
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeneration of controllable gaseous H2S concentrations using microfluidics†
Theodore Christoforidis a,
Tom G. Driverb,
Jalees Rehmancd and
David T. Eddington*a
a,
Tom G. Driverb,
Jalees Rehmancd and
David T. Eddington*a
aDepartment of Bioengineering, University of Illinois at Chicago, Chicago, Illinois 60607, USA. E-mail: dte@uic.edu
bDepartment of Chemistry, University of Illinois at Chicago, Chicago, Illinois 60607, USA
cDepartment of Medicine, University of Illinois at Chicago, Chicago, Illinois 60607, USA
dDepartment of Pharmacology, University of Illinois at Chicago, Chicago, Illinois 60607, USA
First published on 23rd January 2018
Abstract
Hydrogen sulfide (H2S) plays an important role as an intercellular and intracellular signaling molecule, yet its targets are not well understood. As a molecule it easily evaporates and it is hard to acquire stable concentration for in vitro studies, constituting a major problem for the field to identify its downstream targets and function. Here we develop a microfluidic system that can provide consistent and controllable H2S levels in contrast to the current method of delivering large bolus doses to cells. The system relies on the permeability of H2S gas through a polydimethylsiloxane thin membrane. A hydrogen sulfide donor, sodium hydrosulfide, is perfused in the microchannels below the gas permeable membrane and gaseous H2S diffuses across the membrane, providing a stable concentration for up to 5 hours. Using electrochemical sensors within 3 ppm range, we found that H2S concentration was dependent on two parameters, the concentration of H2S donor, sodium hydrosulfide and the flow rate of the solution in the microchannels. Additionally, different H2S concentration profiles can be obtained by alternating the flow rate, providing an easy means to control the H2S concentration. Our approach constitutes a unique method for H2S delivery for in vitro and ex vivo studies and is ideally suited to identify novel biological processes and cellular mechanisms regulated by H2S.
Introduction
Hydrogen sulfide (H2S) is a colorless gas with a pungent smell of ‘rotten eggs’. In humans, hydrogen sulfide (H2S) is known to be a toxic gas that is lethal in high concentrations over 500 ppm. However, emerging research demonstrates that H2S is generated endogenously in low concentrations and serves as a signalling molecule which regulates cellular homeostasis.1–3 H2S, thus, belongs to the family of ‘gasotransmitters’ along with nitric oxide (NO) and carbon monoxide (CO). Additionally, its biological role is evident in plants and bacteria, either as a bi-product or a power source.2,4–9H2S is endogenously produced in mammals mainly by three enzymes, cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE or CGL) and 3-mercaptopyruvate sulfur transferase (3-MST) which are located in different organs.3 H2S is additionally produced non-enzymatically, but