
A pilot study on clinical pharmacokinetics and preclinical pharmacodynamics of (+)-epicatechin on cardiometabolic endpoints†
Aldo
Moreno-Ulloa
 ab,
Nayelli
Nájera-García
c,
Marcela
Hernández
c,
Israel
Ramírez-Sánchez
ac,
Pam R.
Taub
a,
Yongxuan
Su
d,
Ernesto
Beltrán-Partida
e,
Guillermo
Ceballos
c,
Sundeep
Dugar
f,
George
Schreiner
f,
Brookie M.
Best
h,
Theodore P.
Ciaraldi
ag,
Robert R.
Henry
ag and
Francisco
Villarreal
*a
ab,
Nayelli
Nájera-García
c,
Marcela
Hernández
c,
Israel
Ramírez-Sánchez
ac,
Pam R.
Taub
a,
Yongxuan
Su
d,
Ernesto
Beltrán-Partida
e,
Guillermo
Ceballos
c,
Sundeep
Dugar
f,
George
Schreiner
f,
Brookie M.
Best
h,
Theodore P.
Ciaraldi
ag,
Robert R.
Henry
ag and
Francisco
Villarreal
*a
aUniversity of California, San Diego, School of Medicine, USA. E-mail: fvillarr@ucsd.edu; Fax: +(858) 534-0522; Tel: +(858) 534-3630 Tel: +(858) 534-1317
bDepartamento de Innovación Biomédica, Centro de Investigación Científica y de Educación Superior de Ensenada (CICESE), Baja California, Mexico
cEscuela Superior de Medicina del Instituto Politécnico Nacional, Sección de Posgrado, México City, Mexico
dUniversity of California, San Diego, Chemistry and Biochemistry, USA
eUniversidad Autónoma de Baja California, Facultad de Odontología, Mexicali, BC, Mexico
fCardero Therapeutics, Inc., USA
gSan Diego VA Health Care System, USA
hUniversity of California, Skaggs School of Pharmacy and Pharmaceutical Sciences, San Diego, USA
First published on 15th November 2017
Abstract
We reported that (−)-epicatechin can stimulate mitochondria biogenesis and improve metabolism. However, preliminary studies indicate that the (+) stereoisomer form may be more potent. We evaluated in a preliminary manner, the pharmacokinetics (PK) and initial safety analysis of (+)-epicatechin ((+)-Epi) in healthy and pre-diabetic subjects. Using a mouse model of diet-induced obesity and insulin resistance, we also evaluated the metabolic effects of (+)-Epi vs. (+)-catechin (Cat) to determine class effects. In the Phase I PK study, subjects were provided a single incremental oral dose of (+)-Epi (10, 30 or 100 mg). For the PD study, subjects were provided a single 30 mg dose per day for 7 days. Blood samples were collected and safety measures were performed. Incremental doses of (+)-Epi increase the half-life of blood metabolites from 1.2–4.9 h. The compound was well tolerated and no adverse effects were reported. Seven day dosing of pre-diabetic subjects led to tendencies for reductions in circulating levels of tumor necrosis factor-α and monocyte chemoattractant protein-1, which returned to baseline by 7 days after treatment. In animals, 2 weeks of oral dosing (0.003, 0.01, 0.03, 0.1 and 0.3 mg kg−1 day−1) dose dependently improved metabolism-related endpoints (weight gain, glucose, cholesterol, triglyceride, with thresholds as low as 0.01 mg kg−1 day−1). Cat yielded no effects at 0.1 mg kg−1 day−1. Results indicate that (+)-Epi evidences a favorable PK and safety profile. Using a pre-clinical model, the compound positively modulates metabolism, which may link to mitochondrial effects. Effects are not due to general antioxidant actions, as Cat yielded no effects.
1. Introduction
Worldwide, there is an alarming increase in the incidence of obesity and as a consequence in the number of subjects with multiple metabolic alterations such as hyperglycemia, hypertriglyceridemia and hypercholesterolemia.1,2 These metabolic disarrangements, which are frequently accompanied by chronic low-grade systemic inflammation,3 increase the risk for the development of cardiovascular and metabolic diseases such as atherosclerosis and type 2 diabetes mellitus. A successful change in these worrisome trends will require the implementation of educational programs on proper nutrition and lifestyle changes such as exercise. However, in many cases the use of complementary strategies such as pharmacotherapy will be necessary so as to effectively correct such cardiometabolic (CM) alterations and minimize the risk of developing chronic diseases. However, many of the pharmacological compounds currently in use to treat cardiovascular and metabolic diseases (in particular, when used over the long term) carry adverse effects.4–6 Hence, there is the need to identify safe and effective agents that may aid in the prevention and/or treatment of such CM alterations.There is ample evidence indicating that the consumption of flavanol containing foods favorably impacts multiple CM endpoints.7–11 (−)-Epicatechin ((−)-Epi) is part of a group of 4 flavanol stereoisomers that differ in the configuration of their carbons 2 and 3 (chiral carbons), based on its structural sequential numeration.12 (−)-Epi fully mimics the effects of flavanol containing foods such as cocoa as it exerts positive metabolic effects in cultured cells,13,14 animal models15,16 and humans.17–19 In contrast to (−)-Epi, which is readily found in cacao, green tea and in fruits such as grapes, the natural occurrence of (+)-epicatechin ((+)-Epi) is much more limited as it has been only reported in guaraná seeds and as a consequence of processing (i.e., epimerization) of green tea and cacao.20,21 In preliminary studies, we reported that (+)-Epi appears to evidence greater in vitro and in vivo efficacy and/or potency as compared to (−)-Epi in stimulating metabolism related endpoints.22 However, no studies have examined the pharmacokinetic (PK) profile of (+)-Epi using single ascendant or multiple doses in humans, as well as associated safety and clinical pharmacodynamics (PD). Published studies have focused mainly on (+)-Epi metabolite characterization and evaluated limited pre-clinical PD.23
In the present study, we report on the PK, partial PD and initial safety analysis of (+)-Epi as implemented in healthy and pre-diabetic subjects. Using a mouse model of diet-induced obesity and insulin resistance we also provide PD evidence for the metabolic effects of (+)-Epi and contrast results with those of (+)-catechin (Cat) to determine if the observed effects correspond to class actions.
2. Methods
2.1. Study compounds
(+)-Epi was synthesized by a GMP-certified facility (Syngene, Bangalore, India) and provided by Cardero Therapeutics Inc. (Los Altos, CA). The substance was tested in a GMP analytical lab using appropriate qualified high-performance liquid chromatography (HPLC) methods for impurities, including chirality testing. Release specifications required >90% purity, <5% of the enantiomer and 5% of Cat. It was also tested for other characteristics typical in GMP materials (identity by 1H NMR, IR; water content by Karl Fischer titration; ethanol content by Gas chromatography; and the general USP tests of residue on ignition and heavy metals). Based on test results, a Certificate of Analysis was generated and a %-content by weight is calculated. Stability testing was performed after storage at 16–42 °C. The powdered test material was found to be stable for at least 12 months at the temperatures tested. Purity of the (+)-Epi (>98%) was established by 1H-NMR and HPLC-Mass Spectrometry (MS), while HPLC analysis revealed (−)-Epi content to be <1%, far exceeding release specifications. Microbiological tests for aerobic microorganisms, yeast and molds were negative (<10 cfug−1). Tests for E. coli, Pseudomonas aeruginosa, Salmonella species and Staphylococcus aureuswere also negative. (+)-Epi was encapsulated by the VA San Diego Healthcare System (VASDHS) research pharmacy service. The same material in powdered form was dissolved in sterile water to gavage animals with (+)-Epi at the doses tested. For animal studies using Cat, the compound was purchased from Sigma-Aldrich Chemicals Inc. (C1251) and purity was reported by a Certificate of Analysis as ≥98%. As above, powdered Cat was dissolved in water for animal gavaging purposes.2.2. Human studies-recruitment of participants
Volunteers were recruited to participate in the study through word of mouth and advertisements. Prior to enrollment, a physician obtained detailed health information and performed a brief physical examination. A complete blood count (CBC) and electrocardiogram (ECG) was performed in all volunteers. Vital signs (heart rate and blood pressure), and laboratory analysis for hematology, urinalysis, electrolytes, renal and liver function tests were also performed in all volunteers.Inclusion criteria were: Healthy or pre-diabetic (see below) subjects based on screening results, 21–75 years of age, male or female. If female, they must be either postmenopausal or test negative for pregnancy at screening and on the day of the procedure (women on estrogen therapy were included) and be able to give informed consent to the procedures. If female of childbearing potential, they must practice and be willing to continue to practice appropriate birth control during the entire duration of the study. If subjects are on medication, their stable use for 4 weeks prior to screening. The presence of pre-diabetes as determined by the criteria defined by the American Diabetes Association as follows:24 body mass index (BMI) > 27 kg m−2, impaired fasting glucose (IFG, fasting glucose = 100–125 mg dL−1) and elevated HbA1c (5.7–6.4%), each in the absence of other risk factors for diabetes, and had no abnormalities in laboratory tests and ECG.
Exclusion criteria were: type 2 diabetes, pregnancy, clinical significant abnormalities in liver or kidney function (>3× upper limit of normal), determined in the last 6 months by a certified clinical laboratory, blood pressure >160 mmHg systolic and >100 mmHg diastolic, medications – thiazolidinediones, any-steroids, anti-depressants, weight loss drugs and for other diseases that may influence carbohydrate metabolism. Testing was performed in accordance with the guidelines on good clinical practice and within ethical standards for human experimentation as established by the Declaration of Helsinki. The Institutional Review Board on human research of the VASDHS approved the human protocol and amendments and each subject provided written informed consent before participating in the study. The study was registered at Clinical Trials.gov (NCT02330276).
