
Photophysical properties of wavelength-tunable methylammonium lead halide perovskite nanocrystals†
Daniel J.
Freppon
,
Long
Men
,
Sadie J.
Burkhow
,
Jacob W.
Petrich
,
Javier
Vela
* and
Emily A.
Smith
*
The Ames Laboratory, U.S. Department of Energy, and Department of Chemistry, Iowa State University, Ames, Iowa 50011-3111, USA. E-mail: esmith1@iastate.edu; vela@iastate.edu
First published on 25th November 2016
Abstract
We present the time-correlated luminescence of isolated nanocrystals of five methylammonium lead mixed-halide perovskite compositions (CH3NH3PbBr3−xIx) that were synthesized with varying iodide and bromide anion loading. All analyzed nanocrystals had a spherical morphology with diameters in the range of 2 to 32 nm. The luminescence maxima of CH3NH3PbBr3−xIx nanocrystals were tuned to wavelengths ranging between 498 and 740 nm by varying the halide loading. Both CH3NH3PbI3 and CH3NH3PbBr3 nanocrystals exhibited no luminescence intermittency for more than 90% of the 250 s analysis time, as defined by a luminescence intensity three standard deviations above the background. The mixed halide CH3NH3PbBr0.75I0.25, CH3NH3PbBr0.50I0.50, and CH3NH3PbBr0.25I0.75 nanocrystals exhibited luminescence intermittency in 18%, 4% and 26% of the nanocrystals, respectively. Irrespective of luminescence intermittency, luminescence intensities were classified for each nanocrystal as: (a) constant, (b) multimodal, (c) photobrightening, and (d) photobleaching. Based on their photophysics, the CH3NH3PbBr3−xIx nanocrystals can be expected to be useful in a wide-range of applications where low and non-intermittent luminescence is desirable, for example as imaging probes and in films for energy conversion devices.
Introduction
Luminescent semiconductors are used in a variety of applications from microscopy probes1–4 to thin film energy capture and conversion devices.5–19 There is increasing interest in luminescent methylammonium lead halide perovskites (CH3NH3PbX3, X = halide) due to their low cost, broad absorption as well as their charge transport diffusion lengths that range from ∼100 nm to greater than 175 μm.20–24 In energy capture and conversion films, large exciton diffusion lengths remove the need for a transport layer in perovskite-based solar cells, thus simplifying the device architecture. Single-crystalline perovskites, as opposed to polycrystalline films, could further increase the efficiency of energy capture and conversion devices. Further, CH3NH3PbX3 nanocrystals are being evaluated for use in light emitting diodes,25 and as lasing materials.26In addition to the energy capture and conversion applications outlined above, there are other uses for non-intermittent, photostable, as well as tunable nanocrystals. For example, as imaging probes the nanocrystal absorption and emission wavelengths should match wavelength criteria for the optical setup, so tunability can be advantageous. Tuning the luminescence wavelength of CsPbX327–29 as well as CH3NH3PbX330–33 perovskite nanocrystals across the visible spectrum has been reported.
Despite their promise, organometal halide perovskites have been reported to suffer from low photostability.34,35 Superoxide (O2−), generated from the photoreaction of perovskites and oxygen, can accelerate their degradation to PbI2, I2 and CH3NH2.36 Perovskite films degrade upon exposure to 457 nm light with a power density of 15 mW cm−2 or higher.37,38 Laser illumination with 500 W cm−2 was shown to cause photodamage to CH3NH3PbI3 films within several seconds, leading to a decrease in luminescence.39 However, little photobleaching (i.e., decreasing luminescence) or photobrightening (i.e., increasing luminescence) was observed for CH3NH3PbI3 nanocrystals with wire, rod, and dot morphologies using mercury lamp illumination.40 Matsumoto et al. and Manshor et al. report changes in surface morphology and reduced device efficiency for CH3NH3PbI3-based films when exposed to both light and humidity, but claim the devices remain stable when exposed to light only.41,42
When illuminated with visible light, many types of semiconductor materials show luminescence intermittency (i.e., blinking), when the level of radiative decay drops below the background or other threshold intensity.40,43–56 Luminescence intermittency has also been studied for CH3NH3PbX3 materials40,50,51,54 although the mechanism is still under debate.34,35,38,39 One reported mechanism of luminescence intermittency in a dense CH3NH3PbBr3 nanocrystal film is non-radiative Auger-like recombination of electrons and holes due to the accumulation of charge.51 In closely packed nanocrystals, charge accumulates at the nanocrystal surface leading to intermittency; whereas, no intermittency was measured for well-dispersed nanocrystals. On the other hand, Tachikawa et al. reported intermittency in single CH3NH3PbBr3 nanocrystals and postulated it is the result of charge trapping by surface lead sites reducing the probability of a recombination event.54 They estimate 1 to 4 trap sites per 10 to 50 nm nanocrystal. The halide composition affected the trap density; an order of magnitude lower trap site density was measured in CH3NH3PbI3 films grown in the presence of chloride compared to films in the absence of chloride.57 In a CH3NH3PbI3 film, a deep trap state 0.16 eV above the valence band was measured, and calculations showed this trap might arise from iodine defects.58
Zheng et al. reported differences in bulk and nanosized crystals of CH3NH3PbBr3.59 In bulk CH3NH3PbBr3, filling of trap sites was dependent on the photon density, and luminescence lifetimes increased with higher photon densities. Compared to bulk, the luminescence lifetime of CH3NH3PbBr3 nanocrystals was not as dependent on photon densities; 30% of the excitation photons underwent trap-free recombination; trap states were more likely to be present on the surface than in the volume of the crystal; and trap lifetimes could be short. Tian et al. showed that light illumination removes traps leading to photobrightening in CH3NH3PbI3 films, and that the size of CH3NH3PbI3 crystallites (nanometers to microns) within the film had an effect on the rate of photobrightening.60
Here, we report data from three luminescence techniques used to measure the photophysics of CH3NH3PbBr3−xIx nanocrystals to gain a fundamental understanding of how composition affects their luminescence and, thus, their continued and potential usability in energy conversion and imaging applications. Ensemble luminescence measurements show the nanocrystal emission wavelengths are tunable across the visible wavelength region by varying the ratio of iodide and bromide salts in the synthetic loading. Due to heterogeneous crystal formation, individual nanocrystal photophysical properties vary with the type and number of defect and trap sites. Nanocrystal microscopy measurements of the luminescence intensity over time reveal the temporal dynamics (e.g., intermittency, flickering), as well as heterogeneity in the photophysical properties. Finally, nanocrystal microspectroscopy enables the full luminescence spectrum of isolated nanocrystals to be collected in order to monitor the stability of the emission peak over time.