is considered negligible in comparison to the enzymatic production.10 In 1996 H2S was firstly introduced as a signalling molecule and since then, H2S has attracted increased attention for its potential therapeutic effects in mammals. For example, it has been found that H2S is cytoprotective with antioxidant properties,11 vasodilatory,12,13 angiogenetic,14,15 and neuromodulatory,16 and its repertoire of biological functions is steadily expanding.1,2,17–19
Hydrogen sulfide gas can be diluted in liquid at a ratio 1 mg L−1 = 717 μM [STP]. In an aqueous solution hydrogen sulfide follows the equilibrium:
| H2S ⇌ H⊕ + HS⊖ ⇌ H⊕ + S2− |
With pKa varying depending on the temperature. At room temperature, the pKa for the left equilibrium is almost 7 while the pKa for the second equilibrium varies in different studies which is over 12. Hydrogen sulfide in its protonated form evaporates from the solution in a gaseous form, a phenomenon that causes the pH to increase. Therefore, the equation is constantly driven to the left, diminishing the concentration of H2S in the solution.20,21
Using different measurement techniques, a discrepancy in in vivo H2S concentrations has been reported which is mostly attributed to the techniques themselves1,22 with older studies reporting elevated H2S endogenous concentrations.23,24 Additionally, for in vivo measurements it is assumed that H2S is rapidly metabolized within a tissue, thus the concentrations vary depending on the measuring location.20,25 Thus the reported physiological concentrations vary from the submicromolar range to 200 μM.26
A key challenge for H2S research is typical H2S donors provide H2S molecules in an unstable and uncontrolled manner. In most of the studies, hydrogen sulfide is generated by inorganic salts, either sodium hydrosulfide (NaSH) or sodium sulfide (Na2S), or by purging a solution with gaseous H2S. When these solutions are added in vivo or in cell cultures, a certain concentration of H2S is achieved, which sometimes can reach cytotoxic levels.27,28 This concentration of H2S is achieved for seconds as it degrades with time mostly by volatilization, and by oxidation in a pH manner, where oxidation increases for physiological pH.2,25,29 DeLeon et al. showed the significance of this fact for three different cases, a cell culture plate, a myograph bath, and a Langendorff isolated heart apparatus. It is shown that the concentration of H2S in a Na2S solution declines rapidly with a half-life in the order of minutes. This loss of H2S is attributed mostly to volatilization and minimally to oxidation.25
