
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCurrent research on anti-breast cancer synthetic compounds
Jia Liu†
ab,
Bian Mingab,
Guo-Hua Gong†c,
Di Wangab,
Gui-Lan Bao*ab and
Li-Jun Yu*ab
aMedicinal Chemistry and Pharmacology Institute, Inner Mongolia University for Nationalities, No. 22 Holin He Street, Tongliao, Inner Mongolia 028002, People's Republic of China. E-mail: 306502773@qq.com; xiaopang1224@126.com; 690922191@qq.com; bgl-6891@163.com; tl_ylj@163.com; Fax: +86-139-4875-0571; Tel: +86-139-4875-0571
bInner Mongolia Key Laboratory of Mongolian Medicine Pharmacology for Cardio-Cerebral Vascular System, Inner Mongolia University for Nationalities, No. 22 Holin He Street, Tongliao, Inner Mongolia 028002, People's Republic of China
cFirst Clinical Medical of Inner Mongolia University for Nationalities, Tongliao, Inner Mongolia 028002, People's Republic of China
First published on 24th January 2018
Abstract
Breast cancer (BC) is the most common cancer for females and its incidence tends to increase year by year. Currently, the backbone of therapy for BC is mainly chemotherapy, however its toxicity in normal cells and acquired tumor resistance to the drug used are considered as the main barriers. Therefore, there is still an urgent need for the development of more effective and safer anti-BC agents. Based on previous reference documents in recent years, this review covers the work reported on the anti-BC compounds which are classified according to the structures. This review summarized 185 significant anti-BC compounds which are classified by functional groups according to the animal model data, although there would be some limitations with the data. This review highlights the properties of new compounds endowed with promising anti-BC properties, which may be proven to be more effective and selective, and possibly free of unwanted side effects. The reviewed compounds represent an interesting possibility to overcome BC and to reduce the percentage of patients with a poor response to drug therapy.
1. Introduction
Cancer is one of the crucial public health problems in the world. The corresponding incidence and mortality statistics shows that it is growing in the economically developing and developed countries.1,2 Breast cancer (BC) is the most frequently diagnosed cancer and the leading cause of cancer death among females, accounting for 23% of the total cancer cases and 14% of the cancer deaths, and is now also the leading cause of cancer death among females in economically developing countries.2 Environmental factors and dietary habits are the primary source of BC induction with some secondary factors like virus-mediated genetic disturbances and many more.3 During the development of cancer, multiple signaling cascades get deregulated which result in increased cell proliferation, cell survival, as well as the emergence of resistance towards several anticancer drugs. The deregulation mainly recognized in BC is the inactivation of tumor suppressor genes and hyperactivation of proto-oncogenes.4 BC can be described as a heterogeneous group of neoplasms originating from the epithelial cells and it as a complex disease has posed a great challenge to the field of medicine and immunology.5 As a result, it is evident that more effective therapy is needed to treat these malignancies.For the treatment of BC, fulvestrant, lapatinib, eribulin mesylate, pertuzumab, everolimus and numerous other agents have been approved by the FDA for various BC subtypes treatments. The occurrence of resistance that is faced by these drugs has restricted their use, and we still need some alternates for a full proof treatment option against BC. Despite extensive research and rapid progress in cancer treatment, there is a need to develop a new group of anticancer agents targeting BC cells.6 Nowadays a large number of potent bioactive entities originating from natural sources as well as derived by synthetic methodology exist as potent anticancer agents that can be used for BC chemotherapy.7 This review summarizes the recent discovery of newly synthesized anti-breast cancer compounds that further promote our understanding of new synthetic compounds and provide a basis for further research in the future.
