
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium nanoparticles induce autophagy and autophagic flux blockade in Hela cells†
Li Zhanga,
Xuerui Chena,
Jianzhou Wua,
Shiping Dingb,
Xu Wangc,
Qunfang Lei*a and
Wenjun Fang *a
*a
aDepartment of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: fwjun@zju.edu.cn; qflei@zju.edu.cn; Fax: +86-571-88981416
bSchool of Medicine, Zhejiang University, Hangzhou 310058, China
cHangzhou Medical College, No. 481 Binwen Road, Hangzhou 310053, China
First published on 23rd January 2018
Abstract
Autophagy is a lysosome-based degradative pathway associated with cancer. As a novel class of autophagy activator, nanoparticles (NPs) have been recently found to have potential applications in clinical therapy. Palladium nanoparticles (PdNPs), which have unique physical and chemical properties, have been used in biosensing and biological imaging. In the present study, size-dependent PdNPs-induced autophagy and autophagic flux blockade in Hela cells were investigated. By monitoring the transformation of autophagosome marker protein LC3, the intensity of fluorescence labeling and the quantity of autophagosomes, autophagosome accumulation with increasing concentration and varying size of PdNPs was observed. The slowed degradation of autophagy substrate p62 and long-lived proteins together with the impairment of lysosomes indicates that PdNPs treatment results in a decrease of the degradation capability of lysosomes and blockade of autophagic flux. In this work, PdNPs were found to affect autophagosome accumulation in two ways. One is led by autophagy activated through the mTOR signaling pathway at low concentration, and another is dominated by autophagic flux blockade resulting from lysosome impairment at high concentration. Autophagy in Hela cells could be effectively regulated by controlling the concentration and size of PdNPs; this provides an important reference for future applications of PdNPs in biomedicine.
1. Introduction
Autophagy is a lysosome-based degradative pathway to maintain cellular homeostasis and achieve cellular update;1 in this process, harmful proteins and damaged organelles are wrapped in double-membrane vesicles to form autophagosomes. The autophagosomes then fuse with lysosomes to form hybrid organelles named autolysosomes. The wrapped intracellular contents and the inner membranes of the autolysosomes finally degrade while releasing energy.2 Usually, autophagy is an essential approach to aid cell survival in an ever-changing environment.3 When cells have obtained sufficient external nutrients, the formation of autophagosomes occurs at a low rate and few autophagosomes accumulate in the cells. The mammalian target of rapamycin, mTOR, which plays a decisive role in negative regulation of autophagy, participates in the initiation of autophagy.4,5 Once autophagy is triggered by inhibiting the mTOR signaling pathway, expression of the mTOR activity marker phospho-p70 S6 kinase (P-S6K) is inhibited. When cells are under stress conditions, such as starvation, autophagy is activated through the mTOR signaling pathway, causing a significant accumulation of autophagosomes.6 However, increasing numbers of studies show that autophagy is strongly associated with diseases such as cancer, infection, and neurodegenerative diseases.7,8 Therefore, to manipulate the function of autophagy more effectively, it is highly desirable to gain deep insight into the mechanism of autophagy.During the autophagy process, LC3 is first deacetylated and then escapes from the nucleus to bind with phosphatidylethanolamine (PE). Eventually, it is transformed into LC3-II, which is a prerequisite to form the autophagosome membrane.9 The level of LC3-II is widely used to measure accumulation of autophagosomes. Autophagy is a dynamic process where autophagosome formation is balanced by turnover upon delivery to lysosomes. The autophagosomes accumulate when the formation rate of autophagosomes is greater than the degradation rate. Thus, accumulation of autophagosomes is a result of both the formation and degradation of autophagosomes. Autophagy induction affects the formation of autophagosomes, while the blockade of autophagic flux affects the degradation of autophagosomes.10 Lysosomes, as a degradation organelle, also play an important role in autophagy. Lysosome impairment may undermine their fusion with autophagosomes and lysosomal degradation, which further leads to blockade of autophagic flux. Thus, additional measurements, such as monitoring the degradation of the autophagy-specific substrate sequestosome 1 (p62) and detecting lysosome impairment, are needed to determine whether the autophagic flux is blocked.
Compared with bulk materials, nanomaterials have advantages of optical, thermal, mechanical and magnetic properties due to scale effects.11 In the wake of the rapid development of synthesis techniques, diverse metal nanoparticles (NPs) with unique and outstanding properties have been successfully synthesized and gradually applied in various areas, especially in the life sciences for biosensing,12,13 cell imaging,14 drug delivery,15–17 and clinical treatment.18,19 For example, NPs for targeted delivery have unique capability to localize delivery of drugs to designated tissues.20 Combining medicines with functionalized NPs may decrease their intrinsic toxicities and improve their therapeutic efficacy.21