Recently, several compounds that slowly release hydrogen sulfide have been reported. The compound most commonly used is GYY 4137 which is commercially available. GYY 4137 releases hydrogen sulfide slowly for several minutes, but the production rate cannot be controlled and the release increases with time, therefore, high concentrations of H2S can be achieved after a few days.30 The only study that partial control of the release of hydrogen sulfide is shown in a pH-controlled manner using the slow releasing chemical JK-1–JK-5.31 Using this method, the concentration of H2S can be controlled by changing the pH, which unfortunately is not applicable for biological applications. Additionally, it is reported that other impurities of the donor or the chemical properties of the donor itself might implicate the already complicated phenomenon.27,32 For example, impure NaSH can be a source of solid sulfur in astrocyte studies with a developing Ca2+ influx due to sulfur.33 The combination of these factors constitute a major problem for the field and thus the exact mechanism of H2S activity is still unclear.29,32
Recently, Faccenda et al. showed that H2S penetrates through a thin polydimethylsiloxane (PDMS) membrane allowing a continuous measurement in which the solutions are not influenced by impurities of the donor as they cannot penetrate the PDMS membrane. In this study the donor, either Na2S or CSE, is inserted in a well and the H2S molecules that have diffused through the 100 μm membrane can be measured with different methods.34
Microfluidic devices offer new approaches to stably generate environment gas landscapes such as defined stable oxygen concentration environments for both cell cultures and brain tissue ex vivo studies. These devices rely on a microfluidic network that is bonded to a thin PDMS membrane (Fig. 1). Previously our lab has published many studies using similar devices to deliver controlled oxygen landscapes to cells and tissues35–41 Similarly, we found that these devices can be used to establish environments of different concentrations of hydrogen sulfide. As the solution of NaSH is introduced in the microfluidic network, the gaseous H2S diffuses across the PDMS membrane and a steady state concentration is achieved. This method delivers H2S in the reservoir for a long period of time (hours), in comparison to a conventional method, where a solution of NaSH is directly injected in the reservoir and diminishes rapidly (minutes) (Fig. 2). The established concentration of a specific pH NaSH solution flowing in the microfluidic network provides hydrogen sulfide depending on two main factors: the concentration of the NaSH solution and the flow rate of the liquid. In order to quantify the method, we measured the mean value of H2S in the reservoir for 5 hours and we found a linear relationship with the flow rate (Fig. 3). Additionally, the concentration of hydrogen sulfide can be modulated by alternating the flow rate (Fig. 4). Finally, different microfluidic networks of different areas were compared in order to further validate the phenomenon (Fig. 5). Using these microfluidic devices different concentrations of H2S are achieved where only pure gaseous H2S molecules are provided in the system and can be controlled.
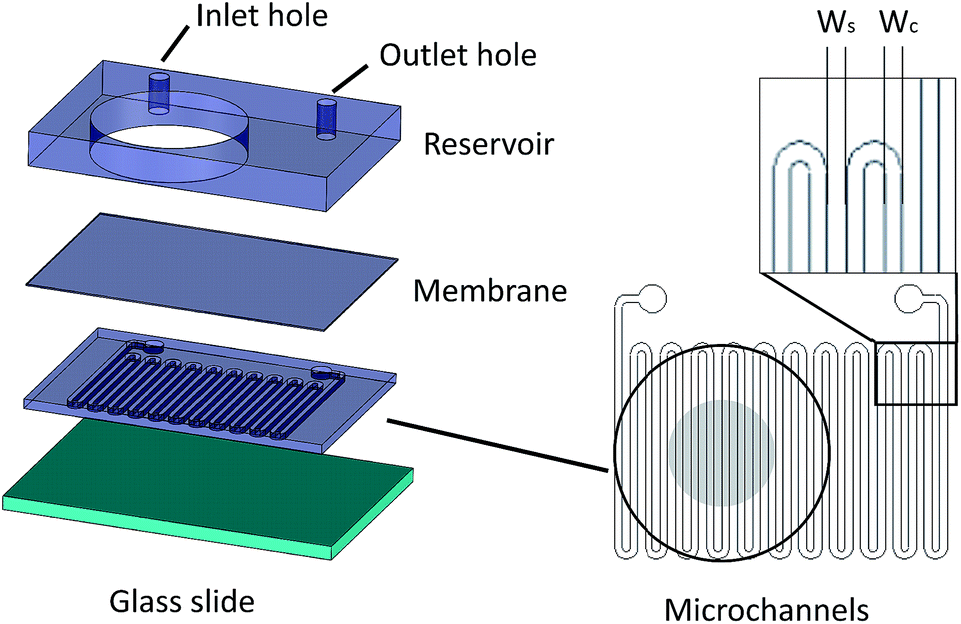 | ||