2. The quinoline functional group
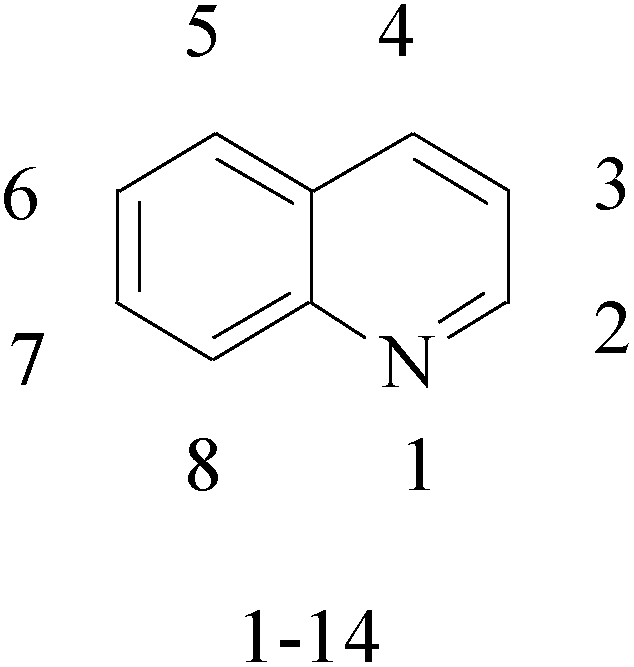
Quinoline is nitrogen-containing heterocyclic aromatic compound. Pharmacologically active substances display a broad range of biological activity. Quinoline has been found to possess anti-malarial, antibacterial, antifungal, anticonvulsant, anti-inflammatory and analgesic activity.8–12 Quinoline derivatives are the pharmacologically important heterocycles which have been studied extensively for their anticancer properties.Bharathkumar et al.,13 prepared a novel type of quinoline-based estrogen receptor alpha (ERα) ligands were tested for their bioactivity against ERα-positive and ERα-negative cell lines. The strongest compound 1 displayed significant cytotoxicity against MCF-7 and HepG2 cells with an half maximal inhibitory concentration (IC50) value of 6 and 11 μM, respectively (Table 1).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 6,8-
O, 6,8-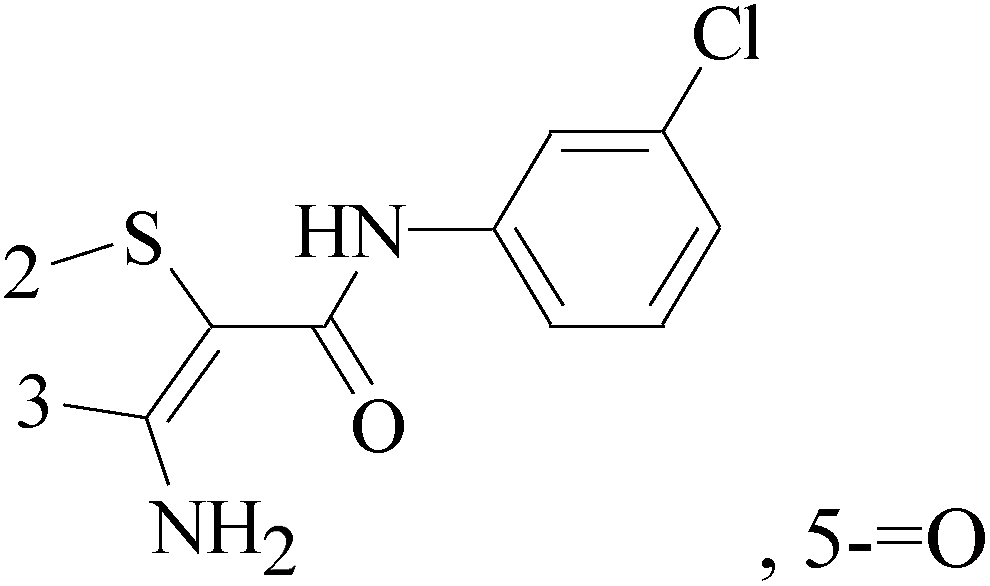
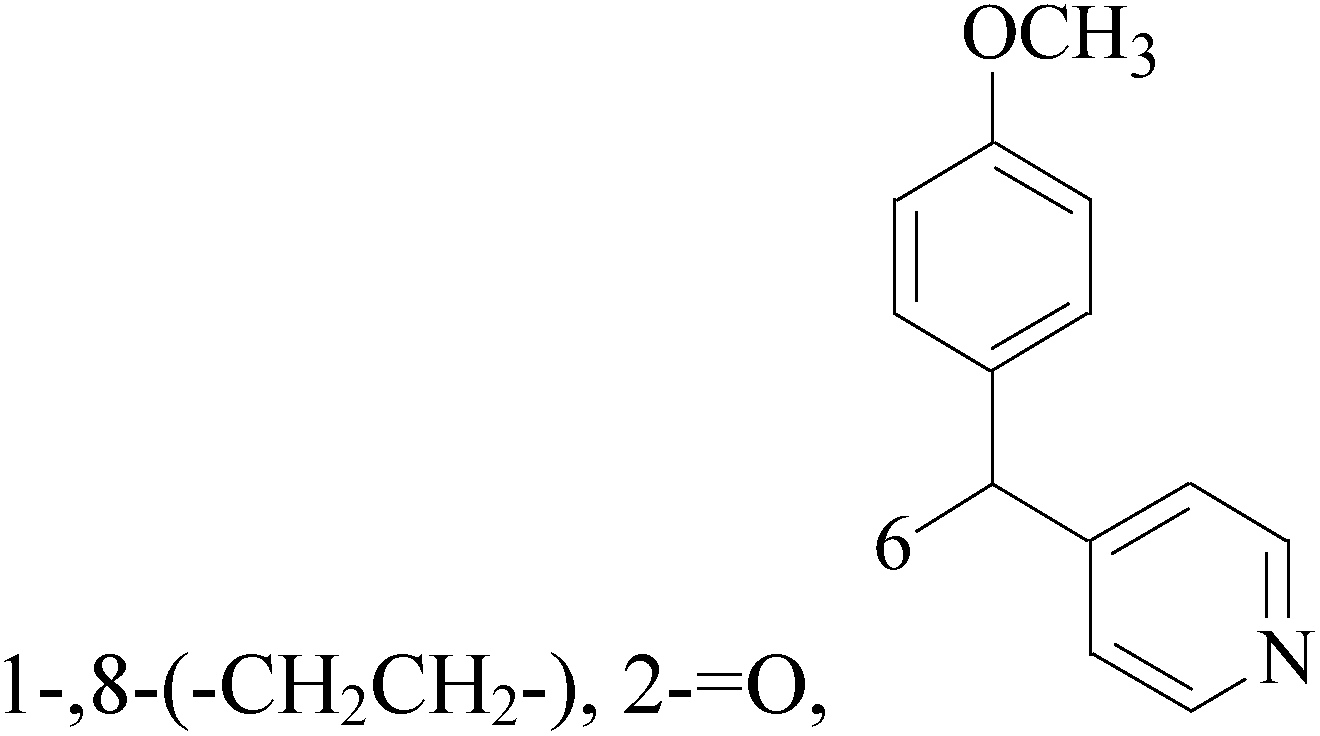
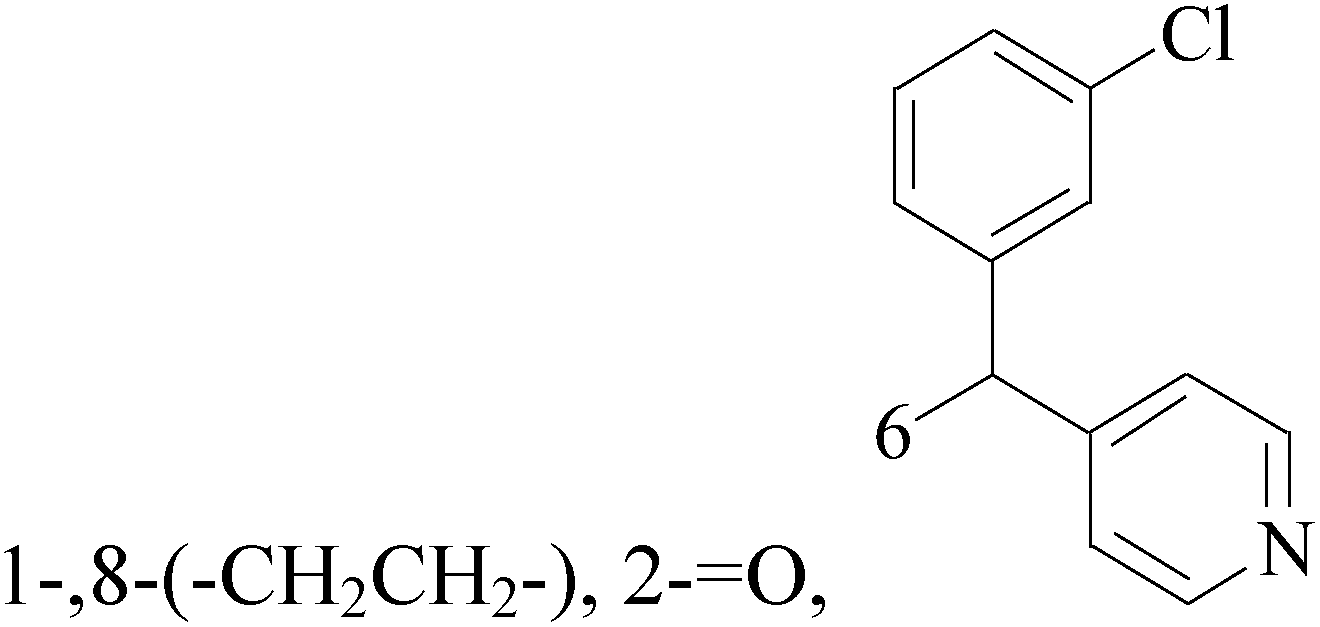
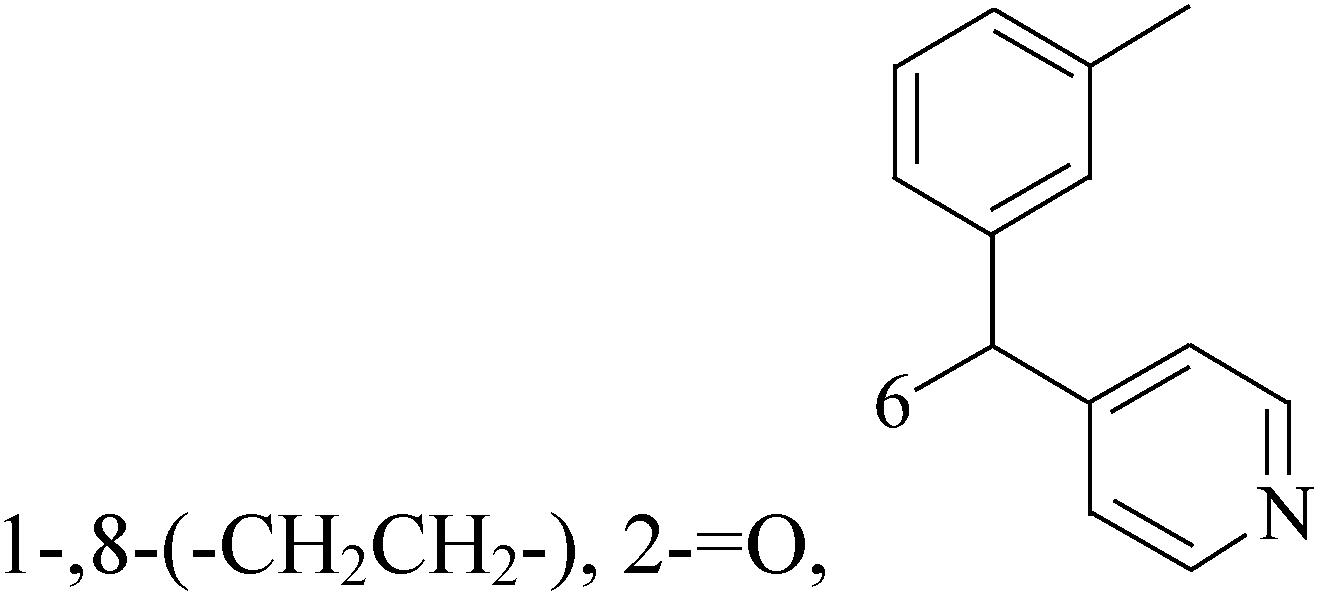
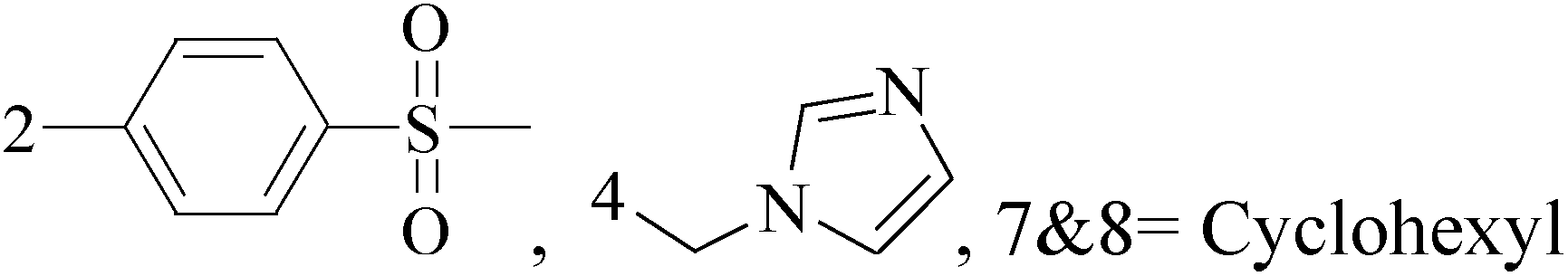
A new group of 4-(imidazolylmethyl)quinoline derivatives were designed and synthesized as selective cyclooxygenase (COX-2) inhibitors and in vitro anti-BC agents. In anti-breast cancer screening, only one compound 2 (IC50 < 5 μM) was identified as the most potent and selective COX-2 inhibitor as well as the most cytotoxic agent against MCF-7 cells (Table 1).14
A series of novel 6,8-dibromo-2-aryl-2,3-dihydroquinolin-4(1H)-ones have been synthesized and evaluated in vitro (in MCF-7 BC cell lines). Compounds 3–6 exhibited potent growth inhibition of 50% (GI50) and total growth inhibition (TGI) values compared with reference standard (Table 1).15
3-Amino-N-(3-chlorophenyl)-5-oxo-5,6,7,8-tetrahydrothieno[2,3-b]quinoline-2-carboxamide (compound 7) as a putative phosphoinositide specific-phospholipase C-γ enzyme inhibitor, affected the proliferation, morphology and migration of a host of breast cancer cell lines, and arrests cell cycle in the G2/M phases (Table 1).16
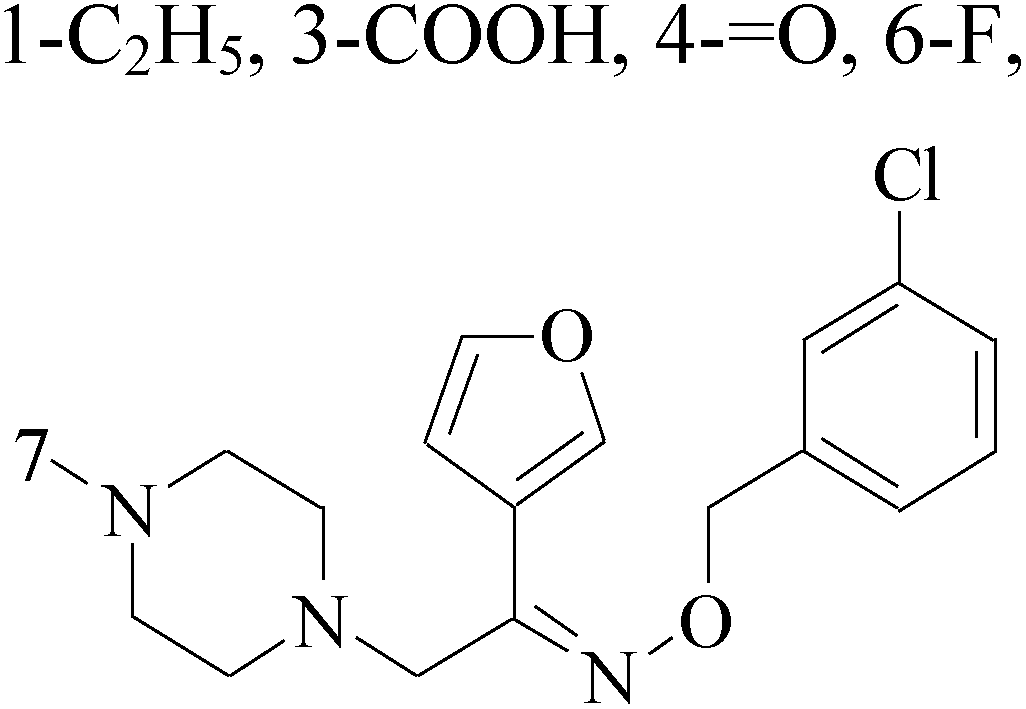
A series of pyridinylmethyl substituted 1,2,5,6-tetrahydro-pyrrolo[3,2,1-ij]quinolin-4-ones were designed and synthesized as a novel strategy for BC with elevated cardiovascular diseases. The compromise of this conflict led to compounds 8–11 as potent and selective dual inhibitors of aromatase (CYP19) and aldosterone synthase (CYP11B2), especially compound 11, which exhibited IC50 values of 32 and 41 nM, respectively, and a high selectivity toward 17α-hydroxylase-17,20-lyase and 11β-hydroxylase.17 Through the approach of combining important structural features of CYP19 and CYP11B2 inhibitors to design dual inhibitors, compounds 12 and 13 were obtained as selective dual inhibitors with IC50 values around 50 and 20 nM toward CYP19 and CYP11B2, respectively, similar to that of fadrozole as a reference (Table 1).18
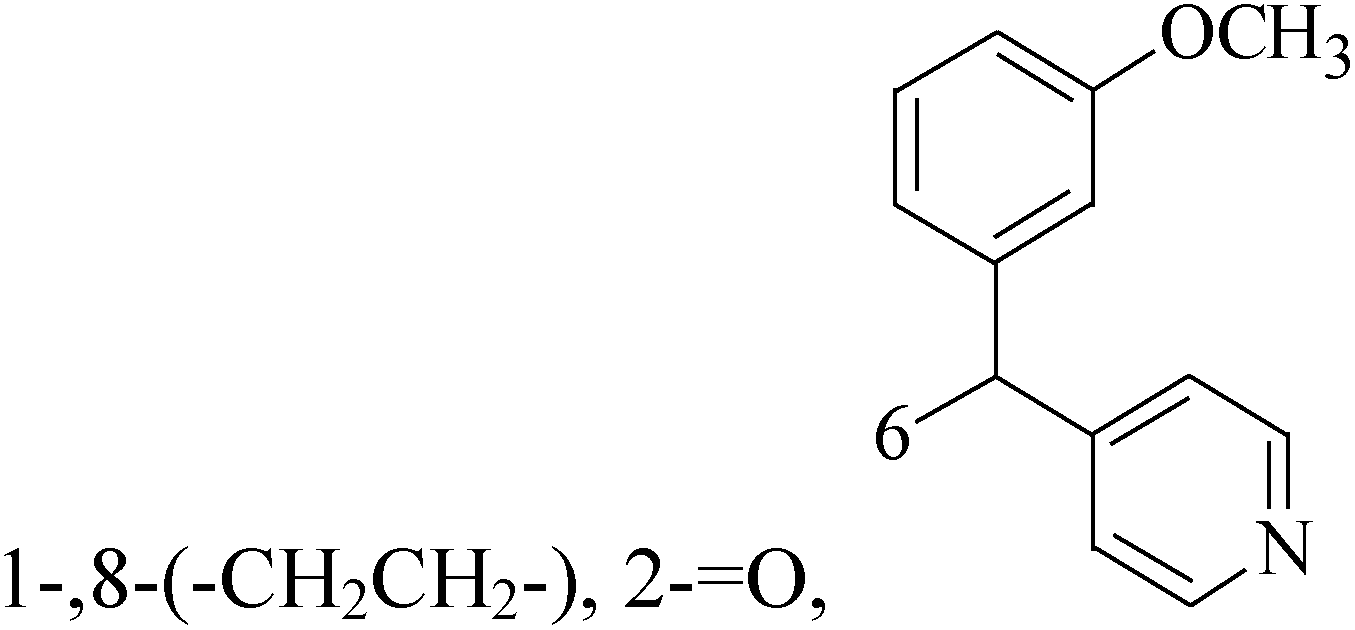
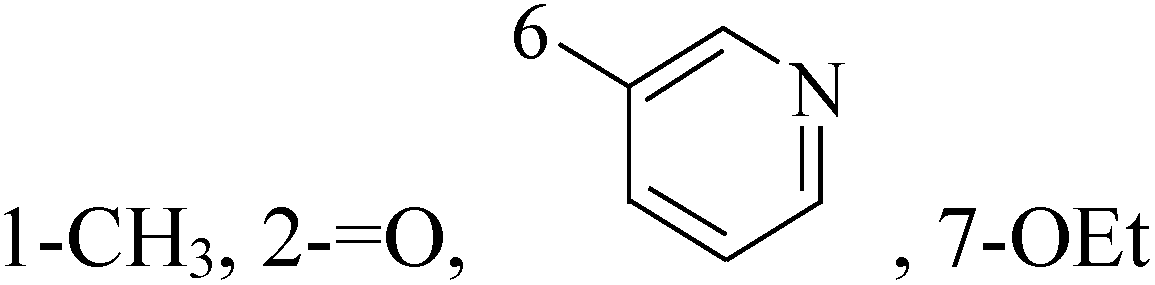
Mohammadhosseini et al.,19 reported synthesis and cytotoxic activity evaluation of a new series of N-pipearzinyl quinolones containing N-2-(furyl-2 or 3-yl)-2-(chlorobenzyloxyimino)ethyl moiety. Preliminary screening indicated that compound 14 with IC50 values of 3.03, 11.9 and 2.2 for MCF-7, MDA-MB-231 and T47D, respectively demonstrated significant growth inhibitory potential against all evaluated cell lines (Table 1).
