DOI:
10.1039/C7RA12701D
(Paper)
RSC Adv., 2018,
8, 7330-7339
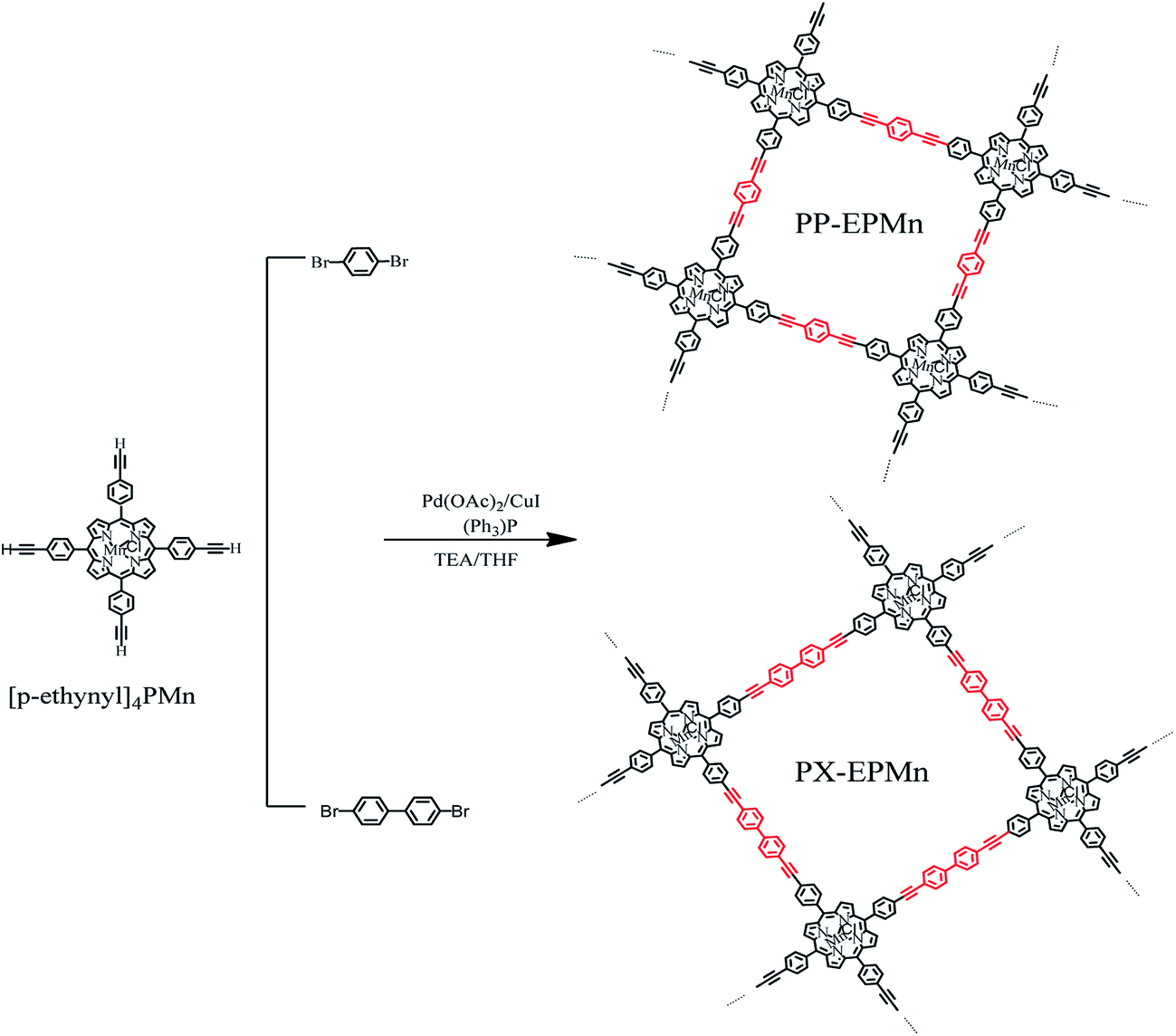
Porous metalloporphyrinic nanospheres constructed from metal 5,10,15,20-tetraksi(4′-ethynylphenyl)porphyrin for efficient catalytic degradation of organic dyes†
Received
23rd November 2017
, Accepted 8th February 2018
First published on 15th February 2018
Abstract
Novel conjugated metalloporphyrin polymers (PP-EPMn and PX-EPMn) were synthesized by the Sonogashira coupling reaction with Mn(III)5,10,15,20-tetraksi(4′-ethynylphenyl)porphyrin ([p-ethynyl]4PMn) as building block, and with phenylene and xenyl groups as bridges, respectively. Fine nanospheres were obtained through control of the reaction conditions. The two polymer nanospheres were characterized in detailed by BET, FE-SEM, HR-TEM, FT-IR, UV-Vis and XPS, revealing that they were highly ordered, highly developed microporous and super-conjugated metalloporphyrin polymers with large surface areas and uniform pore sizes. Their electrochemical behaviors and hydrophilicity were also investigated. With the synergistic effects of porous nanosphere structure and conjugated covalent structure, PP-EPMn and PX-EPMn showed excellent catalytic activity and reusability in the catalytic degradation of methylene blue (3,7-bis(dimethylamino)-5-phenothiazinium chloride) in aqueous solution. The good hydrophilicity of the polymer nanospheres promoted the catalytic degradation. In particular, unlike Fenton degradation systems that require acidic conditions, PP-EPMn degraded methylene blue significantly better under alkaline conditions, which implied a good prospect in treating industrial methylene blue dye wastewater.
1. Introduction
Porphyrin and its derivatives, as an important class of biological compounds in the nature, have been widely used in solar cells, biosensors, catalytic reactions and gas adsorption due to their characteristic ring structures of conjugated double bonds.1–6 In recent years, metalloporphyrins have been employed as biomimetic catalysts in a variety of oxidation reactions such as the degradation of organic dyes.7,8 But in fact, monomer metalloporphyrins have limitations in practice application.9–11 For example, some water-soluble metalloporphyrins with peripheral groups (COOH, N(CH3)3, SO3) have been used to treat organic dyes in water. However, they cannot be recycled as they are hard to separate from reaction solutions.12–15 Some other metalloporphyrins, although with high catalytic oxidation activities, cannot dissolve in water, which limits the catalytic performance in aqueous solution, especially in the degradation of water-soluble organic dyes. Some new immobilized monomer metalloporphyrin composites with outstanding catalytic performances in aqueous solution, such as carbon-nanotube–porphyrin, C60–porphyrin, graphene oxide–porphyrin and TiO2–porphyrin composites, have been synthesized.16–25 However, the bonds between porphyrins and solid support surfaces are unstable, so the activities of supported metalloporphyrins decline soon in reuse.
Conjugated microporous and mesoporous polymers (CMPs) are a new type of covalent polymers with novel structures as well as high thermostability and porosity, some of which exhibit superb catalytic performances in non-polar reactions.26–29 However, it is difficult to synthesize CMPs constructed from metalloporphyrins as building blocks, as we all know that porphyrin rings are hard to be polymerized directly by covalent linkages because of the bulky steric hindrance between porphyrin molecules. Therefore, metalloporphyrin CMPs have rarely been reported.30–32 Based on the disadvantages of metalloporphyrins and their immobilized composites in aqueous reaction and the advantages of metalloporphyrin CMPs for catalysis, we are interested in whether we can synthesize conjugated micro- and mesoporous polymers with monomer metalloporphyrins as building blocks to apply them in the catalytic degradation of water-soluble organic dyes. Since it is rather difficult to control the morphology of porphyrin polymers, most currently available conjugated polymers have sheet structures. In this study, the morphology of metalloporphyrin CMPs was regulated by selecting appropriate reaction conditions, metalloporphyrin templates and bridging groups.
Mn(III)5,10,15,20-Tetraksi(4′-ethynylphenyl)porphyrin ([p-ethynyl]4PMn) can connect another small organic molecule through its terminal alkyne on the para-way.33,34 So we chose small organic molecule 1,4-dibromobenzene and 4,4′-dibromobiphenyl as the precursor bridges. The formation of xenyl or phenylene between porphyrin molecules can weaken the steric hindrance, and the rotation of alkynyl group and rigidity of xenyl and phenylene groups can modulate the morphology of polymer products during polymerization. So we try to synthesize two kinds of organic conjugated porphyrin polymers with nanosphere features, PP-EPMn and PX-EPMn, which were bridged by phenylene and xenyl groups respectively. Methylene blue is a typical organic dye in waste water and has usually been selected as a model of hard-degradable organic dyes. So with methylene blue as degradation model, the catalytic performance of the two polymers was studied under mild conditions in this work, and excellent results were produced.