Palladium nanoparticles (PdNPs), the applications of which mainly focus on catalysis,22,23 possess great potential in biomedicine.24 Numerous efforts have been made to fully exploit this material. For example, PdNPs are utilized in dental materials and surgical instruments.25 PdNPs conjugated with antibodies can be used for multiple ultrastructural immunolabeling.26 An n-alkylamine-stabilized PdNPs-based electrode has also been fabricated as an amperometric glucose biosensor.27 Moreover, palladium-based materials such as dithiocarbamate Pd(II) complexes28 and quinolinic acid Pd(II) complexes29 exhibit high antitumor activity against cisplatin-resistant cancer cells. Self-assembled palladium cages have been designed as prospective delivery systems for anticancer drugs.30 However, to discover potential applications of PdNPs in the pharmaceutical industry, an in-depth exploration of the impact of PdNPs on biological systems is necessary. Recently, it has been found that metal NPs can promote the expression of autophagic proteins;31,32 they have also been proved to be a novel class of autophagy activator.33 The effects of autophagy on cells can be either cyto-protective or cyto-destructive; these effects are related to the size and dose of the NPs.34 Thus, we studied three sizes of cubic PdNPs synthesized through a stable and well shape-controlled method, aiming to determine a plausible mechanism of its biological functions in autophagy. Dose- and size-dependent autophagy experiments on Hela cells treated with PdNPs were performed. Autophagy induction and lysosome impairment were further investigated to provide fundamental information for developing biological and medical applications of PdNPs.
2. Materials and methods
2.1. Preparation of PdNPs
The PdNPs were prepared using K2PdCl4 as the precursor.35 Briefly, K2PdCl4 (163 mg, 25 mmol L−1), polyvinylpyrrolidone (PVP, 278 mg, 125 mmol L−1) and KBr (0.38 g, 160 mmol L−1; 0.8 g, 336 mmol L−1; 1.5 g, 630 mmol L−1 were used for 6 nm, 12 nm and 20 nm PdNPs, respectively) were added to 15 mL of ultra-pure water (Millipore Q3 system), and they were pre-heated in a 100 mL flask at 85 °C for 5 min before adding 5 mL of aqueous solution of ascorbic acid (AA, 150 mg, 42.6 mmol L−1). After stirring for 3 h, the mixture was cooled to room temperature. The product was collected by diluting the resulting solution with acetone and centrifuging (5000 rpm) for 10 min. The black precipitations were washed five times with ultra-pure water and acetone to remove remaining PVP. The residual acetone was vaporized at room temperature. Finally, the products were stored in a glass vessel with 10 mL of ultra-pure water. Before treating the cells, the PdNPs were diluted with cell culture medium and sterilized by UV light for 30 min.2.2. Characterization of PdNPs
Micrographs of the synthesized PdNPs were recorded on a transmission electron microscope (TEM) (Hitachi, HT 7700, Japan). The PdNPs were examined after suspension in ethanol and deposition onto a copper grid coated with an ultra-thin carbon film. Then, the grids were dried under vacuum at 50 °C overnight, and the shapes and sizes of the PdNPs were observed with TEM. The hydrodynamic sizes, zeta potentials and polydispersity indices (PDI) of the PdNPs in water and Dulbecco's modified Eagle's medium (DMEM) were measured by dynamic light scattering (DLS) (Malvern, ZEN 3600, Britain). The PdNP systems were diluted to 1 mg mL−1 with ultra-pure water or DMEM.2.3. Cell culture and co-incubation with PdNPs
The Hela cell line, HEK293 cell line and HepG2 cell line were obtained from the Institute of Immunology, School of Medicine, Zhejiang University. The Hela cell line and HEK293 cell line were cultured in DMEM (Gibco BRL, Grand Island, NY) containing 10% fetal calf serum (FCS) (Gibco BRL, Grand Island, NY). The HepG2 cell line was cultured in RPMI-1640 (Gibco BRL, Grand Island, NY) containing 10% FCS. For starvation conditions, cells were cultured in DMEM (or RPMI-1640) only. Cells were incubated in a humidified atmosphere containing 5% CO2 at 37 °C. The PdNP systems were freshly prepared from stock solution and sonicated for 5 min before addition to cell cultures.2.4. Cell viability assay
Succinate dehydrogenase in the mitochondria of living cells can reduce exogenous 3-(4,5-dimethylthiazol-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) to blue-violet crystalline formazan.36 Briefly, cells were plated onto 96 multiwell plates (Costar, Corning, NY) at a density of 1.0 × 104 cells per well. After incubation with different concentrations of PdNPs ranging from 0.1 to 50 μg mL−1 at 37 °C for 12, 24, 48 and 72 h, the medium was removed and replaced with 0.5 mg mL−1 MTT. The cells were incubated at 37 °C for 4 h, and the MTT reaction product formazan was dissolved in DMSO before the OD value at the wavelength of 570 nm was measured by a microplate reader (Shimazdu, UV-2540, Japan). The cell viability was obtained by taking the ratio of the OD value of each sample to that of the control group.2.5. Microscopic analysis of Hela cells
Hela cells were incubated with 0.1, 0.5, 1.0, 5.0, and 10.0 μg mL−1 PdNPs for 24 h. Then, the cells were collected and centrifuged at 667 g for 5 min. The cell pellets were fixed with 2.5% glutaraldehyde in phosphate buffered saline (PBS) at pH 7.4 for 4 h. The samples were washed with 0.1 M PBS for about 15 min, postfixed in 1% OsO4 for 1 h, and washed again with D2O. The samples were subsequently dyed with 2% uranyl acetate for 30 min and dewatered with a gradient of different concentrations of aqueous ethanol solution (50, 70, 90, and 100 vol%) and acetone. The cell samples were infiltrated, embedded and polymerized to obtain ultra-thin slices; they were then observed by transmission electron microscope (JEOL JEM-1200EX, Japan).2.6. Cell morphology and autophagosome observation
A confocal laser scanning microscope (CLSM) was used to observe cell morphologies and the formation of autophagosomes. The green fluorescent dye dansylcadaverine (MDC) can specifically accumulate in autophagosomes to detect their presence. The cells were seeded on a 6-well plate with round coverslips at a density of 1.5 × 105 cells per well for overnight growth. Then, the cells were incubated with 0.1, 0.5, 1.0, 5.0, and 10.0 μg mL−1 PdNPs for 24 h. In the negative control experiment, the cells were incubated with an autophagy inhibitor, 3-methyladenine (3-MA), for 24 h. Then, the cells were fixed with PBS solution containing 4% paraformaldehyde for 5 min. The cells were dyed with 50 μmol L−1 MDC (Sigma-Aldrich, USA) for 15 min. Finally, the fluorescence was observed by CLSM (ZEISS, LSM710NLO, Germany).2.7. Determination of cellular uptake of PdNPs