Experimental
Materials
Lead(II) iodide (PbI2, 99%), lead(II) bromide (PbBr2, ≥98%), methylamine (CH3NH2, 33 wt% in ethanol), N,N-dimethylformamide (DMF, 99.8%, anhydrous) and n-octylamine (CH3(CH2)7NH2, 99%) were purchased from Sigma-Aldrich; hydroiodic acid (ACS, 55–58%) and hydrobromic acid (ACS, 47.0–49.0%) from Alfa-Aesar; acetonitrile (99.9%) and toluene (99.9%) from Fisher Scientific. All chemicals were used as received.Synthesis of methylammonium lead(II) mixed-halide nanocrystals
Nanocrystal ensemble characterization
XRD was measured using Cu Kα radiation on a Rigaku Ultima diffractometer. Solution extinction (absorption plus scattering) spectra were measured with a photodiode array Agilent 8453 UV-Vis spectrophotometer. Steady-state photoluminescence (PL) spectra were measured using a Horiba-Jobin Yvon Nanolog scanning spectrofluorometer.Nanocrystal luminescence microscopy
CH3NH3PbBr3−xIx nanocrystals were diluted to a tenth (/10) in toluene (∼0.1 mM, 50 μL) and sonicated for 90 minutes before drop casting onto a glass microscope coverslip (Carlson Scientific, Peotone, IL, USA). Nanocrystal luminescence microscopy was performed using an upright microscope (Nikon Eclipse 80i, Melville, NY, USA). A mercury lamp was used for excitation (XCite 120 PC, EXFO Photonic Solutions Inc., Quebec City, Canada). Excitation and emission filters were from Omega Optical (Brattleboro, VT, USA), unless noted otherwise: 500 ± 5 nm excitation and 535 ± 7.5 nm emission filters were used for the CH3NH3PbBr3, CH3NH3Pb(Br0.75I0.25)3, and CH3NH3Pb(Br0.50I0.50)3 samples; 500 ± 5 nm excitation and 615 ± 15 nm emission filters were used for the CH3NH3Pb(Br0.25I0.75)3 sample; 510 ± 5 nm excitation and 730 ± 40 nm emission filters (Semrock, Inc., Lake Forest, IL, USA) were used for the CH3NH3PbI3 sample. A 100× Plan Apo, 1.49 numerical aperture oil-immersion objective was used and PL images were collected in the epi-direction using a charged-coupled device (CCD) camera (Photometrics Evolve, Tucson, AZ, USA) with a 250 ms exposure time and zero gain. Each PL movie consists of 1000 images collected sequentially for a total analysis time of 250 s. The signal-to-noise ratio was at least 2 at the start of each movie, and a diffraction-limited point spread function was measured for each analyzed nanocrystal.Threshold calculation
ImageJ was used to analyze the PL movies. For each nanocrystal analyzed, the nanocrystal's luminescence intensity and a background value were quantified in each of the 1000 frames using the Z-axis profile function in ImageJ. The most intense pixel for the spheroid nanocrystals was used to plot the nanocrystal luminescence intensity over the entire 1000 frames (i.e., over time). The background in each frame was measured approximately 8 pixels away from the center of each nanocrystal. The average background value (μbg) and background standard deviation (σbg) were calculated across all 1000 frames, and was used to calculate a threshold: threshold = μbg + 3σbg. Nanocrystals were considered intermittent in luminescence if their intensity fell below the threshold for more than 25 s over the entire movie (i.e., 10% of the collection time).Nanocrystal microspectroscopy
The suspended CH3NH3PbI3 nanocrystals prepared from precursor were diluted to a tenth (/10) in toluene (∼0.1 mM, 50 μL) and sonicated for 60 min before drop casting onto a glass microscope coverslip (Carlson Scientific, Peotone, IL). A lab-built optical microscope based on a DM IRBE platform (Leica, Wetzlar, Germany) with 532 nm laser excitation (Sapphire SF 532 nm, Coherent, Santa Clara, CA, USA) was used to collect emission spectra as a function of illumination time. A 100× HCX PL APO, 1.49 numerical aperture oil-immersion objective (Leica) was used to achieve a laser spot with a diameter of 440 nm. The excitation power density at the sample was 1 × 105 W cm−2. Photoluminescence was collected from the epi-direction and focused onto a HoloSpec f/1.8i spectrograph (Kaiser Optical Systems, Ann Arbor, MI, USA) equipped with a broad range grating (HFG-650, Kaiser Optical Systems) and then directed to a CCD (Newton 940, Andor Technology, Belfast, UK). A series of 2500 spectra were collected for 0.1 s each. Spectra were then analyzed using IGOR Pro 6.34 batch fit (Wavemetrics, Lake Oswego, OR) and 3D images of the fits shown in Fig. 5 were plotted in Matlab 2016a (Mathworks, Natick, MA).Results and discussion