| Fig. 1 The microfluidic device assembly. The microchannel layer is bonded on a thin H2S permeable membrane. The adjacent side of the membrane is bonded to the reservoir layer that the inlet and outlet holes of the microchannels are punched. On the right, the microchannels in a serpentine shape have a width Wc which are separated by a spacing with width Ws. The reservoir is outlined with the black circle while the grey circle in the middle denotes the measuring region of each sensor. | ||
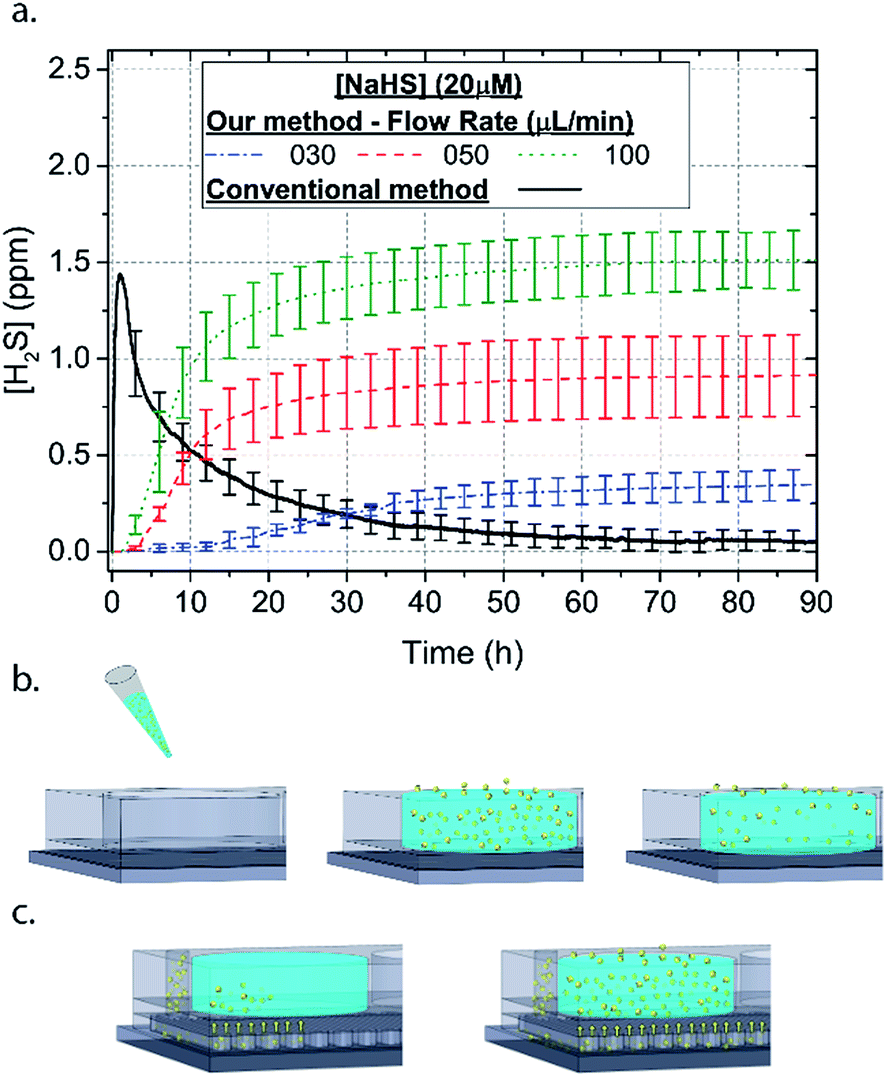 | ||
| Fig. 2 (a) The concentration using the microfluidic method is increasing with time until it reaches a stable concentration. The NaSH solution introduced in the syringe pump is 20 μM and different concentrations can be achieved by flowing the NaSH solution at different rates. Conventionally, dispensing a 20 μM NaSH PBS solution in a well the concentration spikes up in the beginning and it is totally lost within approximately 70 minutes. (b) Graphical representation of the conventional introduction of H2S in a cell culture well plate. The H2S molecules are (c) graphical representation of the microfluidic method of H2S. The H2S molecules are introduced in the well as the solution is flown within the microchannels. | ||
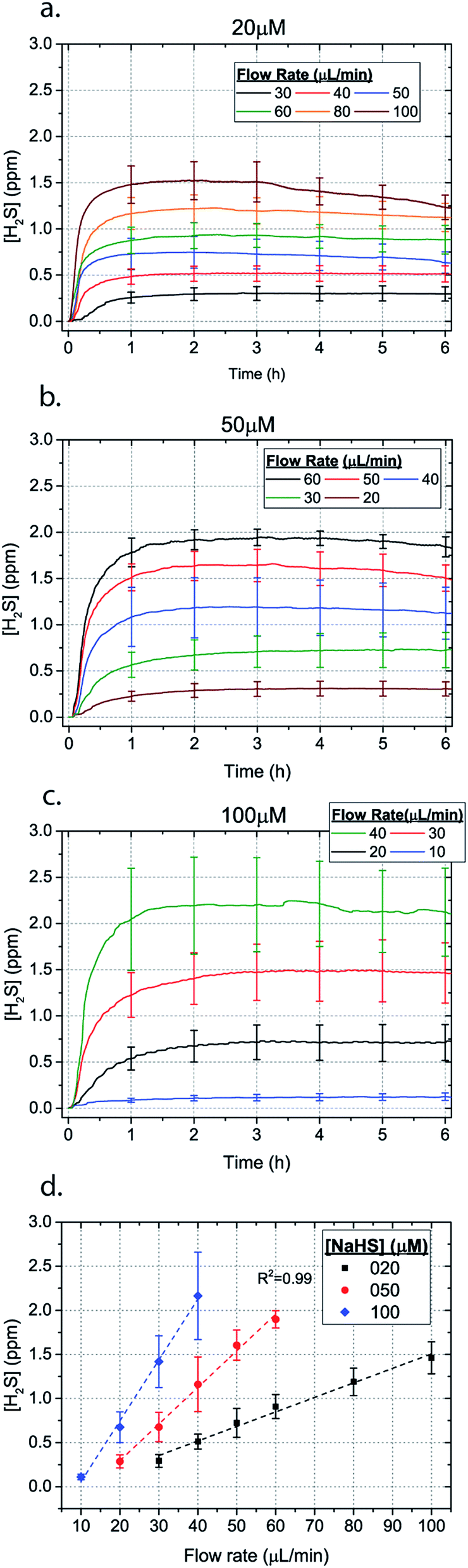 | ||
| Fig. 3 H2S concentration for different flow rates for (a) 20 μM (b) 50 μM and (c) 100 μM NaSH (error bars shown every hour). The average between 1–6 hours for different flow rates is depicted in (d). | ||
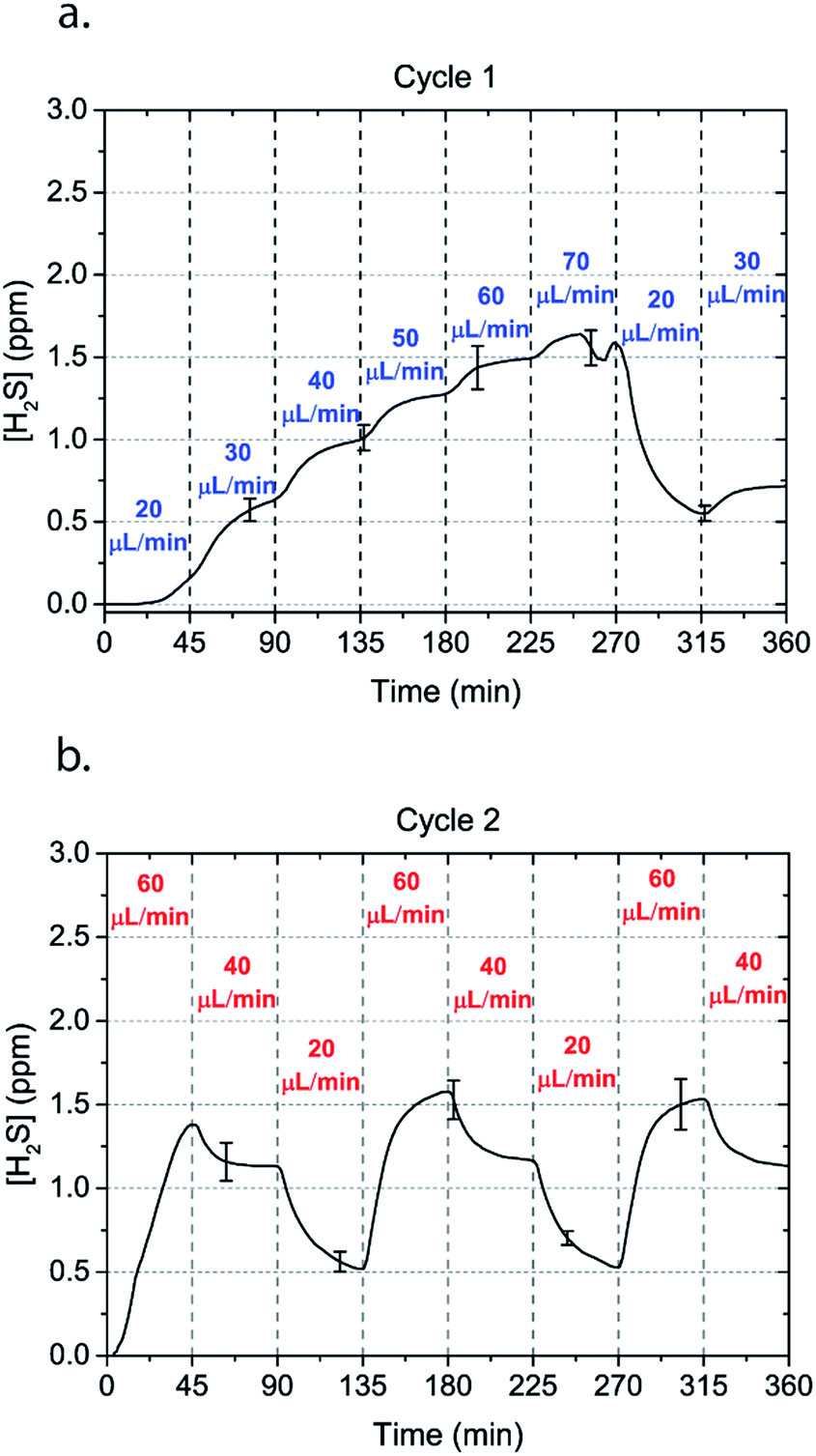 | ||
| Fig. 4 Alternating H2S concentrations can be obtained by alternating the flow rate of a 50 μM NaSH solution in the microchannels. Two cycles of flow rates with an interval of 45 minutes are shown. | ||
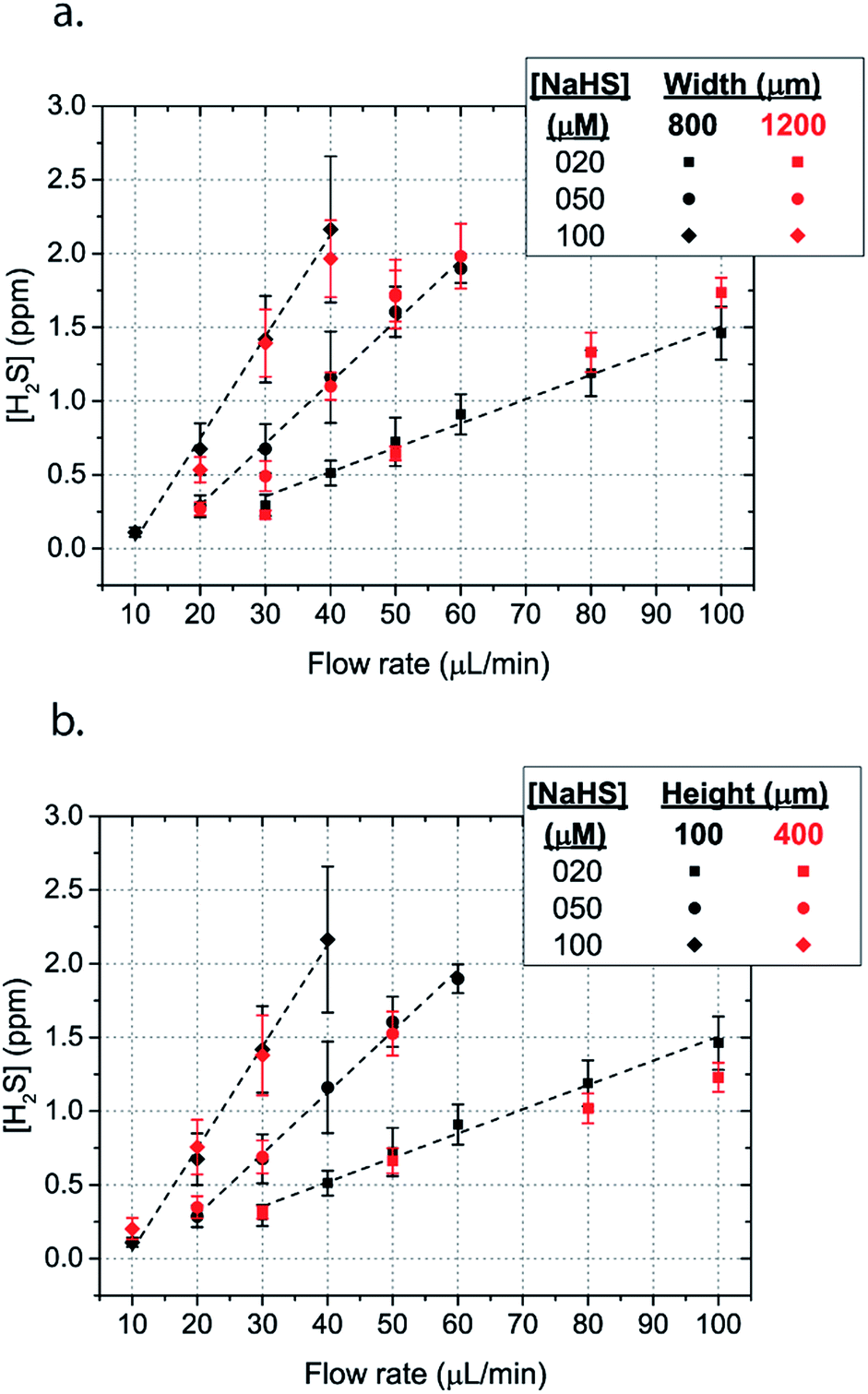 | ||
| Fig. 5 Mean H2S concentration in the steady concentration for different flow rates for a serpentine with (a) wide microchannels (width = 1200 μm, spacing = 200 μm and height = 100 μm) (b) high height microchannels (width = 1200 μm, spacing = 800 μm and height = 400 μm) in comparison to the model serpentine microchannels (width = 800 μm, spacing = 800 μm and height = 100 μm). | ||
Methods & materials
Fabrication of microfluidic device
The microfluidic device consists of three different layers, the microfluidic channels superimposed by a 100 μm thick PDMS membrane and a reservoir, where liquid/media/solution can be introduced for different studies. The microchannels are fabricated using typical soft lithography techniques. Specifically, photosensitive polymer SU-8 resist was spun on a silicon wafer and exposed to UV light under a photomask (Fineline Imaging, Colorado Springs, CO). The design was then developed and polydimethylsiloxane (PDMS) (Sylgard 184, Dow Corning, Midland, MI) was poured on top followed by curing at 85 °C for two hours. The 100 μm PDMS membrane was fabricated by spinning uncured PDMS at 800 rpm and then curing at 85 °C for 45 minutes. A PDMS slab of 6.5 mm thickness was punched with 22.6 mm diameter holes that serve as potential liquid reservoirs, and 1 mm holes that served as inlet and outlet holes for the microchannels. The microfluidic channels were bonded on the PDMS membrane and the PDMS piece was bonded on top of the membrane using Corona plasma treatment (Electro Technic Products, Chicago, IL).Solution preparation