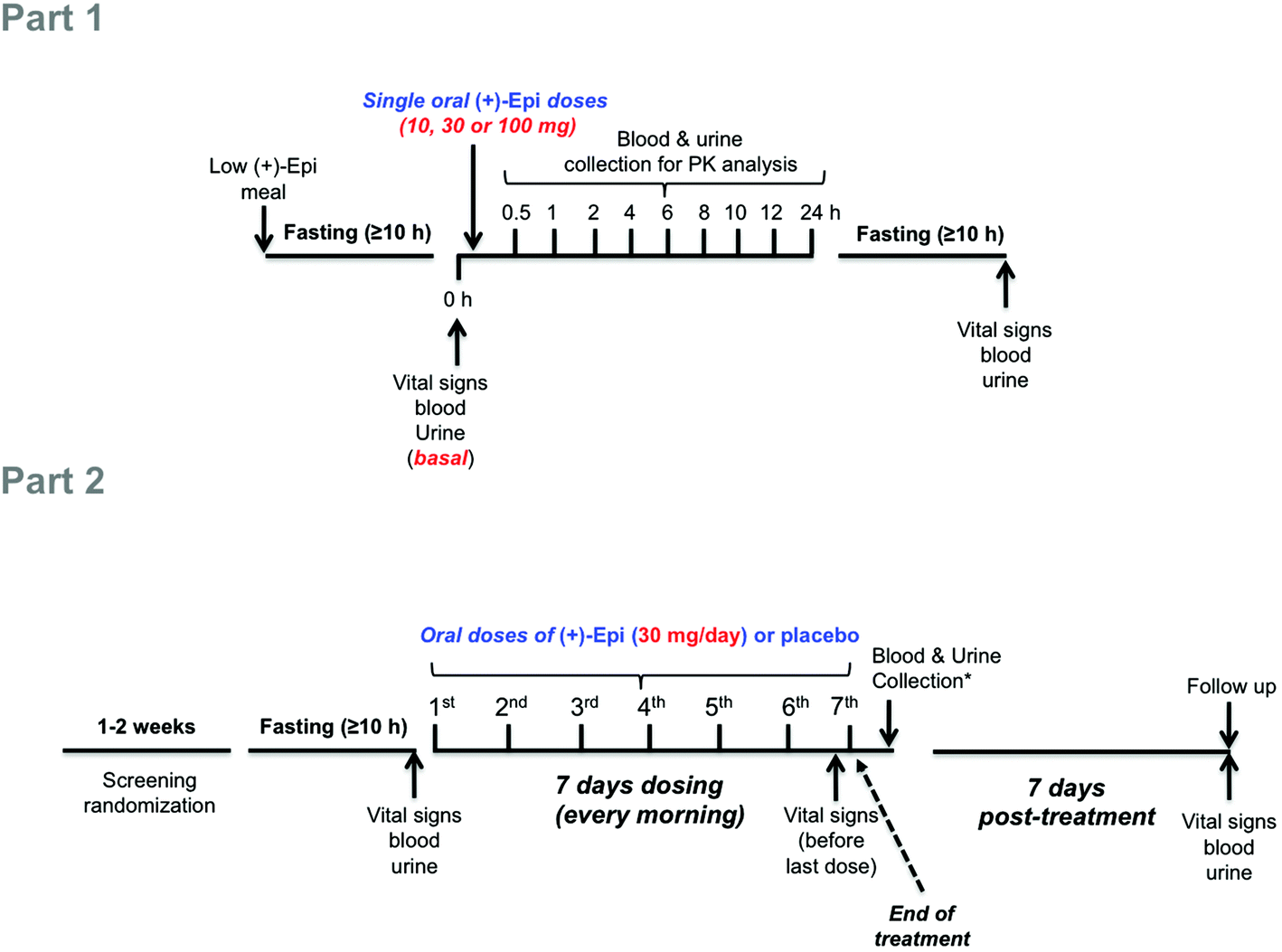 | ||
| Fig. 1 Schematic illustration of study design. Abbreviations: h, hours; (+)-Epi, (+)-epicatechin; PK, pharmacokinetics. *Blood and urine were collected ∼1 hour after last dose (7th). | ||
In the Part 2 of the study, using a double-blind, randomized, placebo-controlled trial we examined as primary endpoints the safety and tolerability of (+)-Epi dosing for 7 days. Studies were only implemented in pre-diabetic subjects. Subjects were given either (+)-Epi (30 mg day−1, SID, n = 10) or placebo (n = 5) and instructed to take the capsule each morning before breakfast for 7 days (Fig. 1, Part 2). This dose was selected on the basis of two sets of previous results, (1) (−)-Epi PK in humans were we use 100 mg day−1(ref. 25) and, (2) Our preliminary studies in mice evidencing an equivalency in metabolic effects between 1 mg kg−1 day−1 of (−)-Epi to 0.3 mg kg−1 day−1 of (+)-Epi.22 As a secondary endpoint, we examined the effects on blood glucose, insulin and HOMA-IR2, lipids, and selected markers of inflammation. Assessments were performed at baseline (≥10 h fasting and before 1st dose administration), after 7 days of treatment (≥10 h fasting and ∼1 h after 7th dose administration) (end of treatment) and at follow-up 7 days post-treatment (≥10 h fasting) (end of study). Since (−)-epicatechin and other flavanols are components of many foods, subjects were provided dietary guidelines by staff to be followed during the study. The dietary plan was designed to provide a low flavanol intake.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and the supernatant was transferred to an eppendord tube and evaporated to dryness using a SpeedVac Concentrator SVC200H (Savant) at room temperature. The dried samples were dissolved in 100 μL of 5% acetonitrile in water with 0.1% formic acid. Either 5.0 μL, 10.0 or 20 μL of the solution was injected into a HPLC column. In the absence of available standards, we implemented a modification of a published method by Roura et al. to identify and quantify each (+)-Epi metabolite based on their fragmentation pattern and accurate mass by using HPLC-tandem MS (MS/MS) and HPLC-high resolution-MS, respectively.26 The equipments utilized were a Thermo LCQdeca mass spectrometer and an Agilent 1260 Infinity Binary HPLC coupled with a 6230 Accurate-Mass Time-of-Flight-MS (TOF-MS). Electrospray ionization (ESI) in the negative ion mode was used in both equipments. However, the Agilent 6230 Accurate-Mass TOF-MS was used for metabolites detection and quantification since it provided better detection sensitivity. Details of the HPLC method are as follows: mobile phase A: 2.5% Acetonitrile in water with 0.1% formic acid; mobile phase B: acetonitrile with 0.1% formic acid. LC gradient: 5% B to 95% B in 10 min, back to 5% B in one min, and hold at 5% B for 7 min. A Shiseido CAPCELL PAK C-18 column (MGIII, 2 mm x 50 mm, 3 μm) with guard column was used for separation at a flow rate of 300 μL per minute. Agilent MassHunter workstation was used for data acquisition and analysis. Due to the lack of (+)-Epi metabolites standards Taxifolin and known amounts of (+)-Epi (aglycone standard) were spiked in blank plasma to quantify the concentration of identified (+)-Epi metabolites as described previously.25 The recovery efficiency of (+)-Epi (55.07 ± 2.9%) was considered for the quantification of (+)-Epi and metabolites. The performance of the method was monitored using quality control samples in the same matrix on every day of plasma sample analyses.
000 rpm and the supernatant was transferred to an eppendord tube and evaporated to dryness using a SpeedVac Concentrator SVC200H (Savant) at room temperature. The dried samples were dissolved in 100 μL of 5% acetonitrile in water with 0.1% formic acid. Either 5.0 μL, 10.0 or 20 μL of the solution was injected into a HPLC column. In the absence of available standards, we implemented a modification of a published method by Roura et al. to identify and quantify each (+)-Epi metabolite based on their fragmentation pattern and accurate mass by using HPLC-tandem MS (MS/MS) and HPLC-high resolution-MS, respectively.26 The equipments utilized were a Thermo LCQdeca mass spectrometer and an Agilent 1260 Infinity Binary HPLC coupled with a 6230 Accurate-Mass Time-of-Flight-MS (TOF-MS). Electrospray ionization (ESI) in the negative ion mode was used in both equipments. However, the Agilent 6230 Accurate-Mass TOF-MS was used for metabolites detection and quantification since it provided better detection sensitivity. Details of the HPLC method are as follows: mobile phase A: 2.5% Acetonitrile in water with 0.1% formic acid; mobile phase B: acetonitrile with 0.1% formic acid. LC gradient: 5% B to 95% B in 10 min, back to 5% B in one min, and hold at 5% B for 7 min. A Shiseido CAPCELL PAK C-18 column (MGIII, 2 mm x 50 mm, 3 μm) with guard column was used for separation at a flow rate of 300 μL per minute. Agilent MassHunter workstation was used for data acquisition and analysis. Due to the lack of (+)-Epi metabolites standards Taxifolin and known amounts of (+)-Epi (aglycone standard) were spiked in blank plasma to quantify the concentration of identified (+)-Epi metabolites as described previously.25 The recovery efficiency of (+)-Epi (55.07 ± 2.9%) was considered for the quantification of (+)-Epi and metabolites. The performance of the method was monitored using quality control samples in the same matrix on every day of plasma sample analyses.
2.3. Animal model
Animal handling followed Institutional Guidelines and National Official Standards of Mexico. The institutional review committee approved the animal protocol. Male CD-1 mice (5 weeks old) were purchased from Harlan Laboratories (Mexico). Animals (25–30 g of body weight) were housed under controlled temperature and humidity, with 12 h light/dark cycles. Mice were provided a standard diet (Lab Diets, Mexico) and water ad libitum during the initial acclimatization period. After acclimatization, a group of mice were placed on a high-fat diet (HFD, 60% kcal from fat) (product 58Y1, Lab Diets, Mexico) until they gained >20% of their body weight and reached average blood glucose levels of ≥200 mg dL−1 (∼6 weeks under HFD) when compared to animals under standard chow.:1. The detector and injector temperatures were 230 and 250 °C, respectively. Determination of fatty acids was verified by comparison of retention times of test samples with those of reference standards and internal standardization.