The cells were incubated with 0.1, 0.5, 1.0, 5.0, and 10.0 μg mL−1 PdNPs for 24 h and counted with a blood corpuscle counting meter. The cell samples were digested with 0.5 mL aqua regia. Then, 0.5 mL aqueous ammonia was slowly added until the solution became colorless and clear. The samples were filtrated with 0.22 μm filters after being diluted with 0.2% HNO3 to 10 mL and were subsequently analyzed using inductively coupled plasma mass spectrometry (ICP-MS) (Thermo Elemental X7, Thermo Fisher Scientific Inc., USA). The concentrations of palladium in the acid-digested cell solutions were directly measured. The number of PdNPs per cell was calculated according to the number of PdNPs converted from the concentration of palladium in solution and the total number of cells in solution. The cellular uptake efficiency was evaluated by taking the ratio of the number of internalized PdNPs and the total number of PdNPs initially present in the culture medium. Two sets of experiments to determine the effects of temperature and adenosine triphosphate (ATP) on the cellular uptake of PdNPs were performed to investigate the mechanism of entry of PdNPs into Hela cells. In one experiment, Hela cells were cultured with 10.0 μg mL−1 PdNPs at 4 °C for 1 h. In another experiment, Hela cells were pre-incubated with 10.0 mmol L−1 NaN3 for 30 min before incubation with 10.0 μg mL−1 PdNPs at 37 °C for 1 h. In the control experiment, cells were cultured with 10.0 μg mL−1 PdNPs at 37 °C for 1 h. Then, the values of the number of PdNPs per cell were determined by ICP-MS.2.8. Western blot assay
Lysis buffer was added to the PdNPs-treated cells to extract the total protein. The target proteins LC3-I, LC3-II, p62, p70S6K and p-p70S6K were separated by SDS-PAGE (Kaiji Biotechnology Co. Ltd., Nanjing, China), and transferred to nitrocellulose filter membranes, which were blocked with 5% non-fat milk and incubated with primary antibody overnight. The corresponding secondary antibody diluted with blocking solution was added, followed by coloration with enhanced chemiluminescent (ECL) reagent. Between each step, the membrane was washed three times with Tris-buffered saline containing 0.1% Tween (TBST). The protein loading control was GAPDH expression.2.9. Long-lived protein degradation assay
Hela cells were incubated in growth medium containing [14C]-valine for 24 h and washed with PBS three times. The cells were incubated in growth medium containing unlabeled valine for 2 h to degrade short-lived proteins, which are primarily degraded through the proteasome pathway. Then, the cells were cultured in growth medium, DMEM, DMEM containing 10.0 μg mL−1 PdNPs or DMEM containing 20 μM chloroquine. After 24 h incubation, the collected cell samples were precipitated overnight by addition of 10% trichloroacetic acid (TCA). The samples were centrifuged at 4 °C, 3111g for 10 min. The pellets were washed twice with cold 10% TCA and dissolved with 0.2 mol L−1 NaOH for 2 h. The radioactivity of the pellet was measured by a scintillation counter (Beckman, LS6500). Autophagic degradation was calculated as a percentage of the chloroquine-treated sample.2.10. Evaluation of lysosomal acidity
Hela cells were treated with different concentrations of PdNPs for 24 h and washed twice with PBS. The cells were incubated for 60 min with 50 nmol L−1 Lyso-Tracker Red DND-99 dye (Beyotime Biotechnology Co. Ltd., Shanghai, China) and 1 μmol L−1 Lysosensor Green DND-189 dye (Yeasen Biotechnology Co. Ltd., Shanghai, China), respectively, and fixed with 4% paraformaldehyde for 5 min. The obtained cell samples were observed with confocal microscopy. Meanwhile, fluorescence intensity analysis was performed on the cells stained with Lysosensor Green DND-189 dye by flow cytometry. The reversible dissociation of the V1 protein from vacuolar H+-ATPase was determined by confocal microscopy.2.11. Statistical analysis
All data are presented as the mean value ± standard deviation (SD) by at least three independent parallel experiments. Statistical significance was determined using Student's t-test for comparison between the means or using one-way analysis of variance with a post hoc Dunnett's test. Differences were considered significant when p < 0.05. Images are representative of three or more experiments.3. Results
3.1. Characterization of the synthesized PdNPs
The PdNPs were synthesized using K2PdCl4 as the precursor in the presence of PVP, AA, and KBr, which were applied as stabilizer, reducing agent and capping agent, respectively. PdNPs with three sizes (6 nm, 12 nm, and 20 nm) were obtained by changing the KBr ratio. TEM images of the PdNPs are presented in Fig. 1a, showing that the three PdNPs are monodisperse cubes with statistical particle sizes of 6.3 ± 1.9 nm, 11.9 ± 2.3 nm, and 20.4 ± 1.4 nm, respectively (Fig. 1b). Characteristic data of the three PdNPs in water and DMEM are listed in Table 1. The hydrodynamic sizes of the PdNPs in both water and DMEM are similar. Compared with the TEM results, the precipitation or agglomeration of PdNPs and the hydration shell formed outside the PdNPs in solution resulted in PdNPs with relatively large sizes, as measured by DLS. The small values of the polydispersity index (PDI), which is associated with the homogeneity of the particle population,37 indicate that the three PdNPs were well-dispersed in water and DMEM. The surface charges of the three PdNPs are negative (Table 1); this is considered to be advantageous to reduce plasma protein bioadhesion, resulting in good stability of the PdNPs in solution.38 After 24 h incubation in growth medium, the PdNPs remained well dispersed and retained the same morphologies as the newly prepared PdNPs (Fig. S1†).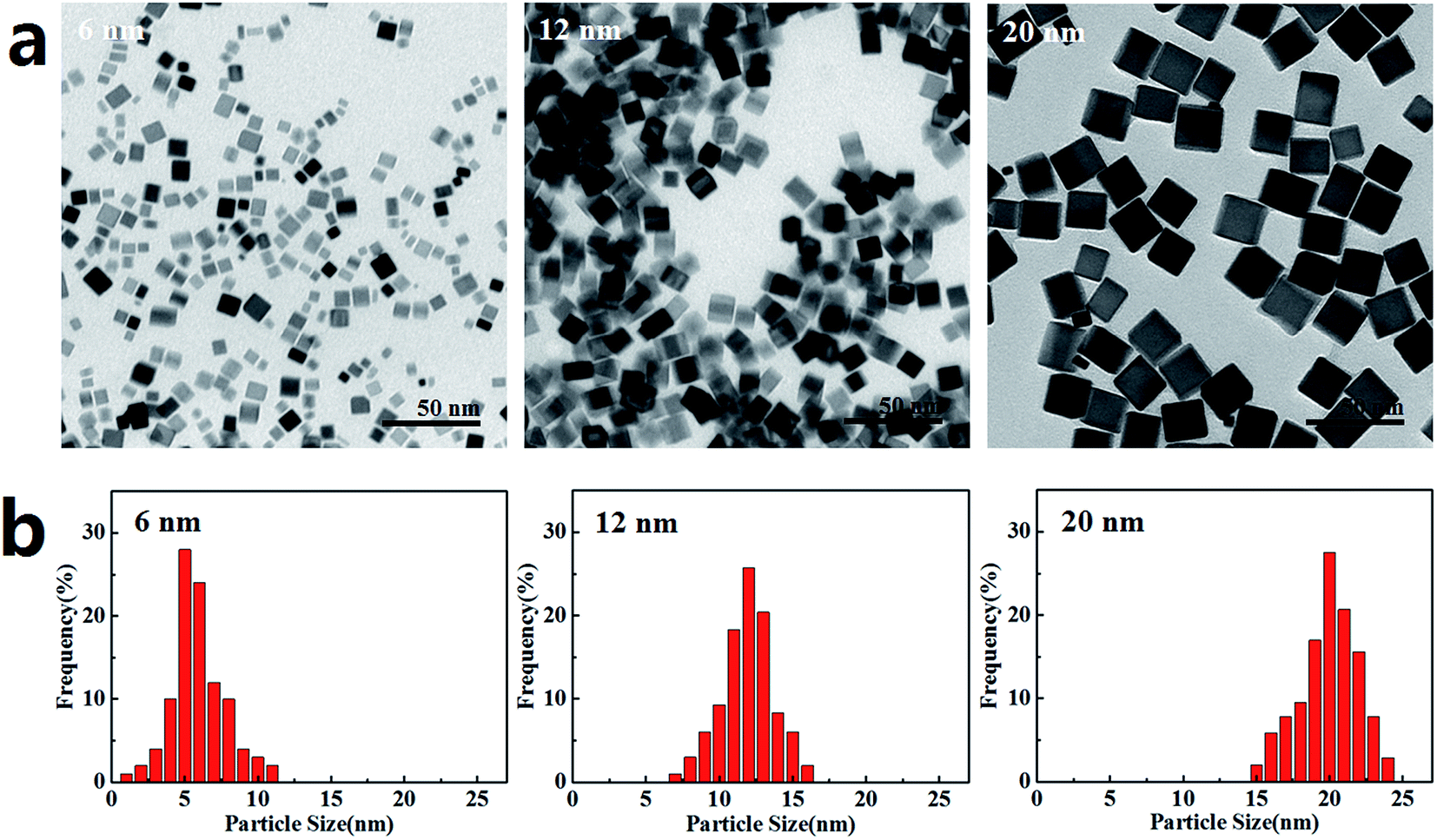 | ||
| Fig. 1 TEM characterization of PdNPs. (a) Images of PdNPs: cubic in shape and well-dispersed with consistent sizes (scale bar, 50 nm). (b) Particle size distributions of the three PdNPs. Mean sizes are 6.3 ± 1.9 nm, 11.9 ± 2.3 nm, and 20.4 ± 1.7 nm, respectively. | ||
| NP | Morphology | Size (nm) | Dispersant | Hydrodynamic size (nm) | Polydispersity index | Zeta potential (mV) |
|---|---|---|---|---|---|---|
| 6 nm Pd | Cubical | 6.3 ± 1.9 | Water | 47.14 ± 0.8 | 0.210 ± 0.01 | −2.67 ± 0.48 |
| DMEM | 57.43 ± 0.4 | 0.151 ± 0.02 | −2.09 ± 0.32 | |||
| 12 nm Pd | Cubical | 11.9 ± 2.3 | Water | 84.56 ± 1.3 | 0.209 ± 0.01 | −4.03 ± 0.69 |
| DMEM | 84.10 ± 1.2 | 0.149 ± 0.03 | −2.51 ± 0.45 | |||