Five CH3NH3PbBr3−xIx nanocrystal compositions were prepared with x = 0, 0.75, 1.50, 2.25 and 3. Throughout, the presented formulas refer to the synthetic loading not a measured nanocrystal composition.61 To control particle size, we used octylammonium halides (CH3(CH2)7NH3X) as surfactants, which have been shown to passivate the surface and terminate/truncate further crystal growth.40 All synthetic loadings produced primarily spherical nanocrystals, as shown by transmission electron microscopy (ESI,† Fig. S1). The perovskites are nanocrystalline as evident by the peak broadness of their powder XRD patterns (ESI,† Fig. S2). The XRD pattern shows a major peak at 30.2 degrees for CH3NH3PbBr3 nanocrystals, which is typically assigned to the cubic crystal structure, and a peak at 28.7 for CH3NH3PbI3 nanocrystals, which is typically assigned to the tetragonal crystal structure. According to the experimental patterns of CH3NH3PbBr3−xIx (0.75, 1.5, 2.25), all three nanocrystalline samples are cubic. The reported experimental XRD patterns of CH3NH3PbI3 and CH3NH3PbBr3 indicate the nanocrystals grow mainly along the [110] direction.40 This preferred orientation is the main reason behind the differences observed between the experimental and standard XRD patterns (ESI,† Fig. S2), which were collected from bulk single crystals.The CH3NH3PbBr3−xIx nanocrystals absorbed a broad range of visible wavelengths (Fig. 1). The luminescence spectra show a range in their wavelength of maximum emission with halide loading (Fig. 1). As the iodide content increased from 0 (CH3NH3PbBr3) to 100% (CH3NH3PbI3), the luminescence maximum shifted from 498 to 740 nm. The shift in luminescence maximum is not linear with halide loading (Fig. 2), which is consistent with previous reports.30,62
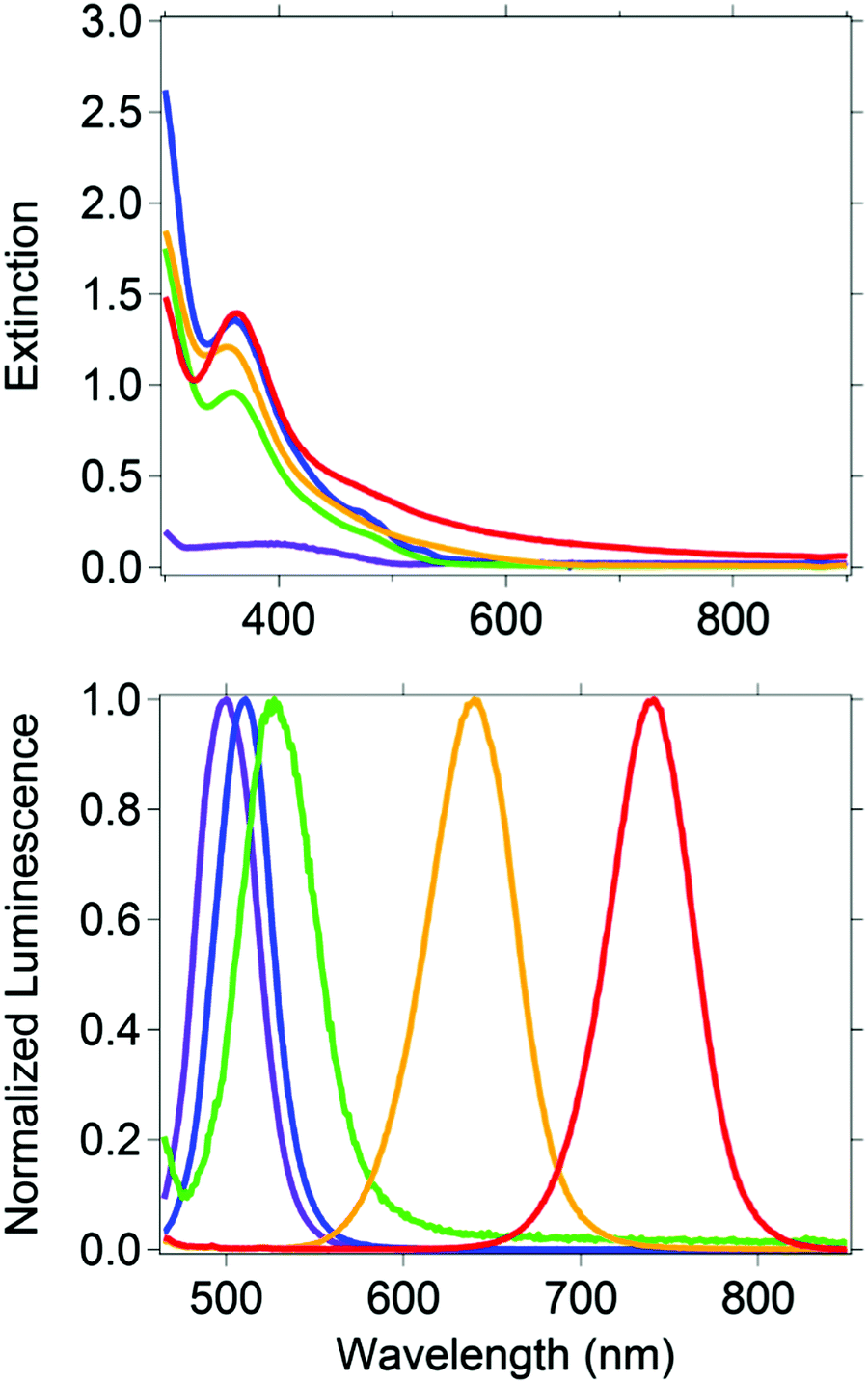 | ||
| Fig. 1 (top) Extinction (absorption plus scattering) and (bottom) normalized emission (λexc = 375 nm) spectra of CH3NH3PbBr3 (purple), CH3NH3Pb(Br0.75I0.25)3 (blue), CH3NH3Pb(Br0.50I0.50)3 (green), CH3NH3Pb(Br0.25I0.75)3 (orange), CH3NH3PbI3 (red). Extinction and emission spectra were collected in toluene as the solvent. | ||
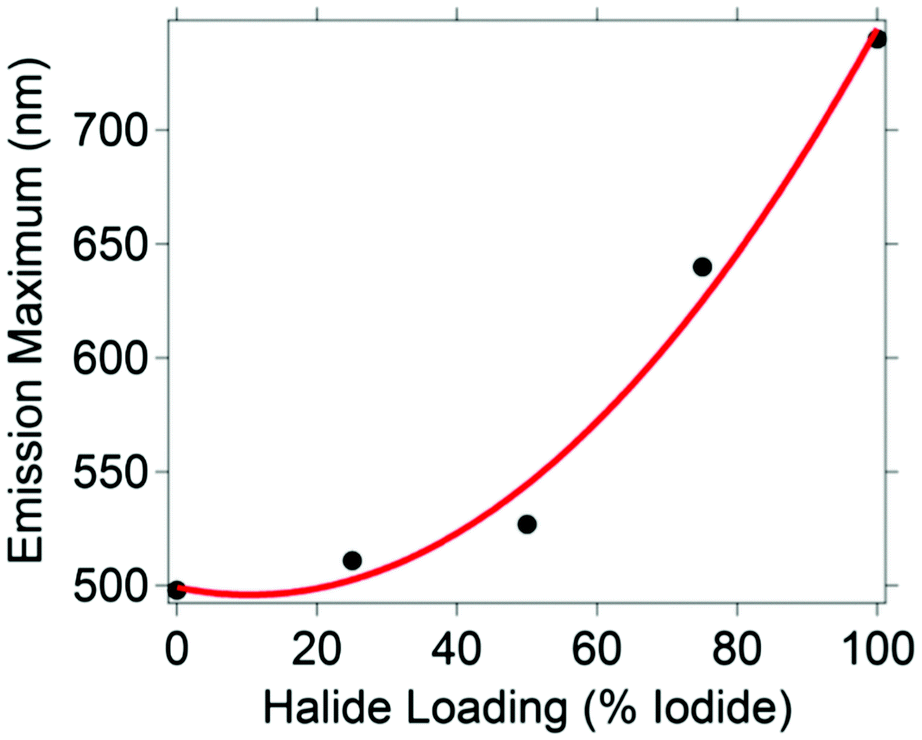 | ||
| Fig. 2 Emission maximum wavelength versus iodide loading for the (0%) CH3NH3PbBr3, (25%) CH3NH3Pb(Br0.75I0.25)3, (50%) CH3NH3Pb(Br0.50I0.50)3, (75%) CH3NH3Pb(Br0.25I0.75)3, and (100%) CH3NH3PbI3 nanocrystals. The third-order polynomial fit line is shown in red. The equation for the best fit of the maximum emission wavelength (λmax) versus percent iodide loading (x) is: λmax = −9 × 10−5x3 + 0.04x2 − x + 501. The R2 value for the fit is 0.99. | ||
The luminescence intensity as a function of time was analyzed for up to 50 nanocrystals from each halide loading. The CH3NH3(Br0.25I0.75)3 nanocrystals were more dilute relative to the other compositions over replicate syntheses and the same sample preparation method only allowed 39 nanocrystals to be measured. An average luminescence intensity across the 250 s analysis time was calculated for each nanocrystal; histograms of the background subtracted values are shown in ESI,† Fig. S3 for each halide loading. The absolute values on the X-axis cannot be compared across different compositions as the optical throughput of the instrument varies with wavelength (e.g., filter bandwidth, etc.). The shapes of the distributions, however, can be compared. The mixed halide compositions of CH3NH3Pb(Br0.75I0.25)3, CH3NH3Pb(Br0.50I0.50)3, and CH3NH3Pb(Br0.25I0.75)3 had more outliers with intensities 13 to 230× higher than the intensity with the highest frequency compared to the single halide CH3NH3PbBr3 and CH3NH3PbI3 nanocrystals.