2.4. Statistical analysis
Statistical analysis was performed using Prism Graph Pad version 6.0. Laboratory results from repeated dosing on study day 7 were compared using paired t-tests. Effects of (+)-Epi on BP were evaluated using a mixed models approach. PK parameters were calculated using non-compartmental analysis and Software Prism Graph Pad version 6.0. For PK analysis, concentration time curves were constructed and the AUC determined. Maximal plasma concentration (Cmax) and time of maximal concentration (Tmax) was determined for (+)-Epi and each of the detected metabolites. Natural log transformed data and non-linear regression analysis was used to determine the plasma elimination constant (kel) and plasma half-life (T1/2). Dose proportionality was assessed by calculating the dose-normalized AUC. Metabolite to parent drug AUC ratios were calculated for each metabolite. For the animal studies results are expressed as mean ± SEM. ANOVA test with Tukey's post-test was used for the analysis of statistical differences. A non-parametric analysis was used when appropriated. A p < 0.05 was considered statistically significant.3. Results
3.1. Human Studies
Baseline characteristics of the subjects who received a single capsule of (+)-Epi are presented in Table 1. Subjects were well matched but for those who received the 10 mg dose were younger than the individuals receiving the other doses. Characteristics of the subjects who received (+)-Epi or placebo for 7 days are presented in Table 2. Subjects were well matched but for the placebo group having lower TNFα levels at baseline.| (+)-Epi dose | 10 mg | 30 mg | 100 mg |
|---|---|---|---|
| Data is expressed as mean ± SEM. (Years) is in range. (+)-Epi-(+)-epicatechin; BMI-body mass index; FG-fasting glucose; FI-fasting insulin; HOMA-IR2, homeostatic model assessment-insulin resistance.*p < 0.05. | |||
| N (M/F) | 4 (3/1) | 4 (2/2) | 4 (3/1) |
| Age (years) | 46 ± 9* (28–70) | 60 ± 2 (55–64) | 68 ± 1 (66–70) |
| Systolic blood pressure (mm Hg) | 126 ± 5 | 117 ± 3 | 131 ± 7 |
| Diastolic blood pressure (mm Hg) | 73 ± 3 | 72 ± 2 | 79 ± 3 |
| Heart rate (beats per min) | 70 ± 7 | 66 ± 5 | 54 ± 1 |
| BMI (kg m−2) | 33.9 ± 2.7 | 32.1 ± 2.2 | 31.6 ± 1.3 |
| FG (mM) | 5.0 ± 0.7 | 5.0 ± 0.3 | 5.4 ± 0.2 |
| FI (pM) | 86 ± 14 | 64 ± 14 | 91 ± 21 |
| HOMA-IR2 | 1.87 ± 0.30 | 1.40 ± 0.38 | 1.99 ± 0.47 |
| (+)-Epi | Placebo | |||
|---|---|---|---|---|
| Parameter | Baseline | 7 day | Baseline | 7 day |
| Data is expressed as mean ± SEM. (+)-Epi-(+)-epicatechin; BMI – body mass index; HbA1c – glycated hemoglobin; FG – fasting glucose; FI – fasting insulin; HOMA-IR – homeostatic model assessment-insulin resistance; TG – triglycerides; FFA – free fatty acids; HDL – high density lipoprotein cholesterol; LDL – low density lipoprotein cholesterol; HsCRP – high-sensitivity C – reactive protein; TNFα – tumor necrosis factor alpha; MCP1 – monocyte chemoattractant protein. *p < 0.05 vs. (+)-Epi group, †p < 0.05 post-treatment vs. baseline. | ||||
| n (F/M) | 10 (1/9) | 5 (2/3) | ||
| Age (year) | 61 ± 3 | 60 ± 3 | ||
| BMI (kg m−2) | 33.4 ± 1.3 | 32.1 ± 0.6 | ||
| HbA1c (%) | 5.91 ± 0.12 | nd | 5.84 ± 0.14 | nd |
| FG (mM) | 6.03 ± 0.22 | 5.72 ± 0.25 | 6.02 ± 0.40 | 5.42 ± 0.19 |
| FI (pM) | 93 ± 12 | 86 ± 11 | 121 ± 34 | 78 ± 18 |
| HOMA-IR | 2.07 ± 0.27 | 1.90 ± 0.25 | 2.65 ± 0.74 | 1.70 ± 0.38 |
| TG (mg dL−1) | 138 ± 22 | 127 ± 26 | 132 ± 44 | 109 ± 21 |
| FFA (mM) | 0.34 ± 0.05 | 0.38 ± 0.06 | 0.32 ± 0.08 | 0.55 ± 0.10† |
| HDL (mg dL−1) | 44.8 ± 2.7 | 47.0 ± 3.0 | 51.6 ± 4.5 | 53.2 ± 5.4 |
| LDL (mg dL−1) | 113.8 ± 9.0 | 111.6 ± 10.4 | 123.6 ± 24.7 | 124.0 ± 17.6 |
| Cholesterol (mg dL−1) | 186.3 ± 7.2 | 184.0 ± 8.5 | 201.6 ± 30.0 | 198.8 ± 23.7 |
| HsCRP (pg mL−1) | 13.42 ± 7.83 | 7.38 ± 5.07 | 5.97 ± 3.74 | 8.42 ± 5.44 |
| TNFα (pg mL−1) | 7.90 ± 1.10 | 6.69 ± 0.62 | 4.13 ± 0.36* | 4.48 ± 0.58 |
| MCP1 (pg mL−1) | 432 ± 66 | 380 ± 42 | 347 ± 17 | 387± 38 |
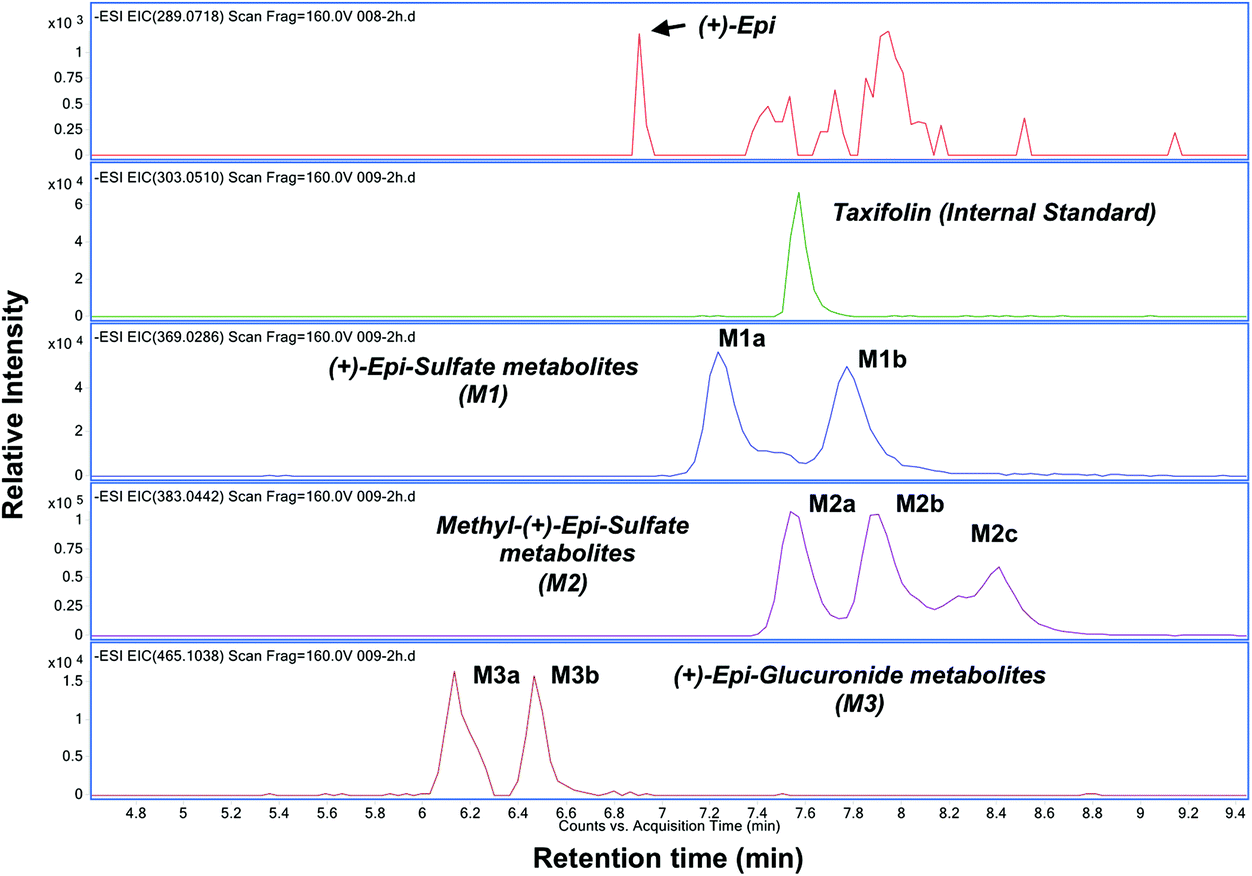 | ||
| Fig. 2 Representative chromatogram used for the pharmacokinetic analysis of (+)-epicatechin ((+)-Epi) metabolites. Extracted ion chromatogram (EIC) for (+)-Epi and its metabolites in a representative plasma sample (2 h post consumption of (+)-Epi 100 mg) obtained by using high-resolution negative ion mode electrospray ionization-time-of-flight mass spectrometry. Peaks; (+)-Epi (289.0718 m/z); Taxifolin (303.0510 m/z); (+)-Epi-glucuronides (two peaks; 465.1034 m/z and 465.1032 m/z); methyl-(+)-Epi-sulfate (three peaks; 383.0440 m/z, 383.0440 m/z and 383.0438 m/z) and (+)-Epi-sulfate (two peaks; 369.0281 m/z and 369.0277 m/z). The accurate masses of their [M − H]− peaks are in parentheses. | ||
The PK parameters are summarized in Tables 3 and 4. Free (+)-Epi and its metabolites (i.e., glucuronides, sulfates and methyl sulfates) appeared rapidly in plasma following oral administration with a high inter-individual variability (Fig. 3A and B). The shape of the natural log transformed plasma concentration curves was consistent with first order kinetics for all doses (data not shown). The most abundant (+)-Epi metabolites were in the methyl-sulfated form at all doses (Fig. 3A). The maximum concentration of (+)-Epi and its metabolites was seen at 1 h following consumption and similar amongst them (Fig. 3A). However, free (+)-Epi was only confidently detected (above LOD) at 1 h of post-consumption in the group receiving 30 and 100 mg of (+)-Epi. Similarly to free (+)-Epi, its glucuronide metabolites were below the LOQ for most time points at the dose of 10 mg. Area under the curve (AUC) was calculated for each group of metabolites at the different doses (10, 30 and 100 mg), whereby, an increased out of proportion for AUC values (non-linear) was observed when comparing same metabolites across doses (Table 3). There were significant differences in the half-life of each (+)-Epi metabolite at the different doses, which ranged from 1.2–4.9 h (Table 3).