3. The quinazoline or quinazolinone functional groups
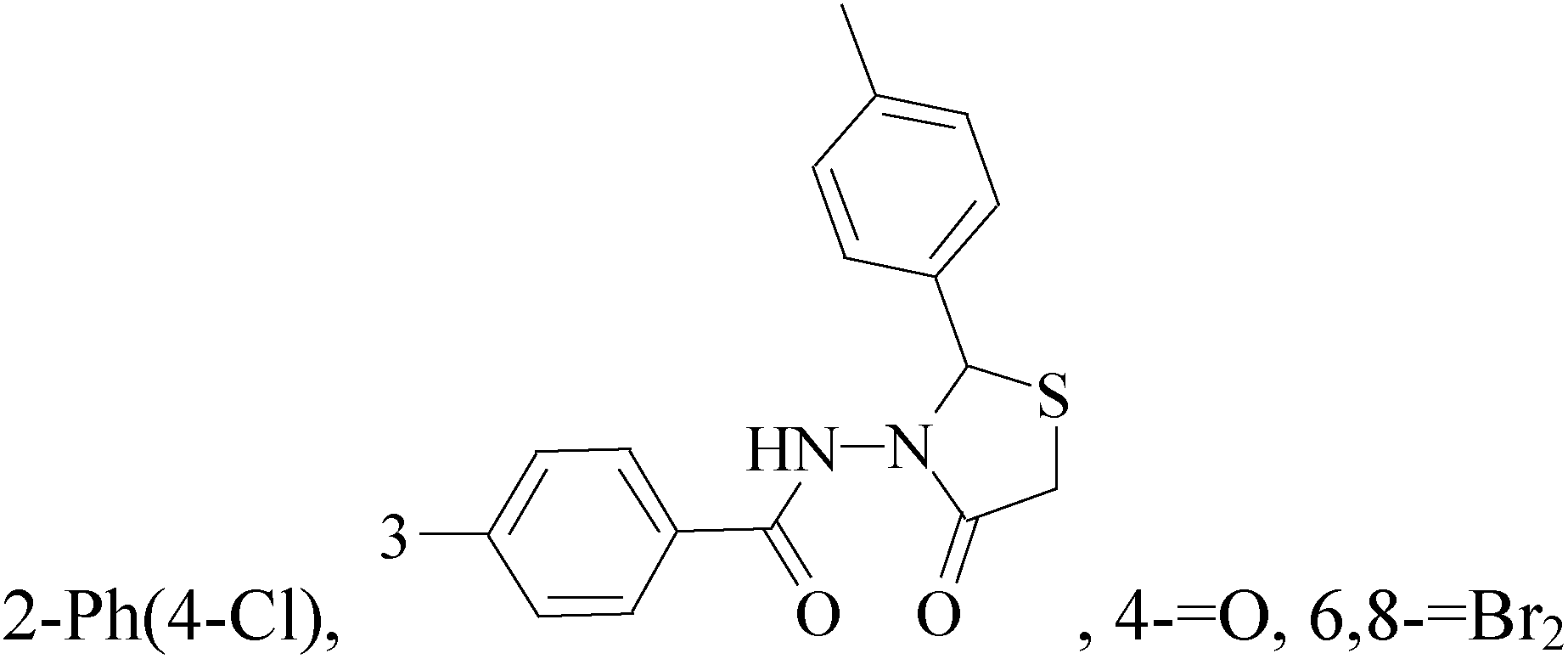
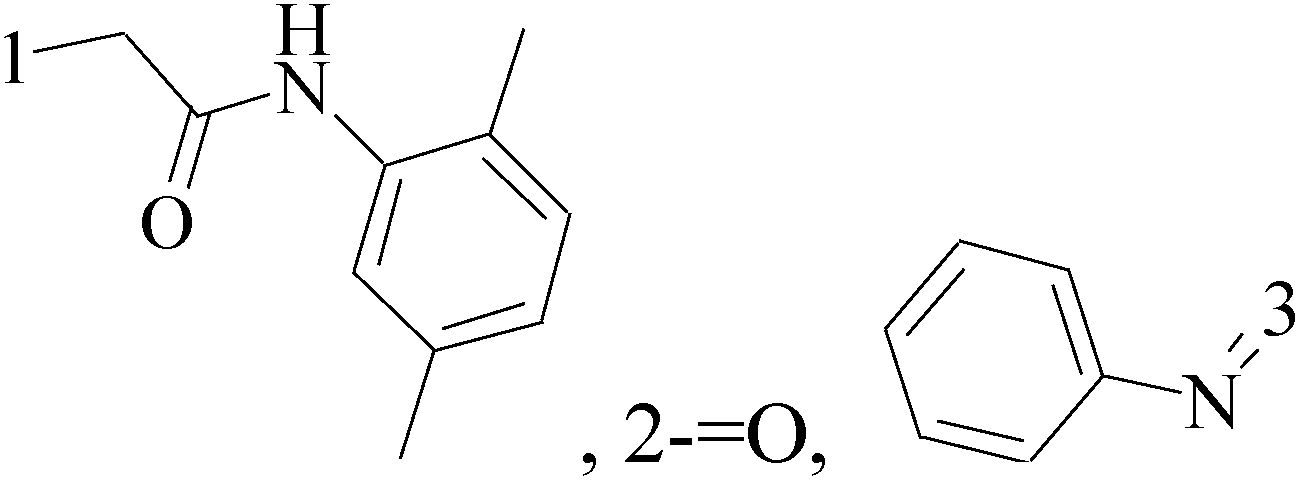
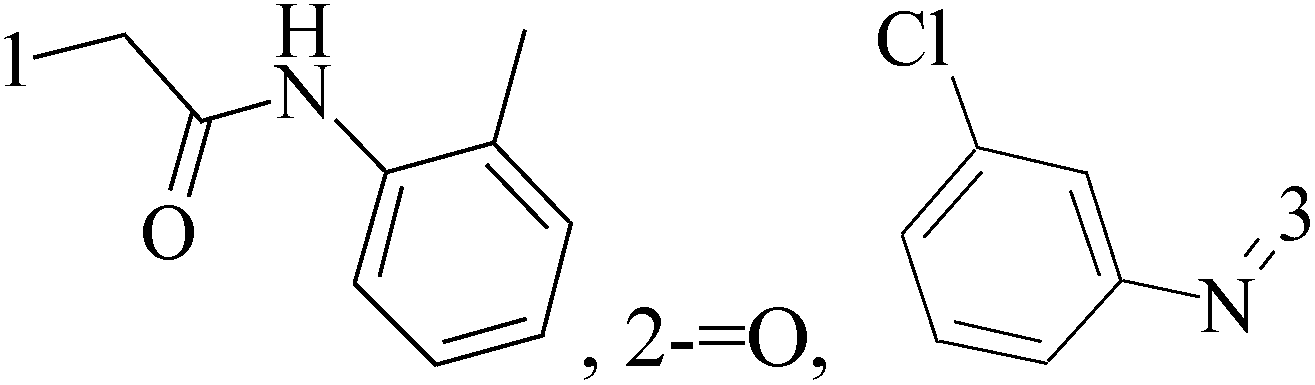
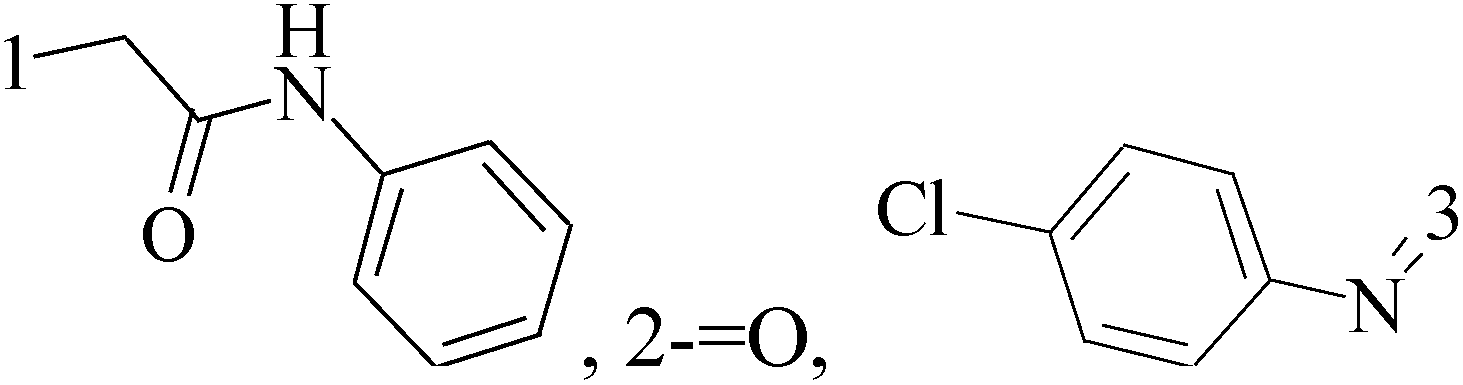
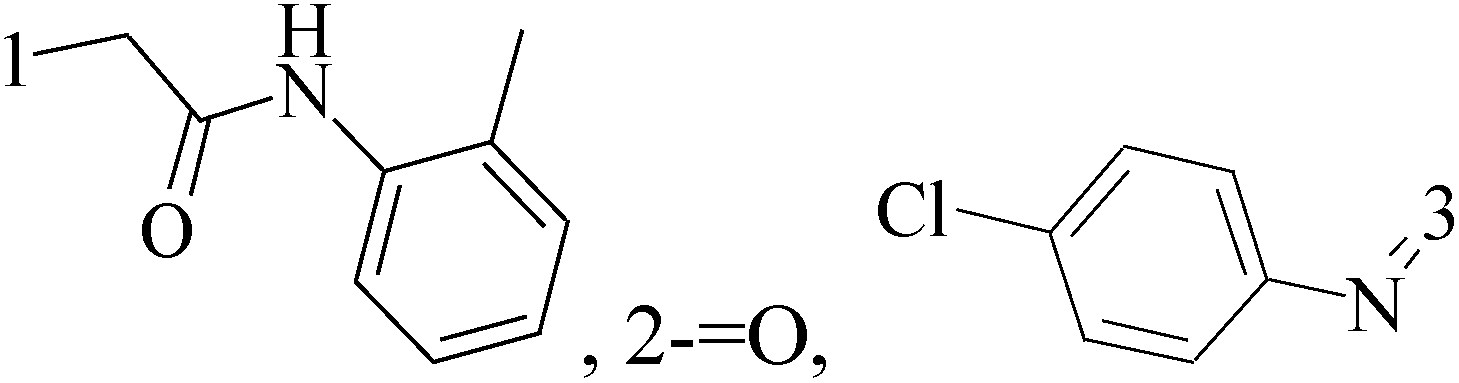
It is well known that quinazoline derivatives have a wide range of biological activities such as anticonvulsant, antibacterial, antiviral, antifungal, anticancer, analgesic and COX-2 inhibitors.20–25 Several quinazoline derivatives have been approved by FDA as anticancer drugs such as erlotinib, lapatinib, gefitinib and caneratinib.6,8-Dibromo-2-(4-chlorophenyl)-quinazolin-4-one linked directly to oxadiazole, pyrazole or through amide linkage to thiazolidinone were synthesized, and evaluated their anti-BC cell line (MCF-7) activity using doxorubicin as a reference drug. The most active compounds of the hybrid molecules between quinazolin-4-one and thiazolidinone are compounds 15–18 (IC50 = 3–9 μM). In vitro screening of these four compounds against EGFR tyrosine kinases demonstrated inhibitory activity range 54–77.2% (Table 2).26
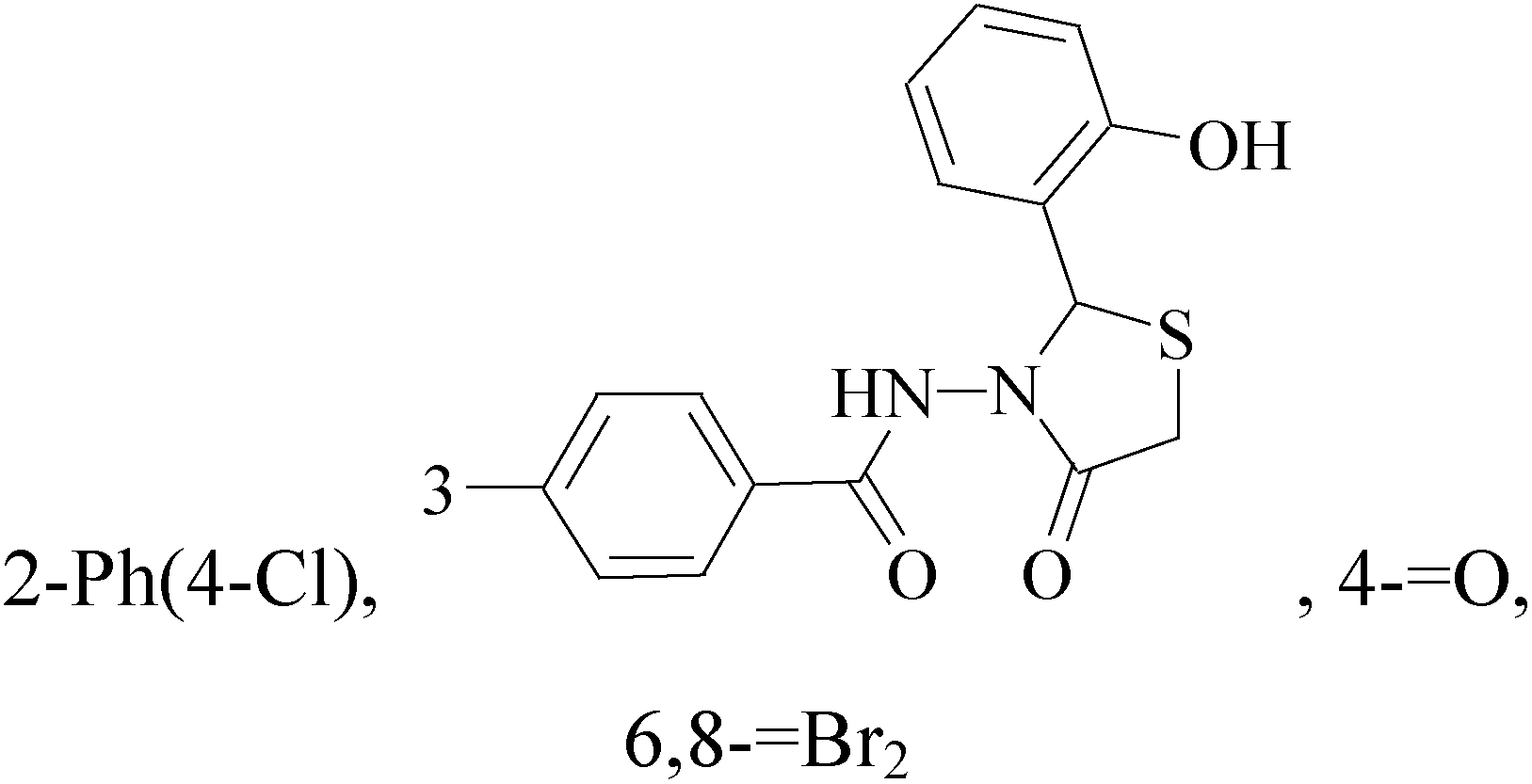
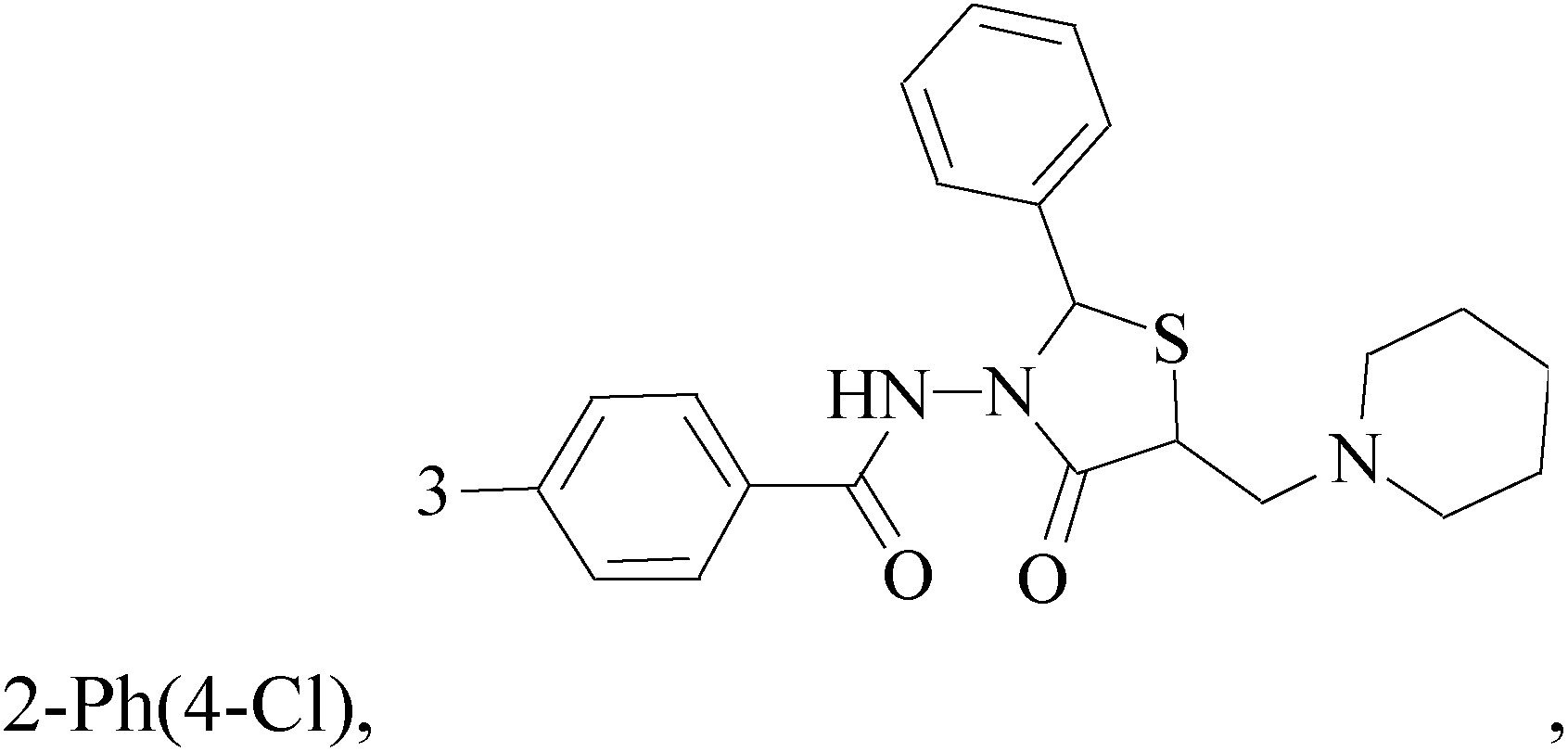
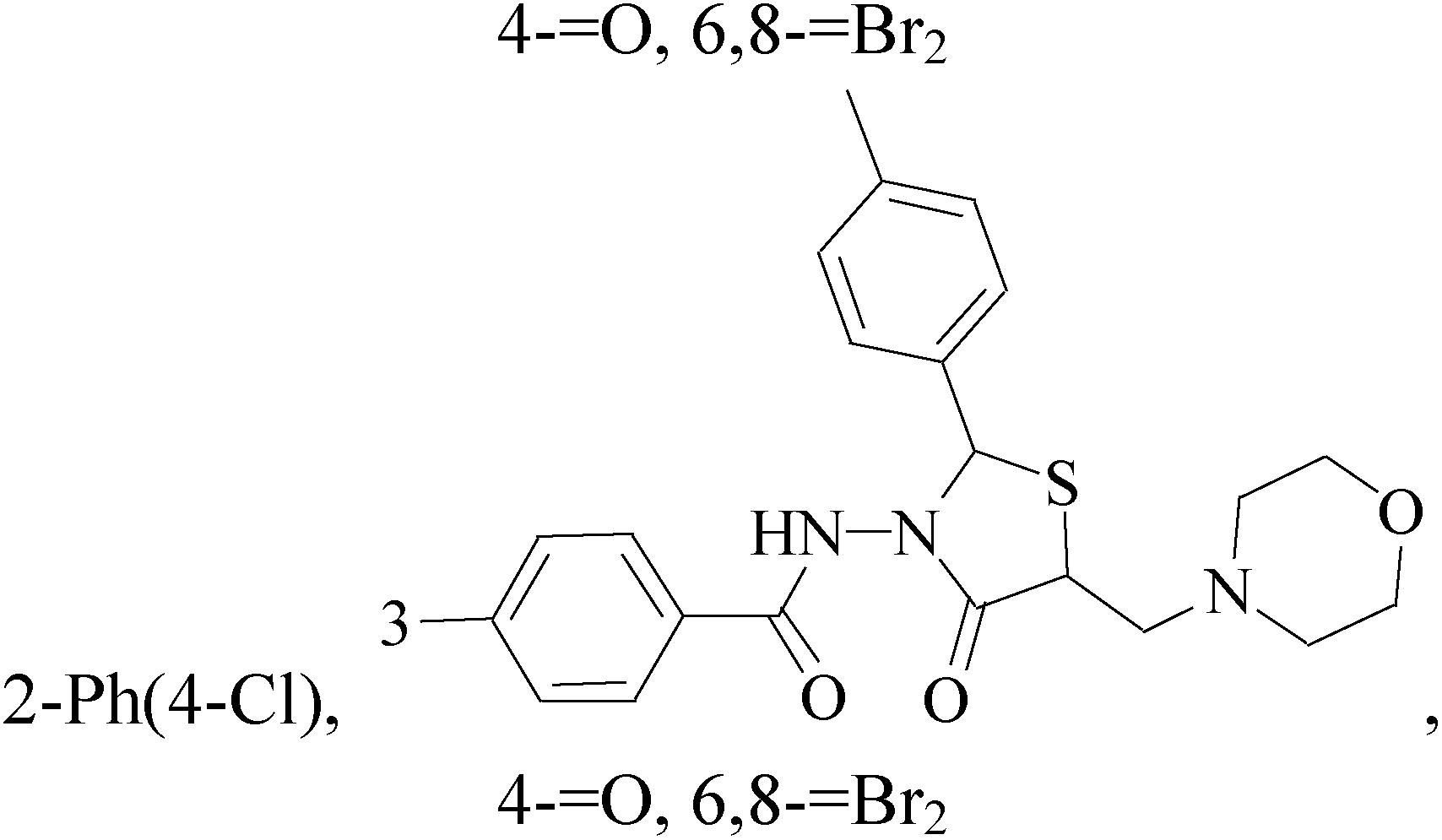
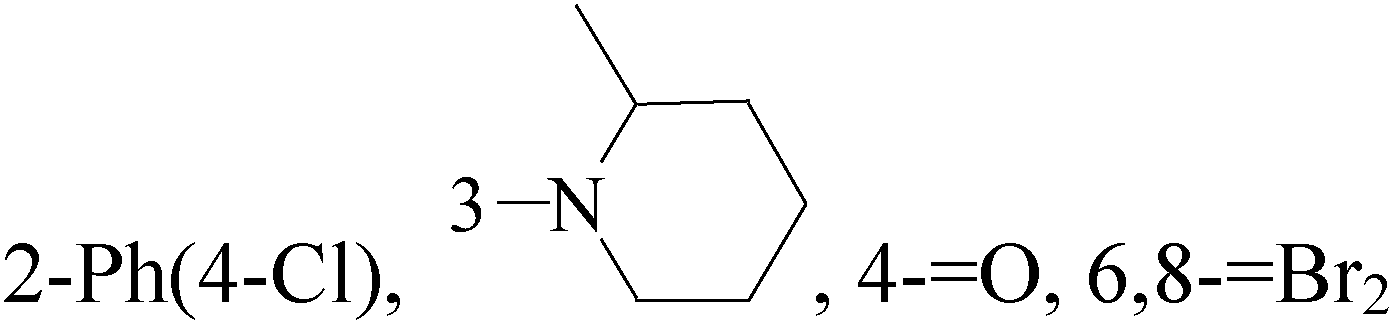