The single halide nanocrystals exhibit no luminescence intermittency as defined by a luminescence intensity that is above the calculated threshold for distinguishing luminescence from the background more than 90% of the analysis time (Table 1). The percentage of CH3NH3Pb(Br0.75I0.25)3, CH3NH3Pb(Br0.50I0.50)3, and CH3NH3Pb(Br0.25I0.75)3 nanocrystals that exhibit luminescence intermittency is 18, 4, and 26%, respectively. We hypothesize that the fraction of mixed halide nanocrystals that exhibit intermittency have crystal defects or trap states. When electrons become trapped, radiative recombination of excitons cannot occur, providing periods of no measured luminescence. Further, this leads to the assumption that functional trap states are absent in the single halide nanocrystals. The single halide perovskites, CH3NH3PbI3 and CH3NH3PbBr3, have quantum yields of 1.5% and 44% respectively. These quantum yields are higher than the mixed halide perovskite nanocrystals (0.02 to 1.3%), which is consistent with the hypothesis that increased crystal defects or trap states are responsible for blinking in the mixed halide perovskites.
| Sample | Number of nanocrystals analyzed | Percentage nanocrystals “ON”a | Percentage time above thresholdb |
|---|---|---|---|
| a The nanocrystals are considered to be in an “ON” state if the luminescence intensity is above the calculated threshold at least 90% of the analysis time. b Based on a total time of 250 s × number of nanocrystals analyzed. | |||
| CH3NH3PbBr3 | 50 | 100 | 100 |
| CH3NH3Pb(Br0.75I0.25)3 | 50 | 82 | 93 |
| CH3NH3Pb(Br0.50I0.50)3 | 50 | 96 | 99 |
| CH3NH3Pb(Br0.25I0.75)3 | 39 | 74 | 89 |
| CH3NH3PbI3 | 50 | 100 | 100 |
In addition to the percentage of nanocrystals that exhibit intermittency, it is also useful to consider the percentage of time the nanocrystals were above the threshold over the total analysis time (Table 1). The intensities of the CH3NH3PbBr3 and CH3NH3PbI3 nanocrystals are always above the threshold over the entire 208 minutes (i.e., 50 nanocrystals analyzed for 250 s each). For the intermediate halide loadings, the percentage of non-intermittent nanocrystals and the percentage of the analysis time the nanocrystals are above the threshold are correlated. The latter, however, is always higher than the former. For example, 74% of the CH3NH3Pb(Br0.25I0.75)3 nanocrystals are not intermittent and are above the threshold 89% of the analysis time.
The luminescence signal over time was categorized into four different luminescence behaviors irrespective of the threshold or intermittency (Table 2). The behaviors are: (a) constant intensity, (b) multimodal intensity, (c) photobrightening, and (d) photobleaching. In order to be classified as constant luminescence intensity, shown in Fig. 3a, the slope of the line connecting the intensity at 0 s and 250 s was between −0.2 and 0.2 s−1. The small fluctuations in intensity of a nanocrystal classified as constant luminescence are due to instrument noise; this produces an intensity histogram with a Gaussian distribution (Fig. 3a′). The CH3NH3Pb(Br0.75I0.25)3 and CH3NH3Pb(Br0.50I0.50)3 nanocrystals were the only compositions to exhibit type a constant intensities with populations of 28% for both compositions (Table 2).
| Sample | Number of nanocrystals analyzed | Type a: constant intensity (%) | Type b: multimodal behavior (%) | Type c: photo-brightening (%) | Type d: photo-bleaching (%) |
|---|---|---|---|---|---|
| CH3NH3PbBr3 | 50 | 0 | 70 | 4 | 26 |
| CH3NH3Pb(Br0.75I0.25)3 | 50 | 28 | 38 | 22 | 12 |
| CH3NH3Pb(Br0.50I0.50)3 | 50 | 28 | 18 | 8 | 46 |
| CH3NH3Pb(Br0.25I0.75)3 | 39 | 0 | 0 | 5 | 95 |
| CH3NH3PbI3 | 50 | 0 | 18 | 8 | 74 |
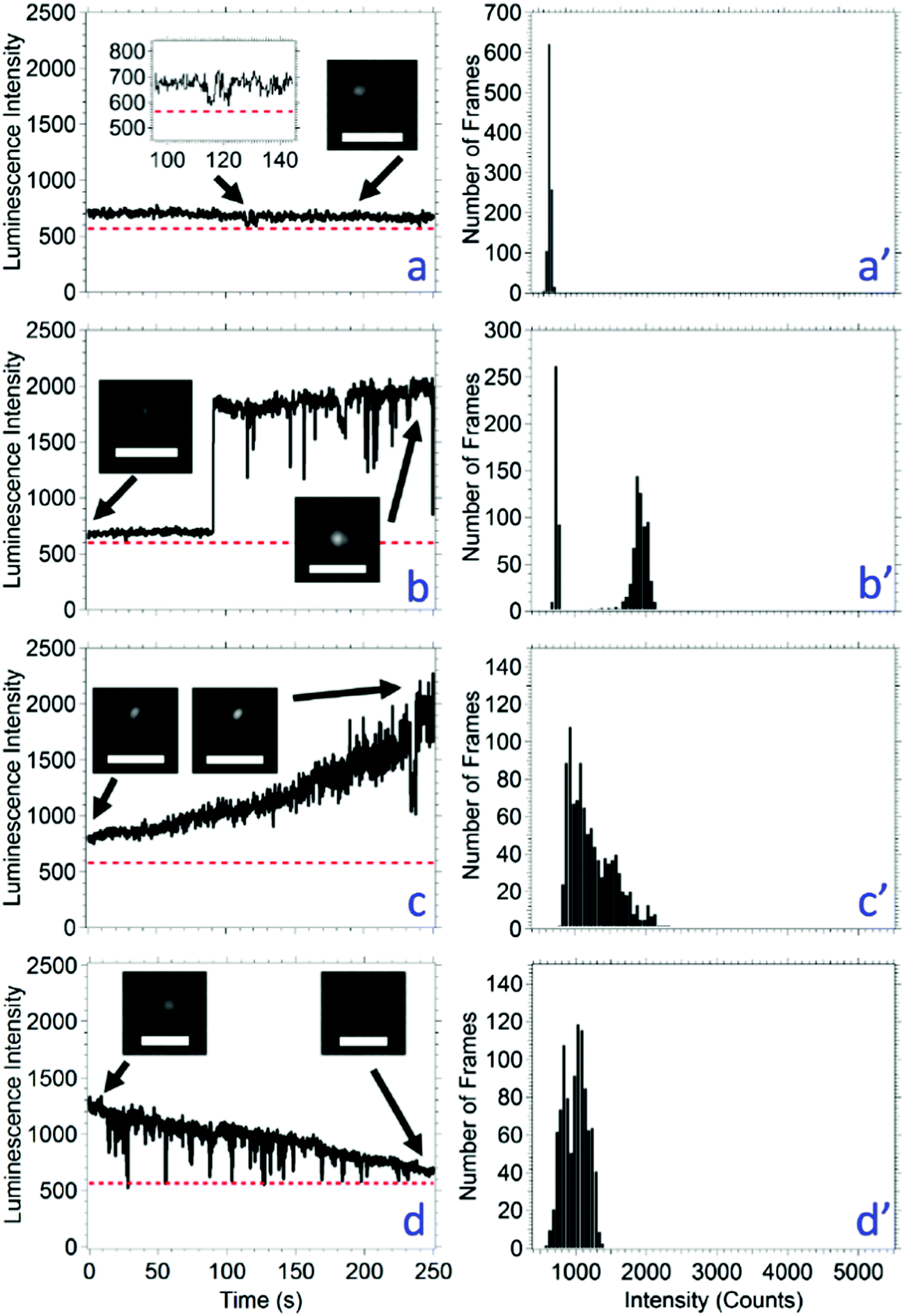 | ||
| Fig. 3 Representative luminescence intensity versus time graphs for CH3NH3Pb(Br0.75I0.25)3 nanocrystals. The dotted red line is the threshold for distinguishing nanocrystal luminescence from the background. The panels represent four of the photophysical properties measured for all the perovskite compositions. The time-correlated luminescence intensity is found on the left. Histograms of the corresponding intensities are found on the right. Type (a) is representative of nanocrystals that only show fluctuations in luminescence intensity due to instrument noise. Type (b) is representative of nanocrystals that exhibit a multimodal luminescence intensity profile, in this case being bimodal, a low intensity and a high intensity or vice versa. Type (c) is representative of nanocrystals with luminescence intensities that photobrighten over time (a slope greater than 0.2 s−1). Type (d) is representative of nanocrystals with luminescence intensities that photobleach over time (a slope smaller than −0.2 s−1). Figures on the right show the intensity histograms for all 250 s. Each histogram corresponds to the same letter (e.g. a′ corresponds to a). The insets show the image at selected time points, as noted by the arrow. In all cases, the images show a nanocrystal intensity that can be differentiated from the background. The scale bar in each image is 2 μm and the acquisition time is 250 ms. | ||