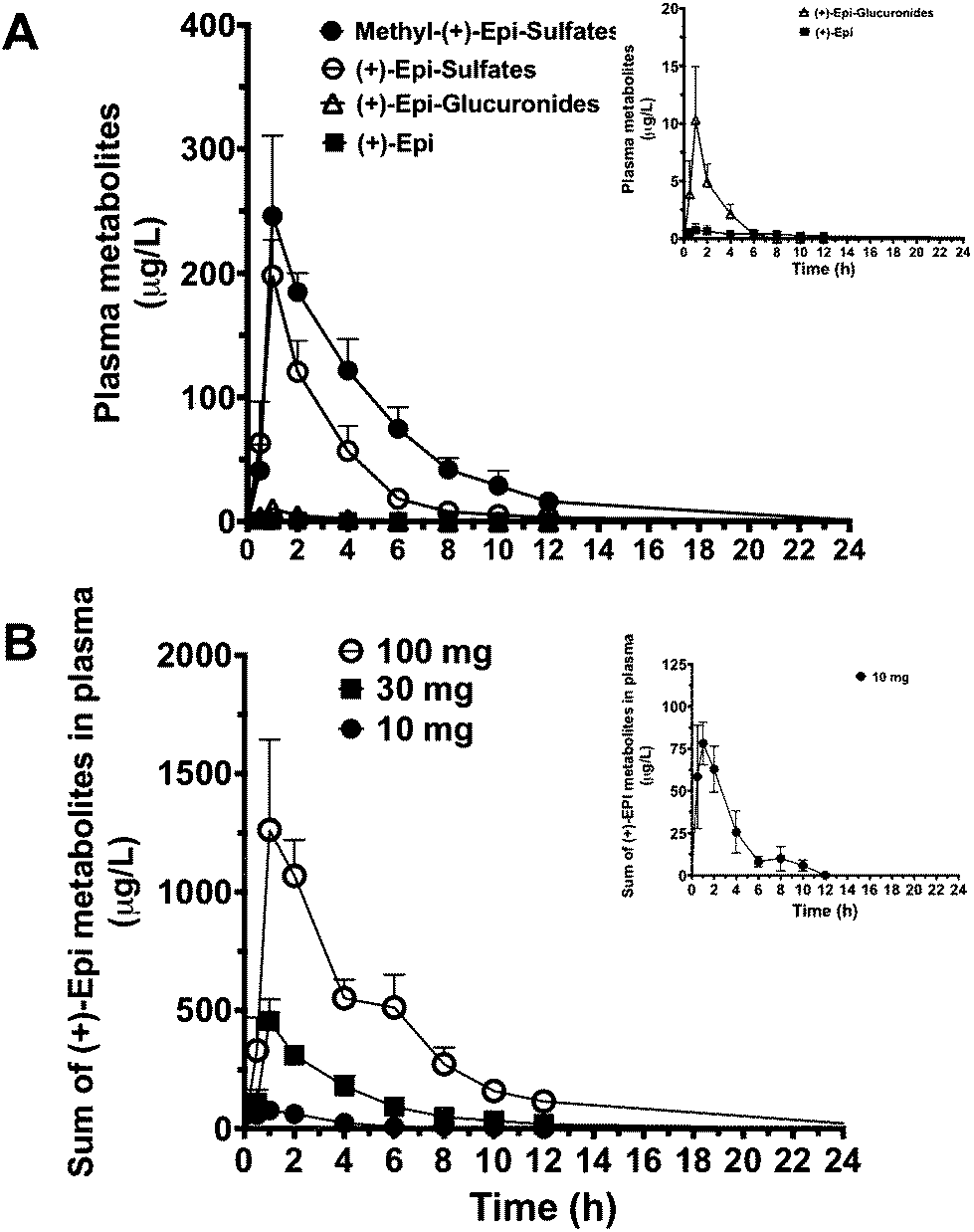 | ||
| Fig. 3 (+)-Epicatechin ((+)-Epi) and metabolite profile curves. (A) Concentration vs. time profile of (+)-Epi and its metabolites after oral administration of single 30 mg dose (n = 4). For a better appreciation, the graph in the upper right corner shows the concentration vs. time profile of (+)-Epi-glucuronide metabolites due to its low plasma abundance. (B) Area under the curve (AUC0–24h) of the sum of (+)-Epi metabolites after the oral administration of 10, 30 and 100 mg (+)-Epi single doses in pre-diabetic individuals (n = 4 per dose). For a better appreciation, the graph in the upper right corner shows the AUC0–24h of 10 mg (+)-Epi dose. (+)-Epi-sulfate is the sum of two metabolites. (+)-Epi-glucuronide is the sum of two metabolites. Methyl-(+)-Epi-Sulfate is the sum of three metabolites. Results are expressed as means with their standard errors. | ||
| 10 mg | 30 mg | 100 mg | ||||
|---|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | Mean | SEM | |
| Data is expressed as mean and SEM. Cmax, maximum plasma concentration; tmax, time to reach Cmax; AUC, area under the plasma concentration–time curve; Kel, terminal elimination constant; t1/2, terminal elimination half life. ND, not determined.a Tmax is median and range. | ||||||
| (+)-Epi | ||||||
| C max (μg L−1) | 0.4 | 0.1 | 0.7 | 0.5 | 3.6 | 2.9 |
|
T
maxa (h) |
1.0 | 0–10.0 | 1.0 | 0.5–4 | 1.0 | 0.5–10 |
| K el (h−1) | ND | ND | ND | |||
| t 1/2 (h) | ND | ND | ND | |||
| AUC0–24 (μg L−1 h−1) | 4.6 | 1.9 | 6.9 | 3.8 | 14.2 | 5.5 |
| AUC0-inf (μg L−1 h−1) | ND | ND | ND | |||
| (+)-Epi-glucuronide | ||||||
| C max (μg L−1) | 0.9 | 0.42 | 10.3 | 4.6 | 52.4 | 24.1 |
|
T
maxa (h) |
1.0 | 0.5–2.0 | 1.0 | 1.0 | 1.0 | 1–2 |
| K el (h−1) | ND | 0.3 | 0.06 | 0.2 | 0.03 | |
| t 1/2 (h) | ND | 2.3 | 0.5 | 2.6 | 0.2 | |
| AUC0–24 (μg L−1 h−1) | 1.5 | 0.5 | 22.0 | 9.1 | 185.8 | 68.2 |
| AUC0-inf (μg L−1 h−1) | ND | ND | 30.1 | 9.4 | 200.6 | 70.7 |
| Methyl-(+)-Epi-sulfate | ||||||
| C max (μg L−1) | 41.5 | 7.9 | 246.0 | 64.8 | 841.1 | 290.2 |
|
T
maxa (h) |
1.5 | 1.0–2.0 | 1.5 | 1.0–2.0 | 1.5 | 1.0–6.0 |
| K el (h−1) | 0.4 | 0.1 | 0.2 | 0.04 | 0.18 | 0.03 |
| t 1/2 (h) | 2.3 | 0.6 | 3.6 | 0.9 | 4.3 | 1.2 |
| AUC0–24 (μg L−1 h−1) | 176 | 40.6 | 1137 | 93.3 | 4866 | 393.5 |
| AUC0-inf (μg L−1 h−1) | 199.4 | 55.6 | 1199 | 115.8 | 5059 | 492.4 |
| (+)-Epi-sulfate | ||||||
| C max (μg L−1) | 36.3 | 19.3 | 197.9 | 28.9 | 365.1 | 67.3 |
|
T
maxa (h) |
0.75 | 0.5–2.0 | 1.0 | 1.0 | 1.0 | 1–2 |
| K el (h−1) | 0.7 | 0.2 | 0.2 | 0.06 | 0.2 | 0.05 |
| t 1/2 (h) | 1.2 | 0.4 | 4.9 | 2.7 | 4.1 | 1.7 |
| AUC0–24 (μg L−1 h−1) | 101.2 | 19.6 | 563.7 | 79.6 | 1599 | 268.9 |
| AUC0-inf (μg L−1 h−1) | 116.8 | 23.1 | 580.0 | 76.8 | 1684 | 310.9 |
| Single dose of (+)-Epi | ||||||
|---|---|---|---|---|---|---|
| 10 mg | 30 mg | 100 mg | ||||
| Parameters | Mean | SEM | Mean | SEM | Mean | SEM |
| C max, maximum plasma concentration; tmax, time to reach Cmax; AUC, area under the plasma concentration–time curve; Kel, terminal elimination constant; t1/2, terminal elimination half life.a tmax is median and range.b In parenthesis, concentrations are expressed in umol L−1 units.c In parenthesis, SEM of umol L−1 units. | ||||||
| C max (μg L−1) | 78.2 (0.207)b | 12.6 (0.03)c | 455.0 (1.2) | 93.5 (0.24) | 1262.5 (3.3) | 379.7 (0.98) |
|
T
maxa (h) |
1.0 | 0.5–2.0 | 1.0 | 1.0–2.0 | 1.5 | 1.0–6.0 |
| K el (h−1) | 0.44 | 0.15 | 0.21 | 0.04 | 0.19 | 0.03 |
| t ½ (h) | 2.04 | 0.5 | 3.7 | 0.9 | 4.2 | 1.2 |
| AUC0–24 (μg L−1 h−1) | 282.9 | 56.6 | 1730 | 144.1 | 6666 | 675.3 |
| AUC0-inf (μg L−1 h−1) | 307.1 | 70.07 | 1804 | 154.3 | 6904 | 818.3 |
After 7 days of treatment with 30 mg capsules, the basal concentration of flavanol and its metabolites was 133.4 ± 57.8 μg L−1 (0.35 ± 0.153 μmol L−1) in the (+)-Epi group compared to baseline levels of 15.3 ± 2.9 μg L−1 (0.039 ± 0.007 μmol L−1) in the same group before (+)-Epi dosing. No change in basal concentrations of flavanol and metabolites was observed in the placebo group before (12.2 ± 2.0 μg L−1 [0.031 ± 5.1 μmol L−1]) and after 7 days dosing (15.0 ± 2.9 μg L−1 [0.036 ± 0.007 μmol L−1]). It is worth mentioning that (+)-Epi is not commonly present in the human diet and because we did not use chiral chromatography, basal flavanol and metabolites concentrations, before dosing with either (+)-Epi or placebo could not be assigned specifically to a particular flavanol isomer. However, the higher basal flavanol and metabolites levels after 7 days of (+)-Epi dosing suggest the presence of (+)-Epi metabolites.