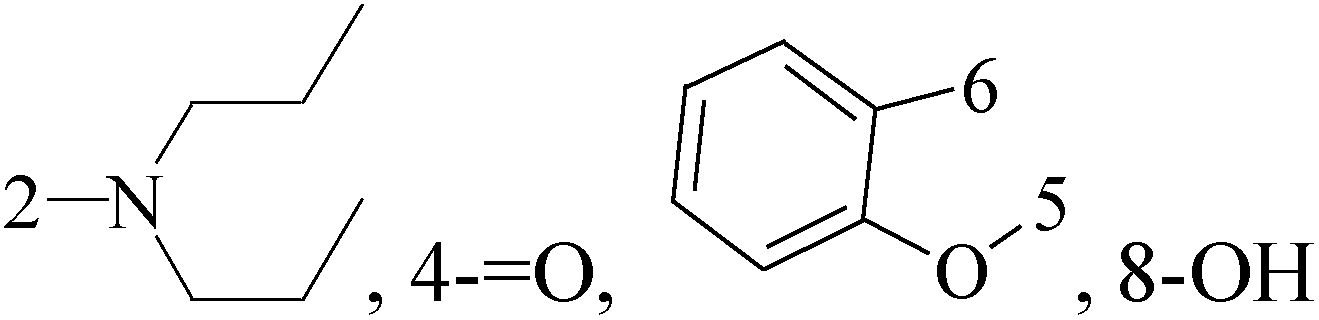
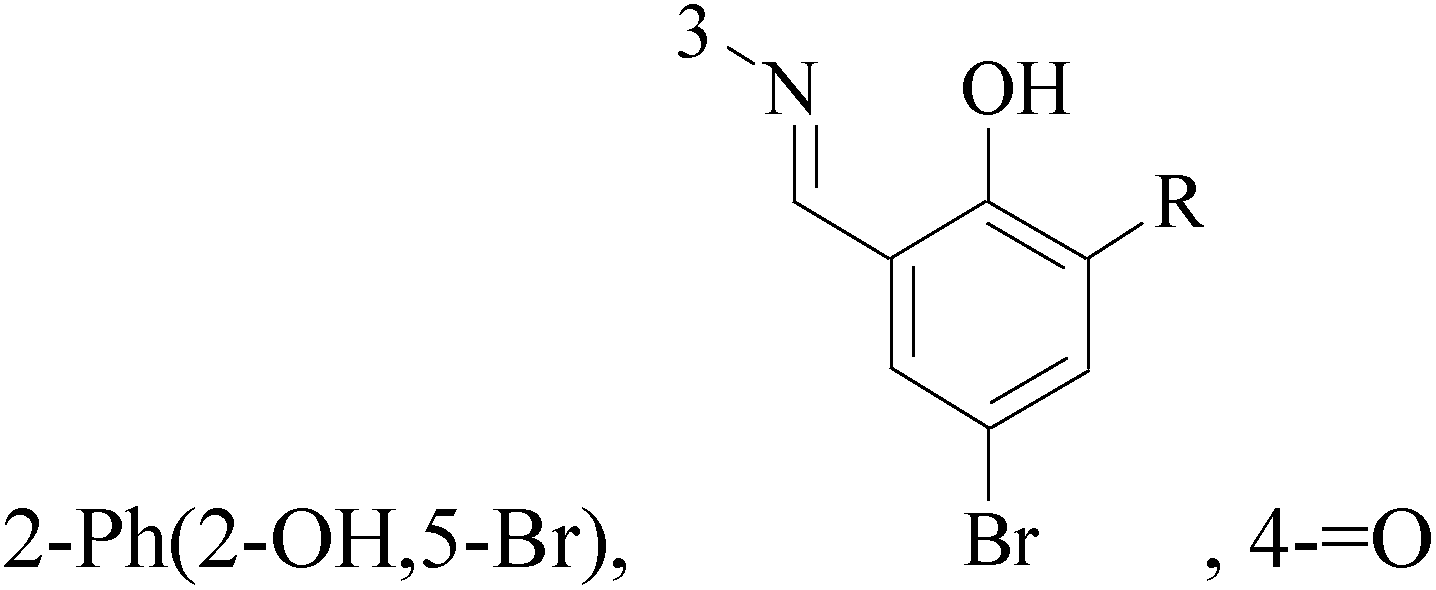
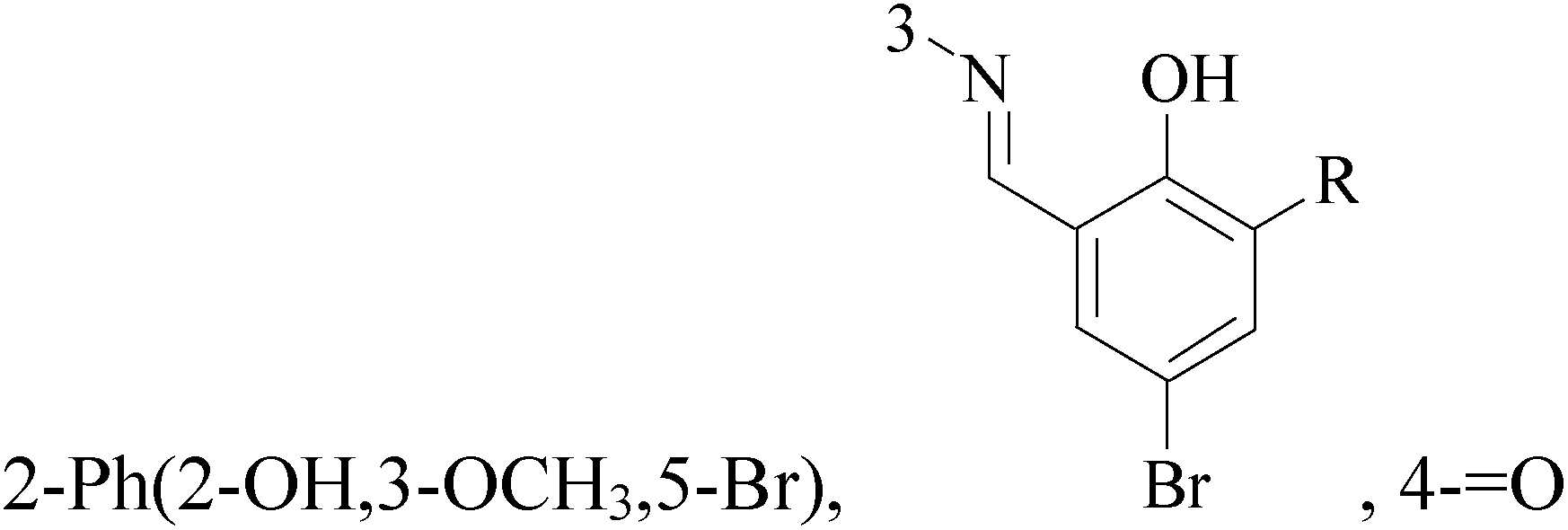
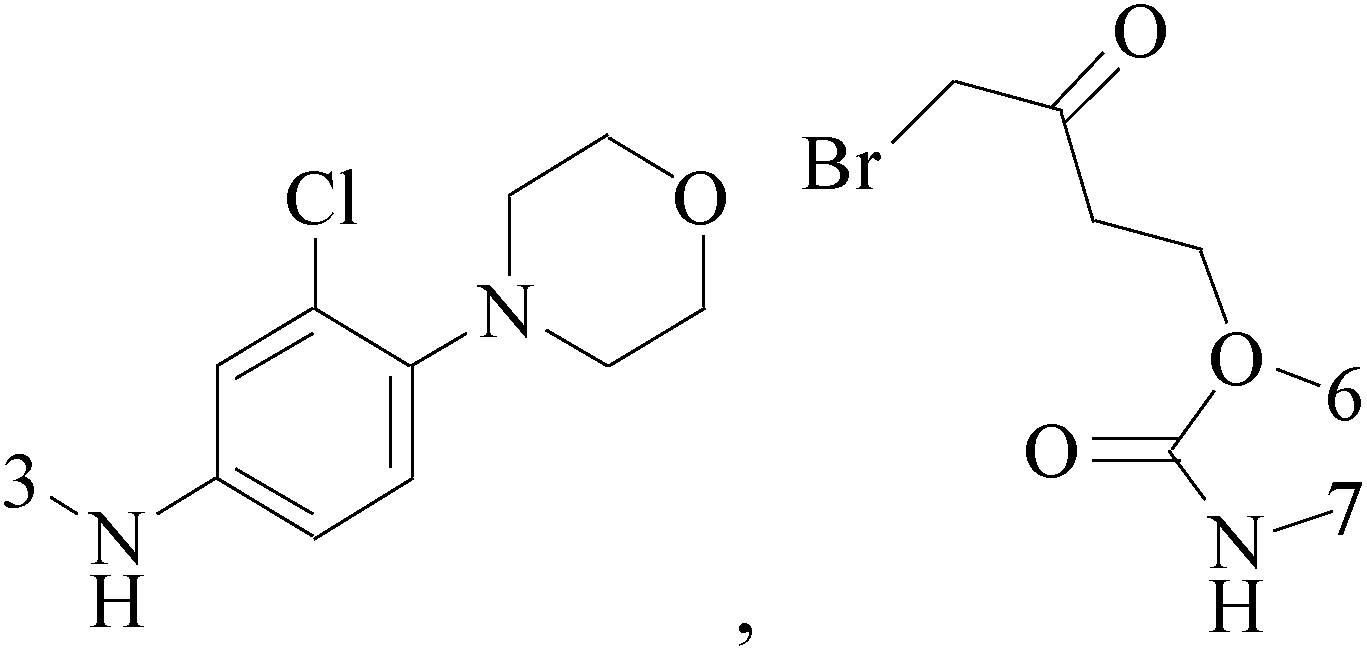
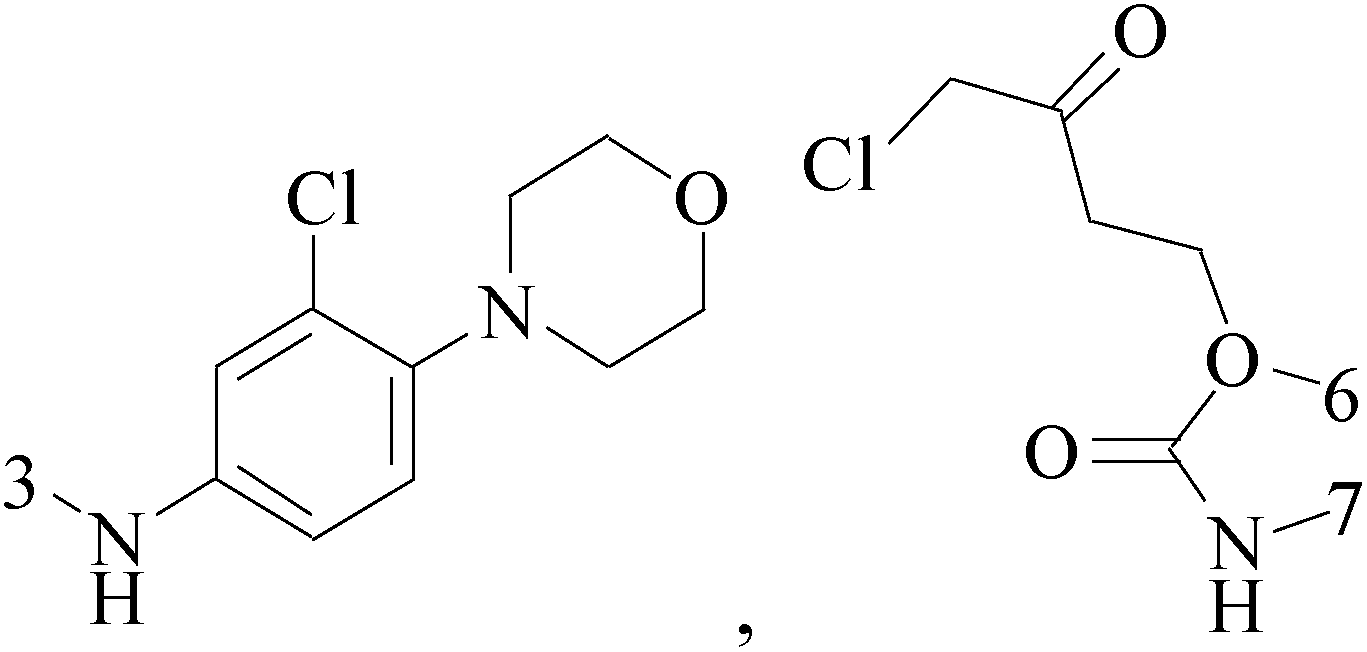
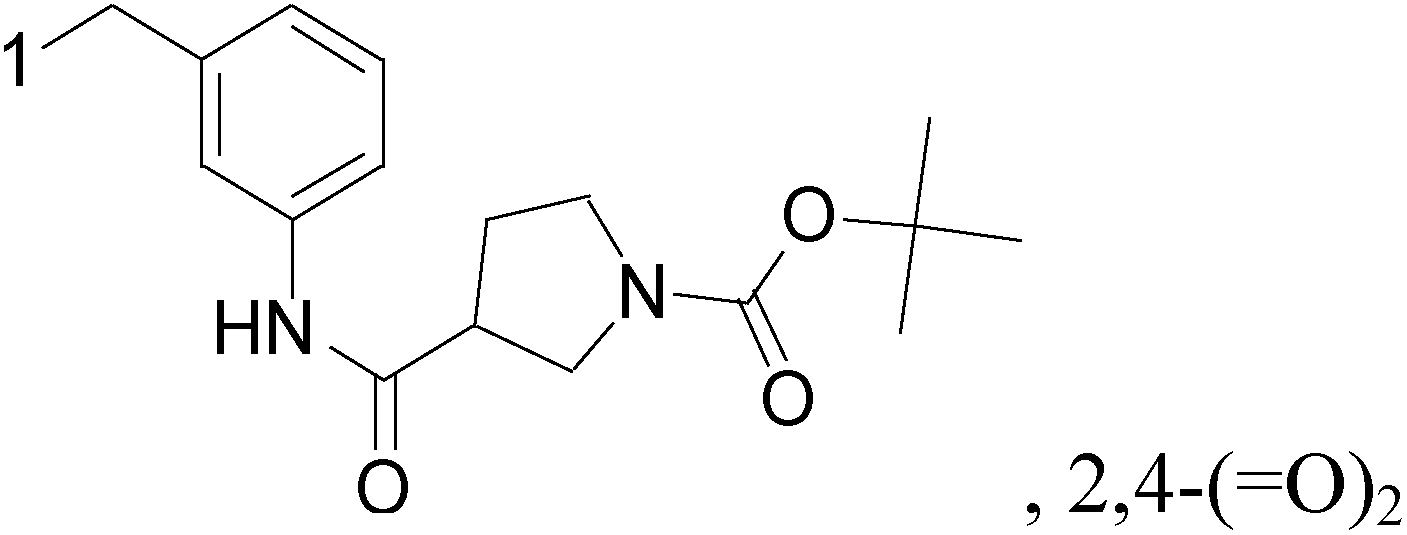
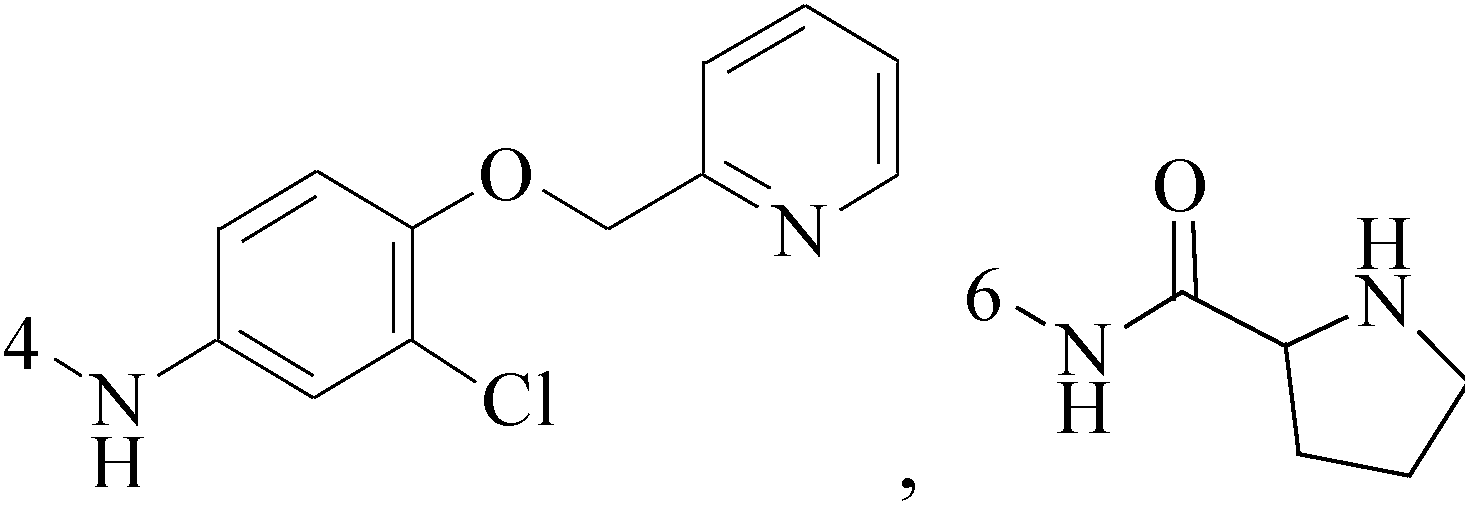
A new series of 6,8-dibromo-2-(4-chlorophenyl)quinazolin-4(3H)-one derivatives were synthesized and tested in vitro against human BC cell line (MCF-7). Compounds 19 and 20 exerted a powerful cytotoxic effect against MCF-7 with a very low IC50 (1.5 and 4.7 μM), compared to doxorubicin (2.5 μM) (Table 2).27
A series of unknown 3-(alkyl(dialkyl)amino)benzofuro[2,3-f]quinazolin-1(2H)-ones has been synthesized as new ellipticine analogs. The cytotoxic activities of all the synthesized compounds, tested in ERα+, ERα− and tamoxifen-resistant BC cell lines, revealed that DPA-HBFQ-1 (compound 21) was the most active compound via up-regulating the cell cycle regulators p53 and p21Cip1//WAF1 and selectively inhibiting the human topoisomerase II (Table 2).28
Two new synthesized and characterized quinazoline Schiff bases 22 and 23 showed a remarkable antiproliferative effect for MCF-7 human BC cell line, with an IC50 value of 6.246 mM and 5.910 mM, respectively, through inducing apoptosis pathway, activating caspases-3/7, -8, and -9 and inhibiting NF-B translocation (Table 2).29