2. Experimental section
2.1. Materials and characterizations
Bromobenzaldehyde (99.77%, Aladdin Reagents (Shanghai) Co., Ltd.), palladium acetate (47.5% Pd), triphenylphosphine, copper iodide, H2O2 (30%), pyrrole (redistilled before use) and other reagents were all analytical grades and purchased from J&K Chemicals. THF was distilled over sodium and benzophenone ketyl under argon before use. [p-Ethynyl]4PMn was synthesized according to documented procedures and characterized by IR, UV-Vis, 1H-NMR and MS spectroscopies (ESI†).35
2.2. Synthesis of alkynylporphyrin conjugated organic polymers PP-EPMn and PX-EPMn
The steps of synthesizing PP-EPMn by 1,4-dibromobenzene were as follows:
A mixture of [p-ethynyl]4PMn (80.0 mg, 0.1 mmol), 1,4-dibromobenzene (56.64 mg, 0.24 mmol), triphenylphosphine (105.0 mg, 0.4 mmol), palladium acetate (22.4 mg, 0.1 mmol) and copper iodide (38.1 mg, 0.2 mmol) was added into a 100 mL three-necked flask, and air was degassed by three cycles of argon gas pumping-inflation. Air-removed THF (6 mL) and triethylamine (4 mL) were then injected into the three-necked flask under stirring and refluxed at 76 °C for two hours. Finally, the mixture was poured into plenty of aqueous KI solution after being cooled to room temperature. The solid crude product was then collected by filtration, and carefully washed with water, THF, methanol and trichloromethane in turn. It was also washed by Soxhlet extraction with water, THF, methanol and trichloromethane for 24 h, respectively, and then dried in vacuum, giving a dark green solid (PP-EPMn) with a yield of 88.9% (Scheme 1).
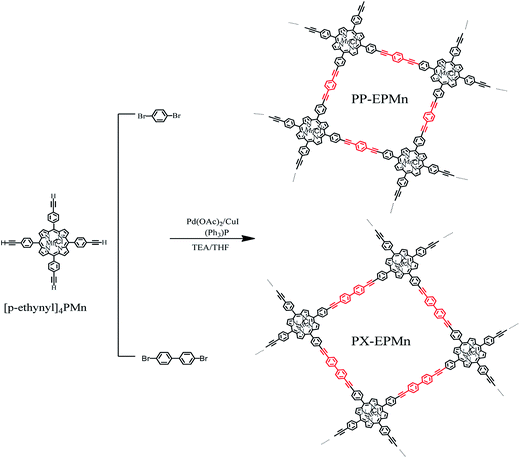 |
| | Scheme 1 Synthesis of nanoporous polymer PP-EPMn and PX-EPMn. | |
PX-EPMn was synthesized by using 4,4′-dibromobiphenyl as bridge. The steps were similar to those for PP-EPMn, but the reaction time must be extended to three hours, with the yield of only 70.0%.
2.3. Degradation of methylene blue
Aqueous methylene blue solution (10 mg L−1, 50 mL) was put in a flask, after pH adjustment, the reaction was carried out at room temperature (25 °C) with magnetic stirring when a certain amount of PP-EPMn or PX-EPMn and H2O2 solution were added into the flask. The mixture was sampled at fixed time intervals and rapidly centrifuged, from which the supernatants were collected and tested by UV-Vis spectroscopy. The absorbance of the characteristic absorption peak at 664 nm was measured to calculate the corresponding reduction rate of methylene blue.
3. Results and discussion
3.1. Characterizations of PP-EPMn and PX-EPMn
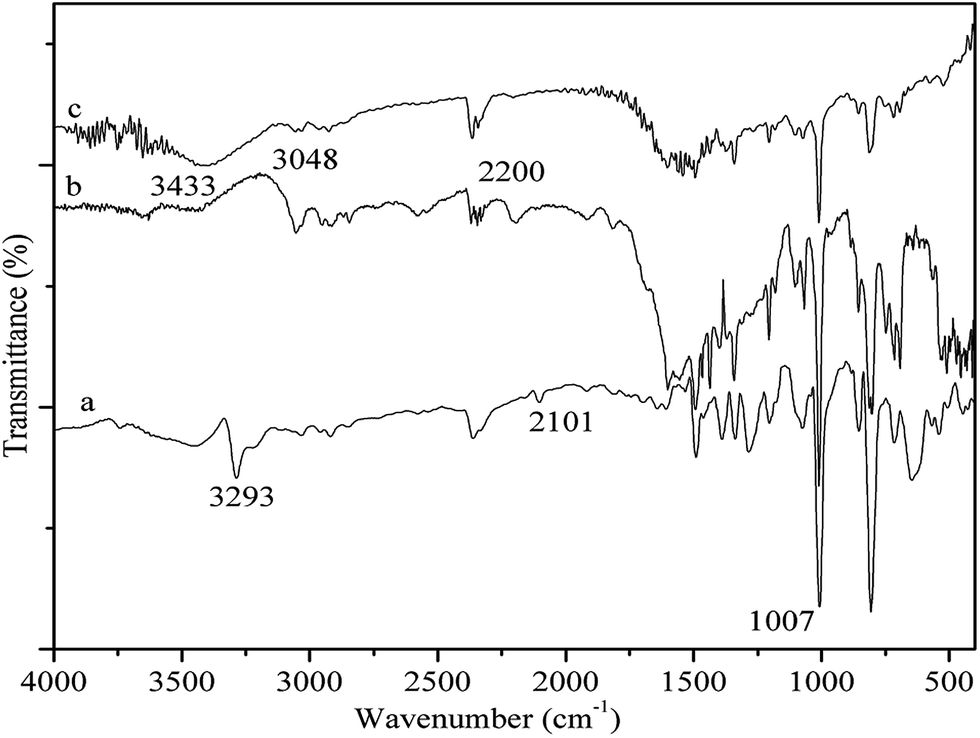
As shown in Fig. 1, the IR spectra of PP-EPMn (b) and PX-EPMn (c) are obviously different from that of [p-ethynyl]4PMn (a), suggesting that the polymerization reaction occurred indeed. The peak at 3293 cm−1 in the spectrum of [p-ethynyl]4PMn is assigned to C–H stretching of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H, which disappears after formation of PP-EPMn and PX-EPMn due to the consumption of C–H. The characteristic peak of –CC– at 2101 cm−1 attenuates and even disappears because the structures of PP-EPMn and PX-EPMn are highly symmetric. Meanwhile, the peak bathochromically shifts to 2200 cm−1. Besides, the peak at 3048 cm−1 belonging to C–H stretching vibration of phenylene and xenyl is strengthened, which means there are phenylene and xenyl in PP-EPMn and PX-EPMn. On the other hand, the IR spectra of PP-EPMn and PX-EPMn exhibit a characteristic N–Mn vibration band at 1009 cm−1, which is the same as that of [p-ethynyl]4PMn (1007 cm−1). The bands at 1599 cm−1 correspond to C
C–H, which disappears after formation of PP-EPMn and PX-EPMn due to the consumption of C–H. The characteristic peak of –CC– at 2101 cm−1 attenuates and even disappears because the structures of PP-EPMn and PX-EPMn are highly symmetric. Meanwhile, the peak bathochromically shifts to 2200 cm−1. Besides, the peak at 3048 cm−1 belonging to C–H stretching vibration of phenylene and xenyl is strengthened, which means there are phenylene and xenyl in PP-EPMn and PX-EPMn. On the other hand, the IR spectra of PP-EPMn and PX-EPMn exhibit a characteristic N–Mn vibration band at 1009 cm−1, which is the same as that of [p-ethynyl]4PMn (1007 cm−1). The bands at 1599 cm−1 correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching in phenyls, and that at 1493 cm−1 represents CC stretching in porphyrin. Thus, [p-ethynyl]4PMn had already been introduced into the porous structures of PP-EPMn and PX-EPMn in which manganese ion was still stably coordinated.
C stretching in phenyls, and that at 1493 cm−1 represents CC stretching in porphyrin. Thus, [p-ethynyl]4PMn had already been introduced into the porous structures of PP-EPMn and PX-EPMn in which manganese ion was still stably coordinated.
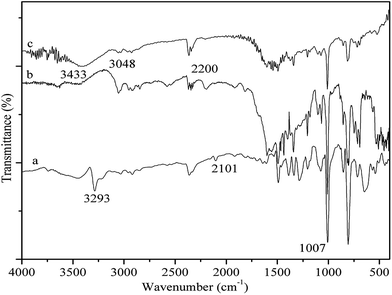 |
| | Fig. 1 FT-IR spectra of [p-ethynyl]4PMn (a), PP-EPMn (b) and PX-EPMn (c). | |
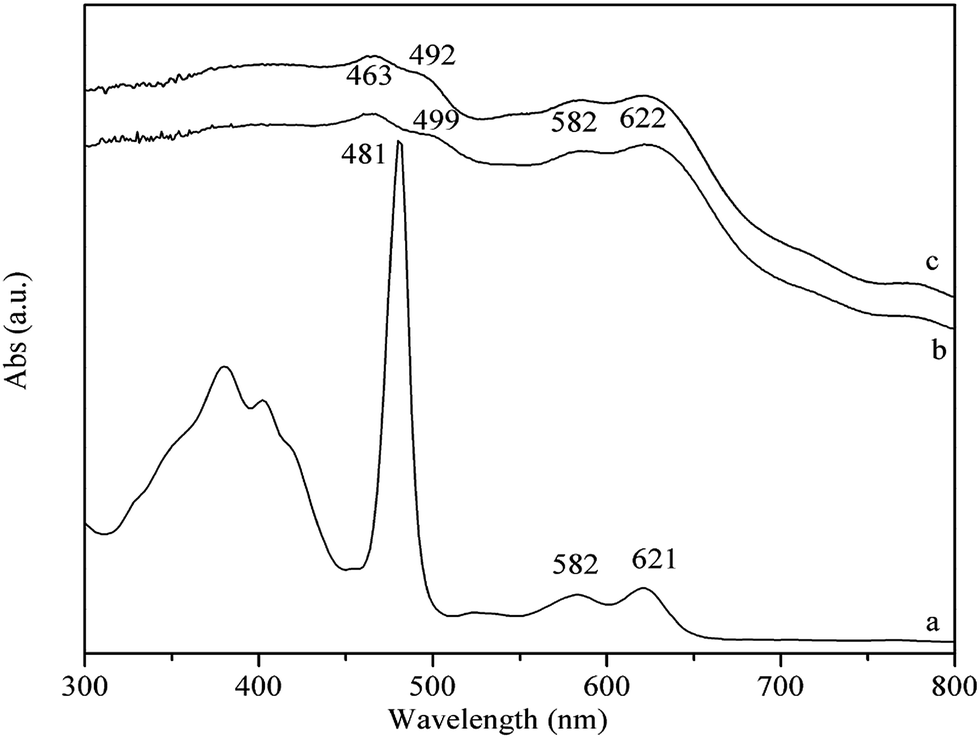
The UV-Vis spectra of PP-EPMn and PX-EPMn solid powders and that of [p-ethynyl]4PMn in CHCl3 solution are exhibited in Fig. 2. In curve (a), a strong absorption band at 481 nm is the Soret band of [p-ethynyl]4PMn, and those at 582 and 621 nm are the Q-bands. PP-EPMn and PX-EPMn (curves b, c) also show similar typical UV-Vis spectra to that of [p-ethynyl]4PMn, i.e. there are Soret bands at 463, 499 nm and Q-bands at 582, 622 nm in PP-EPMn, and Soret bands at 463, 492 nm and Q-bands at 582, 622 nm in PX-EPMn. Compared with [p-ethynyl]4PMn monomer, the Soret band in PP-EPMn splits into 463, 499 nm and that in PX-EPMn splits into 463, 492 nm because of the conjugated bridges increasing the molecular connectivity and the electronic couplings of polymers.