| (+)-Epi | Placebo | |||
|---|---|---|---|---|
| Parameter | Baseline | Day 7 | Baseline | Day 7 |
| Data is expressed as mean ± SEM. (+)-Epi-(+)-epicatechin; AAT – alanine aminotransferase; AST – aspartate aminotransferase; AP – alkaline phosphatase; EGFR – estimated glomerular filtration rate; BUN – blood urea nitrogen. | ||||
| White cell count (m3) | 6.9 ± 0.4 | 7.3 ± 0.7 | 5.8 ± 0.8 | 5.9 ± 0.8 |
| Hemoglobin (g dL−1) | 14.5 ± 0.3 | 14.6 ± 0.2 | 14.2 ± 0.5 | 14.3 ± 0.6 |
| Hematocrit (%) | 44.5 ± 0.8 | 42.7 ± 0.5 | 41.2 ± 1.1 | 41.4 ± 1.2 |
| Sodium (Meq L−1) | 138.4 ± 0.8 | 138.1 ± 0.5 | 138.6 ± 0.7 | 137.6 ± 0.8 |
| Potassium (Meq L−1) | 4.1 ± 0.1 | 4.1 ± 0.1 | 4.3 ± 0.4 | 4.2 ± 0.1 |
| Bicarbonate (Meq L−1) | 25.7 ± 0.6 | 25.9 ± 0.8 | 26.8 ± 1.3 | 25.7 ± 0.9 |
| Total protein (g dL−1) | 6.6 ± 0.1 | 6.3 ± 0.3 | 6.6 ± 0.2 | 6.8 ± 0.4 |
| AST (U L−1) | 23.0 ± 1.8 | 23.0 ± 1.2 | 24.0 ± 1.6 | 26.8 ± 1.3 |
| AAT (U L−1) | 26.2 ± 2.1 | 26.1 ± 2.4 | 25.0 ± 2.8 | 28.2 ± 2.9 |
| AP (U L−1) | 59.0 ± 8.1 | 59.0 ± 6.4 | 71.8 ± 11.4 | 73.8 ± 12.7 |
| Creatinine (mg dL−1) | 0.96 ± 0.05 | 0.91 ± 0.04 | 0.94 ± 0.09 | 0.89 ± 0.10 |
| EGFR | 79.2 ± 4.8 | 83.1 ± 5.0 | 78.2 ± 7.5 | 79.6 ± 9.1 |
| BUN (mg dL−1) | 15.7 ± 1.5 | 13.9 ± 1.1 | 10.4 ± 1.3 | 7.8 ± 0.2 |
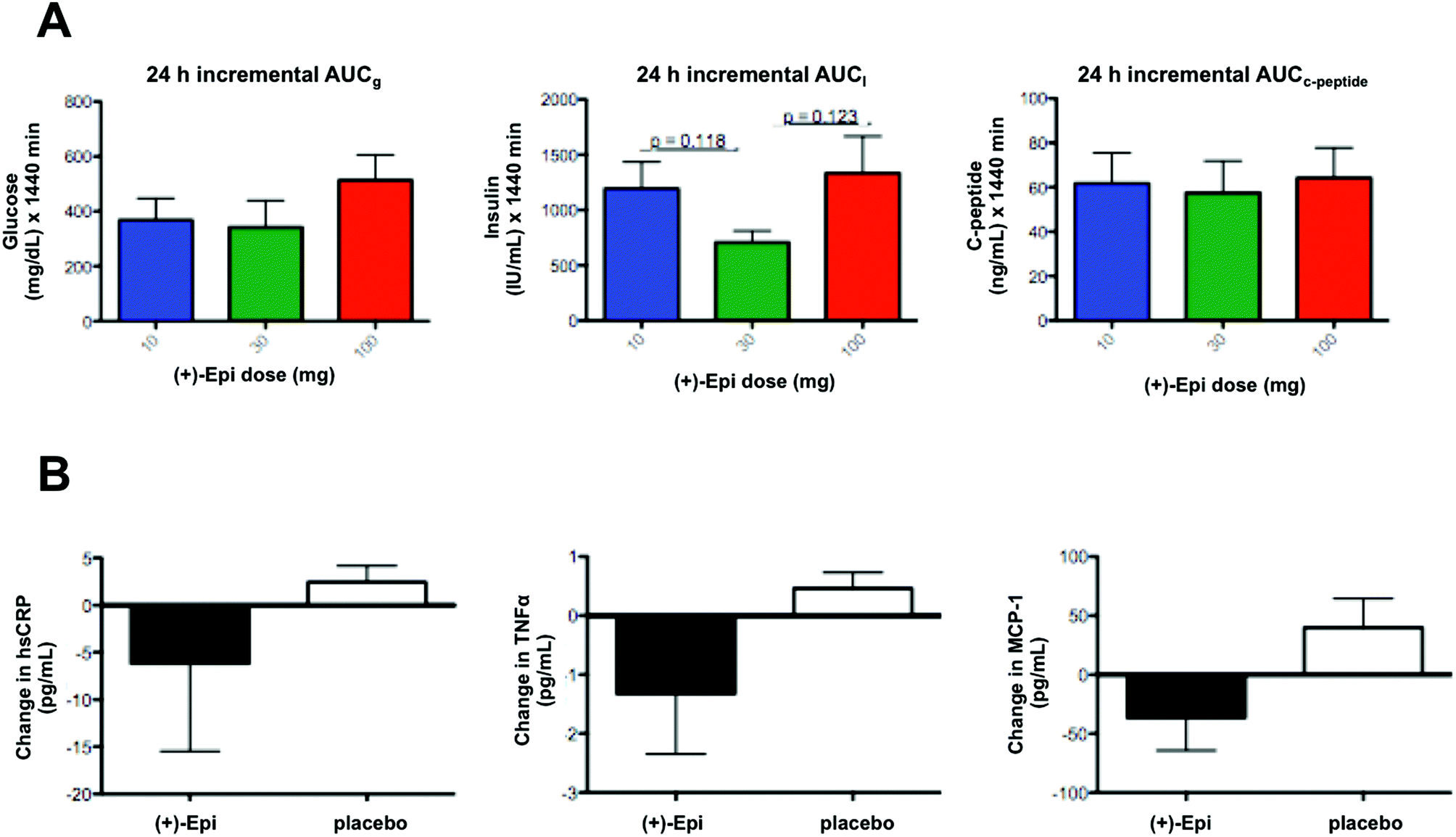 | ||
| Fig. 4 Pharmacodynamics of escalated and multiple dosages (+)-Epicatechin ((+)-Epi) orally administered in pre-diabetic individuals. (A) Dose-escalation (10, 30 and 100 mg) study of (+)-Epi on glycemic control as determined by the total excursions in serum glucose (AUCg), insulin (AUCI) and c-peptide (AUCc-peptide) over a period of 24 h after single-doses (+)-Epi administration. (B) Effects of 7 day (+)-Epi or placebo dosing on markers of systemic inflammation. Blood high-sensitivity C-reactive protein (hsCRP), tumor necrosis factor alpha (TNFα) and monocyte chemoattractant protein (MCP-1) levels were measured before and after treatment with (+)-Epi or placebo and plotted as net changes. All subjects were provided the same 3 standardized macronutrient content meals and an evening snack over the day of the study. Data is expressed as mean ± SEM. | ||
In regards to markers of systemic inflammation, there were no significant differences. However, tendencies were noted for MCP-1 and TNFα levels to fall with (+)-Epi treatment (Fig. 4B), especially compared to the placebo group. Furthermore, there were tendencies for both MCP-1 (p = 0.12) and TNFα (p = 0.082) levels to rise at 7 days after stopping (+)-Epi treatment.