Two novel series of oxazolo[4,5-g]quinazolin-2(1H)-one derivatives as EGFR/HER2 dual inhibitors were synthesized and subjected to pharmacological evaluation. Compounds 24 and 25 with IC50 value of 2.3 μM and 0.47 μM respectively had well exhibition to the EGFR and HER2 inhibition activity and excellent anti-proliferation activity against human lung adenocarcinoma cell line (A549) and human BC cell line (SK-Br3) comparing with lapatinib (Table 2).30
A novel series of 1-benzyl-quinazoline-2,4(1H,3H)-dione derivatives were designed and synthesized as human poly(ADP-ribose)polymerase-1 (PARP-1) inhibitors. Compound 26 was a promising PARP-1 and PARP-2 inhibitor and could selectively kill the BC cells MX-1 (IC50 = 4.59 μM) and MDA-MB-468 with mutated BRCA1/2 and PTEN, respectively, compared with homologous recombination proficient cell types such as BC cells MDA-MB-231 (Table 2).31
Jang et al.,32 discovered novel quinazoline analogues that had high potency and selectivity for BC cells. Derivative 27 (IC50 = 8.8 × 10−3 μM) was the most potent, with high selectivity for SKBr-3 over A431 cells by inhibiting EGFR and HER2 expressing (Table 2).