If the intensity of a nanocrystal varied between two or three relatively constant values for a duration of at least 10 s with the transition between intensities occurring within 500 ms (i.e., two times the acquisition time), this is classified as type b multimodal behavior, shown in Fig. 3b. The resulting intensity histogram is either a bimodal or trimodal distribution (Fig. 3b′). The number of nanocrystals that have a multimodal intensity profile decreased with the amount of bromide loading from CH3NH3PbBr3 to CH3NH3Pb(Br0.25I0.75). The CH3NH3PbI3 nanocrystals do not fit this trend with 18% of nanocrystals exhibiting this behavior. Across all nanocrystal compositions, 72 exhibited multimodal behavior, and this was the second most common behavior. Eight of the 72 total where trimodal (only found in the CH3NH3PbBr3 and CH3NH3Pb(Br0.75I0.25)3 compositions) and 64 were bimodal. A possible mechanism for type b photophysics is considered: given the nanocrystals are smaller than the diffraction limit of light, it is possible that a bimodal luminescence intensity arises from nanocrystal dimers or aggregates. Considering a bimodal distribution, the most common type b behavior, and a possible nanocrystal dimer, the high luminescence state would represent times when both nanocrystals are emitting; whereas, the low intensity state would represent times when only one nanocrystal is emitting. Statistically, there is a possibility that both nanocrystals are not emitting, and the intensity would drop below the threshold. This possibility increases as the duration of the low intensity state increases; however, an intensity below the threshold is never measured in this population. One may argue that the lower intensity state is one where the nanocrystal is not luminescent, and that an incorrect threshold has been applied. However, the image inset in Fig. 3b clearly shows that the low intensity state is higher than the background. A low intensity, or gray state, has been reported for quantum dots, including ZnS capped CdSe63 and CdS capped CdSe.64,65 The emitting low intensity state in CH3NH3PbBr3−xIx nanocrystals may be a gray state, although the mechanism for the gray state may be unique and requires further study.
In up to 22% of the population, the luminescence intensity of a nanocrystal increased over the analysis time. If the slope of the intensity between the data points at time 0 s and 250 s was greater than 0.2 s−1, then the nanocrystal was considered to be photobrightening, shown in Fig. 3c. As expected, photobrightening results in an intensity histogram with an asymmetric distribution (Fig. 3c′). Photobrightening has been reported in CH3NH3PbBr3 nanocrystals and CH3NH3PbI3 films by Tachikawa et al. and deQuilettes et al., respectively.54,66 One mechanism reported for photobrightening is a photoinduced trap reduction caused by the movement of halides when illuminated with light. As the halides are transported about the nanocrystalline lattice, vacancies that make up trap sites are filled. Considering all nanocrystal compositions as an aggregate, this is the least common behavior.
Each nanocrystal composition produced a population where the luminescence intensity of a nanocrystal decreased over the analysis time. If the slope of the intensity between the data points at time 0 s and 250 s was less than −0.2 s−1, then the nanocrystal was considered to be photobleaching, shown in Fig. 3d. Photobleaching generally results in an intensity histogram that skews to lower values (Fig. 3d′). Photobleaching was most common (74–95%) in the nanocrystals containing high amounts of iodide (i.e., CH3NH3Pb(Br0.25I0.75)3 and CH3NH3PbI3). We hypothesize that in cases of photobleaching, the nanocrystal lattice begins to decompose leading to higher non-radiative recombination events, which has also been observed in CsPbX3 nanocrystals52 and CdSe/ZnS quantum dots.67 A small number of nanocrystals exhibited photobleaching as well as short periods of lower intensity up to 10 s in duration (i.e., below the 50 s used to categorize a nanocrystal as exhibiting a bimodal distribution). Each nanocrystal was analyzed for 250 s; it is possible that the percentage of nanocrystals with type d photobleaching behavior increases as the analysis time increases.
Flickering is defined here as less than 500 ms (i.e., two times the acquisition time) periods of a drop in luminescence intensity. Flickering is observed in types b, c, and d luminescence behaviors. Examples of flickering are shown in Fig. 3b and d. In some instances of flickering, the luminescence intensity goes below the threshold, which represent brief intermittent events. For the events that do not go below the threshold, a faster acquisition rate may reveal whether these events represent intermittent events. A faster acquisition time of 20 ms was used to measure the time correlated luminescence intensity of all nanocrystal compositions; results for one composition are shown in Fig. 4 and for the other compositions in ESI,† Fig. S4. A majority of the flickering measured with a 20 ms acquisition time remain above the threshold, revealing a mechanism faster than 20 ms is responsible for a majority of these events.