A solution of distilled water was purged with nitrogen for at least 1 hour in an aspirator bottle with a bottom hose outlet of 1 L (Cole Parmer, Vernon Hills, IL). As soon as the sodium hydrosulfide (NaSH) (Sigma Aldrich) was added to the solution, the bottle was corked with a two-hole rubber stopper, and 1/4′′ barb male luer lock fittings attached to a large bore stopcock were placed in the holes of the stopper. The solution was extracted from the bottle by opening a stopcock attached on a tube connected to the bottom hose outlet. This minimizes air introduced into the bottle, minimizing both volatilization and oxidation of H2S gas. The solution was then titrated into 220 mL of a weak sulfuric acid solution (2 μL in 220 cc of DI water) keeping the pH level below 3.7, within a 250 cc glass bottle sealed similarly with a rubber stopper. Each specific concentration of NaSH solution was loaded to the syringes via the luer lock stopcock. For each experiment a fresh solution was prepared as the concentration of H2S in this solution diminishes over time.Experimental setup
NaSH solution was loaded in plastic 60 mL syringes (60 cc Luer Lock BD, Fischer Scientific, Hanover Park, IL). A syringe pump (PHD 2000, Harvard Apparatus, Holliston, MA) was used to drive the fluid through a needle connected with a thin Tygon Tubing (Tygon Microbore Tubing, 0.020′′ × 0.060′′ OD, Cole Parmer, Vernon Hills, IL). The outer diameter of the tubing is bigger than the inlet and outlet holes, securing a leak free flow. All the experiments were carried out in ambient air under a fume hood.Validation
The detection of gaseous H2S concentration on top of the membrane was achieved using electrochemical sensors (Alphasense, Essex, UK). The electrical signal is acquired at a sampling rate of 1 Hz with a data acquisition system (FS-1608, Measurement Computing, Norton, MA) using DAQami software. The sensors were calibrated using premixed gases of H2S in nitrogen (Cal Gas Direct Incorporated, Huntington Beach, CA) (Fig. S1†).Results and discussion
In order to characterize our method, the electrochemical sensors were placed on top of the membrane of our microfluidic devices and H2S gas concentration was measured. The microfluidic network consists of a serpentine of rectangular cross section of 800 μm width and 100 μm height, while the microfluidic channels were 800 μm apart for most of the studies aside from those investigating other microchannel geometries. It is shown that the concentration of H2S increases when NaSH solution is introduced in the microfluidic network. The flow rate