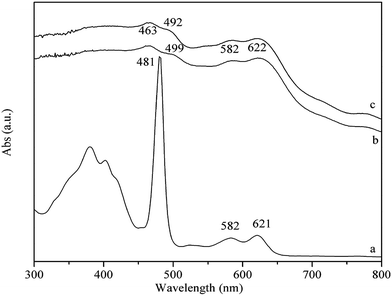 |
| | Fig. 2 UV-Vis spectra of [p-ethynyl]4PMn (a; CHCl3 solution), PP-EPMn (b; powder sample), and PX-EPMn (c; powder sample). | |
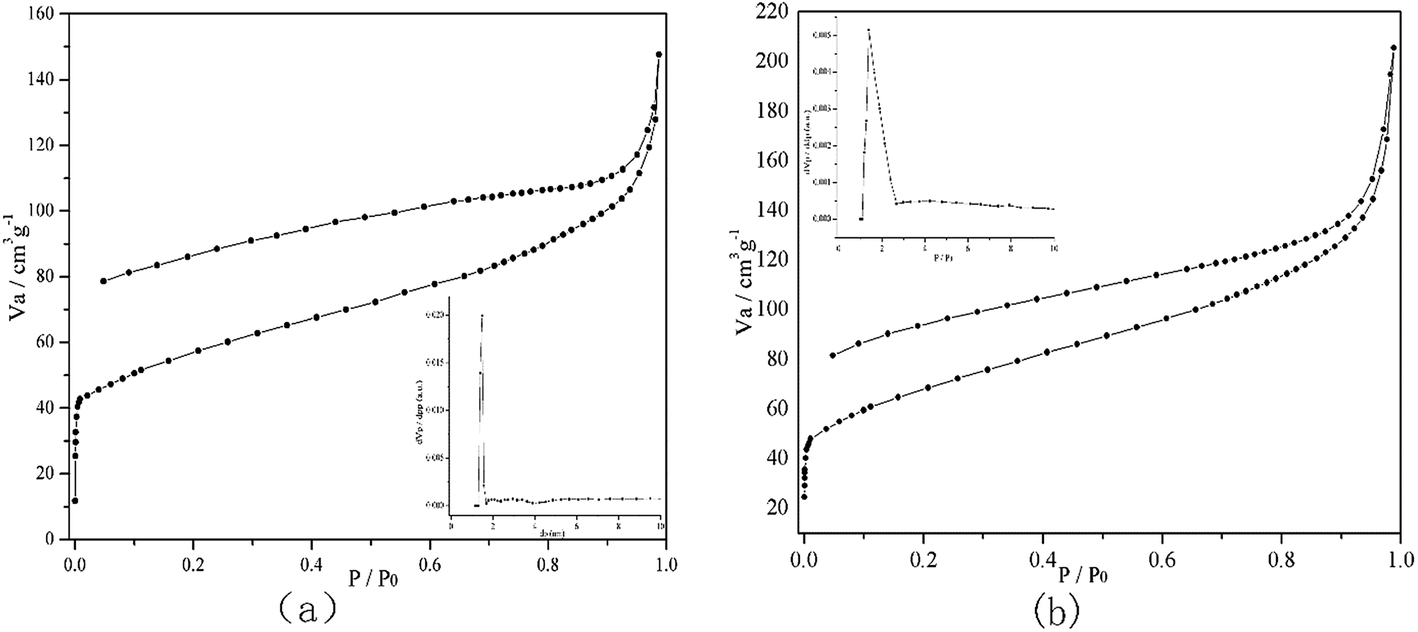
PP-EPMn and PX-EPMn were conjugated metalloporphyrin polymers with large BET surface areas and porous structures. At 77 K, they displayed typical type-III nitrogen sorption isotherm curves along with strong adsorptions at low pressure (P/P0 < 0.1) (Fig. 3), suggesting micropores and mesopores coexisted in the framework. The BET surface area of PP-EPMn was as high as 420 m2 g−1, of which the microporous area (about 194 m2 g−1) accounted for 46.2%. Meanwhile, the BET surface area of PX-EPMn was 480 m2 g−1, of which the microporous area (about 204 m2 g−1) accounted for 42.5% (Table 1). Fig. 3 also shows that there are a large number of micropores in PP-EPMn and PX-EPMn frameworks. The main pore diameters of PP-EPMn and PX-EPMn were about 1.48 nm and 1.37 nm respectively, and the contributions to pore volume were 44.76% and 41.52% respectively. The pore structures of PP-EPMn and PX-EPMn also verified porphyrin polymerization. The pore diameter of PP-EPMn slightly exceeded that of PX-EPMn because the bridge of phenylene was not strictly coplanar with porphyrin ring during polymerization, leading to distortion and interpenetration between the polymer mesh and chain and forming micropores. The non-coplanarity between xenyl and porphyrin ring was stronger than that between phenylene and porphyrin ring, so PX-EPMn had more obvious distortion and interpenetration as well as smaller pore diameter than those of PP-EPMn. Moreover, due to the larger molecular size of xenyl than that of phenylene, the pore size distribution of PX-EPMn was more decentralized than that of PP-EPMn.
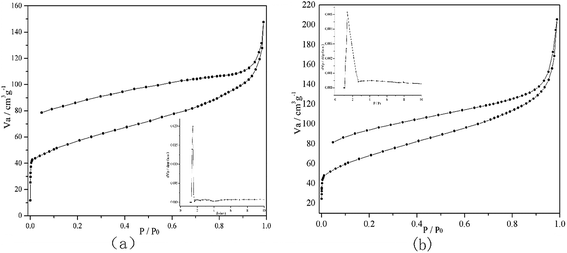 |
| | Fig. 3 Nitrogen adsorption and desorption isotherm profiles and pore diameter examination of PP-EPMn (a) and PX-EPMn (b) at 77 K. | |
Table 1 Surface properties of PP-EPMn and PX-EPMna
| |
SBET (m2 g−1) |
Smicro (m2 g−1) |
Vtotal (cm3 g−1) |
Vmicro (cm3 g−1) |
| SBET: total surface area; Smicro: micropore surface area; Vtotal: total pore volume; Vmicro: micropore volume. |
| PP-EPMn |
420 |
194 |
0.42 |
0.188 |
| PX-EPMn |
480 |
204 |
0.619 |
0.257 |
| [p-Ethynyl]4PMn |
16.2 |
2.0 |
2.0 × 10−4 |
— |
FE-SEM images (Fig. 4a and b) show that PP-EPMn and PX-EPMn are composed of nanosized round spheres. The diameters of PP-EPMn nanospheres ranged from 200 nm to 400 nm, whereas those of PX-EPMn nanospheres with a more uniform diameter were mostly 150 nm. The diameter of PP-EPMn nanospheres was slightly larger than that of PX-EPMn, corresponding to the higher distortion degree of polymer mesh and chain during polymerization. Therefore, with increasing distortion degree, PX-EPMn became more compact during polymerization, forming nanospheres with smaller diameters. TEM images (Fig. 4c and e) clearly demonstrate spherical structures of the materials, and the sizes of spheres are the same as those observed by SEM. The surface of PX-EPMn was much rougher than that of PP-EPMn because the spheres surface of PP-EPMn was will-proportioned, and PX-EPMn was more uneven. TEM revealed that PP-EPMn nanospheres had smooth edges but PX-EPMn ones had obvious light and dark areas. In addition, HE-TEM images (Fig. 4d and f) present that PP-EPMn and PX-EPMn have the same nanoporous structures, but the porous channel of the former is slightly larger.