3.2. Animal model
Fig. 5A summarizes results observed on the effects of treatment on weight gain. Over the course of the 2 weeks treatment, control animals gained ∼4 g (from 25 to 29 g on average). Animals treated with Cat evidenced no significant differences in weight vs. controls. There were no differences in 24 h food intake between groups (control 4.5 ± 0.2 g, (+)-Epi 4.4 ± 0.3 g and Cat 4.4 ± 0.3 g). Fat content in feces (38 ± 1.6 mg g−1 and 31 ± 5 mg g−1, obese and obese plus (+)-Epi highest dose) was not significantly different (p = 0.207). With (+)-Epi a dose dependent reduction in weight gain was observed. Such reduction became significant at 0.01 mg kg−1 and reached a maximum at the highest dose used (0.3 mg kg−1) where animals only gained in average 0.4 g of weight. Fig. 5B illustrates the effects observed on blood glucose levels. Whereas Cat had no effect, with (+)-Epi a reduction in levels was observed which became significant at 0.01 mg kg−1 and reached a plateau with 0.03–0.3 mg kg−1. Fig. 5C illustrates changes observed in triglyceride levels. Cat had no effects whereas a significant dose dependent reduction in triglyceride levels was observed at the lowest dose used of (+)-Epi (∼220 mg dL−1 at 0.003 mg kg−1) reaching normal levels ∼140 mg dL−1 with 0.1 and 0.3 mg kg−1. In cholesterol (Fig. 5D), Cat also evidenced no effect. With (+)-Epi while no effect on cholesterol levels was seen at the lowest dose, at the 0.01 mg kg−1 and higher doses reductions on cholesterol levels were observed that reached a plateau at ∼150 mg dL−1.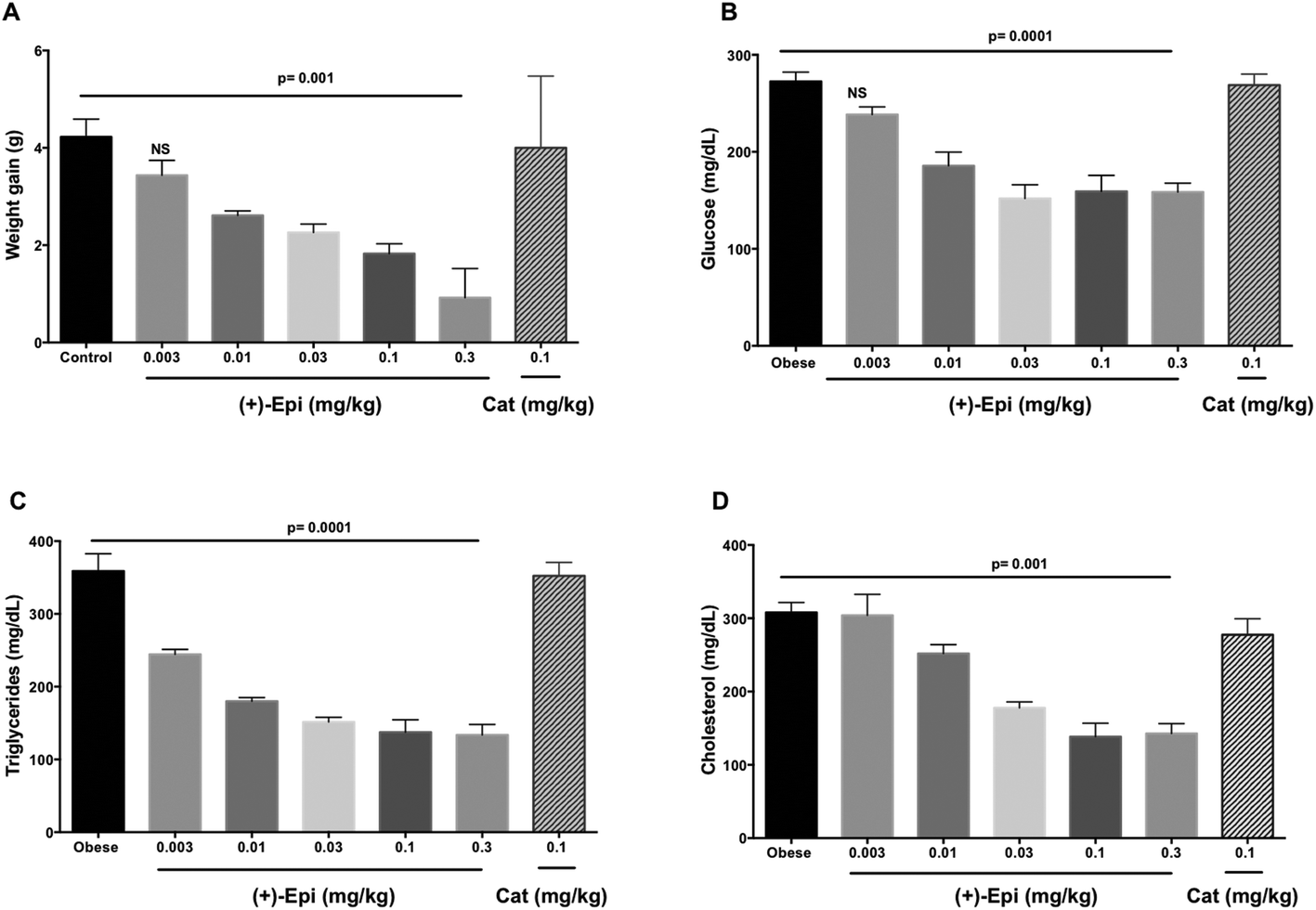 | ||
| Fig. 5 Effects of (+)-epicatechin ((+)-Epi) and (+)-catechin (Cat) on (A) body weight gain, (B) glucose, (C) triglyceride and (D) cholesterol serum levels in a high-fat diet (HFD) fed-mouse model. When animals gained >20% of body weight animals were randomly assigned to the following groups; (1) control obese group, receiving vehicle (water) only (n = 12), (2), (+)-Epi at 0.003, 0.01, 0.03, 0.1 or 0.3 mg kg−1 day−1 (n = 10 per dose) or (3), Cat at 0.1 mg kg−1 day−1 (n = 10). Flavanols were administered by daily oral gavage for 15 days while continuing with the HFD. Data is expressed as mean ± SEM. | ||
As noted in Fig. 6A, the assessment of triglyceride levels in adipose tissue yielded no differences between Cat and control whereas animals treated with (+)-Epi displayed dose dependent decreases that reached a plateau at 0.03 mg kg−1 (∼100 triglyceride [mg] per g of adipose tissue). In liver tissue (Fig. 6B), parallel observations were noted in triglyceride levels. Cat had no effect vs. (+)-Epi which yielded significant reductions from ∼37 to ∼20 triglyceride (mg) per g of liver tissue levels.
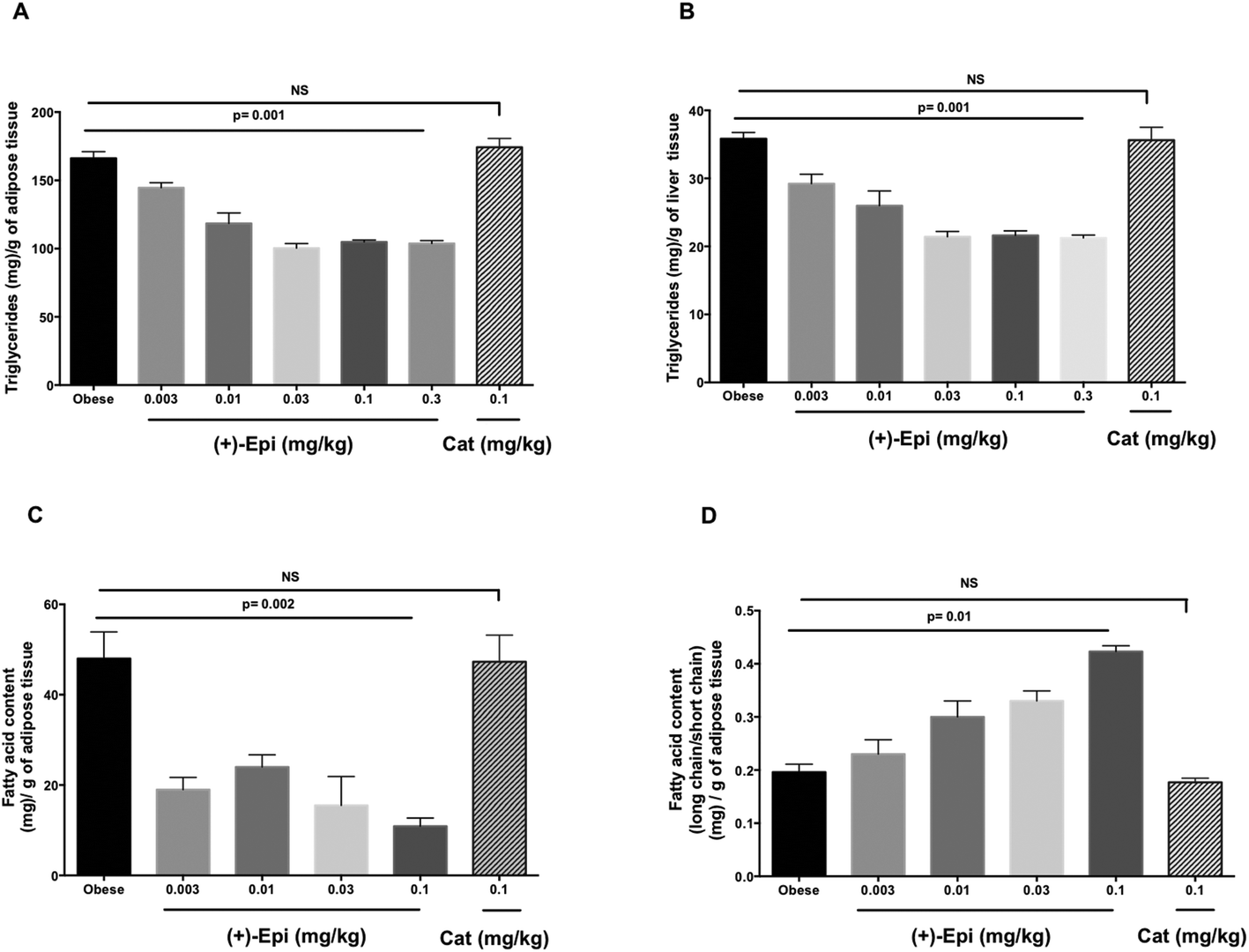 | ||
| Fig. 6 Effects of (+)-epicatechin ((+)-Epi) and (+)-catechin (Cat) on (A) adipose triglyceride, (B) hepatic triglyceride, (C) total adipose fatty acids and (D) adipose long chain/short chain fatty acids in a high-fat diet (HFD) fed-mouse model. When animals gained >20% of body weight animals were randomly assigned to the following groups; (1) control obese group receiving vehicle (water) only (n = 12), (2), (+)-Epi at 0.003, 0.01, 0.03, 0.1 or 0.3 mg kg−1 day−1 (n = 10 per dose) and (3), Cat at 0.1 mg kg−1 day−1 (n = 10). Flavanols were administered by daily oral gavage for 15 days while continuing with the HFD. Data is expressed as mean ± SEM. NS, non significant. | ||
Fig. 6C reports on the assessment of fatty acid content of adipose tissue with treatments. The use of Cat evidenced no effects whereas (+)-Epi yielded notable reductions (from ∼48 to ∼10 fatty acid content (mg) per g of adipose tissue levels at the highest dose tested). As shown in Fig. 6D, the assessment of long/short chain fatty acid content in adipose tissue demonstrated no effect for Cat vs. (+)-Epi, whereby a dose dependent effect was noted that peaked at (+)-Epi 0.1 mg kg−1 dose.