| 20 nm Pd | Cubical | 20.4 ± 1.7 | Water | 105.37 ± 2.3 | 0.250 ± 0.02 | −7.41 ± 1.74 |
| DMEM | 102.13 ± 1.5 | 0.214 ± 0.01 | −3.48 ± 0.66 |
3.2. Cytotoxicity of PdNPs
The cell viability of Hela cells incubated with the PdNPs was measured by MTT assay, and the results are presented in Fig. 2a. After 24 h incubation, the cell viability was closely related to the size of the PdNPs. A significant decline was observed with exposure to PdNPs of increasing size; the 20 nm PdNPs showed the highest cytotoxicity. Additionally, when the PdNPs concentration increased, the cell viability decreased. For the 6 nm PdNPs, the cell viability showed a slight decrease over the range of experimental concentrations. For the 12 nm and 20 nm PdNPs, the cell viability decreased sharply; the half maximal inhibitory concentrations (IC50) are about 35 μg mL−1 and 12 μg mL−1, respectively. The trend of cell viability of HepG2 cells is similar to that of Hela cells; however, in general, the values were higher than those of Hela cells. HEK293 cells maintained vigorous vitality at the investigated concentrations of PdNPs (Fig. 2b). After 12 h, 48 h or 72 h incubation, the cell viabilities showed the same trend as that for 24 h incubation (Fig. S2†); meanwhile, the cytotoxicity difference caused by particle size was not significant. In summary, the concentration range from 0.1 μg mL−1 to 10.0 μg mL−1 was chosen as the experimental concentration, and 24 h was selected as the optimum incubation time in the following experiments.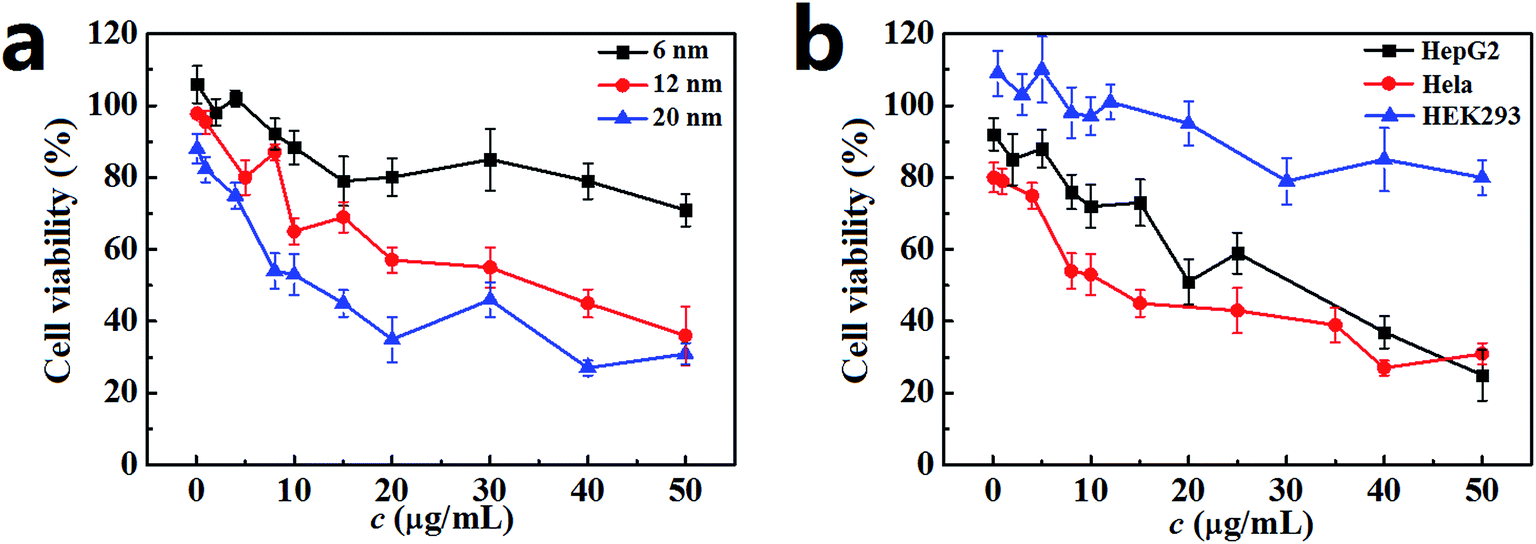 | ||
| Fig. 2 Cell viability after incubation with PdNPs for 24 h. (a) Cell viability for Hela cells after incubation with 6 nm, 12 nm and 20 nm PdNPs. (b) Cell viability for HepG2 cells, Hela cells and HEK293 cells after incubation with 20 nm PdNPs. The PdNPs concentrations ranged from 0.1 μg mL−1 to 50 μg mL−1. Data are presented as mean ± SD of five independent measurements. | ||
3.3. Size-dependent cellular uptake of PdNPs
The activation of autophagy initiated by NPs is closely related to the number of PdNPs internalized in cells. It has also been proved that the uptake of NPs by cells is strongly dependent upon particle size.39,40 Thus, the relationship between the particle size and cellular uptake of PdNPs in Hela cells was analyzed. The bright-field confocal microscopy images clearly show that the PdNPs aggregated in the cytoplasm and that more PdNPs accumulated in Hela cells with increasing particle size (Fig. 3a). Further evidence of the size-dependent uptake of PdNPs in Hela cells was obtained by TEM ultrastructural analysis (Fig. 3b). It was confirmed that the cellular uptake of PdNPs has a significant positive dependence upon the particle size in the culture medium. 6 nm PdNPs were occasionally observed in treated cells, while a large number of 20 nm PdNPs were distributed in the cytoplasm. In the high magnification images of the boxed regions, the increased uptake of PdNPs caused by changes in particle size is more obvious. Interestingly, there was a large increase in double-membrane autophagosome-like structures upon treatment with PdNPs, and most of the internalized PdNPs were encapsulated by the autophagosome-like vesicles.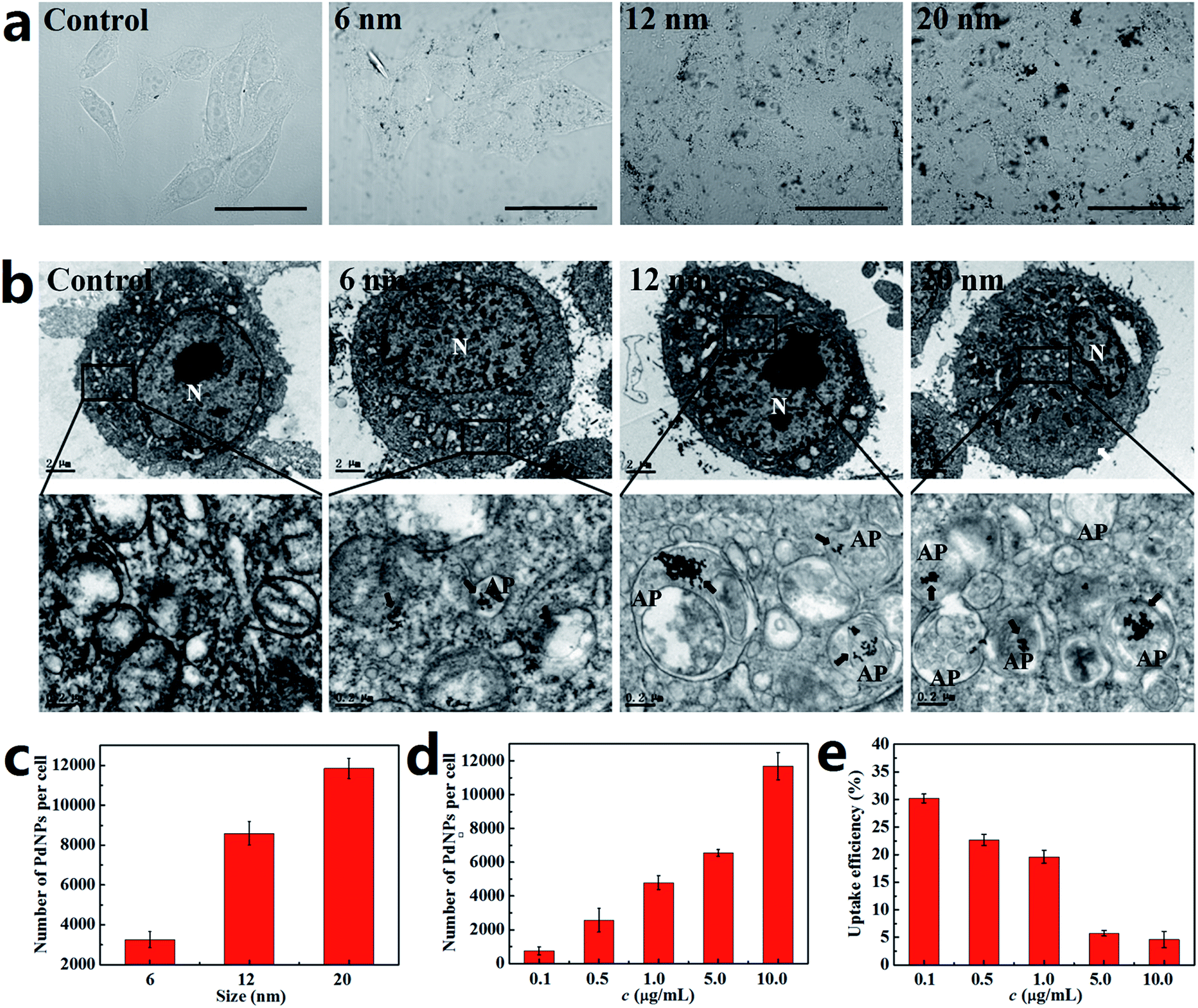 | ||
| Fig. 3 Size-dependent cellular uptake of PdNPs. Cells were cultured with PdNPs for 24 h. (a) Bright-field images acquired by laser scanning confocal microscopy (scale bar, 30 μm). (b) TEM images of the uptake of PdNPs in cells (scale bar, 2 μm). The panels below are high-magnification images of the boxed regions (scale bar, 0.2 μm). N, nuclei; AP, autophagosome; black arrows indicate PdNPs. (c) The number of PdNPs per cell when treated with 10.0 μg mL−1 PdNPs with different sizes. (d) The number of PdNPs per cell when treated with different concentrations of 20 nm PdNPs. (e) The uptake efficiency of PdNPs in Hela cells. Data are presented as mean ± SD of five independent measurements. | ||
To quantify the relationship between particle size and cellular uptake, ICP-MS measurements was performed, and the results are presented in Fig. 3c. Upon treatment with the 6 nm, 12 nm and 20 nm PdNPs, the numbers of PdNPs per cell were 3260, 8600 and 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 850, respectively. Moreover, the number of PdNPs per cell increased from 756 to 11680 with increasing concentration from 0.1 to 10.0 μg mL−1 (Fig. 3d), which shows that the cellular uptake of PdNPs is also closely related to their concentration. After incubation with 10.0 μg mL−1 PdNPs, the uptake data of PdNPs in Hela cells, HepG2 cells and HEK2 cells were compared (Fig. S3†). The results show that the uptake in HepG2 cells is analogous to that in Hela cells, while the uptake in HEK293 cells occurs at a relatively low level.
850, respectively. Moreover, the number of PdNPs per cell increased from 756 to 11680 with increasing concentration from 0.1 to 10.0 μg mL−1 (Fig. 3d), which shows that the cellular uptake of PdNPs is also closely related to their concentration. After incubation with 10.0 μg mL−1 PdNPs, the uptake data of PdNPs in Hela cells, HepG2 cells and HEK2 cells were compared (Fig. S3†). The results show that the uptake in HepG2 cells is analogous to that in Hela cells, while the uptake in HEK293 cells occurs at a relatively low level.
The cellular uptake efficiency decreased with increasing concentration of PdNPs (Fig. 3e), which indicates that PdNPs enter cells through receptor-mediated endocytosis. Thus, we further investigated the uptake mechanism of PdNPs to determine whether the uptake of PdNPs was due to receptor-mediated endocytosis. Because receptor-mediated endocytic efficiency is related to the culture temperature and energy supply, the cells were incubated at 4 °C instead of 37 °C or pre-treated with NaN3 to create an ATP-depleted environment. Significant decreases in the uptake of PdNPs at low temperature and in the ATP-depleted environment were found (Fig. S4†). The decreases were 50.7% and 60.3%, respectively, for 6 nm PdNPs-treated cells. For 12 nm PdNPs-treated cells, the decreases were 67.8% and 57.0%, respectively, and for 20 nm PdNPs-treated cells, the decreases were 71.8% and 62.3%, respectively.