 |
| | Fig. 4 FE-SEM images of PP-EPMn (a) and PX-EPMn (b). TEM images of PP-EPMn (c, d) and PX-EPMn (e, f). | |
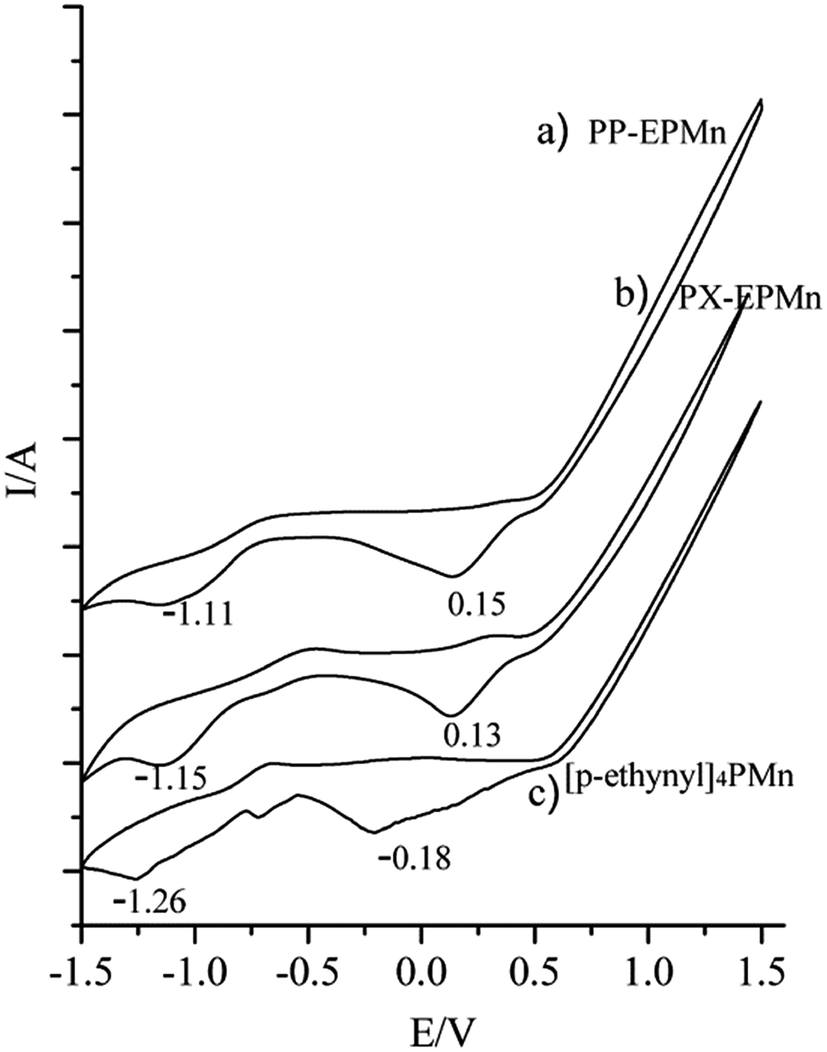
Cyclic voltammograms show that the first reduction potentials of PP-EPMn and PX-EPMn significantly shift to more positive values compared with that of [p-ethynyl]4PMn (from −0.18 V to 0.13 V and 0.15 V respectively, Fig. 5), indicating that porphyrin monomers were bridged by phenylene and xenyl groups on the four directions of porphyrin rings. Over 310 mV decrease in the reduction potential can be attributed to the good electron transfer properties of porphyrin networks in PP-EPMn and PX-EPMn. As a conjugated bridge between porphyrin monomers, phenylene ethylene was advantageous to wide-range flow of electrons and delocalization. As a result, Mn3+ ions in the middle of porphyrin rings were further activated to form active valence ions Mn2+, which enhanced the catalytic activities of polymers in the oxidation reactions.
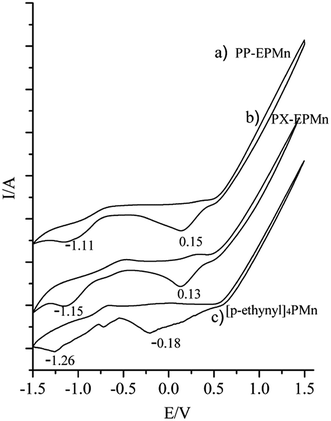 |
| | Fig. 5 Cyclic voltammetry curves of PP-EPMn (a), PX-EPMn (b) on the glass carbon electrodes in 0.1 mol L−1 (CH3CH2)4NBr CH2Cl2 solution; cyclic voltammetry curves of [p-ethynyl]4PMn (c) in CH2Cl2, 0.1 mol L−1 (CH3CH2)4NBr. Scan rate 50 mV s−1. | |
3.2. Catalytic degradation of methylene blue
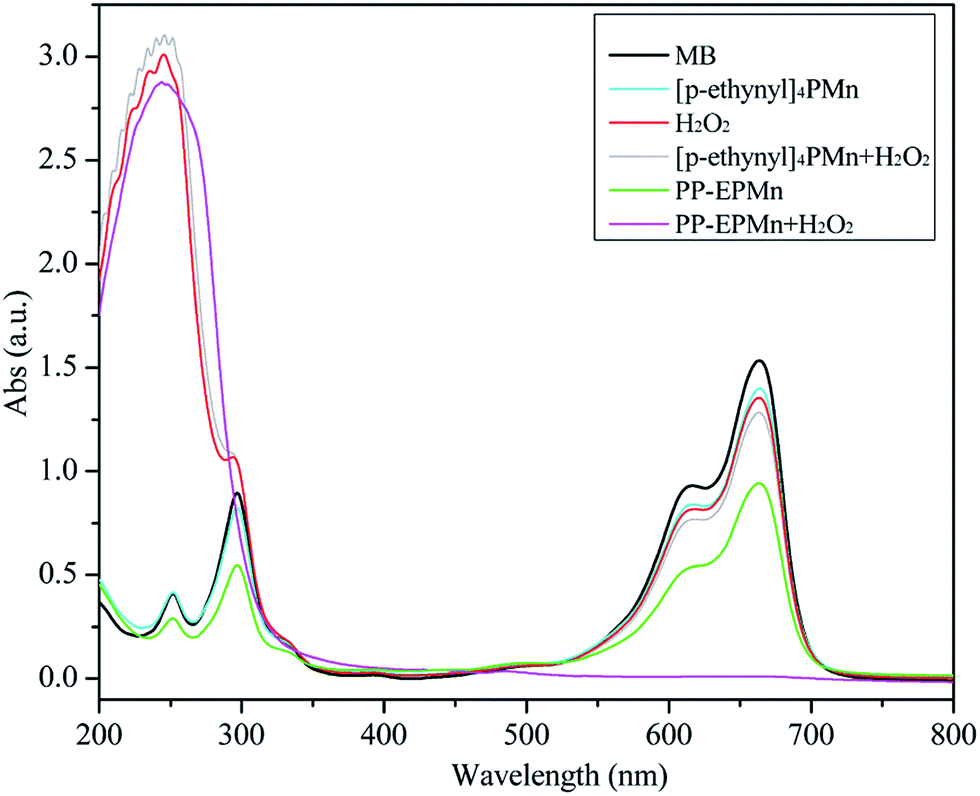
To study the catalytic effects of PP-EPMn and PX-EPMn on methylene blue oxidizing degradation, comparative tests were carried out in the presence of hydrogen peroxide, porphyrin, polymers, porphyrin and hydrogen peroxide, or polymers and hydrogen peroxide, respectively. The results are summarized in Table 2, and the UV-Vis spectra of methylene blue after 3 h of catalytic oxidation with different catalysts are shown in Fig. 6.
Table 2 Efficiency of various catalysts in degradation of methylene bluea
| Entry |
Catalyst |
Degradation (%) |
| Methylene blue, 50 mL (10 mg L−1); catalyst, 4 mg; H2O2, 1 mL; T = 25 °C; pH = 7; time = 3 h. |
| 1 |
H2O2 |
12.5 |
| 2 |
[p-Ethynyl]4PMn |
9.5 |
| 3 |
[p-Ethynyl]4PMn + H2O2 |
15.5 |
| 4 |
PP-EPMn |
38.2 |
| 5 |
PP-EPMn + H2O2 |
99.6 |
| 6 |
PX-EPMn + H2O2 |
99.5 |
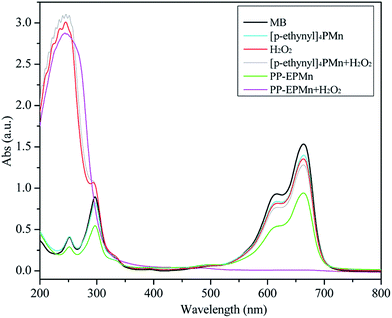 |
| | Fig. 6 UV-Vis spectroscopy of methylene blue after 3 h catalytic oxidation used different catalysts. Reaction conditions: methylene blue, 50 mL (10 mg L−1); catalyst, 4 mg; H2O2, 1 mL; T = 25 °C; pH = 7; time = 3 h. | |
A small amount of methylene blue (12.5%) was degraded only in the presence of hydrogen peroxide and without catalysts (Table 2). In the presence of [p-ethynyl]4PMn or even coexisting hydrogen peroxide, the oxidation did not exceed 15.5%, indicating that it failed to effectively degrade methylene blue. Additionally, 38.2% of methylene blue was degraded by PP-EPMn in the absence of H2O2, which was markedly increased to 99.5% after 1 mL of H2O2 was added. This result is also apparent from Fig. 6 that the characteristic absorption peak of methylene blue at 664 nm disappears after reaction in the presence of H2O2 and PP-EPMn. We also detected the reaction mixture by ion chromatograph and NO3−, SO42− and Cl− were found. These suggest the complete degradation of methylene blue.