4. Discussion
In this study for the first time, we report on the PK profile of single ascending doses of (+)-Epi (up to 24 h after dosing) a flavanol that is not commonly found in human-based foods. Due to the absence of authentic (+)-Epi metabolite standards, we employed both HR/high accuracy-MS and Tandem MS to elucidate and quantify (+)-Epi metabolites. Based on our method, we were able to detect various (+)-Epi metabolites in the form of glucuronides, sulfates and methyl-sulfates (with a mass accuracy of <3 ppm). However, we cannot assign the exact position of substituents (i.e., 3′-,4′-, 5- and 7-OH site) in (+)-Epi. Previously, Ottaviani et al., performed two studies and reported on a limited PK analysis of single dose (1.5 mg kg−1) oral administration of (+)-Epi formulated in a cocoa-based, dairy-containing drink matrix in healthy individuals.12,23 Ottaviani et al., demonstrated that (+)-Epi is readily absorbed and undergoes rapid phase 2 metabolism.Using metabolite standards of the stereoisomer (−)-Epi, Ottaviani et al. suggested (+)-Epi-3′-β-D-glucuronide, (+)-Epi-5-sulfate and (+)-Epi-3′-sulfate as circulatory entities after (+)-Epi oral ingestion, whereby the most and less abundant metabolites suggested were (+)-Epi-5-sulfate and (+)-Epi-3′-β-D-glucuronide, respectively. In agreement with them, we detected (+)-Epi-glucuronides in low plasma abundance while (+)-Epi-sulfates were found in higher levels. However, the main metabolites found in this study were in the methyl-sulfated form. Interestingly, when we compare these results to our previous PK study of the stereoisomer (−)-Epi,25 some differences are apparent regarding metabolism. For example, at equal oral (+)-Epi and (−)-Epi doses (100 mg, single dose), glucuronide metabolites appeared in human plasma in higher levels after (−)-Epi ingestion, in agreement with the above mentioned study from Ottaviani et al., whereby a comparison between both isomers human metabolism was also performed.23 Additionally, in this study, the half-life values estimated for (+)-Epi metabolites (range 2.6–4.3 hours, 100 mg dose) are higher than those reported for (−)-Epi metabolites (range 1.2–2.9 hours, 100 mg dose),25 which may suggests that (+)-Epi could be advantageously administered less frequently than (−)-Epi for potential therapeutic purposes, if plasma accumulation is desired.
Results also indicate that the administration of (+)-Epi was well tolerated and did not lead to any significant alterations in safety parameters (e.g. blood pressure and heart rate) or measured laboratory endpoints and was apparently, well tolerated. Noteworthy, this study was statistically underpowered to detect meaningful changes in metabolic and systemic inflammatory endpoints by (+)-Epi treatment in insulin resistant individuals. In this regard, endpoint data variance generated from multiple dosing with (−)-Epi 30 mg day−1, will assist investigators in the calculation of sample size to be used in future studies interested in assessing the impact of treatment on metabolic or inflammatory endpoints.
In our published studies using a HFD rat model, 1 mg kg−1 day−1 of (−)-Epi yielded significant positive effects on CM endpoints including reduced weight gain, glucose and triglyceride blood levels.16 In preliminary studies, using normal mice a comparable effect on metabolic endpoints could be attained with a 0.1–0.3 mg kg−1 day−1 dose of (+)-Epi as with 1 mg kg−1 day−1 of (−)-Epi suggesting a greater efficacy for the former.22 Cell based assays also suggested that (+)-Epi may be effective at even lower concentrations vs. (−)-Epi.28 In order to gain a more comprehensive assessment of (+)-Epi's PD profile, a preclinical trial was implemented to cover a range of 0.003–0.3 mg kg−1 day−1 using 5 different doses. As noted in results, treatment with (+)-Epi (in most of the parameters measured) led to a stepwise improvement in CM endpoints, and effects were not related to changes in food intake or lipid absorption. At a dose of 0.01 mg kg−1 day−1, significant effects were noted in most endpoints, which appeared to stabilize at 0.03–0.1 mg kg−1 day−1 and beyond. These data appear to confirm our preliminary results as to the apparent greater efficacy of this rare flavanol and suggests that future clinical trials may have to implement a dose ranging study to best design a Phase II trial. If these “low” doses prove to be effective, the chances of developing side effects or toxicity will also be reduced.
The apparent difference in efficacy observed between both stereoisomers suggests that the compounds may interact with specific acceptor/receptor molecules to exert their effects. In this regard, we recently reported on the capacity of (−)-Epi to bind and activate a family of G-protein coupled receptors known to bind estrogens (i.e., GPER),29 a receptor involved in metabolism,30 which may hint at GPER as a likely candidate also for (+)-Epi.
As to the possible mechanisms of action in play, we previously reported using the HFD rat model that treatment with 1 mg kg−1 day−1 of (−)-Epi led to a significant increase in adipose tissue and skeletal muscle peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and sirtuin 1 and 3 protein levels.16 These factors are recognized modulators of energy metabolism (e.g., in glucose transport and FA catabolism) and mitochondrial biogenesis.31,32 Increases in protein levels for Transcription Factor A, Mitochondrial (TFAM) and mitofilin, which reflect a positive impact on mitochondria biogenesis and structure, were also reported as well as in the levels of mitochondrial uncoupling proteins such as uncoupling protein 1 (UCP1). In our preliminary studies comparing (+)-Epi and (−)-Epi, we reported on the stimulation in skeletal muscle of upstream signaling pathways that are recognized as key regulators of metabolism such as phosphorylated liver kinase B1 (p-LKB1) and adenosine monophosphate kinase (p-AMPK) as well as in PGC-1α protein levels.22 Comparable levels of stimulation were noted when using 0.1 mg kg−1 (+)-Epi as with 1 mg kg−1 of (−)-Epi. Thus, as we have previously reported the effects of the stereoisomers appear to involve the activation of signaling pathways that are recognized as key in modulating cellular bioenergetics and metabolism.
We previously reported on the lack of capacity of the stereoismer Cat to reduce fructose induced triglyceride synthesis and mitigate mitochondria generated reactive oxygen species in cultured Hepg2 cells.33 This observation contrasted to that noted for (−)-Epi, which was essentially effective in completely blocking fructose-induced effects. The beneficial actions of flavonoids are commonly attributed to their antioxidant actions.34 Since Cat and (−)-Epi are also stereoisomers, their antioxidant potential is equivalent, thus, the use of either type of the flavanols should generate comparable effects. Results from Hepg2 treated cells indicate that Cat failed to exert any effect, essentially discarding such theory. To further address this matter in the in vivo setting, we included in the treatment groups Cat, which is the 2nd most abundant cacao flavanol. As the results again evidence, the effects are dissimilar between (−)-Epi and Cat and further contradict the common theory of how flavonoids exert their antioxidant actions. In fact, we reported that the use of (−)-Epi rich chocolate could effectively modulate antioxidant (e.g. glutathione) and enzyme systems (e.g. superoxide dismutase and catalase) that are responsible for metabolizing (i.e. neutralizing) reactive oxygen species, leading to lesser degrees of oxidative stress.35
Pertaining to the impact that the use of (−)-Epi has on subjects with altered CM endpoints we recently reported on the effects that one-month dosing of (−)-Epi (100 mg day−1, i.e., ∼1 mg kg−1 day−1) had on triglyceride levels.33 Results from the double-blind placebo controlled trial implemented in subjects with hypertriglyceridemia evidenced a significant decrease in triglyceride blood levels. Furthermore, in those subjects that had high glucose levels, a significant decrease in fructosamine was observed, hinting at possible beneficial glucose regulating effects.
On the basis of the (+)-Epi PK and limited PD and efficacy data obtained from this study and the known effects of (−)-Epi it should be possible to design properly powered, long-term, randomized, placebo controlled Phase II trials that are designed to examine (+)-Epi's potential to beneficially impact CM endpoints.
5. Study limitations
This is a pilot study that included a small number of participants, which evaluated the initial PK profile of (+)-Epi and may not fully represent what may be seen in the general population. The method used for (+)-Epi metabolites quantification was based on the aglycone MS response not on individual metabolites, given the absence of authentic available standards. Such approach may under or overestimate true plasma (+)-Epi metabolite values. Due to high inter-individual variability observed in (+)-Epi metabolism as per metabolites AUC0-inf levels (i.e., 95% confidence intervals, 84.08–530 μg L−1, 1313–2295 μg L−1 and 4300–9508 μg L−1 at 10, 30 and 100 mg dose, respectively), the interpretations of PK parameters between single (+)-Epi doses must be done with caution. A crossover study design will allow a more precise comparison on this matter, as (+)-Epi metabolism intra-individual variability is reported as lower than inter-individual variability. The study was also statistically underpowered and of limited duration to detect differences between placebo and (+)-Epi treatment regarding PD outcomes.6. Conclusion
In conclusion, the administration of (+)-Epi leads to a PK profile that indicates that the flavanol is well absorbed and a series of blood metabolites appear in blood with a 2–4 h half-life. The compound is well tolerated and yields no evidence of adverse effects. In animals, the administration of (+)-Epi yields highly significant effects on CM endpoints at very low doses indicating a unique potency and efficacy profile for this flavanol that needs to be taken into consideration in the design of future clinical trials.Funding
The study was supported by NIH R01 AT8310 to Drs Henry and Villarreal, R01 DK98717 (Villarreal) and CONACyT 253769 and Cardero Therapeutics, Inc. (Ceballos). This material is the result of work supported with resources and the use of the facilities at the VASDHCS.Conflicts of interest
Drs Taub and Villarreal are co-founders and stockholders of Cardero Therapeutics, Inc. Dr Schreiner and Dugar are employed by Cardero Therapeutics and Dr Ceballos is a stockholder of the company.References
- M. Aguilar, T. Bhuket, S. Torres, B. Liu and R. J. Wong, J. Am. Med. Assoc., 2015, 313, 1973–1974 CrossRef CAS PubMed.