3.4. PdNPs-induced autophagosome accumulation
A Hela cell line incubated with three sizes of PdNPs was used to examine the induction of autophagy, during which the accumulation of autophagosomes can be directly observed. As can be seen in the TEM images, treatment with PdNPs results in an increase of autophagosome-like structures (Fig. 4a). Autophagic cell death was observed in cells treated with a high concentration (10.0 μg mL−1) of PdNPs. A large number of autophagic vacuoles occupied the cytoplasm, accompanied by uneven distribution of cytoplasm and specialized structure loss in cell membranes (Fig. S5†). The accumulation of green fluorescent puncta induced by PdNPs (Fig. 4b) and the increased LC3-II/LC3-I ratio measured by western blot assay (Fig. 4c and d) further confirm the accumulation of autophagosomes in PdNPs-treated cells. Upon incubation with 10.0 μg mL−1 3-MA, very few green fluorescent puncta were observed (Fig. S6†); this indicates that autophagy is inhibited by 3-MA. Also, with increasing particle size, the autophagosome-like structures and the fluorescent puncta increased in number and size, strongly indicating that size-dependent autophagosome accumulation was induced by the PdNPs. The LC3-II/LC3-I ratios increased from 0.21 to 0.77 (6 nm and 12 nm, respectively) and to 1.33 (20 nm), as shown in Fig. 4d. In addition, at different concentrations of PdNPs, all the ratios of LC3-II/LC3-I showed a tendency to increase with increasing particle size (Fig. S7†). Autophagosome accumulation was also induced in a dose-dependent manner, as shown in Fig. S8.†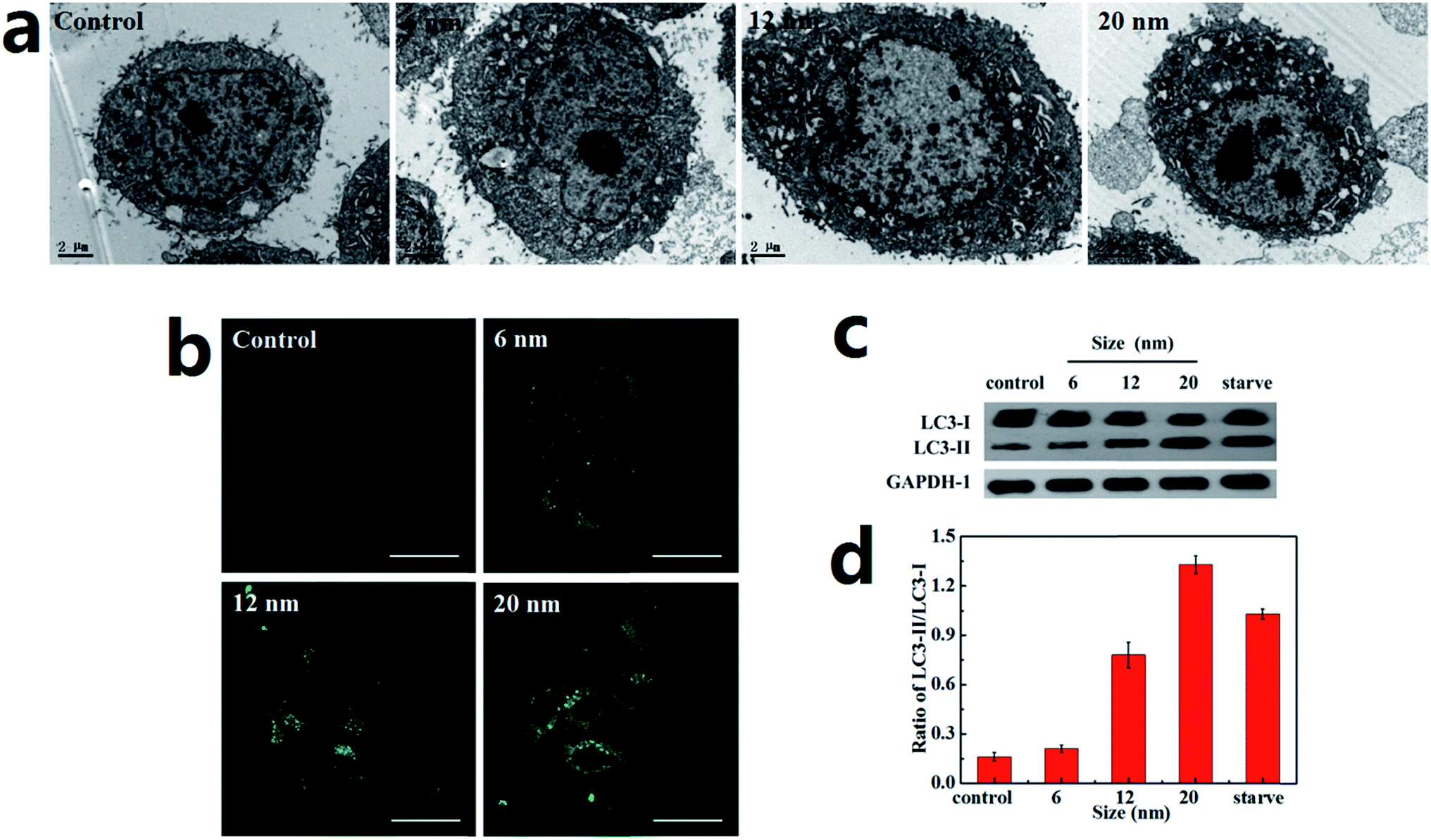 | ||
| Fig. 4 Accumulation of autophagosomes in PdNPs-treated Hela cells. The Hela cells were treated with 10.0 μg mL−1 6 nm, 12 nm, and 20 nm PdNPs for 24 h. (a) Induction of autophagosome-like structure formation by PdNPs (scale bar, 2 μm). (b) Accumulation of green fluorescent puncta in PdNPs-treated cells (scale bar, 30 μm). (c) Western blot assay showing the conversion of the autophagosome marker LC3 from LC3-I to LC3-II. (d) Statistical analysis of the LC3-II/LC3-I ratio. Data are presented as mean ± SD of five independent measurements. | ||
3.5. PdNPs treatment blocks autophagic flux
The content of P-S6K in PdNPs-treated cells was measured to monitor the priming process of autophagy. The decrease of the ratio of p-p70S6K/p70S6K in the PdNPs-treated cells compared with the control group demonstrates that autophagy was activated (Fig. 5a). However, for all three PdNPs, the ratio of p-p70S6K/p70S6K at 10.0 μg mL−1 was higher than that at 0.1 μg mL−1, which indicates that autophagy induction is weakened at a high concentration of PdNPs. Therefore, it can be speculated that the accumulation of a large quantity of autophagosomes in Hela cells after incubation with high concentrations of PdNPs is not only caused by autophagy induction.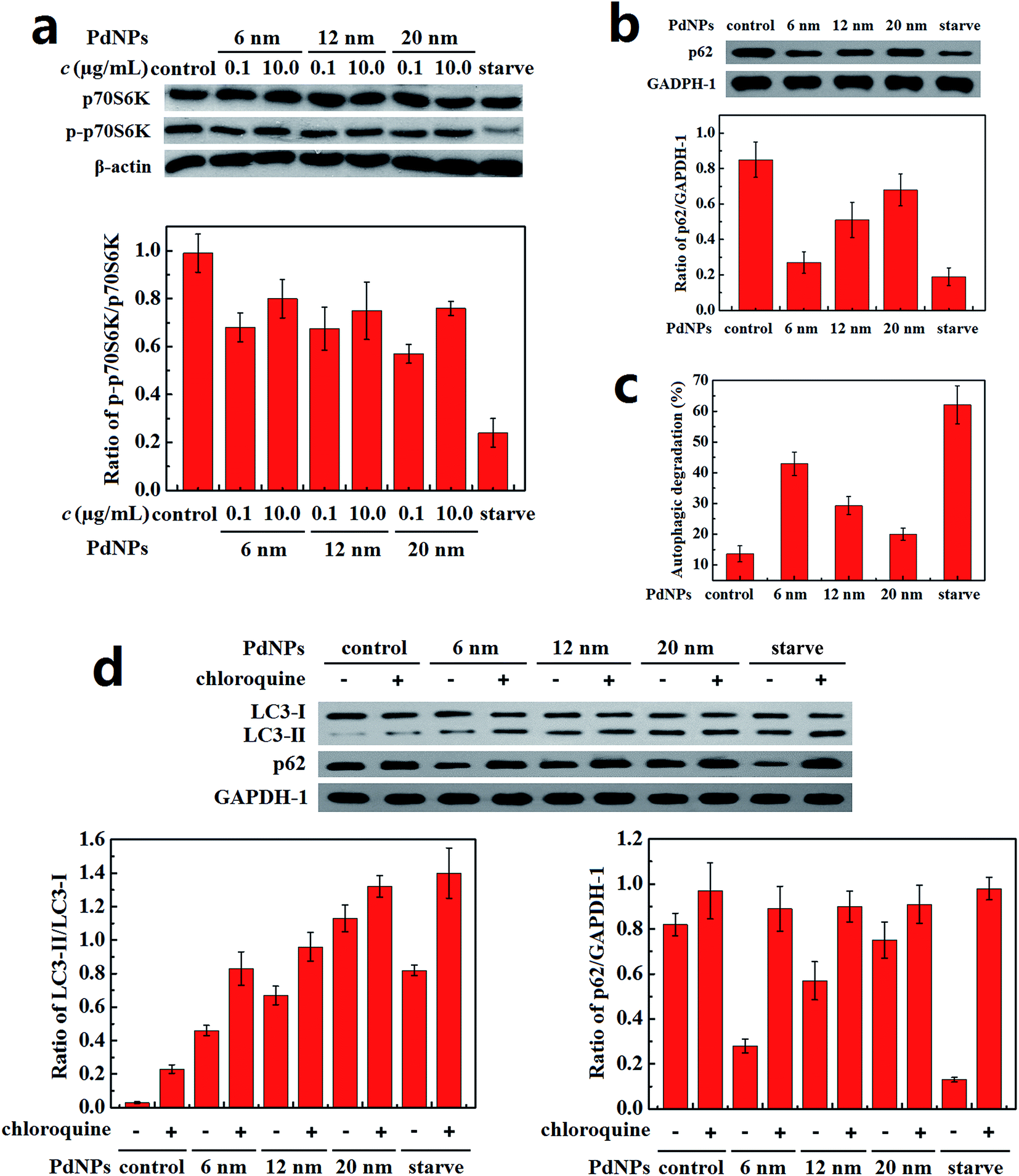 | ||
| Fig. 5 Autophagic flux detection. (a) Effects of PdNPs on the mTOR signaling pathway. (b) Degradation of the autophagy-specific substrate p62 measured by immunoblotting. (c) Autophagic degradation measured by the long-lived protein degradation efficiency. (d) Changes in the ratio of LC3-II/LC3-I and the amount of p62. Cells were cultured as follows: in regular growth medium for 24 h (lane 1); in regular culture medium containing 20 μM chloroquine for 24 h (lane 2); in DMEM containing 10.0 μg mL−1 PdNPs for 24 h (lane 3, lane 5, and lane 7); in DMEM containing 10.0 μg mL−1 PdNPs and chloroquine (lane 6); under starvation conditions for 24 h (lane 4, lane 6, and lane 8); with chloroquine under starvation conditions for 24 h (lane 10). Data are presented as mean ± SD of five independent measurements. | ||
The autophagic flux assay was carried out to confirm whether there a blockade of autophagic flux occurred upon treatment with a high concentration of PdNPs. The effect of PdNPs on the degradation of long-lived proteins can be used to better evaluate the autophagic degradation efficiency. Upon treatment with 6 nm, 12 nm, and 20 nm PdNPs, the amount of p62 increased in turn (Fig. 5b) and the autophagic degradation efficiency decreased successively (Fig. 5c).