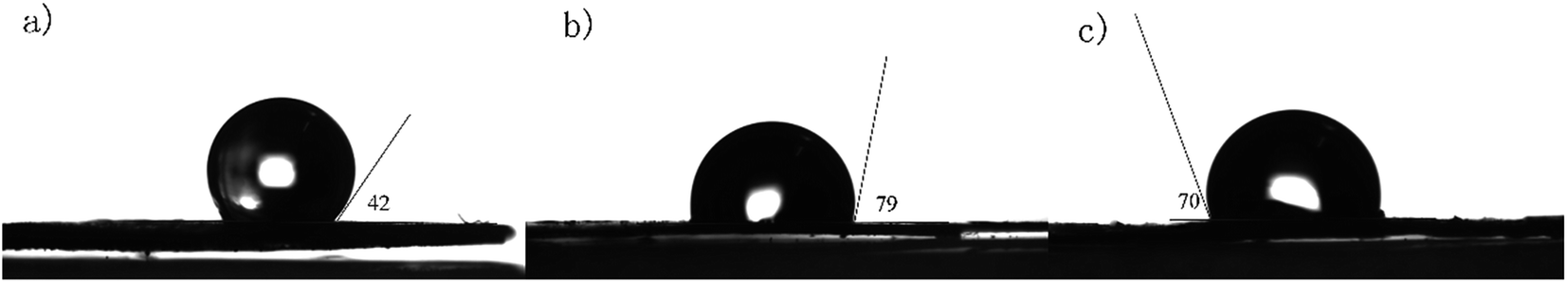
As shown in Fig. 7, unlike highly hydrophobic [p-ethynyl]4PMn which has a contract angle of 138° on the deionized water interface, PP-EPMn and PX-EPMn have the contract angles of about 101° and 110° respectively, suggesting that they were more hydrophilic. The contract angle of PP-EPMn was close to the wettability limit of water (90°), allowing it to contact and to react with dye molecules in water more easily, and its degradation efficiency was higher. This is in accordance with the reports about hydrophobicity affecting the catalytic performance.36 In fact, PP-EPMn dispersed more rapidly and uniformly in aqueous solution in the experimental process. Thus, owing to the synergistic effects of porous nanosphere structure, conjugated covalent structure and good wettability, the catalytic activities of PP-EPMn and PX-EPMn were improved tremendously in the degradation of methylene blue dye.
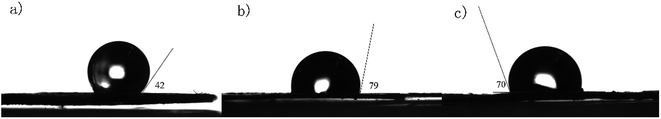 |
| | Fig. 7 Water contact angle of [p-ethynyl]4PMn (a), PP-EPMn (b) and PX-EPMn (c). | |
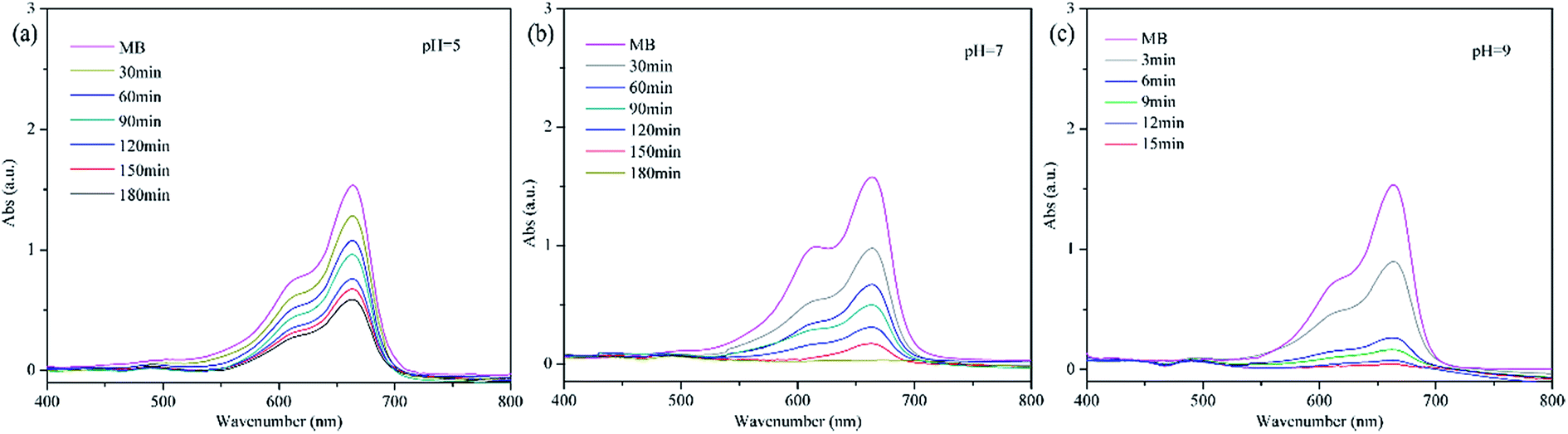
Water-soluble organic dyes are most difficult to deal with among all pollutants in wastewater, the degradation of which is significantly affected by pH, so we evaluated the effects of pH on the catalytic degradation of methylene blue by PP-EPMn (Fig. 8 and 9).
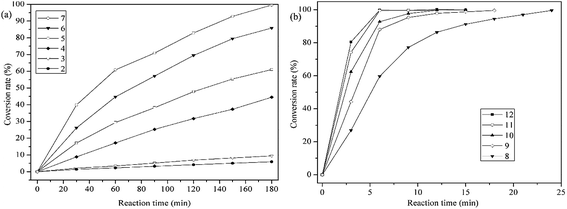 |
| | Fig. 8 Effects of pH on the catalytic oxidation. Reaction condition: methylene blue, 50 mL (10 mg L−1); catalyst, 4 mg; H2O2; T = 25 °C. | |
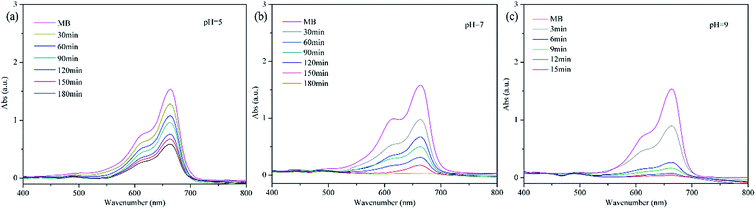 |
| | Fig. 9 Degradation of methylene blue by PP-EPMn at pH = 5, 7, 9. Reaction condition: methylene blue, 50 mL (10 mg L−1); catalyst, 4 mg; H2O2; T = 25 °C. | |
As shown in Fig. 8, methylene blue is hardly degraded at pH < 3, but its degradation rate gradually increases with rising pH within 3 h. The degradation rate of methylene blue reached 59% at pH = 5 (Fig. 9a), increased significantly thereafter (Fig. 8a). Especially, the rate was 99.6% at pH 7 after 3 h (Fig. 9b). Even more gratifying is that PP-EPMn degraded methylene blue better in alkaline solution (Fig. 8b), and the degradation rate remained above 99% with the reaction time greatly reduced. At pH = 9 (Fig. 9c), the degradation rate of methylene blue reached 87% in about 6 min, and reached 99% within 12 min. When pH was increased to 11, the degradation rate reached 99% only within 6 min. Oxomanganese(IV) porphyrin complexes were generated by homolytic –O–O– cleavage of hydroperoxides by manganese(III) porphyrin polymer in acidic condition, which were less reactive intermediates than oxomanganese(V) porphyrin complexes that are generated by heterolytic –O–O– cleavage of hydro peroxides in alkaline condition.37 Hence, PP-EPMn degraded methylene blue better in alkaline condition than in acidic condition.
Actually, printing and dyeing plants generally generate alkaline wastewater. Since traditional Fenton reagents need acidic conditions to treat wastewater, considerable acids are consumed. In contrast, alkaline wastewater can be directly treated by PP-EPMn. On the other hand, only wastewater at pH = 6–9 can be discharged into the environment, according to the national environmental standards. Given that PP-EPMn can function in alkaline wastewater which without cumbersome pH adjustment before being discharged into the environment, it is obviously superior to Fenton reagents. Therefore, PP-EPMn is a suitable catalyst for treating actual neutral or alkaline wastewater.