4. The pyridine functional group
Five and six-membered nitrogen-containing heterocycles are abundant in nature and exhibit diverse and important biological properties.33,34 Pyrazolopyridine derivatives are a significant class of heterocyclic compounds, which exhibit various biological activities including anticancer, anxiolytic, antiviral, antileishmanial and anti-inflammatory activities.35–39 Because of their biological activities, pyridines and substituted pyridines have distinguished themselves as heterocycles of profound chemical and biological significance.40A series of novel 2H-pyrazolo[4,3-c]hexahydropyridine derivatives have been designed, synthesized and evaluated in vitro against two human BC cell lines. Most compounds exhibited good inhibition, and compounds 28 (IC50 = 4.7 μM for MCF-7 and IC50 = 9.3 μM for MDA-MB-231), 29 (IC50 = 2.4 μM for MCF-7 and IC50 = 4.2 μM for MDA-MB-231) and 30 (IC50 = 3.3 μM for MCF-7 and IC50 = 8.6 μM for MDA-MB-231) displayed better inhibitory activity than 5-fluorouracil (IC50 = 4.8 μM for MCF-7 and IC50 = 9.6 μM for MDA-MB-231, respectively) as the control. Moreover, 29 was cytotoxic and able to induced the apoptosis of MCF-7 cells (Fig. 1).41
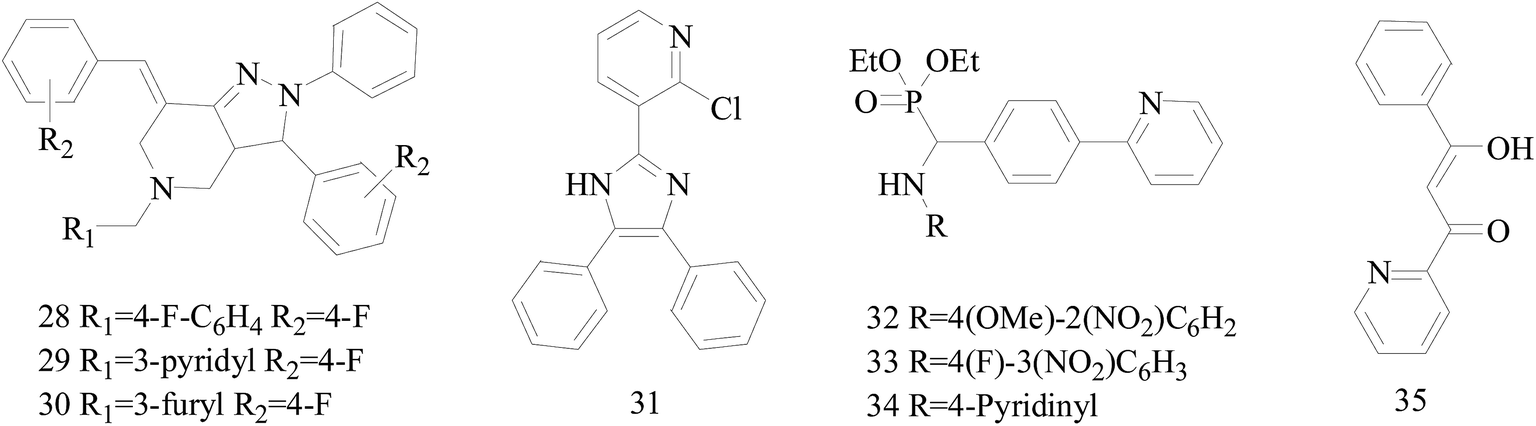 | ||
| Fig. 1 Structure of compounds 28–35. | ||
Mohan et al.,42 reported the synthesis of trisubstituted-imidazoles and identified 2-chloro-3-(4,5-diphenyl-1H-imidazol-2-yl) pyridine (31) as lead cytotoxic agent. The lead compound 31 with IC50 value of 0–50 μM for MCF-7 and MDA-MB-231 suppressed the proliferation of BC cells, decreased the phosphorylation of phosphoinositide-dependent kinase, Akt, Mammalian Target of Rapamycin (mTOR), downregulated the cellular invasion and activated caspases and cleaved PARP to induce apoptosis (Fig. 1).
A series of new diethyl(alkyl/aryl/heteroarylamino)(4-(pyridine-2-yl)phenyl)methylphosphonates were synthesized. The compounds 32–34 exhibited higher cytotoxicity against malignant BC MCF-7 cells via good inhibiting effect on the aromatase enzyme by virtue of their strong binding affinity through H-bonding with the amino acid residues of the enzyme (Fig. 1).43
Radi et al.,44 reported synthesis of novel β-keto–enol derivatives tethered pyrazole, pyridine and furan as new potential antifungal and anti-BC agents. In all compounds tethered pyridine, 35 had the most potent activity against BC with IC50 values of 78.23 μM (Fig. 1).
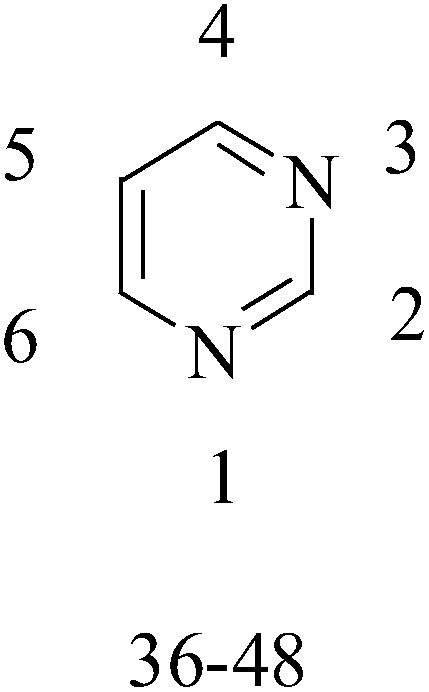
5. The pyrimidine functional group
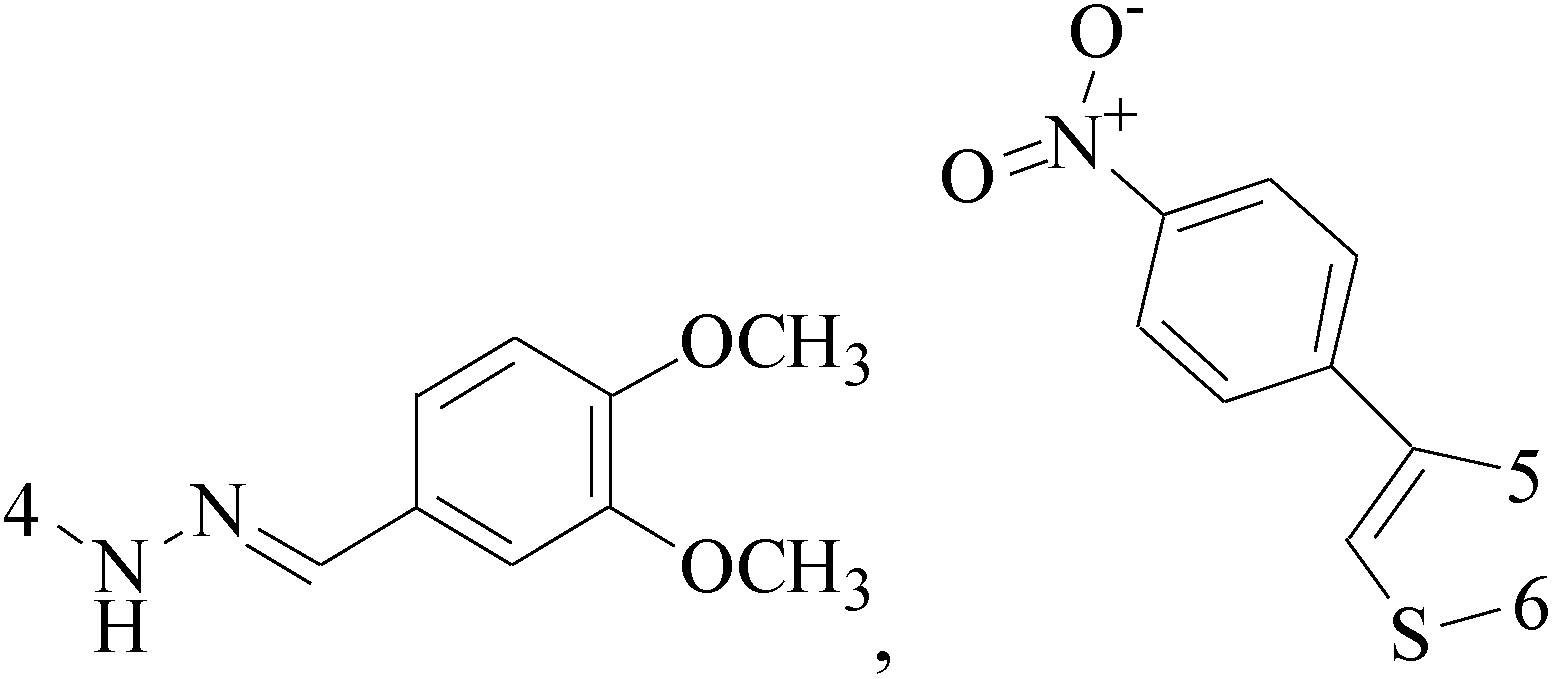