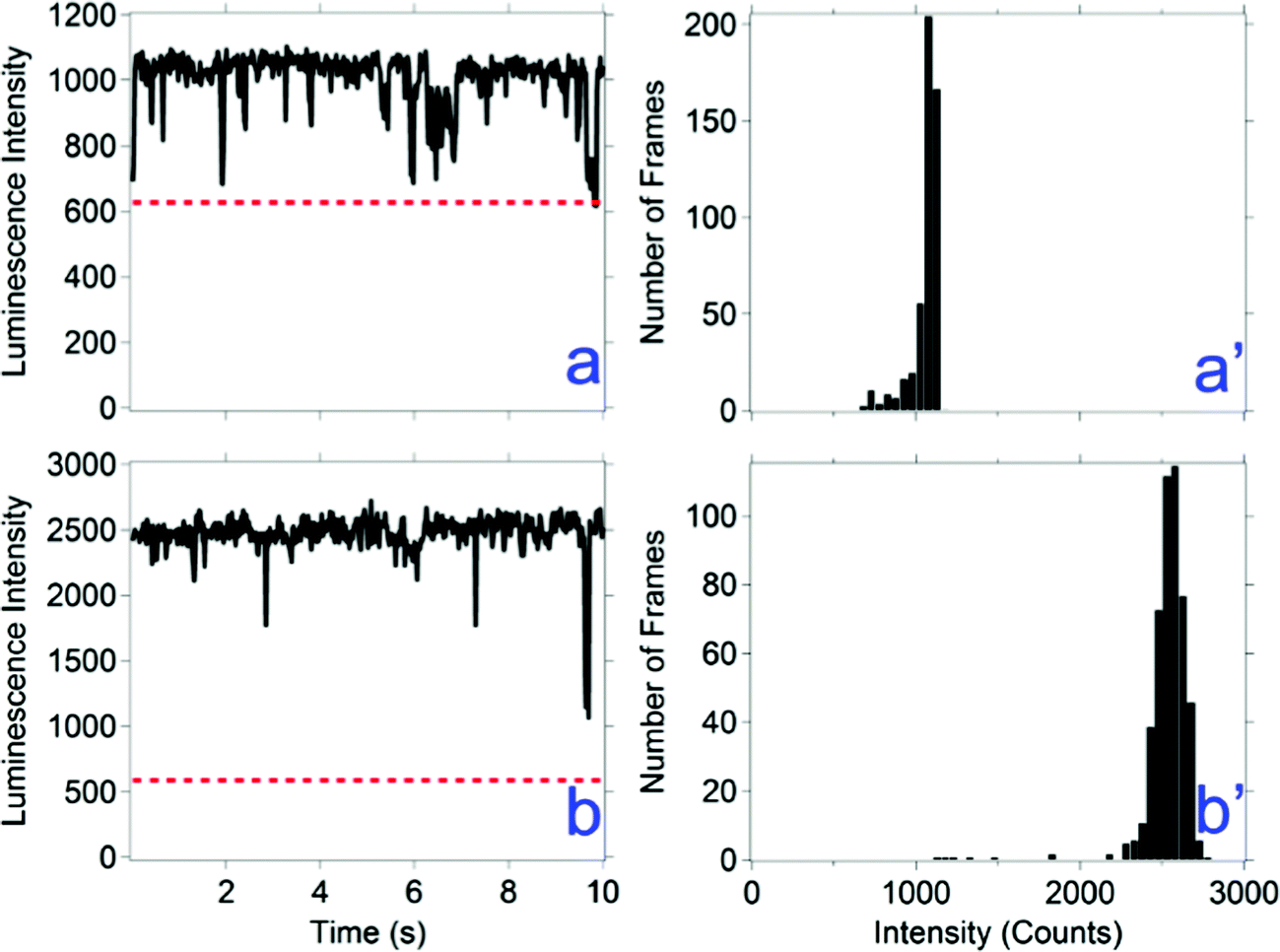 | ||
| Fig. 4 Representative luminescence intensity versus time graphs for CH3NH3PbBr3 nanocrystals. The acquisition time is 20 ms, collected over 10 s. The dotted red line is the threshold for distinguishing nanocrystal luminescence from the background. | ||
In the nanocrystal luminescence microscopy experiments described above, increasing, decreasing or flickering luminescence may be explained by shifting luminescence spectra, which has been measured in similar nanocrystals.31,68 If the luminescence shifts partially, or completely, outside the wavelength range of the filters used to collect the signal, then the luminescence intensity could increase, decrease, or disappear. In order to test for this, the emission spectrum of isolated CH3NH3PbI3 nanocrystals was measured over 250 s under 1 × 105 W cm−2 laser irradiance (Fig. 5). The emission maximum remained constant at 775 nm under continuous illumination with a 100 ms acquisition time. The emission does not shift outside the wavelength region of the filters used to collect the luminescence intensity versus time data. This indicates that the blinking or other varying luminescence intensity states measured for the CH3NH3PbI3 nanocrystals (Table 1) is not the result of shifting luminescence spectra. The instrument setup does not allow the measurement of the other nanocrystal compositions. Finally, this experiment shows that 50% of the nanocrystals exhibit photobrightening and 50% photobleaching under high irradiance, and overall better photostability than many reported in the literature for similar materials.52,69
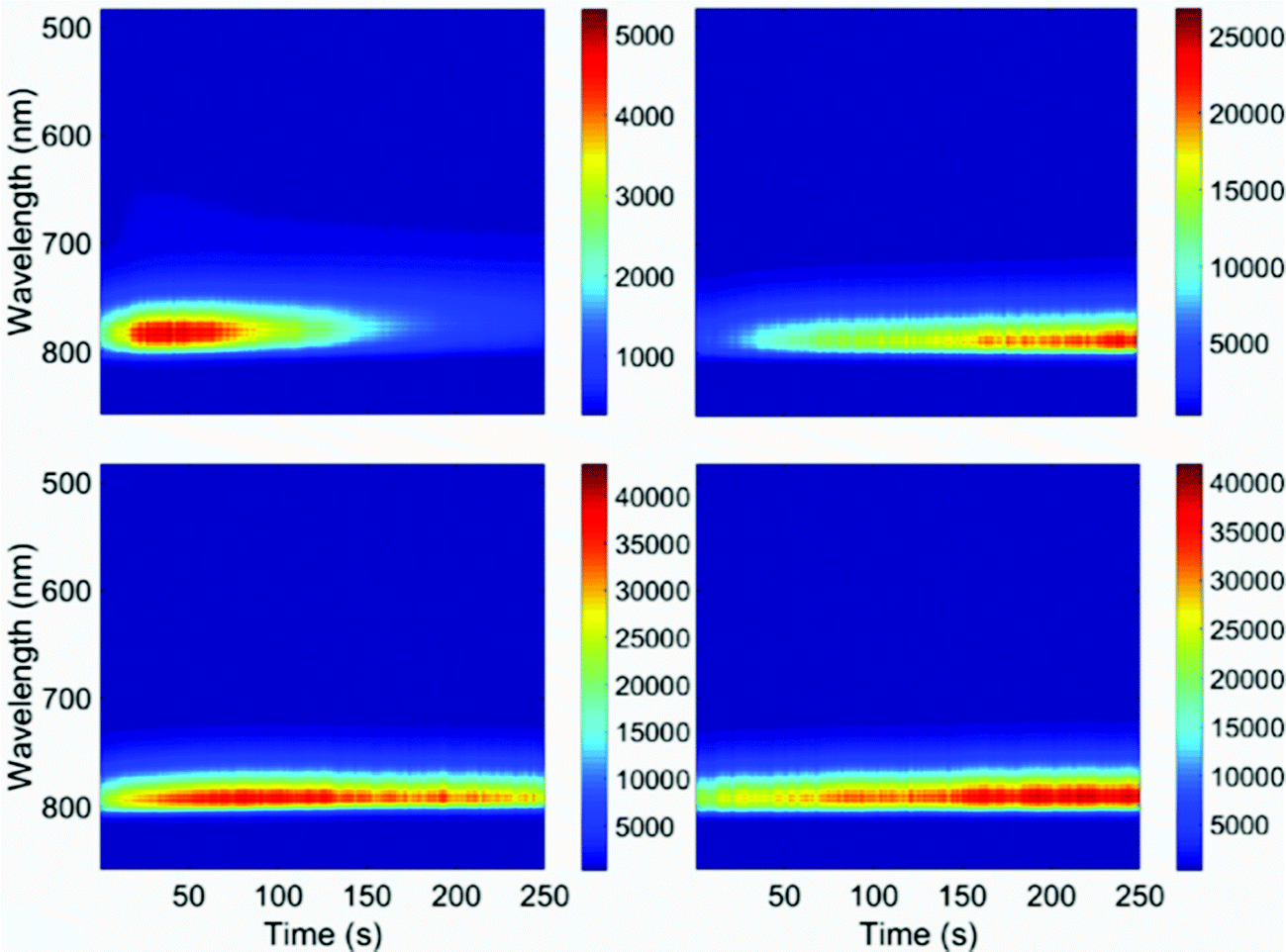 | ||
| Fig. 5 Luminescence spectra of CH3NH3PbI3 nanocrystals over time. Each panel represents a different nanocrystal. The luminescence maximum is constant over time at 775 nm. (This is shifted relative to the ensemble solution data shown in Fig. 1.) The excitation wavelength is 532 nm; the acquisition time is 0.1 s. | ||
Conclusions
The luminescence of a series of methylammonium lead halide nanocrystals has been reported. The wavelength of maximum luminescence was tunable within the range of 498–740 nm by controlling the bromide and iodide loading during nanocrystal synthesis. The CH3NH3PbI3 and CH3NH3PbBr3 nanocrystals exhibited no luminescence intermittency, while 82%, 96%, and 74% of the CH3NH3Pb(Br0.75I0.25)3, CH3NH3Pb(Br0.50I0.50)3, and CH3NH3Pb(Br0.25I0.75)3 nanocrystals exhibited no intermittency, respectively. It is likely that the mixed halide nanocrystals contain iodide-rich and bromide-rich domains when both halides are present in the nanocrystal.38,61 It is also possible that domains form as a result of light-induced segregation. The low percentage of nanocrystals that exhibited intermittency, along with relatively good photostabilities at low (i.e., mercury lamp) and high (i.e., focused laser, up to 1 × 105 W cm−2) photon fluxes, and the emission tunability of the nanocrystals will make these hybrid perovskite nanocrystals useful in a wide range of applications.Acknowledgements
This research is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences through the Ames Laboratory. The Ames Laboratory is operated for the U.S. Department of Energy by Iowa State University under Contract No. DE-AC02-07CH11358.References
- L. A. Lane, A. M. Smith, T. Lian and S. Nie, J. Phys. Chem. B, 2014, 118, 14140–14147 CrossRef CAS PubMed.
- J. Luo, H. Bai, P. Yang and J. Cai, Mater. Sci. Semicond. Process., 2015, 34, 1–7 CrossRef CAS.