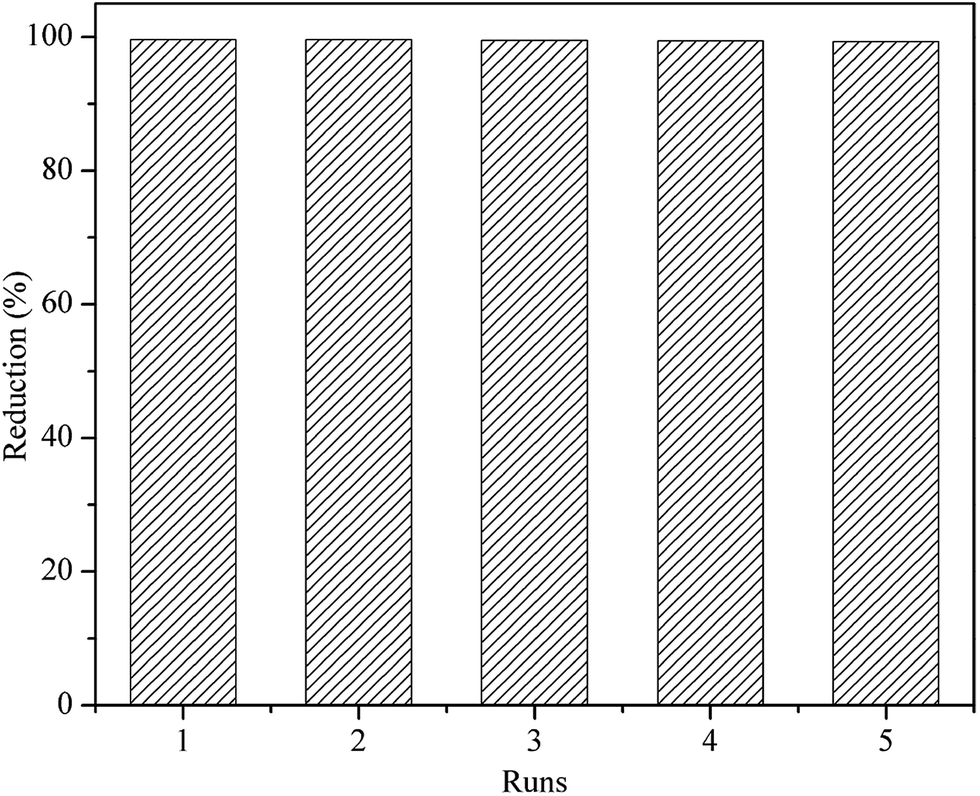
3.3. Catalyst recyclability
Metalloporphyrin was polymerized to special insoluble macromolecular polymers that converted metalloporphyrin catalytic system to heterogeneous catalysis. This is an important way to improve the stability and even catalytic activity of metalloporphyrin, making it easily recovered for recycling in reaction. Fig. 10 shows the percentages of methylene blue degradation catalyzed by PP-EPMn after each recovery cycle. Nearly 99% of methylene blue was reduced in each reaction cycle, suggesting that PP-EPMn remained stable and highly active after 5 runs in the system.
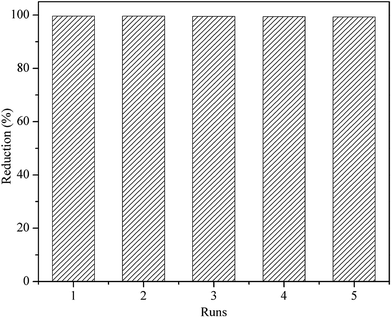 |
| | Fig. 10 The repetitive oxidation of PP-EPMn. | |
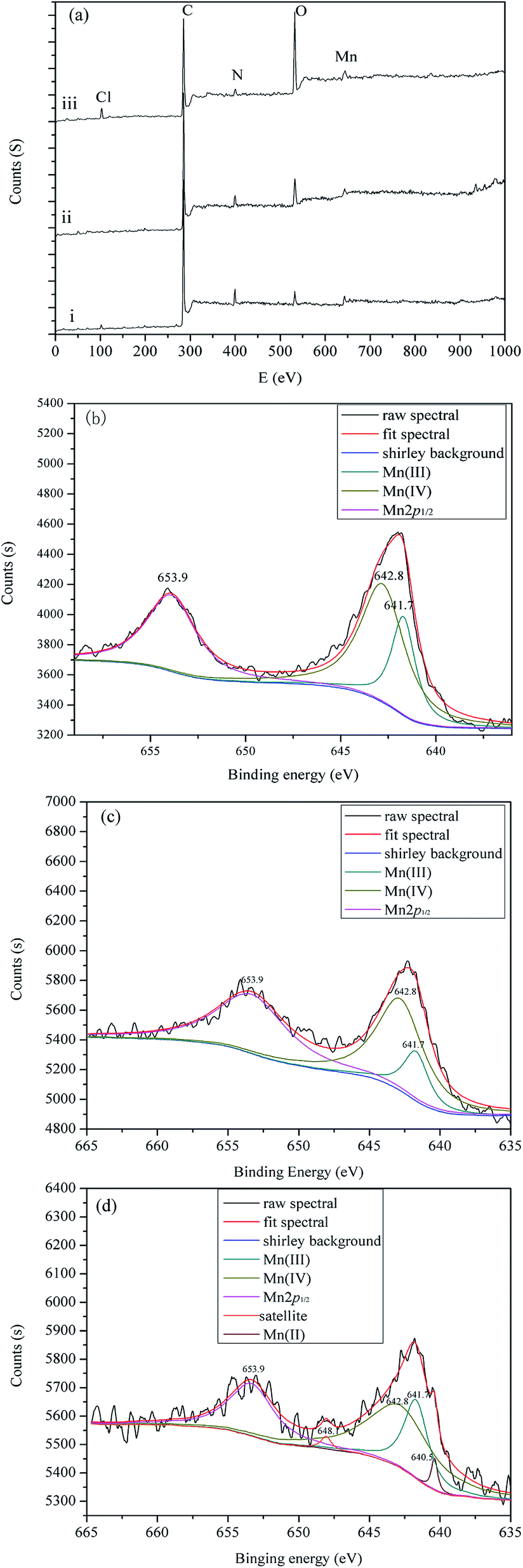
3.4. XPS analysis
As shown in Fig. 11a, a set of peaks corresponding to Cl 2p (197.8 eV), C 1s (284.8 eV), N 1s (398.9 eV), O 1s (532.2 eV) and Mn 2p (643.1 eV) are observed. [p-Ethynyl]4PMn (Fig. 11a, i), PP-EPMn (Fig. 11a, ii) and used PP-EPMn (Fig. 11a, iii) had nearly the same chemical compositions. The ratios of the catalysts are shown in Table 3. Obviously, the proportions of atomic compositions of PP-EPMn and used PP-EPMn did not change which indicated that PP-EPMn was stable and the polymer structure was undamaged after five times of catalytic cycles. In the Mn 2p spectrum of [p-ethynyl]4PMn (Fig. 11b), there were two main peaks, i.e. Mn 2p1/2 at 653.9 eV and Mn 2p3/2 at 641.9 eV. The peak of Mn 2p3/2 can bed convoluted into two peaks, i.e. 641.7 eV for Mn3+ and 642.8 eV for Mn4+. As shown in Fig. 11d, used PP-EPMn also exhibits a Mn3+ peak at 641.7 eV and a Mn4+ peak at 642.8 eV. In the meantime, there is a peak of Mn2+ at 640.5 eV and also a satellite peak at 648.1 eV, indicating that used PP-EPMn had Mn2+. This is the proof to the literature that Mn3+ should be reduced to Mn2+ at first in metalloporphyrin catalysis reactions.38 Besides, the Mn 2p3/2 peak of [p-ethynyl]4PMn (about 641.9 eV) shifted to 642.4 eV in PP-EPMn after polymerization, which revealed that the redox property of PP-EPMn was affected by the hyperconjugation structure in which electron cloud was delocalized from the center of Mn to the bridges and finally promoting the catalytic reaction.39
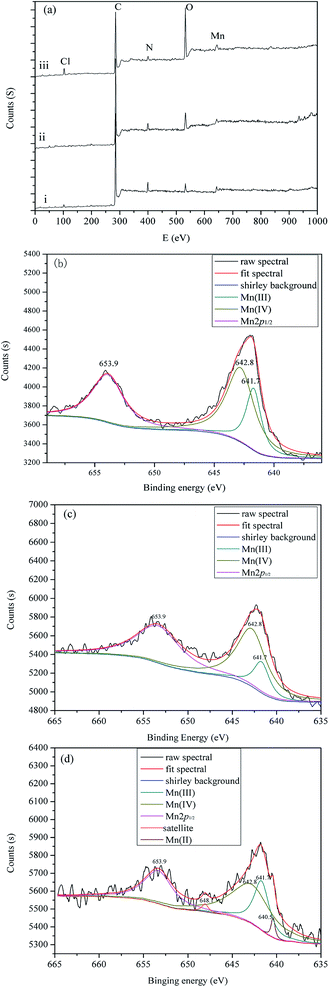 |
| | Fig. 11 XPS analysis of different samples. (a) XPS survey of [p-ethynyl]4PMn (i), PP-EPMn (ii) and used PP-EPMn (iii). (b) Decomposed XPS Mn 2p binding energy spectrum of [p-ethynyl]4PMn catalyst. (c) Decomposed XPS Mn 2p binding energy spectrum of PP-EPMn catalyst. (d) Decomposed XPS Mn 2p binding energy spectrum of used PP-EPMn catalyst. | |
Table 3 The ratio analysis of the peaks in XPS spectra of the catalysts
| Catalysts |
Atomic composition (at%) |
| C |
O |
N |
Cl |
Mn |
| [p-Ethynyl]4PMn |
88.94 |
3.61 |
4.87 |
1.38 |
1.2 |
| PP-EPMn |
84.41 |
9.02 |
4.61 |
0.82 |
1.14 |
| PP-EPMn used |
80.55 |
13.3 |
4.54 |
0.51 |
1.1 |
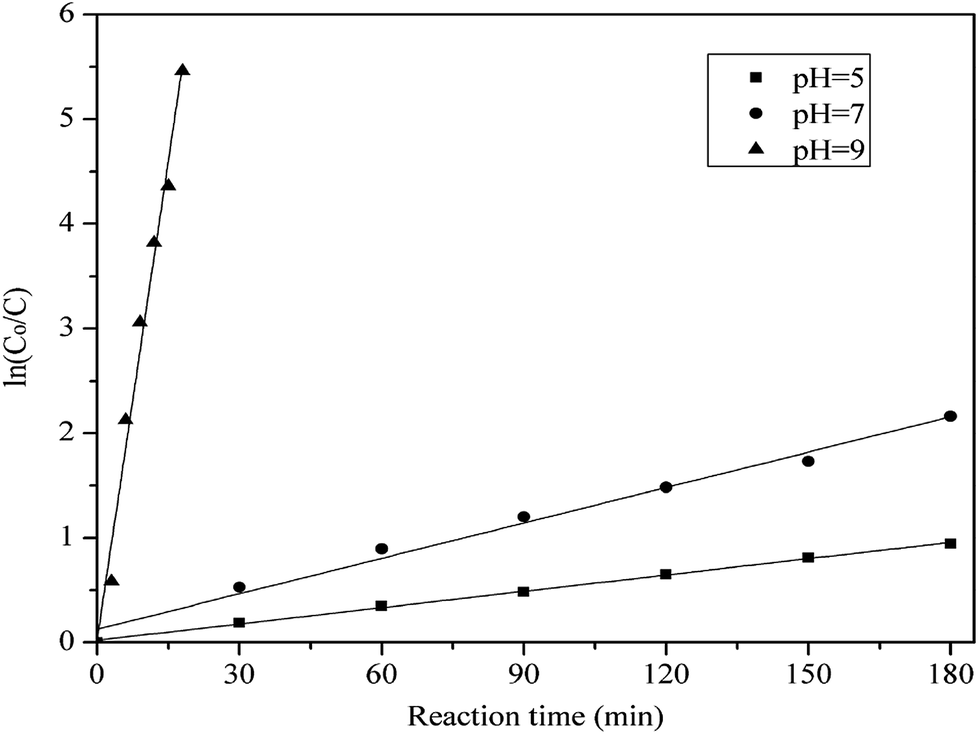
3.5. Reaction kinetics
The reaction kinetics of methylene blue oxidized by hydrogen peroxide with PP-EPMn as catalyst was also investigated. The relationship between the concentration variation of methylene blue (ln(Co/C)) and the reaction time (t) was shown in Fig. 12. It was found that the kinetic model was corresponded to the pseudo-first order at different pH values. The reaction rate constants can be calculated by the following equation, where k is the apparent rate constant at different pH values.