is the main factor that influences the rate of increase of the concentration to reach a steady equilibrium. The levels reach equilibrium within the first hour of the initial flow and remains constant with time (Fig. 2). In comparison, in Fig. 2, H2S concentration of dispensing a 20 μM NaSH PBS solution in an open well is depicted. This measurement was taken by placing the electrochemical sensor over the well supported and sealed by a PDMS piece of the same diameter as the sensors to maintain consistent position and to contain diffused gas in the reservoir. With the conventional method, H2S concentration is diminished within 30 minutes, while with our method the concentration of H2S increases and remains constant. Additionally, using our method three different molarities, 20 μM (Fig. 3a), 50 μM (Fig. 3b), and 100 μM (Fig. 3c) were tested for different flow rates for at least 6 hours. These equilibrium concentrations for each molarity solutions and different flow rates were averaged for 5 hours in the steady H2S concentration (from the first to the sixth hour), shown in Fig. 3d. It is shown that the concentration has a linear relationship (R2 = 0.99) with the flow rate of NaHS solution, while the slope of the line is different for higher concentration solutions of NaHS. In specific the slope is 0.017 ppm μL−1 min−1 for 20 μM, 0.041 ppm μL−1 min−1 for 50 μM, and 0.069 ppm μL−1 min−1 for 100 μM. Error bars show the standard deviation of three different devices. The deviation is likely caused by subtle shifts of the sensor as the reservoirs' diameter was slightly bigger than the sensors' diameter. The concentration of H2S has a linear relationship (R2 = 0.99) with the flow rate of NaSH solution, while the slope of the line correlates with the concentration of NaSH.By simply changing the flow rate of the syringe pump, our method allows for the concentration of H2S to be precisely alternated during the same experiment. As a demonstration of hypothetical H2S exposure experiments, two cycles of different flow rates of a 50 μM NaSH solution are alternated every 45 minutes (Fig. 4). On the first, the flow rate is alternated from 20 to 30 to 40 to 50 to 60 to 70 μL min−1 (Fig. 4a) and on the second, the flow rate is changed from 60 to 40 to 20 μL min−1 (Fig. 4b). At the end of each cycle the flow rate returns to the initial value according to the program that was defined with the syringe pump. For drastic changes in flow rate, the 45 minute interval is not enough for the concentration to reach equilibrium concentration. This occurs in the first cycle when the flow rate returns from 70 μL min−1 to 20 μL min−1 and the concentration of H2S remains above the concentration that corresponds to 20 μL min−1. Similarly, on the second cycle the concentration does not reach equilibrium for the initial flow rate of 60 μL min−1. Nevertheless, this demonstrates the ability to control H2S levels by varying flow rates through the microchannels.