Because LC3-II is produced during the formation of autophagosomes and degraded during lysosomal degradation, the total amount of LC3-II is determined by the balance between LC3-II production and degradation. When the fusion of autophagosomes and lysosomes or lysosomal degradation is inhibited, only the production of LC3-II occurs, dominating the total amount of LC3-II. The amounts of LC3-II with the addition of chloroquine, a lysosome degradation inhibitor, were compared to ensure whether real autophagy induction occurred. Starvation led to a significant induction of autophagy, as expected (Fig. 5d); also, further addition of chloroquine contributed to a great increase of LC3-II, which should have been degraded during starvation-induced autophagy. Meanwhile, in the presence of chloroquine, the LC3-II level in the PdNPs-treated cells increased in a size-dependent manner, indicating enhanced autophagy induction in response to the enlargement of the particle size. On the other hand, in the presence and absence of chloroquine, the difference in the ratio of LC3-II/LC3-I decreased with increasing particle size, which further validates the size-dependent blockade of lysosome degradation. The increase of the LC3-II level after co-treatment with PdNPs and chloroquine shows that the autophagic flux is not blocked entirely upon treatment with PdNPs and that some autophagosomes are still degraded through lysosome degradation. This indicates that the amount of LC3-II in PdNPs-treated cells is determined by the combined effects of real autophagy induction and the blockade of autophagic flux. Moreover, a substantial reduction of p62 level was found under starvation condition; also, chloroquine potently prevented starvation-induced p62 degradation. This also shows that PdNPs can induce the degradation of p62, indicating that PdNPs-induced autophagy occurred. In conclusion, a high concentration of PdNPs causes autophagy induction and autophagic flux blockade in Hela cells, and both these effects are enhanced with increasing particle size.
3.6. Lysosome alkalinization caused by PdNPs treatment
Generally, lysosomes are acidic; their pH is about 4.5.41 Altering lysosome pH can affect their functions; many lysosome inhibitors function in this way. For example, the lysosome inhibitor chloroquine can cause lysosome alkalinization, thereby inhibiting autophagosome degradation and causing autophagosome accumulation.42 Indeed, the endocytosis of PdNPs by lysosomes can be clearly observed in the TEM images (Fig. S9†). In this work, the following series of experiments were designed to determine if PdNPs cause alkalinization of lysosomes.Lyso-Tracker Red DND-99 is used to label and monitor acidic lysosomes in Hela cells. The quantity of lysosomes showed no significant change, while the size of the lysosomes increased upon treatment with larger PdNPs (Fig. 6a). It was confirmed that the dysfunction of lysosomal degradation can lead to lysosome enlargement. Thus, treatment with PdNPs weakens lysosomal degradation capability in a size-dependent manner, resulting in the blockade of autophagic flux.
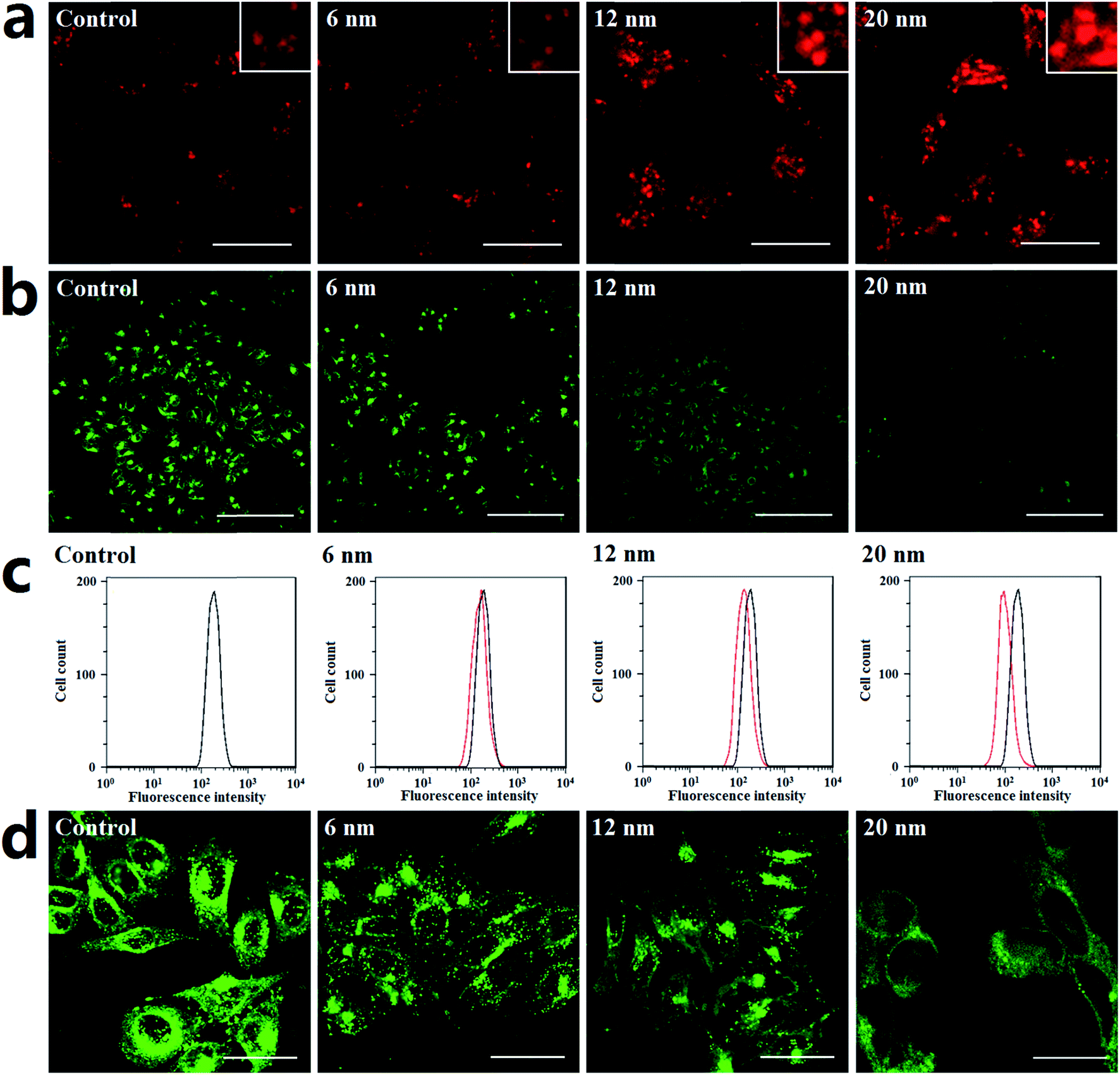 | ||
| Fig. 6 Lysosome alkalinization in PdNPs-treated cells. (a) Fluorescence images of Hela cells stained with Lyso-Tracker Red DND-99 (scale bar, 30 μm). (b) Fluorescence images of Hela cells stained with Lysosensor Green DND-189 (scale bar, 200 μm). (c) Flow cytometry analysis of lysosome alkalization. (d) Localization of the V1 domain (scale bar, 30 μm). The Hela cells were treated with 6 nm, 12 nm, and 20 nm PdNPs for 24 h. | ||
Lysosomal degradation capability can be affected by altering lysosomal pH. Lysosensor Green DND-189, an acidic probe which is selectively retained in acidic lysosomes, shows a significant pH-dependent increase of fluorescence intensity in response to changes in organelle acidity. Herein, Hela cells were labeled with Lysosensor Green DND-189 to investigate lysosome alkalinization caused by PdNPs. The CLSM (Fig. 6b) and flow cytometry (Fig. 6c) results demonstrate that the lysosomal fluorescence intensity of PdNPs-treated cells was much weaker than that of normal cells, which indicates that alkalinization of lysosomes occurs in PdNPs-treated cells. Lysosome acidification is regulated by vacuolar H+-ATPase, a proton pump which is composed of a membrane-associated Vo domain and a peripherally associated V1 domain.43 Dissociation of the V1 domain affects V-ATPase activity and adversely causes lysosome alkalinization. The fluorescent images in Fig. 6d clearly show that the V1 domain aggregates in a punctuated state on lysosomes in untreated cells and gradually dissociates into the cytoplasm upon addition of PdNPs; this effect increases with increasing particle size. Taken together, at high concentrations, PdNPs cause size-dependent lysosome impairment by dissociating the V1 domain from vacuolar H+-ATPase.