- M. Ng, T. Fleming, M. Robinson, B. Thomson, N. Graetz, C. Margono, E. C. Mullany, S. Biryukov, C. Abbafati, S. F. Abera, J. P. Abraham, N. M. Abu-Rmeileh, T. Achoki, F. S. AlBuhairan, Z. A. Alemu, R. Alfonso, M. K. Ali, R. Ali, N. A. Guzman, W. Ammar, P. Anwari, A. Banerjee, S. Barquera, S. Basu, D. A. Bennett, Z. Bhutta, J. Blore, N. Cabral, I. C. Nonato, J. C. Chang, R. Chowdhury, K. J. Courville, M. H. Criqui, D. K. Cundiff, K. C. Dabhadkar, L. Dandona, A. Davis, A. Dayama, S. D. Dharmaratne, E. L. Ding, A. M. Durrani, A. Esteghamati, F. Farzadfar, D. F. Fay, V. L. Feigin, A. Flaxman, M. H. Forouzanfar, A. Goto, M. A. Green, R. Gupta, N. Hafezi-Nejad, G. J. Hankey, H. C. Harewood, R. Havmoeller, S. Hay, L. Hernandez, A. Husseini, B. T. Idrisov, N. Ikeda, F. Islami, E. Jahangir, S. K. Jassal, S. H. Jee, M. Jeffreys, J. B. Jonas, E. K. Kabagambe, S. E. Khalifa, A. P. Kengne, Y. S. Khader, Y. H. Khang, D. Kim, R. W. Kimokoti, J. M. Kinge, Y. Kokubo, S. Kosen, G. Kwan, T. Lai, M. Leinsalu, Y. Li, X. Liang, S. Liu, G. Logroscino, P. A. Lotufo, Y. Lu, J. Ma, N. K. Mainoo, G. A. Mensah, T. R. Merriman, A. H. Mokdad, J. Moschandreas, M. Naghavi, A. Naheed, D. Nand, K. M. Narayan, E. L. Nelson, M. L. Neuhouser, M. I. Nisar, T. Ohkubo, S. O. Oti, A. Pedroza, D. Prabhakaran, N. Roy, U. Sampson, H. Seo, S. G. Sepanlou, K. Shibuya, R. Shiri, I. Shiue, G. M. Singh, J. A. Singh, V. Skirbekk, N. J. Stapelberg, L. Sturua, B. L. Sykes, M. Tobias, B. X. Tran, L. Trasande, H. Toyoshima, S. van de Vijver, T. J. Vasankari, J. L. Veerman, G. Velasquez-Melendez, V. V. Vlassov, S. E. Vollset, T. Vos, C. Wang, X. Wang, E. Weiderpass, A. Werdecker, J. L. Wright, Y. C. Yang, H. Yatsuya, J. Yoon, S. J. Yoon, Y. Zhao, M. Zhou, S. Zhu, A. D. Lopez, C. J. Murray and E. Gakidou, Lancet, 2014, 384, 766–781 CrossRef.
- N. Esser, S. Legrand-Poels, J. Piette, A. J. Scheen and N. Paquot, Diabetes Res. Clin. Pract., 2014, 105, 141–150 CrossRef CAS PubMed.
- E. Ferrannini and R. A. DeFronzo, Eur. Heart J., 2015, 36, 2288–2296 CrossRef PubMed.
- X. Rossello, S. J. Pocock and D. G. Julian, J. Am. Coll. Cardiol., 2015, 66, 1273–1285 CrossRef CAS PubMed.
- C. Buettner, M. J. Rippberger, J. K. Smith, S. G. Leveille, R. B. Davis and M. A. Mittleman, Am. J. Med., 2012, 125, 176–182 CrossRef CAS PubMed.
- B. Buijsse, E. J. Feskens, F. J. Kok and D. Kromhout, Arch. Intern. Med., 2006, 166, 411–417 Search PubMed.
- M. L. McCullough, J. J. Peterson, R. Patel, P. F. Jacques, R. Shah and J. T. Dwyer, Am. J. Clin. Nutr., 2012, 95, 454–464 CrossRef CAS PubMed.
- J. I. Dower, J. M. Geleijnse, P. Hollman, S. S. Soedamah-Muthu and D. Kromhout, Am. J. Clin. Nutr., 2016, 104, 58–64 CrossRef PubMed.
- L. Hooper, P. A. Kroon, E. B. Rimm, J. S. Cohn, I. Harvey, K. A. Le Cornu, J. J. Ryder, W. L. Hall and A. Cassidy, Am. J. Clin. Nutr., 2008, 88, 38–50 CAS.
- P. J. Mink, C. G. Scrafford, L. M. Barraj, L. Harnack, C. P. Hong, J. A. Nettleton and D. R. Jacobs Jr., Am. J. Clin. Nutr., 2007, 85, 895–909 CAS.
- J. I. Ottaviani, T. Y. Momma, C. Heiss, C. Kwik-Uribe, H. Schroeter and C. L. Keen, Free Radical Biol. Med., 2011, 50, 237–244 CrossRef CAS PubMed.
- I. Ramirez-Sanchez, A. Rodriguez, A. Moreno-Ulloa, G. Ceballos and F. Villarreal, Diabetes Vasc. Dis. Res., 2016, 13, 201–210 CrossRef CAS PubMed.
- I. Cordero-Herrera, M. A. Martin, L. Bravo, L. Goya and S. Ramos, Mol. Nutr. Food Res., 2013, 57, 974–985 CAS.
- E. Cremonini, A. Bettaieb, F. G. Haj, C. G. Fraga and P. I. Oteiza, Arch. Biochem. Biophys., 2016, 599, 13–21 CrossRef CAS PubMed.
- G. Gutierrez-Salmean, P. Ortiz-Vilchis, C. M. Vacaseydel, L. Garduno-Siciliano, G. Chamorro-Cevallos, E. Meaney, S. Villafana, F. Villarreal, G. Ceballos and I. Ramirez-Sanchez, Eur. J. Pharmacol., 2014, 728, 24–30 CrossRef CAS PubMed.
- H. Schroeter, C. Heiss, J. Balzer, P. Kleinbongard, C. L. Keen, N. K. Hollenberg, H. Sies, C. Kwik-Uribe, H. H. Schmitz and M. Kelm, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1024–1029 CrossRef CAS PubMed.
- J. I. Dower, J. M. Geleijnse, L. Gijsbers, P. L. Zock, D. Kromhout and P. C. Hollman, Am. J. Clin. Nutr., 2015, 101, 914–921 CrossRef CAS PubMed.
- G. Gutierrez-Salmean, P. Ortiz-Vilchis, C. M. Vacaseydel, I. Rubio-Gayosso, E. Meaney, F. Villarreal, I. Ramirez-Sanchez and G. Ceballos, Food Funct., 2014, 5, 521–527 CAS.
- M. P. Michael Kofink and R. Galensa, Eur. Food Res. Technol., 2006, 225, 569–577 CrossRef.
- M. Kofink, M. Papagiannopoulos and R. Galensa, Molecules, 2007, 12, 1274–1288 CrossRef CAS PubMed.
- G. C. Israel Ramirez-Sanchez, A. Moreno-Ulloa, T. P. Ciaraldi, R. R. Henry and F. Villarreal, presented in part at the American Diabetes Association 74th Scientific Sessions, San Francisco, CA, June 14, 2014 Search PubMed.
- J. I. Ottaviani, T. Y. Momma, G. K. Kuhnle, C. L. Keen and H. Schroeter, Free Radical Biol. Med., 2012, 52, 1403–1412 CrossRef CAS PubMed.
- A. American Diabetes, Diabetes Care, 2017, 40, S11–S24 CrossRef PubMed.
- C. F. Barnett, A. Moreno-Ulloa, S. Shiva, I. Ramirez-Sanchez, P. R. Taub, Y. Su, G. Ceballos, S. Dugar, G. Schreiner and F. Villarreal, Food Funct., 2015, 6, 824–833 CAS.
- E. Roura, C. Andres-Lacueva, O. Jauregui, E. Badia, R. Estruch, M. Izquierdo-Pulido and R. M. Lamuela-Raventos, J. Agric. Food Chem., 2005, 53, 6190–6194 CrossRef CAS PubMed.
- K. F. Blom, J. Am. Soc. Mass Spectrom., 1998, 9, 789–798 CrossRef CAS.
- G. C. Israel Ramirez-Sanchez, A. Moreno-Ulloa, T. P. Ciaraldi, R. R. Henry and F. Villarreal, American Diabetes Association, 2014 Search PubMed.
- A. Moreno-Ulloa, D. Mendez-Luna, E. Beltran-Partida, C. Castillo, G. Guevara, I. Ramirez-Sanchez, J. Correa-Basurto, G. Ceballos and F. Villarreal, Pharmacol. Res., 2015, 100, 309–320 CrossRef CAS PubMed.
- G. Sharma and E. R. Prossnitz, Methods Mol. Biol., 2016, 1366, 489–502 CAS.
- R. Ventura-Clapier, A. Garnier and V. Veksler, Cardiovasc. Res., 2008, 79, 208–217 CrossRef CAS PubMed.
- M. N. Sack and T. Finkel, Cold Spring Harbor Perspect. Biol., 2012, 4 CAS , a013102.
- G. Gutierrez-Salmean, E. Meaney, M. A. Lanaspa, C. Cicerchi, R. J. Johnson, S. Dugar, P. Taub, I. Ramirez-Sanchez, F. Villarreal, G. Schreiner and G. Ceballos, Int. J. Cardiol., 2016, 223, 500–506 CrossRef PubMed.
- R. J. Nijveldt, E. van Nood, D. E. van Hoorn, P. G. Boelens, K. van Norren and P. A. van Leeuwen, Am. J. Clin. Nutr., 2001, 74, 418–425 CAS.
- I. Ramirez-Sanchez, P. R. Taub, T. P. Ciaraldi, L. Nogueira, T. Coe, G. Perkins, M. Hogan, A. S. Maisel, R. R. Henry, G. Ceballos and F. Villarreal, Int. J. Cardiol., 2013, 168, 3982–3990 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C7FO01028A |
| This journal is © The Royal Society of Chemistry 2018 |