacities; up to 28 (20 bar), 37 (30 bar), 46 (40 bar) and 55 (50 bar), are among the highest ever reported for porous carbons. The high gravimetric CO2 uptake of the mesoporous carbons in combination with their packing density (0.25–0.4 g cm−3) results in colossal volumetric uptake (in g l−1) of up to 480 (20 bar), 640 (30 bar), 780 (40 bar) and 930 (50 bar). At 30 bar, the mesoporous carbons can hold more than 10 times the CO2 in a pressurized cylinder, and at 50 bar can store up to 470 cm3 cm−3. We have shown that the all-round pre combustion CO2 capture performance (gravimetric and volumetric uptake, and selectivity for CO2) of the mesoporous carbons is significantly higher than that of the best carbons to date, and outperforms that of benchmark MOF materials. Due to their mesoporosity, the carbons are highly suited, in terms of the CO2 adsorption capacity and selectivity over H2, as materials for hydrogen purification under syngas flow conditions.Acknowledgements
This research was funded by the University of Nottingham.References
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130 CAS.
- K. Z. House, A. C. Baclig, M. Ranjanc, E. A. van Nierop, J. Wilcox and H. J. Herzog, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20428 CrossRef CAS PubMed.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796 CrossRef CAS PubMed.
- H. J. Schellnhuber, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20277 CrossRef CAS PubMed.
- K. S. Lackner, S. Brennan, J. M. Matter, A.-H. A. Park, A. Wright and B. van der Zwaan, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13156 CrossRef CAS PubMed.
- T. C. Drage, C. E. Snape, L. A. Stevens, J. Wood, J. Wang, A. I. Cooper, R. Dawson, X. Guo, C. Satterley and R. Irons, J. Mater. Chem., 2012, 22, 2815 RSC.
- G. T. Rochelle, Science, 2009, 325, 1652 CrossRef CAS PubMed.
- S. Wang, S. Yan, X. Ma and J. Gong, Energy Environ. Sci., 2011, 4, 3805 CAS.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42 CAS.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438 CrossRef CAS.
- J. P. Marco-Lozar, M. Kunowsky, F. Suarez-Garcia, J. D. Carruthers and A. Linares-Solano, Energy Environ. Sci., 2012, 5, 9833 CAS.
- G.-P. Hao, W.-C. Li, D. Qian and A.-H. Lu, Adv. Mater., 2010, 22, 853 CrossRef CAS PubMed.
- J. Silvestre-Albero, A. Wahby, A. Sepulveda-Escribano, M. Martinez-Escandell, K. Kaneko and F. Rodriguez-Reinoso, Chem. Commun., 2011, 47, 6840 RSC.
- M. Sevilla and A. B. Fuertes, J. Colloid Interface Sci., 2012, 366, 147 CrossRef CAS PubMed.
- M. R. Hudson, Q. L. Queen, J. A. Mason, D. W. Fickel, R. F. Lobo and C. M. Brown, J. Am. Chem. Soc., 2012, 134, 1970 CrossRef CAS PubMed.
- J. Mere, M. Clausse and F. Meunier, Ind. Eng. Chem. Res., 2008, 47, 1209 Search PubMed.
- R. V. Siriwardane, M. S. Shen, E. P. Fisher and J. Losch, Energy Fuels, 2005, 19, 1153 CrossRef CAS.
- Z. J. Zhang, Z. Z. Yao, S. C. Xiang and B. L. Chen, Energy Environ. Sci., 2014, 7, 2868 CAS.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed.
- B. Adeniran and R. Mokaya, Nano Energy, 2015, 16, 173 CrossRef CAS.
- N. Balahmar, A. C. Mitchell and R. Mokaya, Adv. Energy Mater., 2015, 5, 1500867 CrossRef.
- W. Sangchoom and R. Mokaya, ACS Sustainable Chem. Eng., 2015, 3, 1658 CrossRef CAS.
- A. Almasoudi and R. Mokaya, J. Mater. Chem. A, 2014, 2, 10960 CAS.
- C. Robertson and R. Mokaya, Microporous Mesoporous Mater., 2013, 179, 151 CrossRef CAS.
- E. Haffner-Staton, N. Balahmar and R. Mokaya, J. Mater. Chem. A, 2016, 4, 13324 CAS.
- B. Adeniran and R. Mokaya, J. Mater. Chem. A, 2015, 3, 5148 CAS.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765 CAS.
- G. Srinivas, J. Burress and T. Yildirim, Energy Environ. Sci., 2012, 5, 6453 CAS.
- N. P. Wickramaratne and M. Jaroniec, J. Mater. Chem. A, 2013, 1, 112 CAS.