- M. Fernández-Suárez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929–943 CrossRef PubMed.
- A. Orte, J. M. Alvarez-Pez and M. J. Ruedas-Rama, ACS Nano, 2013, 7, 6387–6395 CrossRef CAS PubMed.
- P. Patel and D. Mitzi, MRS Bull., 2014, 39, 768–769 CrossRef.
- H. J. Snaith, J. Phys. Chem. Lett., 2013, 4, 3623–3630 CrossRef CAS.
- N.-G. Park, J. Phys. Chem. Lett., 2013, 4, 2423–2429 CrossRef CAS.
- S. D. Stranks, P. K. Nayak, W. Zhang, T. Stergiopoulos and H. J. Snaith, Angew. Chem., Int. Ed., 2015, 54, 3240–3248 CrossRef CAS PubMed.
- E. Edri, S. Kirmayer, D. Cahen and G. Hodes, J. Phys. Chem. Lett., 2013, 4, 897–902 CrossRef CAS PubMed.
- E. Edri, S. Kirmayer, M. Kulbak, G. Hodes and D. Cahen, J. Phys. Chem. Lett., 2014, 5, 429–433 CrossRef CAS PubMed.
- S. De Wolf, J. Holovsky, S.-J. Moon, P. Löper, B. Niesen, M. Ledinsky, F.-J. Haug, J.-H. Yum and C. Ballif, J. Phys. Chem. Lett., 2014, 5, 1035–1039 CrossRef CAS PubMed.
- E. L. Unger, E. T. Hoke, C. D. Bailie, W. H. Nguyen, A. R. Bowring, T. Heumuller, M. G. Christoforo and M. D. McGehee, Energy Environ. Sci., 2014, 7, 3690–3698 CAS.
- T. C. Sum and N. Mathews, Energy Environ. Sci., 2014, 7, 2518–2534 CAS.
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, Angew. Chem., 2014, 126, 11414–11417 CrossRef.
- L. Zuo, Z. Gu, T. Ye, W. Fu, G. Wu, H. Li and H. Chen, J. Am. Chem. Soc., 2015, 137, 2674–2679 CrossRef CAS PubMed.
- L. K. Ono, S. R. Raga, S. Wang, Y. Kato and Y. Qi, J. Mater. Chem. A, 2015, 3, 9074–9080 CAS.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- A. S. Stender, K. Marchuk, C. Liu, S. Sander, M. W. Meyer, E. A. Smith, B. Neupane, G. Wang, J. Li, J.-X. Cheng, B. Huang and N. Fang, Chem. Rev., 2013, 113, 2469–2527 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- C. Wehrenfennig, M. Liu, H. J. Snaith, M. B. Johnston and L. M. Herz, Energy Environ. Sci., 2014, 7, 2269–2275 CAS.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- Y. Fu, F. Meng, M. B. Rowley, B. J. Thompson, M. J. Shearer, D. Ma, R. J. Hamers, J. C. Wright and S. Jin, J. Am. Chem. Soc., 2015, 137, 5810–5818 CrossRef CAS PubMed.
- G. Li, Z.-K. Tan, D. Di, M. L. Lai, L. Jiang, J. H.-W. Lim, R. H. Friend and N. C. Greenham, Nano Lett., 2015, 15, 2640–2644 CrossRef CAS PubMed.
- H. Zhu, Y. Fu, F. Meng, X. Wu, Z. Gong, Q. Ding, M. Gustafsson, M. Trinh, S. Jin and X. Zhu, Nat. Mater., 2015, 14, 636–642 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 16008–16011 CrossRef CAS PubMed.
- Q. A. Akkerman, V. D'Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS PubMed.
- D. M. Jang, K. Park, D. H. Kim, J. Park, F. Shojaei, H. S. Kang, J.-P. Ahn, J. W. Lee and J. K. Song, Nano Lett., 2015, 15, 5191–5199 CrossRef CAS PubMed.
- D. Di, K. P. Musselman, G. Li, A. Sadhanala, Y. Ievskaya, Q. Song, Z.-K. Tan, M. L. Lai, J. L. MacManus-Driscoll, N. C. Greenham and R. H. Friend, J. Phys. Chem. Lett., 2015, 6, 446–450 CrossRef CAS PubMed.
- F. Zhang, H. Zhong, C. Chen, X.-g. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- S. Pathak, N. Sakai, F. Wisnivesky Rocca Rivarola, S. D. Stranks, J. Liu, G. E. Eperon, C. Ducati, K. Wojciechowski, J. T. Griffiths, A. A. Haghighirad, A. Pellaroque, R. H. Friend and H. J. Snaith, Chem. Mater., 2015, 27, 8066–8075 CrossRef CAS.
- T. A. Berhe, W.-N. Su, C.-H. Chen, C.-J. Pan, J.-H. Cheng, H.-M. Chen, M.-C. Tsai, L.-Y. Chen, A. A. Dubale and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 323–356 CAS.