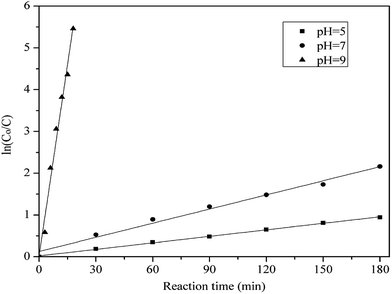 |
| | Fig. 12 ln(Co/C) versus reaction time for the reduction of methylene blue in the presence of PP-EPMn at pH = 5, 7, 9. Co and C are the initial and transient concentrations of methylene blue during the reaction, respectively. Reaction conditions: methylene blue, 50 mL (10 mg L−1); catalyst, 4 mg PP-EPMn; H2O2; T = 25 °C. | |
When pH = 5, 7, 9, the value of k was calculated as 4.26 × 10−3 min−1, 1.14 × 10−2 min−1, 0.308 min−1, respectively. So PP-EPMn had higher degradation efficiency under alkaline condition (pH = ≥7).
3.6. Plausible reaction mechanism
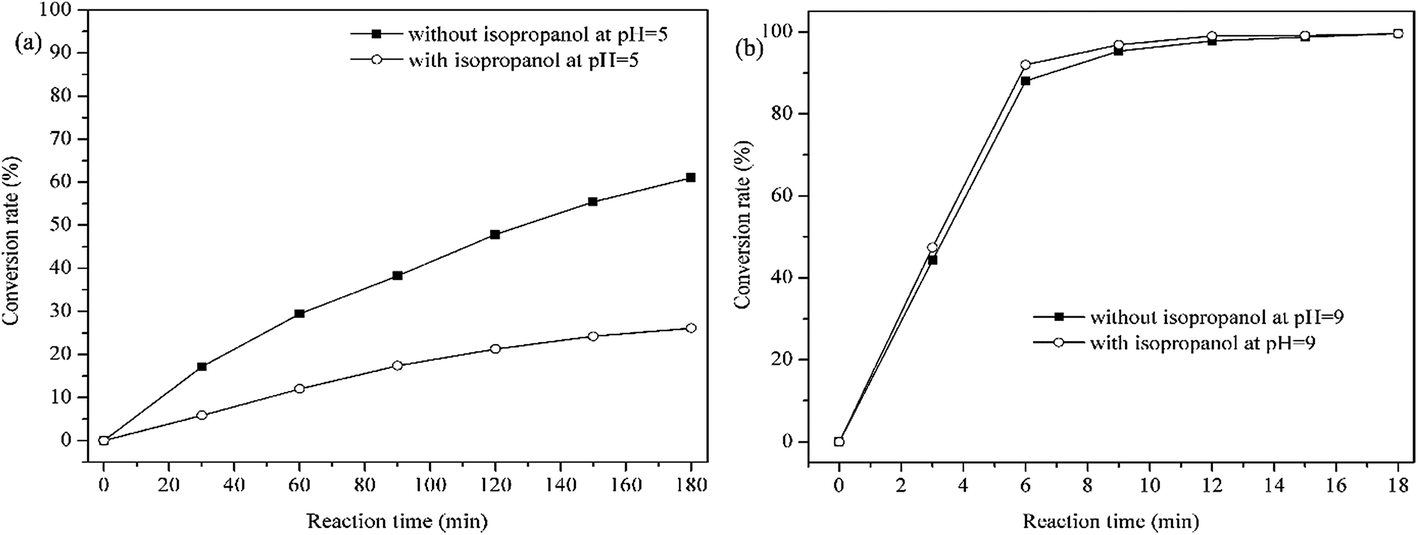
The efficiency of methylene blue decomposition is determined by two competitive processes of hydrogen peroxide cleavages, i.e. heterolytic and homolytic cleavages of –O–O– bond in manganese(III)-coordinated hydrogen peroxide molecule.37,40 The former process produces a ˙OH radical, while the latter process generates a oxomanganese(V) porphyrin intermediate which is active for methylene blue decomposition.39 The main oxidative species in catalytic process can be detected through radical trapping experiments. As shown in Fig. 13b, when adding isopropanol, a scavenger of hydroxyl radicals,41 little changes were found at pH = 9. However, the catalytic activity of PP-EPMn was greatly inhibited at pH = 5 (Fig. 13a). Accordingly, ˙OH radical would be the main active species at pH = 5, while the oxomanganese(V) porphyrin intermediate was the main active and more efficient species at pH = 9.
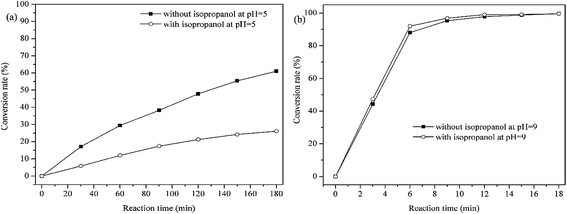 |
| | Fig. 13 Concentration changes of methylene blue in the presence or without isopropanol at pH = 5 and pH = 9. Reaction conditions: methylene blue, 50 mL (10 mg L−1); catalyst, 4 mg PP-EPMn; H2O2; T = 25 °C. | |
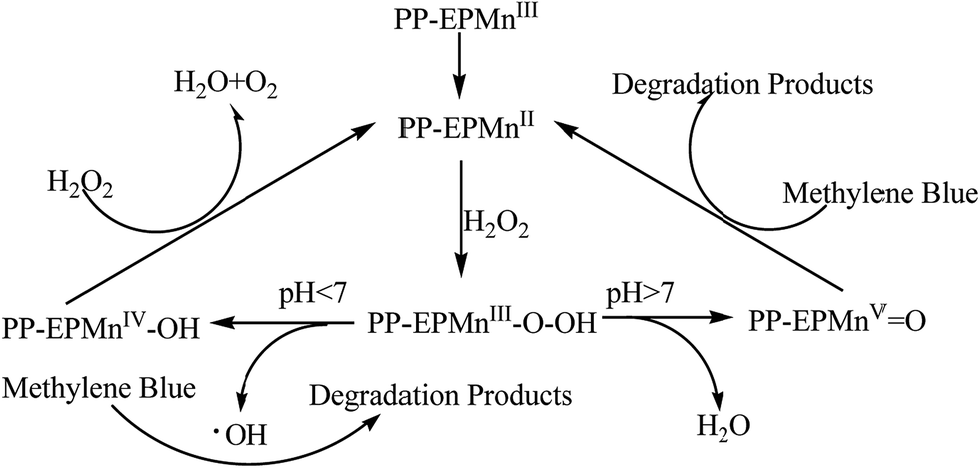
Since methylene blue was completely degraded in a short time in alkaline solution but only partly degraded in a long time in acidic solution, apparently the –O–O– bond cleavage of hydro peroxides was sensitive to the pH of the reaction solution. So we postulated that the –O–O– bond cleavage of H2O2 depended on pH of the reaction solution. –O–O– bond homolysis prevailed at low pH and –O–O– bond heterolysis became predominant at high pH. The plausible reaction mechanism is shown in Scheme 2.