The dimensions of the microchannel and how they related to H2S values in the reservoir were next investigated by changing the geometry of the serpentine microfluidic network. We compared the concentration profiles for microchannels with width ratio Wc/Ws = 800/800, with those of Wc/Ws = 1200/200 for microchannel height of 100 μm, where Wc is the width of the microchannel and Ws is the width of the spacing between the microchannels. Surprisingly, the results are not significantly different for all concentrations of NaSH (Fig. 5a). In addition, the serpentine with Wc/Ws = 800/800 was compared to a 400 μm height and the results were almost identical (Fig. 5a). Thus, we conclude that the only factors that significantly influence H2S concentration are NaSH concentration and the flow rate, indicating that microchannels of different geometries can be used to generate similar concentrations within the given range of dimensions tested.
Conclusion
To date, studying the impact of hydrogen sulfide in biological systems has been stymied by poor control over its concentration in solution. Using our microfluidic approach we show that physiological H2S concentrations of up to 3 ppm can be achieved over many hours. In addition, compared to existing technology (or procedures) which deliver deprotonated HS−, our method uses an acidic solution, which provides a more consistent H2S concentration with time as the solution is separated from the reservoir by a PDMS membrane. By simply changing the flow rate, our microfluidic approach permits control of the H2S concentration within the same experiment. To our knowledge this is the first microfluidic device that allows for the precise and stable control of gasotransmitter levels. This method can be adapted for in vitro and ex vivo studies, and could also enable the use of other gasotransmitters NO and CO, which could be delivered by flowing the gas in the microchannels, either individually or by combining different signalling gases. The use of microfluidic devices to study H2S signalling has the potential to significantly advance the field by providing precise control of its concentration and identify novel H2S-dependent processes in biological systems where current methods of H2S control are lacking.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Science Foundation 1253060, DTE and NIH P01-HL60678.References
- L. Li, P. Rose and P. K. Moore, Annu. Rev. Pharmacol. Toxicol., 2011, 51, 169–187 CrossRef CAS PubMed.
- R. Wang, Physiol. Rev., 2012, 92, 791–896 CrossRef CAS PubMed.
- M. Whiteman, S. Le Trionnaire, M. Chopra, B. Fox and J. Whatmore, Clin. Sci., 2011, 121, 459–488 CrossRef CAS PubMed.
- D. A. Baum and B. Baum, BMC Biol., 2014, 12, 76 CrossRef PubMed.
- E. J. Javaux, Nat. Geosci., 2011, 4, 663–665 CrossRef CAS.
- O. Kabil and R. Banerjee, J. Biol. Chem., 2010, 285, 21903–21907 CrossRef CAS PubMed.
- K. R. Olson and K. D. Straub, Physiology (Bethesda), 2016, 31, 60–72 CAS.
- M. W. Powner, B. Gerland and J. D. Sutherland, Nature, 2009, 459, 239–242 CrossRef CAS PubMed.
- D. Wacey, M. R. Kilburn, M. Saunders, J. Cliff and M. D. Brasier, Nat. Geosci., 2011, 4, 698–702 CrossRef CAS.
- P. K. Moore and M. Whiteman, Chemistry, Biochemistry and Pharmacology of Hydrogen Sulfide, Springer, 2015 Search PubMed.
- L.-F. Hu, M. Lu, P. T. Hon Wong and J.-S. Bian, Antioxid. Redox Signaling, 2010, 15, 405–419 CrossRef PubMed.
- G. Yang, L. Wu, B. Jiang, W. Yang, J. Qi, K. Cao, Q. Meng, A. K. Mustafa, W. Mu, S. Zhang and others, Science, 2008, 322, 587–590 CrossRef CAS PubMed.