4. Discussion
In previous studies, it was suggested that NPs, such as gold and silver NPs, induce accumulation of autophagosomes by impairing lysosomal function and blocking autophagic flux without causing real induction of autophagy.44 Due to the similarity of PdNPs to these NPs, various techniques and methods were applied in this work to study the induction of autophagy by PdNPs. The TEM images, confocal microscopic images and western blot assay together revealed that autophagosomes increasingly accumulated in a PdNPs size-dependent manner. The mTOR signaling pathway assay, autophagic flux assay and lysosome alkalinization detection indicate that both autophagy induction and the blockade of autophagic flux are triggered by treatment with PdNPs.In many cases, the activation of autophagy can be considered to be a cell stress response to the physical properties (rather than the chemical properties or toxicity) of intracellular foreign bodies.45 This explains why the internalized PdNPs are almost all trapped inside autophagosomes, as shown in Fig. 3b. Moreover, NPs induce autophagy in a dispersity-dependent manner,46 and aggregated NPs are more prone to be taken into cells than NPs, with excellent dispersibility. PdNPs with larger particle sizes have higher PDI values, which indicates that larger PdNPs have a propensity to aggregate; in consequence, the size-dependent autophagy induction can be speculated to be mainly due to the increasing amount of internalized PdNPs in cells. The ICP-MS results illustrate a size-dependent increase in cellular uptake of PdNPs and a dose-dependent decrease of uptake efficiency, which are in accordance with the performance of receptor-mediated endocytosis.47 Significant decreases in the uptake of PdNPs can be observed at low temperature or under ATP-depleted conditions. This proves that the PdNPs enter cells through receptor-mediated endocytosis. PdNPs which adsorb proteins on their surfaces are docked by receptors on the cell membrane, then wrapped by the membrane and finally internalized into the cell. The cellular uptake of PdNPs strongly depends on the time of the wrapping process, and the wrapping time is dominated by the thermodynamic driving force for wrapping (the amount of free energy required to drive the NPs into the cell).48 PdNPs must cluster together to obtain sufficient free energy (Fig. S10†); for larger PdNPs, this process occurs much more readily.
Lysosome dysfunction is related to chronic infectious disease, cardiovascular disease, neurodegenerative disease,49 tumor growth and aging. It is believed that the blockade of autophagic flux may result from lysosome impairment. When PdNPs are transferred from autophagosomes to lysosomes during the degradation process, they accumulate in the lysosomes and cause severe damage to lysosome function. Larger PdNPs are more likely to remain in the lysosomes and cause size-dependent lysosome impairment in Hela cells. PdNPs enter cells by endocytosis and complete the entire endocytic pathway. Further, PdNPs can potentially carry therapeutic and diagnostic materials to diseased issues or image targeted issues through this endocytic pathway.
In recent years, metal NPs are considered to possess great capability for disease therapy in the clinic because of their biocompatibility, small size, and tunable surface functionalities. Numerous NPs have been reported to induce autophagy; they have been proposed as autophagy activators50 and are thus considered to have therapeutic potential. It has been demonstrated that abnormal autophagy is relevant to many human diseases, including neurodegenerative diseases, aging and particularly cancer.51 Unlike gold and silver NPs, which induce accumulation of autophagosomes by blockade of autophagic flux, PdNPs cause real autophagy induction in Hela cells and have greater value in autophagy research. Because modulation of autophagy is now believed to have anticancer effects, the size-dependent autophagy induction by PdNPs provides an important reference for the application of NPs in medical therapeutics.
5. Conclusions
Autophagy induction and lysosome impairment were observed to occur in PdNPs-treated cells on the basis of TEM, fluorescence labeling, western blot and flow cytometry analyses. The positively size-dependent uptake of PdNPs leads to size-dependent autophagy induction in Hela cells through the mTOR signaling pathway at low concentrations. Furthermore, the lysosome degradation capability decreases with increasing particle size of PdNPs, which leads to the blockade of autophagic flux. The dose- and size-dependent autophagy activation caused by PdNPs in Hela cells suggests that NP-induced autophagy can be modulated effectively to play an essential role in determining cell survival or death, while latent impairment of lysosomes and autophagic flux is a focus which should be taken into consideration before PdNPs are applied in therapeutics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 21473157, No. 21503190).References
- N. Mizushima, B. Levine, A. M. Cuervo and D. J. Klionsky, Autophagy fights disease through cellular self-digestion, Nature, 2008, 451(7182), 1069–1075 CrossRef CAS PubMed.
- Z. P. Xie and D. J. Klionsky, Autophagosome formation: core machinery and adaptations, Nat. Cell Biol., 2007, 9(10), 1102–1109 CrossRef CAS PubMed.
- H. Abeliovich and D. J. Klionsky, Autophagy in yeast: mechanistic insights and physiological function, Microbiol. Mol. Biol. Rev., 2001, 65(3), 463–479 CrossRef CAS PubMed.
- S. E. Ghayad and P. A. Cohen, Inhibitors of the PI3K/Akt/mTOR pathway: new hope for breast cancer patients, Recent Pat. Anti-Cancer Drug Discovery, 2010, 5(1), 29–57 CrossRef CAS PubMed.
- J. J. Liu, M. Lin, J. Y. Yu, B. Liu and J. K. Bao, Targeting apoptotic and autophagic pathways for cancer therapeutics, Cancer Lett., 2011, 300(2), 105–114 CrossRef CAS PubMed.
- T. P. Neufeld, TOR-dependent control of autophagy: biting the hand that feeds, Curr. Opin. Cell Biol., 2010, 22(2), 157–168 CrossRef CAS PubMed.
- G. Kroemer and M. Jaattela, Lysosomes and autophagy in cell death control, Nat. Rev. Cancer, 2005, 5(11), 886–897 CrossRef CAS PubMed.
- M. Hoyer-Hansen and M. Jaattela, Autophagy – an emerging target for cancer therapy, Autophagy, 2008, 4(5), 574–580 CrossRef PubMed.
- Y. Kabeya, N. Mizushima, T. Uero, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi and T. Yoshimori, LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing, EMBO J., 2000, 19(21), 5720–5728 CrossRef CAS PubMed.
- N. Mizushima, T. Yoshimori and B. Levine, Methods in mammalian autophagy research, Cell, 2010, 140(3), 313–326 CrossRef CAS PubMed.
- N. Lewinski, V. Colvin and R. Drezek, Cytotoxicity of nanoparticles, Small, 2008, 4(1), 26–49 CrossRef CAS PubMed.
- M. Yin, C. Feng, J. Shen, Y. Yu, Z. Xu, W. Yang, W. Knoll and K. Muellen, Dual-responsive interaction to detect DNA on template-based fluorescent nanotubes, Small, 2011, 7(12), 1629–1634 CrossRef CAS PubMed.
- J. Xi, C. Xie, Y. Zhang, L. Wang, J. Xiao, X. Duan, J. Ren, F. Xiao and S. Wang, Pd nanoparticles decorated N-Doped Graphene Quantum Dots@N-Doped carbon hollow nanospheres with high electrochemical sensing performance in cancer detection, ACS Appl. Mater. Interfaces, 2016, 8(34), 22563–22573 CAS.
- M. Yin, J. Shen, R. Gropeanu, G. O. Pflugfelder, T. Weil and K. Muellen, Fluorescent core/shell nanoparticles for specific cell-nucleus staining, Small, 2008, 4(7), 894–898 CrossRef CAS PubMed.
- W. Zhang and C.-H. Tung, Cisplatin cross-linked multifunctional nanodrugplexes for combination therapy, ACS Appl. Mater. Interfaces, 2017, 9(10), 8547–8555 CAS.
- D. R. Nogueira, L. Tavano, M. Mitjans, L. Perez, M. R. Infante and M. P. Vinardell, In vitro antitumor activity of methotrexate via pH-sensitive chitosan nanoparticles, Biomaterials, 2013, 34(11), 2758–2772 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, Impact of nanotechnology on drug delivery, ACS Nano, 2009, 3(1), 16–20 CrossRef CAS PubMed.
- X. Chen and H. J. Schluesener, Nanosilver: a nanoproduct in medical application, Toxicol. Lett., 2008, 176(1), 1–12 CrossRef CAS PubMed.
- M. J. Akhtar, H. A. Alhadlaq, S. Kumar, S. A. Alrokayan and M. Ahamed, Selective cancer-killing ability of metal-based nanoparticles: implications for cancer therapy, Arch. Toxicol., 2015, 89(11), 1895–1907 CrossRef CAS PubMed.
- E. A. Daza, S. K. Misra, A. S. Schwartz-Duva, A. Ohoka, C. Miller and D. Pan, Nano-cesium for anti-cancer properties: an investigation into cesium induced metabolic interference, ACS Appl. Mater. Interfaces, 2016, 8(40), 26600–26612 CAS.
- T. Sadhukha, T. S. Wiedmann and J. Panyam, Enhancing therapeutic efficacy through designed aggregation of nanoparticles, Biomaterials, 2014, 35(27), 7860–7869 CrossRef CAS PubMed.