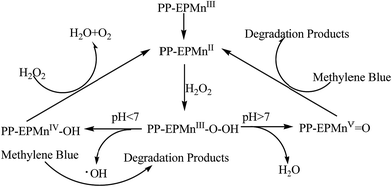 |
| | Scheme 2 The plausible reaction mechanism. | |
In acidic condition, the homolytic cleavage of –O–O– bond in PP-EPMnIII-OOH produces a ˙OH radical, but PP-EPMnIV-OH with a lower activity can decompose H2O2 simultaneously, so methylene blue decomposed slowly and partly. In alkaline solution, however, the heterolytic cleavage of –O–O– bond in PP-EPMnIII-OOH generates an oxomanganese(V) porphyrin intermediate that is active for methylene blue decomposition. As a result, methylene blue was better degraded in alkaline condition.
4. Conclusions
Nanospheres of organic conjugated metalloporphyrin polymers (PP-EPMn and PX-EPMn) were synthesized with [p-ethynyl]4PMn as building block and with phenylene or xenyl groups as bridges for the first time. PP-EPMn and PX-EPMn both had special nanosphere structures and PP-EPMn nanospheres had diameters of 200–400 nm, but PX-EPMn ones had a more uniform diameter of 150 nm. Methylene blue solution was chosen as a model of typical dye-containing wastewater. PP-EPMn and PX-EPMn were applied as catalysts to degrade methylene blue in aqueous solution. The effects of temperature, amount of catalyst, pH and amount of hydrogen peroxidate were assessed in detail (ESI†). Both PP-EPMn and PX-EPMn showed high catalytic efficiencies against methylene blue pollutant through degradation. The two polymers applicable to alkaline wastewater treatment were superior to Fenton reagents which only worked in acidic condition. PP-EPMn and PX-EPMn can be recycled and continuously used, thereby being potentially suitable for industrial methylene blue removal.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to give thanks for the financial support from the National Nature Science Foundation of China (Grant No. 21576074). The authors are also grateful for the financial support of Hunan University.
References
- S. Durot, J. Taesch and V. Heitz, Chem. Rev., 2014, 114, 8542–8578 CrossRef CAS PubMed.
- Q. C. Dong, W. S. Qu, W. Q. Liang, F. F. Tai, K. P. Guo, C. W. Leung, Y. H. Lo and W. Y. Wong, Mater. Chem., 2016, 4, 5010–5018 CAS.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- A. D'Urso, A. Mammana, M. Balaz, A. E. Holmes, N. Berova, R. Lauceri and R. Purrello, J. Am. Chem. Soc., 2009, 131, 2046–2047 CrossRef PubMed.
- M. G. Rabbani, A. K. Sekizkardes, Z. Kahveci, T. E. Reich, R. S. Ding and H. M. A. El-Kaderi, Chem.–Eur. J., 2013, 19, 3324–3328 CrossRef CAS PubMed.
- H. M. Key, P. Dydio, D. S. Clark and J. F. Hartwig, Nature, 2016, 534, 534–537 CrossRef CAS PubMed.
- G. Labut, J. L. Seris and B. Meunier, Angew. Chem., Int. Ed. Engl., 1990, 29, 1471–1473 CrossRef.
- Q. Q. Zhu, M. Igarashi, M. Sasaki, T. Miyamoto, R. Kodama and M. Fukushima, Appl. Catal., B, 2016, 183, 61–68 CrossRef CAS.
- C. L. Hill and R. B. Brown, J. Am. Chem. Soc., 1986, 108, 536–538 CrossRef CAS PubMed.
- I. Dror and M. A. Schlautman, Environ. Toxicol. Chem., 2004, 23, 252–257 CrossRef CAS PubMed.
- W. J. Yang, B. J. Yin, C. C. Guo, Z. Tan and L. Zhang, Chin. J. Catal., 2010, 31, 535–540 CrossRef CAS.
- Q. Y. Liu, R. R. Zhu, Y. L. Jiang, Q. Y. Jia, S. G. Yang, Q. Shao, D. M. Wang and P. Cui, Mater. Sci. Eng., B, 2014, 188, 106–113 CrossRef CAS.
- K. Kalyanasundaram, Inorg. Chem., 1984, 23, 2453–2459 CrossRef CAS.
- M. A. Martí, S. E. Bari, D. A. Estrin and F. Doctorovich, J. Am. Chem. Soc., 2005, 127, 4680–4684 CrossRef PubMed.
- C. Crestini, R. Saladino, P. Tagliatesta and T. Boschi, Bioorg. Med. Chem., 1999, 7, 1897–1905 CrossRef CAS PubMed.
- A. Rezaeifard and M. Jafarpour, Catal. Sci. Technol., 2014, 4, 1960–1969 CAS.
- H. Kim, W. Kim, Y. Mackeyev, G. S. Lee, H. J. Kim, T. Tachikawa, S. k. Hong, S. Lee, J. B. Kim, L. J. Wilson, T. Majima, P. J. J. Alvarez, W. Y. Choi and J. S. Lee, Environ. Sci. Technol., 2012, 46, 9606–9613 CrossRef CAS PubMed.
- H. Ghafuri, S. Rahmani, R. Rahimi and E. Mohammadiyan, RSC Adv., 2016, 6, 62916–62922 RSC.
- M. Moshari, M. Rabbani and R. Rahimi, Res. Chem. Intermed., 2016, 42, 5441–5455 CrossRef CAS.
- M. Wei, J. Wan, Z. W. Hu, Z. Q. Peng and B. Wang, Appl. Surf. Sci., 2016, 377, 149–158 CrossRef CAS.
- C. Wang, J. Li, G. Mele, G. M Yang, F. X. Zhang, L. Palmisano and G. Vasapollo, Appl. Catal., B, 2007, 76, 218–226 CrossRef CAS.
- J. Yoo, N. Park, J. H. Park, J. H. Park, S. Kang, S. M. Lee, H. J. Kim, H. Jo, J. G. Park and S. U. Son, ACS Catal., 2015, 5, 350–355 CrossRef CAS.
- Y. H. Jiang, L. Q. Guo, W. L. Zhang, F. Dai, Y. Yan, F. M. Zhang and H. Y. Lv, Desalin. Water Treat., 2014, 52, 3489–3496 CrossRef CAS.
- X. Guo, Y. Y. Li, D. H. Shen, J. Gan, M. Tian, Z. Gang and Z. G. Liu, Appl. Catal., A, 2012, 413–414, 30–35 CrossRef CAS.
- W. J. Yang, C. C. Guo, Z. Y. Li and N. Y. Tao, J. Porphyrins Phthalocyanines, 2009, 13, 973–979 CrossRef CAS.
- Y. J. Li, B. S. Sun and W. J. Yang, Appl. Catal., A, 2016, 515, 164–169 CrossRef CAS.
- L. Chen, Y. Yang and D. L. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS PubMed.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- Y. J. Li, C. R. Liu and W. J. Yang, New J. Chem., 2017, 41, 8214–8221 RSC.
- J. H. Zhu, Z. W. Tan and W. J. Yang, Macromol. Res., 2017, 25, 792–798 CrossRef CAS.
- Z. W. Tan, J. H. Zhu and W. J. Yang, Catal. Commun., 2017, 94, 60–64 CrossRef CAS.
- Y. J. Li, B. S. Sun, Y. Y. Zhou and W. J. Yang, Appl. Organomet. Chem., 2017, 31, 170–176 Search PubMed.
- K. Ladomenou, V. Nikolaou, G. Charalambidis and A. G. Coutsolelos, Coord. Chem. Rev., 2016, 306, 1–42 CrossRef CAS.
- J. S. Hsiao, B. P. Krueger, R. W. Wagner, T. E. Johnson, J. K. Delaney, D. C. Mauzerall, G. R. Fleming, J. S. Lindsey, D. F. Bocian and R. J. Donohoe, J. Am. Chem. Soc., 1996, 118, 11181–11191 CrossRef CAS.
- R. W. Wagner, T. E. Johnson and J. S. Lindsey, J. Am. Chem. Soc., 1996, 118, 11166–11180 CrossRef CAS.
- Q. Sun, B. Aguila, G. Verma, X. J. Meng, F. S. Xiao and S. Q. Ma, Chem, 2016, 1, 628–639 CAS.
- W. J. Zhang, P. P. Jiang, Y. Wang, J. Zhang and P. B. Zhang, Catal. Sci. Technol., 2015, 5, 101–104 CAS.
- C. C. Guo, M. F. Chu, Q. Liu, Y. Liu, D. C. Guo and X. Q. Liu, Appl. Catal., A, 2003, 246, 303–309 CrossRef CAS.
- W. W. Nam, I. Kim, M. H. Lim, H. J. Choi, J. S. Lee and H. G. Jang, Chem.–Eur. J., 2002, 8, 2067–2071 CrossRef CAS.
- W. W. Nam, H. J. Choi, H. J. Han, S. H. Cho, H. J. Lee and S. Y. Han, Chem. Commun., 1999, 4, 387–388 RSC.
- C. Minero, G. Mariella, V. Maurino, D. Vione and E. Pelizzetti, Langmuir, 2000, 16, 8964–8972 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12701d |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Yingying Li and
Cancheng Guo
*,
Yingying Li and
Cancheng Guo