- W. Zhao, J. Zhang, Y. Lu and R. Wang, EMBO J., 2001, 20, 6008–6016 CrossRef CAS PubMed.
- A. Papapetropoulos, A. Pyriochou, Z. Altaany, G. Yang, A. Marazioti, Z. Zhou, M. G. Jeschke, L. K. Branski, D. N. Herndon, R. Wang and C. Szabó, Proc. Natl. Acad. Sci., 2009, 106, 21972–21977 CrossRef CAS PubMed.
- M.-J. Wang, W.-J. Cai, N. Li, Y.-J. Ding, Y. Chen and Y.-C. Zhu, Antioxid. Redox Signaling, 2009, 12, 1065–1077 CrossRef PubMed.
- H. Kimura, Amino Acids, 2010, 41, 113–121 CrossRef PubMed.
- N. L. Kanagy, C. Szabo and A. Papapetropoulos, Am. J. Physiol.: Cell Physiol., 2017, 312, C537–C549 CrossRef PubMed.
- D. J. Polhemus and D. J. Lefer, Circ. Res., 2014, 114, 730–737 CrossRef CAS PubMed.
- R. Wang, C. Szabo, F. Ichinose, A. Ahmed, M. Whiteman and A. Papapetropoulos, Trends Pharmacol. Sci., 2015, 36, 568–578 CrossRef CAS PubMed.
- M. N. Hughes, M. N. Centelles and K. P. Moore, Free Radical Biol. Med., 2009, 47, 1346–1353 CrossRef CAS PubMed.
- Q. Li and J. R. Lancaster Jr, Nitric Oxide, 2013, 35, 21–34 CrossRef CAS PubMed.
- G. K. Kolluru, X. Shen, S. C. Bir and C. G. Kevil, Nitric Oxide, 2013, 35, 5–20 CrossRef CAS PubMed.
- J. Furne, A. Saeed and M. D. Levitt, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2008, 295, R1479–R1485 CrossRef CAS PubMed.
- N. L. Whitfield, E. L. Kreimier, F. C. Verdial, N. Skovgaard and K. R. Olson, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2008, 294, R1930–R1937 CrossRef CAS PubMed.
- E. R. DeLeon, G. F. Stoy and K. R. Olson, Anal. Biochem., 2012, 421, 203–207 CrossRef CAS PubMed.
- A. Stein and S. M. Bailey, Redox Biol., 2013, 1, 32–39 CrossRef CAS PubMed.
- A. Papapetropoulos, M. Whiteman and G. Cirino, Br. J. Pharmacol., 2015, 172, 1633–1637 CrossRef CAS PubMed.
- C. Szabo and A. Papapetropoulos, Pharmacol. Rev., 2017, 69, 497 CrossRef PubMed.
- K. R. Olson, Antioxid. Redox Signaling, 2012, 17, 32–44 CrossRef CAS PubMed.
- Z. W. Lee, J. Zhou, C.-S. Chen, Y. Zhao, C.-H. Tan, L. Li, P. K. Moore and L.-W. Deng, PLoS One, 2011, 6, e21077 CAS.
- J. Kang, Z. Li, C. L. Organ, C.-M. Park, C. Yang, A. Pacheco, D. Wang, D. J. Lefer and M. Xian, J. Am. Chem. Soc., 2016, 138, 6336–6339 CrossRef CAS PubMed.
- P. Nagy, Z. Pálinkás, A. Nagy, B. Budai, I. Tóth and A. Vasas, Biochimica et Biophysica Acta (BBA)–General Subjects, 2014, 1840, 876–891 CrossRef CAS PubMed.
- H. Kimura, Proc. Jpn. Acad., Ser. B, 2015, 91, 131–159 CrossRef CAS PubMed.
- A. Faccenda, J. Wang and B. Mutus, Anal. Chem., 2012, 84, 5243–5249 CrossRef CAS PubMed.
- M. D. Brennan, M. L. Rexius-Hall, L. J. Elgass and D. T. Eddington, Lab Chip, 2014, 14, 4305–4318 RSC.
- J. F. Lo, E. Sinkala and D. T. Eddington, Lab Chip, 2010, 10, 2394–2401 RSC.
- G. Mauleon, C. P. Fall and D. T. Eddington, PLoS One, 2012, 7, e43309 CAS.
- G. Mauleon, J. F. Lo, B. L. Peterson, C. P. Fall and D. T. Eddington, J. Neurosci. Methods, 2013, 216, 110–117 CrossRef CAS PubMed.
- S. C. Oppegard, K.-H. Nam, J. R. Carr, S. C. Skaalure and D. T. Eddington, PLoS One, 2009, 4, e6891 Search PubMed.
- S. C. Oppegard, A. J. Blake, J. C. Williams and D. T. Eddington, Lab Chip, 2010, 10, 2366–2373 RSC.
- M. L. Rexius-Hall, G. Mauleon, A. B. Malik, J. Rehman and D. T. Eddington, Lab Chip, 2014, 14, 4688–4695 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12220a |
| This journal is © The Royal Society of Chemistry 2018 |