- A. Khalafi-Nezhad and F. Panahi, Size-controlled synthesis of palladium nanoparticles on a silica-cyclodextrin substrate: a novel palladium catalyst system for the heck reaction in water, ACS Sustainable Chem. Eng., 2014, 2(5), 1177–1186 CrossRef CAS.
- J. Mondal, T. Quang Thang, A. Jana, W. K. H. Ng, P. Borah, H. Hirao and Y. Zhao, Size-dependent catalytic activity of palladium nanoparticles fabricated in porous organic polymers for alkene hydrogenation at room temperature, ACS Appl. Mater. Interfaces, 2016, 8(24), 15307–15319 CAS.
- A. Tavangar, P. Premnath, B. Tan and K. Venkatakrishnan, Noble hybrid nanostructures as efficient anti-proliferative platforms for human breast cancer cell, ACS Appl. Mater. Interfaces, 2016, 8(16), 10253–10265 CAS.
- S. E. Letant, J. Herberg, L. N. Dinh, R. S. Maxwell, R. L. Simpson and A. P. Saab, Structure and catalytic activity of POSS-stabilized Pd nanoparticles, Catal. Commun., 2007, 8(12), 2137–2142 CrossRef CAS.
- M. Slouf, E. Pavlova, M. Bhardwaj, J. Plestil, H. Onderkova, A. A. Philimonenko and P. Hozak, Preparation of stable Pd nanoparticles with tunable size for multiple immunolabeling in biomedicine, Mater. Lett., 2011, 65(8), 1197–1200 CrossRef CAS.
- Z. Li, X. Wang, G. Wen, S. Shuang, C. Dong, M. C. Paau and M. M. F. Choi, Application of hydrophobic palladium nanoparticles for the development of electrochemical glucose biosensor, Biosens. Bioelectron., 2011, 26(11), 4619–4623 CrossRef CAS PubMed.
- H. Khan, A. Badshah, G. Murtaz, M. Said, R. Zia-ur, C. Neuhausen, M. Todorova, B. J. Jean-Claude and I. S. Butler, Synthesis, characterization and anticancer studies of mixed ligand dithiocarbamate palladium(II) complexes, Eur. J. Med. Chem., 2011, 46(9), 4071–4077 CrossRef CAS PubMed.
- E. Gao, F. Liu, M. Zhu, L. Wang, Y. Huang, H. Liu, S. Ma, Q. Shi and N. Wang, Synthesis, characterization, DNA interaction, and cytotoxicity of novel Pd(II) and Pt(II) complexes, J. Enzyme Inhib. Med. Chem., 2010, 25(4), 557–564 CrossRef CAS PubMed.
- A. Schmidt, V. Molano, M. Hollering, A. Poethig, A. Casini and F. E. Kuehn, Evaluation of new palladium cages as potential delivery systems for the anticancer drug cisplatin, Chem.–Eur. J., 2016, 22(7), 2253–2256 CrossRef CAS PubMed.
- Y. H. Lee, F. Y. Cheng, H. W. Chiu, J. C. Tsai, C. Y. Fang, C. W. Chen and Y. J. Wang, Cytotoxicity, oxidative stress, apoptosis and the autophagic effects of silver nanoparticles in mouse embryonic fibroblasts, Biomaterials, 2014, 35(16), 4706–4715 CrossRef CAS PubMed.
- J. J. Li, D. Hartono, C. N. Ong, B. H. Bay and L. Y. L. Yung, Autophagy and oxidative stress associated with gold nanoparticles, Biomaterials, 2010, 31(23), 5996–6003 CrossRef CAS PubMed.
- O. Zabirnyk, M. Yezhelyev and O. Seleverstov, Nanoparticles as a novel class of autophagy activators, Autophagy, 2007, 3(3), 278–281 CrossRef CAS PubMed.
- Y. Wang, G. Kaur, Y. Chen, A. Santos, D. Losic and A. Evdokiou, Bioinert anodic alumina nanotubes for targeting of endoplasmic reticulum stress and autophagic signaling: a combinatorial nanotube-based drug delivery system for enhancing cancer therapy, ACS Appl. Mater. Interfaces, 2015, 7(49), 27140–27151 CAS.
- J. Z. Wu, J. Zhao, H. H. Qian, L. Yue, Y. S. Guo and W. J. Fang, Deep insights into the growth pattern of palladium nanocubes with controllable sizes, RSC Adv., 2016, 6(70), 66048–66055 RSC.
- V. R. Lopes, V. Loitto, J. N. Audinot, N. Bayat, A. C. Gutleb and S. Cristobal, Dose-dependent autophagic effect of titanium dioxide nanoparticles in human HaCaT cells at non-cytotoxic levels, J. Nanobiotechnol., 2016, 14(1), 22–35 CrossRef PubMed.
- S. Saremi, F. Atyabi, S. P. Akhlaghi, S. N. Ostad and R. Dinarvand, Thiolated chitosan nanoparticles for enhancing oral absorption of docetaxel: preparation, in vitro and ex vivo evaluation, Int. J. Nanomed., 2011, 6, 119–128 CAS.
- C. Zhang, W. Wang, T. Liu, Y. Wu, H. Guo, P. Wang, Q. Tian, Y. Wang and Z. Yuan, Doxorubicin-loaded glycyrrhetinic acid-modified alginate nanoparticles for liver tumor chemotherapy, Biomaterials, 2012, 33(7), 2187–2196 CrossRef CAS PubMed.
- F. Lu, S.-H. Wu, Y. Hung and C.-Y. Mou, Size effect on cell uptake in well-suspended, uniform mesoporous silica nanoparticles, Small, 2009, 5(12), 1408–1413 CrossRef CAS PubMed.
- F. Osaki, T. Kanamori, S. Sando, T. Sera and Y. Aoyama, A quantum dot conjugated sugar ball and its cellular uptake on the size effects of endocytosis in the subviral region, J. Am. Chem. Soc., 2004, 126(21), 6520–6521 CrossRef CAS PubMed.
- E. S. Trombetta, M. Ebersold, W. Garrett, M. Pypaert and I. Mellman, Activation of lysosomal function during dendritic cell maturation, Science, 2003, 299(5611), 1400–1403 CrossRef CAS PubMed.
- P. S. Tietz, K. Yamazaki and N. F. Larusso, Time-dependent effects of chloroquine on pH of hepatocyte lysosomes, Biochem. Pharmacol., 1990, 40(6), 1419–1421 CrossRef CAS PubMed.
- T. Nishi and M. Forgac, The vacuolar (H+)-ATPases – nature's most versatile proton pumps, Nat. Rev. Mol. Cell Biol., 2002, 3(2), 94–103 CrossRef CAS PubMed.
- X. W. Ma, Y. Y. Wu, S. B. Jin, Y. Tian, X. N. Zhang, Y. L. Zhao, L. Yu and X. J. Liang, Gold nanoparticles induce autophagosome accumulation through size-dependent nanoparticle uptake and lysosome impairment, ACS Nano, 2011, 5(11), 8629–8639 CrossRef CAS PubMed.
- O. Seleverstov, O. Zabirnyk, M. Zscharnack, L. Bulavina, M. Nowicki, J. M. Heinrich, M. Yezhelyev, F. Emmrich, R. O'Regan and A. Bader, Quantum dots for human mesenchymal stem cells labeling. A size-dependent autophagy activation, Nano Lett., 2006, 6(12), 2826–2832 CrossRef CAS PubMed.
- D. T. Huang, H. L. Zhou and J. H. Gao, Nanoparticles modulate autophagic effect in a dispersity-dependent manner, Sci. Rep., 2015, 5, 14361 CrossRef CAS PubMed.
- B. B. Manshian, D. F. Moyano, N. Corthout, S. Munck, U. Himmelreich, V. M. Rotello and S. J. Soenen, High-content imaging and gene expression analysis to study cell-nanomaterial interactions: the effect of surface hydrophobicity, Biomaterials, 2014, 35(37), 9941–9950 CrossRef CAS PubMed.
- S. H. Wang, C. W. Lee, A. Chiou and P. K. Wei, Size-dependent endocytosis of gold nanoparticles studied by three-dimensional mapping of plasmonic scattering images, J. Nanobiotechnol., 2010, 8 DOI:10.1186/1477-3155-8-33.
- L. S. Zhang, R. Sheng and Z. H. Qin, The lysosome and neurodegenerative diseases, Acta Biochim. Biophys. Sin., 2009, 41(6), 437–445 CrossRef CAS PubMed.
- W. Song, M. Shi, M. Dong and Y. Zhang, Inducing temporal and reversible autophagy by nanotopography for potential control of cell differentiation, ACS Appl. Mater. Interfaces, 2016, 8(49), 33475–33483 CAS.
- L. Galan-Acosta, H. G. Xia, J. Y. Yuan and H. Vakifahmetoglu-Norberg, Activation of chaperone-mediated autophagy as a potential anticancer therapy, Autophagy, 2015, 11(12), 2370–2371 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra11400a |
| This journal is © The Royal Society of Chemistry 2018 |