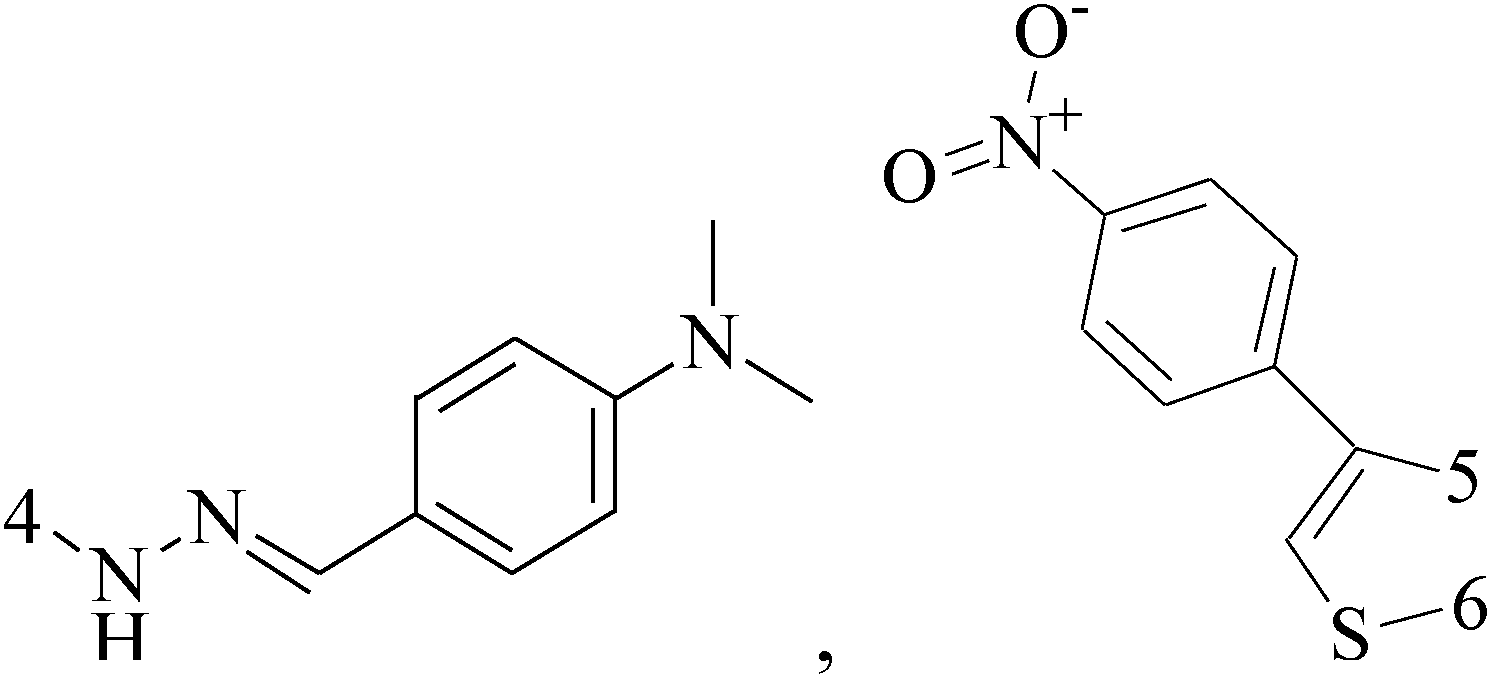
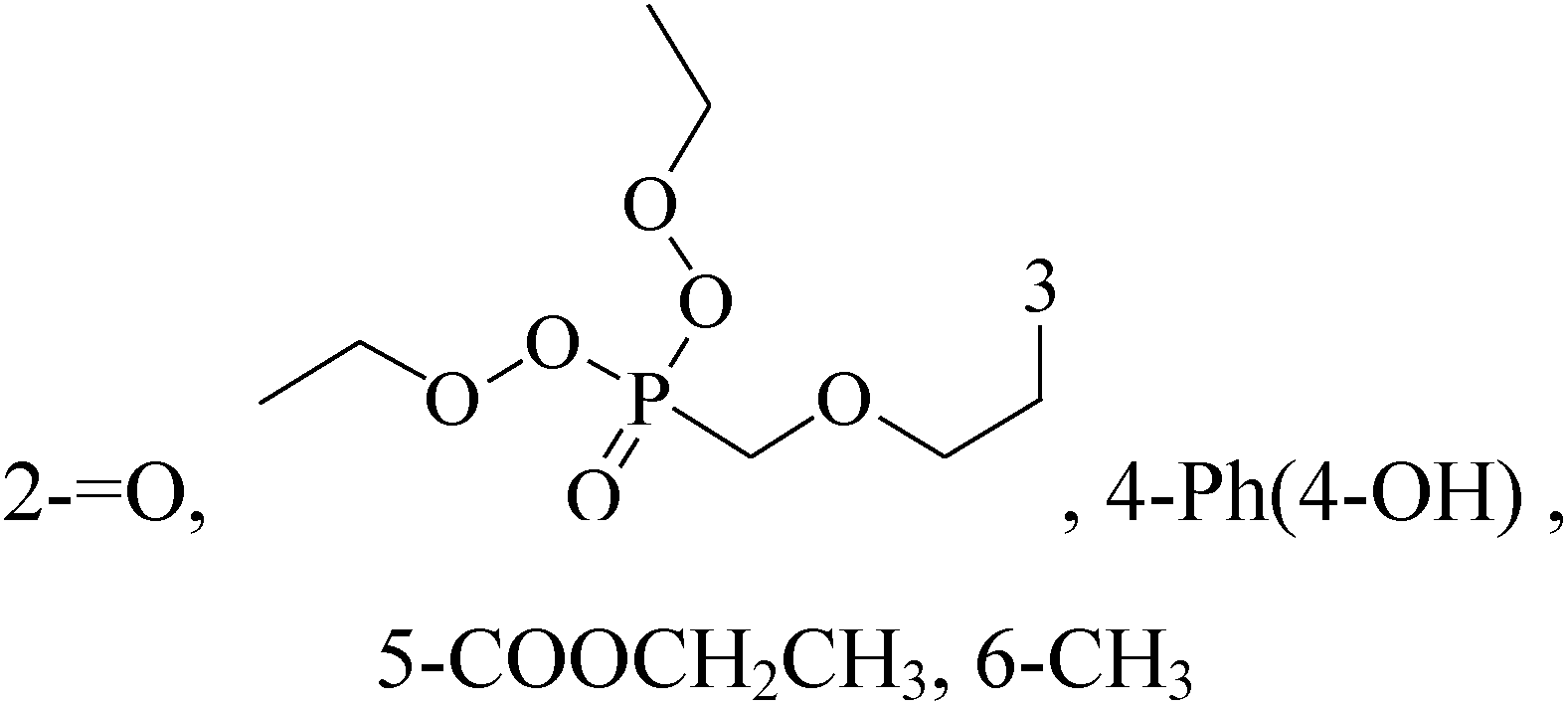
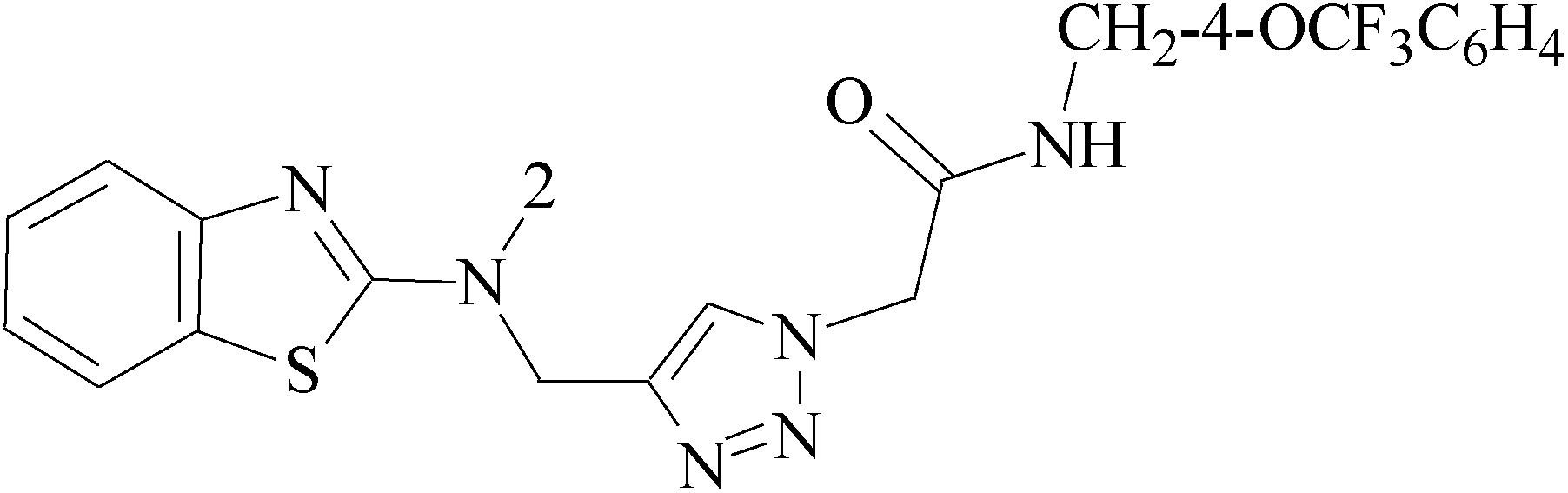
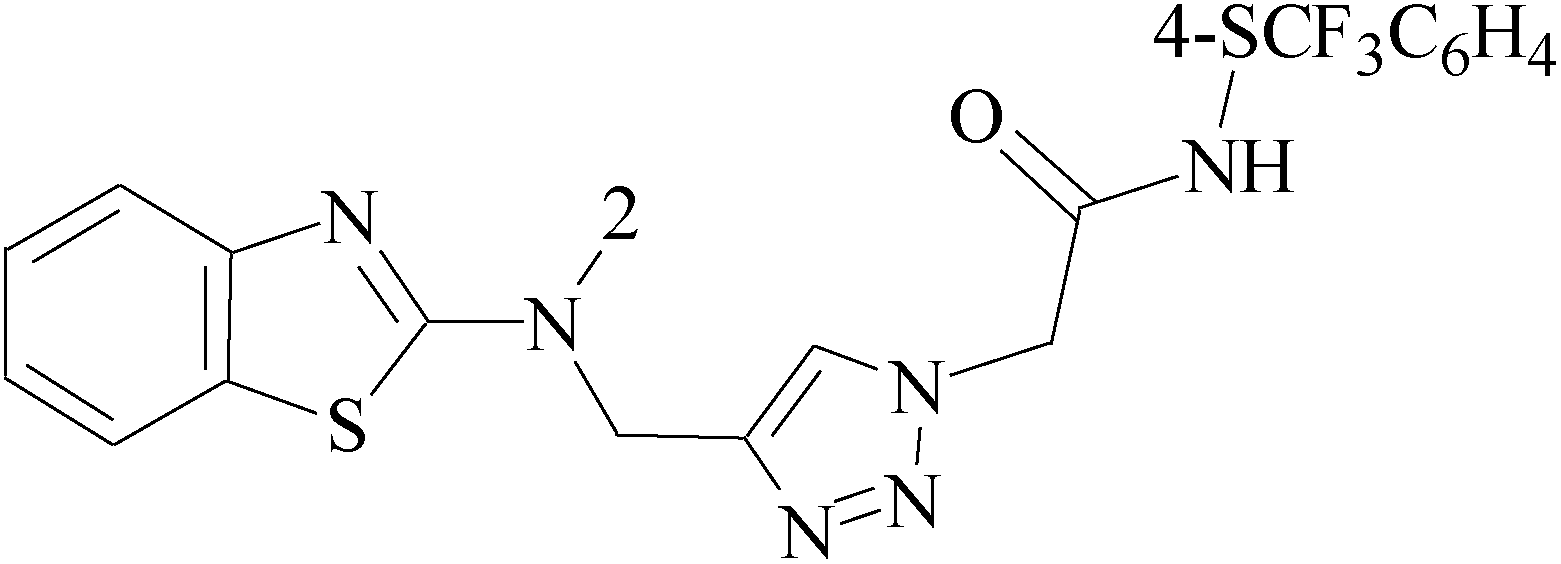
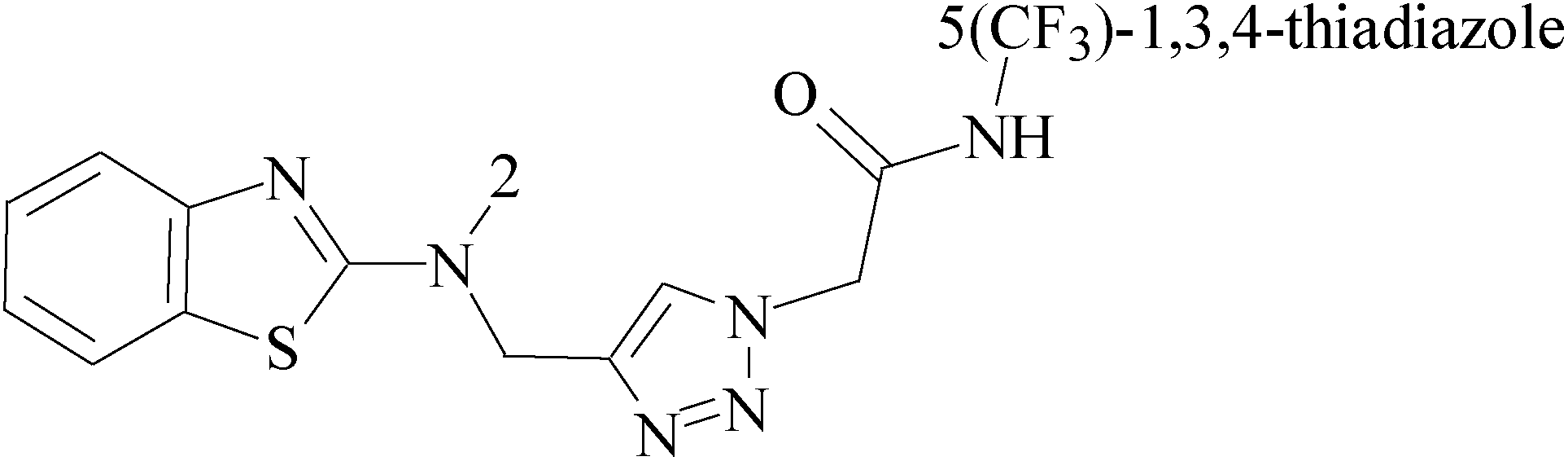
Pyrimidine ring is an aromatic heterocyclic organic compound similar to pyridine. One of the three diazines, six-membered heterocyclics with two nitrogen atoms in the ring, has the nitrogens at positions 1 and 3 in the ring. Pyrimidines, a significant one of the heterocyclic compounds, have a broad spectrum of bioactivities (antibacteria, anticancer and anti-inflammation and the like).45–47It was reported that a series of 5-arylthieno[2,3-d]pyrimidines as anti-BC agents. In all the tested compounds 36 and 37 (Table 3) with IC50 9.8 nM and 10.2 nM respectively were the most active compounds in all the tested compounds, because of the deep interaction of these compounds in the back of ATP binding site and the extra hydrophobic interaction provided by para and meta dimethoxy groups in 36 and para dimethylamino group in 37.48
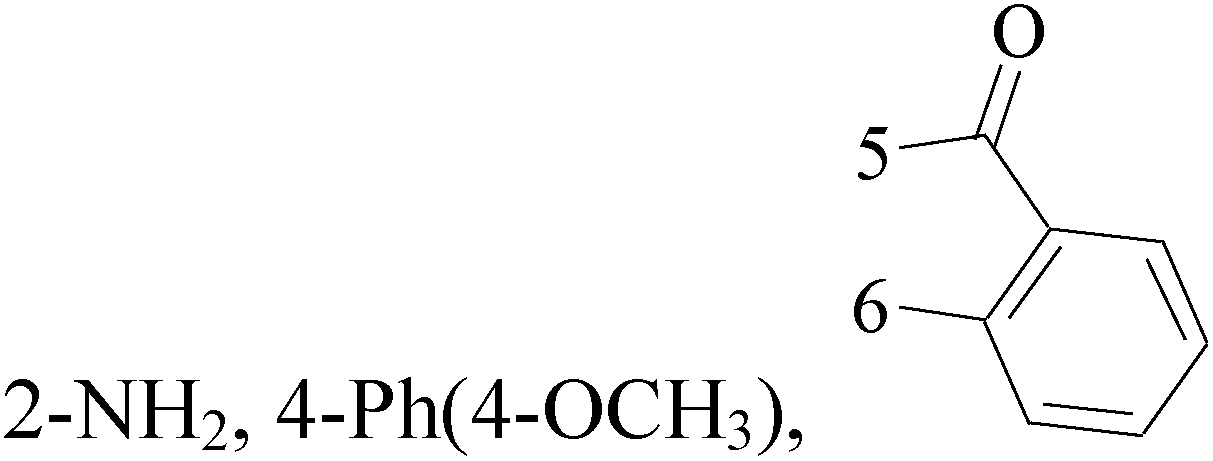
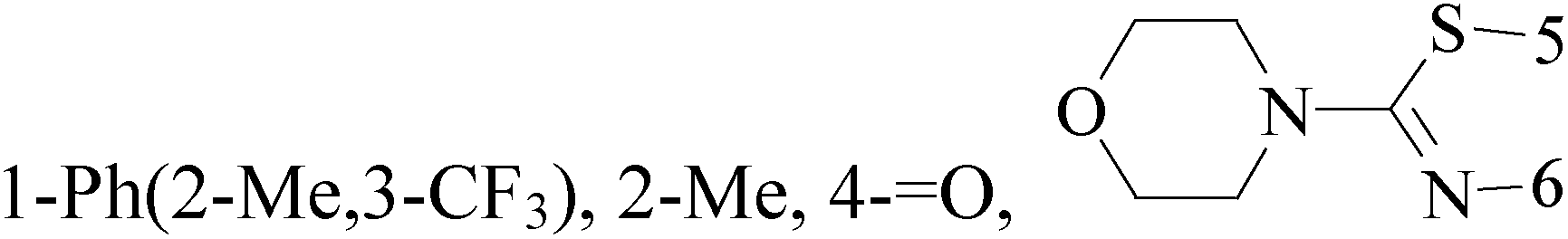
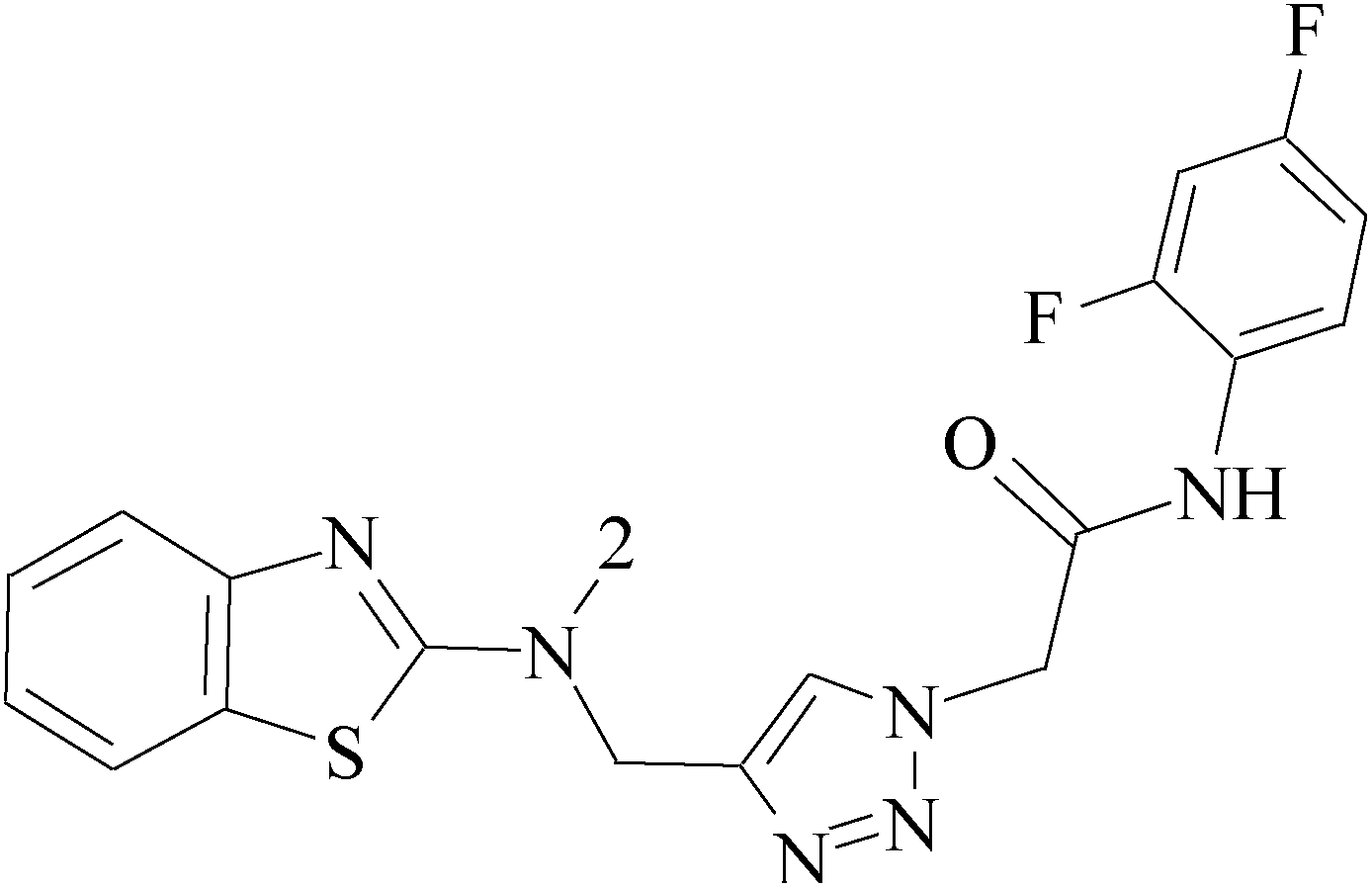
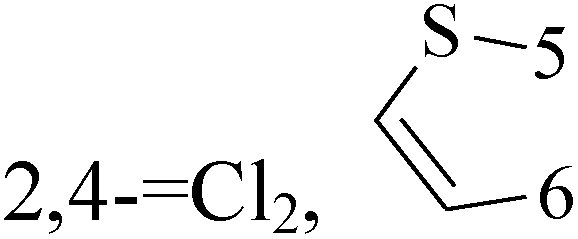
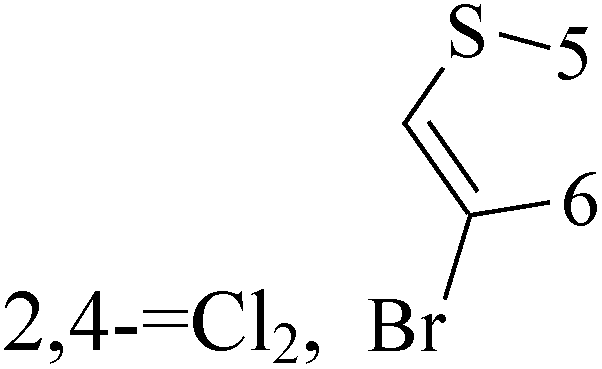
A series of 3-(phenylethynyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine derivatives were designed and synthesized. Compound 38 showed the highest inhibitory potency against the Src kinase and the most potent antiviability activity against the typical TNBC cell line MDA-MB-231 among all the synthesized compounds. Further kinase inhibition assays showed that this compound was a multikinase inhibitor and potently significantly inhibited Src (IC50 = 0.9 nM) and MAPK signaling and markedly induced apoptosis in tumor tissues (Table 3).49
Novel pyrimidine phosphonate molecules were designed so as to inhibit aromatase, a potential target of BC. The ligand–receptor complex of compound 39 showed a best docking score of −15.776 kcal mol−1 among all. Hence, this compound was synthesized and tested in vitro against MDA-MB-231 adenocarcinoma BC cells and it exhibited excellent antiproliferative activity and also induced apoptosis (Table 3).50
The present study describes an alkaline water–ethanol mediated series of combinatorial synthesis of 2-amino-4-phenyl-5-H-indeno[1,2-d]pyrimidine-5-one derivatives. The selected synthesized compounds have been screened against the human BC cell line MCF-7, human colon cancer cell line HT29, and normal viro monkey cell line, out of which compound 40 (GI50 = 22.8 μM) demonstrated significant potency toward human BC cell line (MCF-7) (Table 3).51