- G. Niu, X. Guo and L. Wang, J. Mater. Chem. A, 2015, 3, 8970–8980 CAS.
- N. Aristidou, I. Sanchez-Molina, T. Chotchuangchutchaval, M. Brown, L. Martinez, T. Rath and S. A. Haque, Angew. Chem., Int. Ed., 2015, 54, 8208–8212 CrossRef CAS PubMed.
- S. Ito, S. Tanaka, K. Manabe and H. Nishino, J. Phys. Chem. C, 2014, 118, 16995–17000 CAS.
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC.
- H. Yuan, E. Debroye, K. Janssen, H. Naiki, C. Steuwe, G. Lu, M. l. Moris, E. Orgiu, H. Uji-i and F. De Schryver, J. Phys. Chem. Lett., 2016, 7, 561–566 CrossRef CAS PubMed.
- F. Zhu, L. Men, Y. Guo, Q. Zhu, U. Bhattacharjee, P. M. Goodwin, J. W. Petrich, E. A. Smith and J. Vela, ACS Nano, 2015, 9, 2948–2959 CrossRef CAS PubMed.
- F. Matsumoto, S. M. Vorpahl, J. Q. Banks, E. Sengupta and D. S. Ginger, J. Phys. Chem. C, 2015, 119, 20810–20816 CAS.
- N. A. Manshor, Q. Wali, K. K. Wong, S. K. Muzakir, A. Fakharuddin, L. Schmidt-Mende and R. Jose, Phys. Chem. Chem. Phys., 2016, 18, 21629–21639 RSC.
- M. D. Barnes, A. Mehta, T. Thundat, R. N. Bhargava, V. Chhabra and B. Kulkarni, J. Phys. Chem. B, 2000, 104, 6099–6102 CrossRef CAS.
- J. J. Glennon, W. E. Buhro and R. A. Loomis, J. Phys. Chem. C, 2008, 112, 4813–4817 CAS.
- J. J. Glennon, R. Tang, W. E. Buhro and R. A. Loomis, Nano Lett., 2007, 7, 3290–3295 CrossRef CAS PubMed.
- M. Kuno, D. P. Fromm, A. Gallagher, D. J. Nesbitt, O. I. Micic and A. J. Nozik, Nano Lett., 2001, 1, 557–564 CrossRef CAS.
- M. Kuno, D. P. Fromm, H. F. Hamann, A. Gallagher and D. J. Nesbitt, J. Chem. Phys., 2001, 115, 1028–1040 CrossRef CAS.
- M. Kuno, D. P. Fromm, H. F. Hamann, A. Gallagher and D. J. Nesbitt, J. Chem. Phys., 2000, 112, 3117–3120 CrossRef CAS.
- C. Galland, Y. Ghosh, A. Steinbrück, M. Sykora, J. A. Hollingsworth, V. I. Klimov and H. Htoon, Nature, 2011, 479, 203–207 CrossRef CAS PubMed.
- Y. Tian, A. Merdasa, M. Peter, M. Abdellah, K. Zheng, C. S. Ponseca, T. Pullerits, A. Yartsev, V. Sundström and I. G. Scheblykin, Nano Lett., 2015, 15, 1603–1608 CrossRef CAS PubMed.
- X. Wen, A. Ho-Baillie, S. Huang, R. Sheng, S. Chen, H.-c. Ko and M. A. Green, Nano Lett., 2015, 15, 4644–4649 CrossRef CAS PubMed.
- Y.-S. Park, S. Guo, N. S. Makarov and V. I. Klimov, ACS Nano, 2015, 9, 10386–10393 CrossRef CAS PubMed.
- S. Seth, N. Mondal, S. Patra and A. Samanta, J. Phys. Chem. Lett., 2016, 7, 266–271 CrossRef CAS PubMed.
- T. Tachikawa, I. Karimata and Y. Kobori, J. Phys. Chem. Lett., 2015, 6, 3195–3201 CrossRef CAS.
- K. A. Lidke, B. Rieger, T. M. Jovin and R. Heintzmann, Opt. Express, 2005, 13, 7052–7062 CrossRef PubMed.
- U. Banin, M. Bruchez, A. P. Alivisatos, T. Ha, S. Weiss and D. S. Chemla, J. Chem. Phys., 1999, 110, 1195–1201 CrossRef CAS.
- X. Wu, M. T. Trinh, D. Niesner, H. Zhu, Z. Norman, J. S. Owen, O. Yaffe, B. J. Kudisch and X.-Y. Zhu, J. Am. Chem. Soc., 2015, 137, 2089–2096 CrossRef CAS PubMed.
- H.-S. Duan, H. Zhou, Q. Chen, P. Sun, S. Luo, T.-B. Song, B. Bob and Y. Yang, Phys. Chem. Chem. Phys., 2015, 17, 112–116 RSC.
- K. Zheng, K. Zidek, M. Abdellah, M. E. Messing, M. J. Al-Marri and T. Pullerits, J. Phys. Chem. C, 2016, 120, 3077–3084 CAS.
- Y. Tian, A. Merdasa, E. Unger, M. Abdellah, K. Zheng, S. McKibbin, A. Mikkelsen, T. Pullerits, A. Yartsev, V. Sundström and I. G. Scheblykin, J. Phys. Chem. Lett., 2015, 6, 4171–4177 CrossRef CAS PubMed.
- B. A. Rosales, L. Men, S. D. Cady, M. P. Hanrahan, A. J. Rossini and J. Vela, Chem. Mater., 2016, 28, 6848–6859 CrossRef CAS.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- M. Osborne and A. Fisher, Nanoscale, 2016, 8, 9272–9283 RSC.
- M. D. Lesoine, U. Bhattacharjee, Y. Guo, J. Vela, J. W. Petrich and E. A. Smith, J. Phys. Chem. C, 2013, 117, 3662–3667 CAS.
- A. V. Malko, Y.-S. Park, S. Sampat, C. Galland, J. Vela, Y. Chen, J. A. Hollingsworth, V. I. Klimov and H. Htoon, Nano Lett., 2011, 11, 5213–5218 CrossRef CAS PubMed.
- D. W. deQuilettes, W. Zhang, V. M. Burlakov, D. J. Graham, T. Leijtens, A. Osherov, V. Bulovic, H. J. Snaith, D. S. Ginger and S. D. Stranks, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- W. G. J. H. M. van Sark, P. L. T. M. Frederix, D. J. Van den Heuvel, H. C. Gerritsen, A. A. Bol, J. N. J. van Lingen, C. de Mello Donegá and A. Meijerink, J. Phys. Chem. B, 2001, 105, 8281–8284 CrossRef CAS.
- H. H. Fang, R. Raissa, M. Abdu-Aguye, S. Adjokatse, G. R. Blake, J. Even and M. A. Loi, Adv. Funct. Mater., 2015, 25, 2378–2385 CrossRef CAS.
- B. Luo, Y.-C. Pu, Y. Yang, S. A. Lindley, G. Abdelmageed, H. Ashry, Y. Li, X. Li and J. Z. Zhang, J. Phys. Chem. C, 2015, 119, 26672–26682 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures of the experimental data. Fig. S1–S4. See DOI: 10.1039/c6tc03886g |
| This journal is © The Royal Society of Chemistry 2017 |