- N. P. Wickramaratne and M. Jaroniec, ACS Appl. Mater. Interfaces, 2013, 5, 1849 CAS.
- Z. Zhang, J. Zhou, W. Xing, Q. Xue, Z. Yan, S. Zhuo and S. Z. Qiao, Phys. Chem. Chem. Phys., 2013, 15, 2523 RSC.
- B. Adeniran, E. Masika and R. Mokaya, J. Mater. Chem. A, 2014, 2, 14696 CAS.
- X. Fan, L. Zhang, G. Zhang, Z. Shu and J. Shi, Carbon, 2013, 61, 423 CrossRef CAS.
- H. Wei, S. Deng, B. Hu, Z. Chen, B. Wang, J. Huang and G. Yu, ChemSusChem, 2012, 5, 2354 CrossRef CAS PubMed.
- B. Adeniran and R. Mokaya, Chem. Mater., 2016, 28, 994 CrossRef CAS.
- H. M. Coromina, D. A. Walsh and R. Mokaya, J. Mater. Chem. A, 2016, 4, 280 CAS.
- M. Sevilla, W. Sangchoom, N. Balahmar, A. B. Fuertes and R. Mokaya, ACS Sustainable Chem. Eng., 2016, 4, 4710 CrossRef CAS.
- J. He, J. W. F. To, P. C. Psarras, H. Yan, T. Atkinson, R. T. Holmes, D. Nordlund, Z. Bao and J. Wilcox, Adv. Energy Mater., 2016, 6, 1502491 CrossRef.
- A. S. Jalilov, Y. Li, J. Tian and J. M. Tour, Adv. Energy Mater., 2017, 7, 1600693 CrossRef.
- J. R. Hufton, S. Mayorga and S. Sircar, AIChE J., 1999, 45, 248–256 CrossRef CAS.
- C. Descamps, C. Bouallou and M. Kanniche, Energy, 2008, 33, 874 CrossRef CAS.
- A. Agarwal, L. T. Biegler and S. E. Zitney, Ind. Eng. Chem. Res., 2010, 49, 5066 CrossRef CAS.
- S.-I. Yang, D.-Y. Choi, S.-C. Jang, S.-H. Kim and D.-K. Choi, Adsorption, 2008, 14, 583 CrossRef CAS.
- Z. R. Herm, J. A. Swisher, B. Smit, R. Krishna and J. R. Long, J. Am. Chem. Soc., 2011, 133, 5664 CrossRef CAS PubMed.
- N. Balahmar, A. M. Lowbridge and R. Mokaya, J. Mater. Chem. A, 2016, 4, 14254 CAS.
- F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by powders and porous solids: principles, methodology and applications, Academic Press, San Diego, 1999 Search PubMed.
- M. Sevilla, R. Mokaya and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 2930 CAS.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250 CAS.
- J.-S. M. Lee, M. E. Briggs, T. Hasell and A. I. Cooper, Adv. Mater., 2016, 28, 9804 CrossRef CAS PubMed.
- O. K. Farha, A. O. Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944 CrossRef CAS PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed.
- M. E. Casco, M. Martínez-Escandell, J. Silvestre-Albero and F. Rodríguez-Reinoso, Carbon, 2014, 67, 230 CrossRef CAS.
- G. Srinivas, V. Krungleviciute, Z.-X. Guo and T. Yildirim, Energy Environ. Sci., 2014, 7, 335 CAS.
- B. Ashourirad, A. K. Sekizkardes, S. Altarawneh and H. M. El-Kaderi, Chem. Mater., 2015, 27, 1349 CrossRef CAS.
- Y. Li, T. Ben, B. Zhang, Y. Fu and S. Qiu, Sci. Rep., 2013, 3, 2420 CrossRef PubMed.
- J. Purewal, D. Liu, A. Sudik, M. Veenstra, J. Yang, S. Maurer, U. Muller and D. J. Siegel, J. Phys. Chem. C, 2012, 116, 20199 CAS.
- J. Purewal, D. Liu, J. Yang, A. Sudik, D. Siegel, S. Maurer and U. Mueller, Int. J. Hydrogen Energy, 2012, 37, 2723 CrossRef CAS.
- A. Dailly and E. Poirier, Energy Environ. Sci., 2011, 4, 3527 CAS.
- R. Zacharia, D. Cossement, L. Lafi and R. Chahine, J. Mater. Chem., 2010, 20, 2145 RSC.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 11887 CrossRef CAS PubMed.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Two (2) tables and eleven (11) figures. See DOI: 10.1039/c7se00300e |
| This journal is © The Royal Society of Chemistry 2017 |