A novel thiazolopyrimidinone series of phosphatidylinositol 3-kinase (PI3K)-beta selective inhibitors has been identified. This chemotype has provided an excellent tool compound, 41 (IC50 = 22.8 μM) (Table 3), that showed potent growth inhibition in the PTEN-deficient MDA-MB-468 breast cell under anchorage independent conditions, and it also demonstrated pharmacodynamic effects and efficacy in a PTEN deficient prostate cancer PC-3 xenograft mouse model.52
In the present study, a series of novel triazole linked N-(pyrimidin-2-yl)benzo[d]thiazol-2-amine were synthesized and evaluated for anticancer activity against MCF-7 BC cells. Among the compounds tested, promising compounds 42–45 (Table 3), under the concentration of 3 μM, 3.2 μM, 2.52 μM, 2.12 μM, respectively, caused most remarkable cytotoxicity against MCF-7 BC cells, by inducing apoptosis and affecting the expression of key proteins such as ERK1/2, NF-B and survivin that cause abnormal cell proliferation and up-regulate the activity of caspase-9.53
One compound, 8-cyclopentyl-2-[4-(4-methyl-piperazin-1-yl)-phenylamino]-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidine-6-carbonitrile (46) induced growth arrest of most tumor cell lines, including a panel of BC cell lines, with GI50 values ranging from 0.025 to 2 μM, as a potent inhibitor of cyclin-dependent kinase 4 (CDK4) and AMPK-Related Kinase 5 (ARK5) (Table 3).54
Halogenated thieno[3,2-d]pyrimidines (47 and 48) with IC50 value of 9 μM and 5.9 μM respectively are both toxic to the TNBC cell model MDA-MB-231 at low-micromolar concentrations, but that only for compound 47 can selectively triggers mitotic arrest (Table 3).55
6. The imidazole functional group
Imidazole and its derivatives are a class of 5-membered heterocyclic structure having two non-adjacent nitrogen atoms. Recent studies revealed that the substituted imidazole derivatives have attracted much attention due to their broad spectrum of pharmacological activities such as anti-inflammatory, antifungal, anticonvulsant.56–58A series of novel aminosubstituted xantheno[1,2-d]imidazole derivatives have been designed and synthesized and their antiproliferative activity has been evaluated against human breast MDA-MB-231 cell line. Compounds 49–52 with IC50 value of 46 μM, 28 μM, 16 μM and 18 μM respectively (Table 4) exhibited the highest antiproliferative activity of all the compounds tested.59
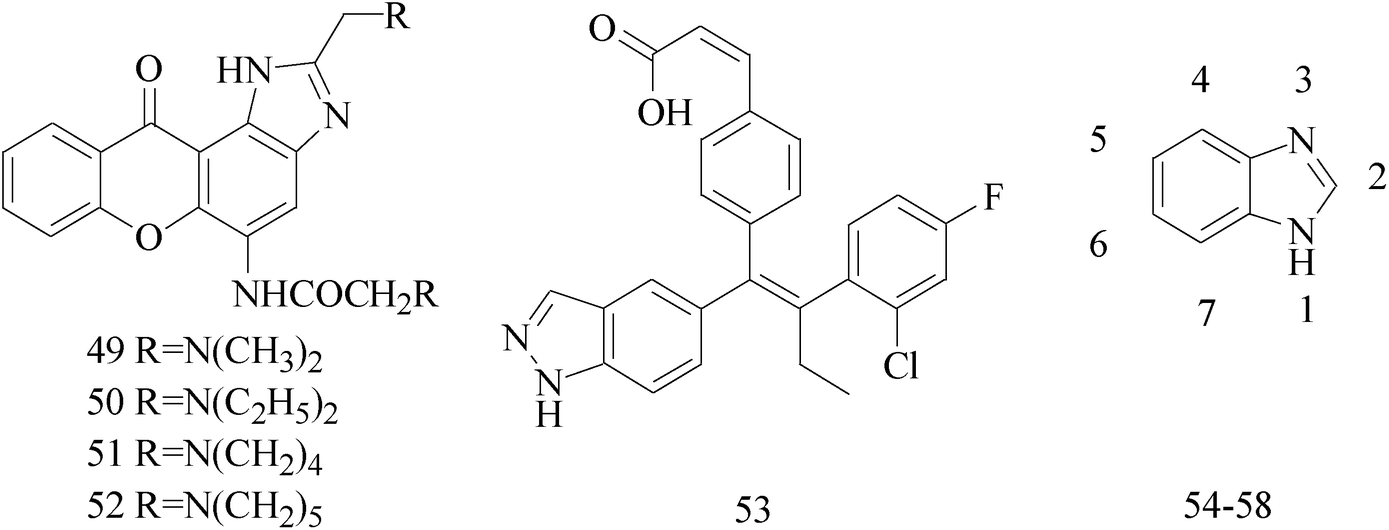
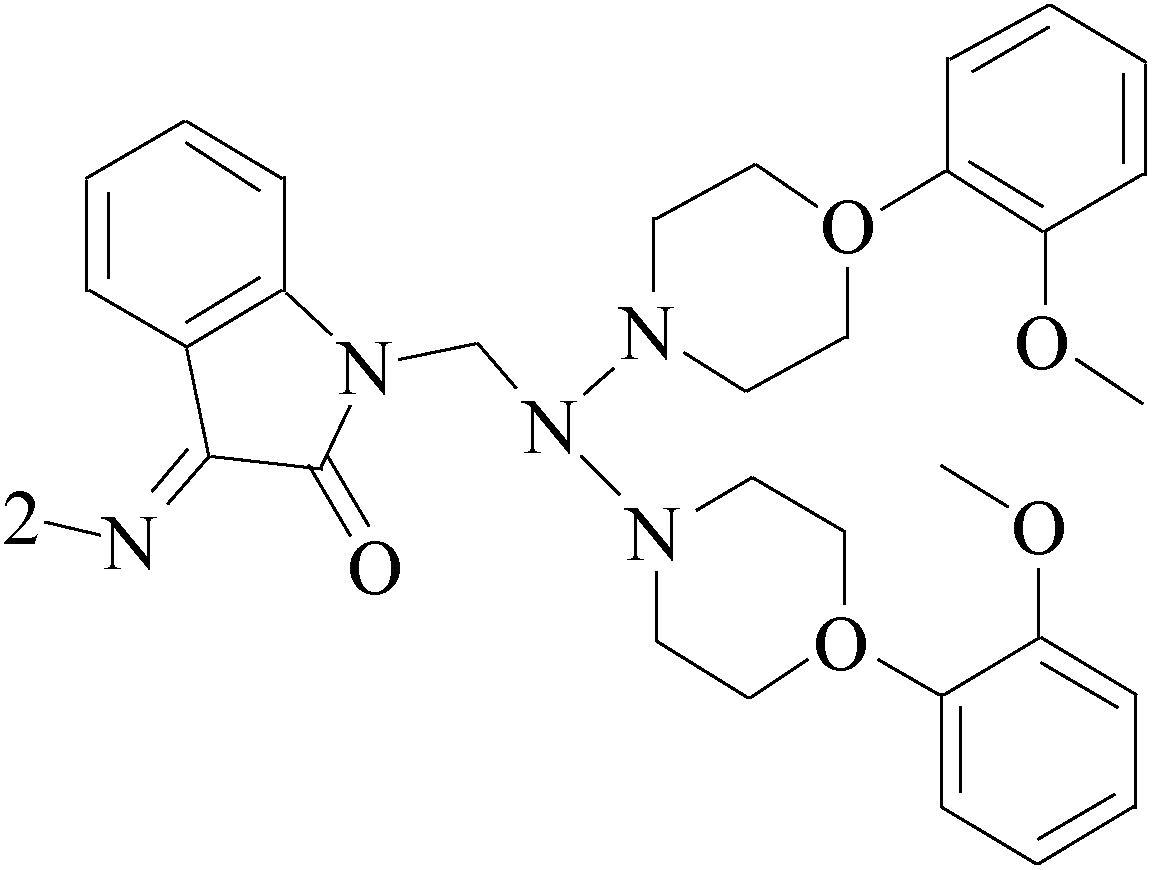
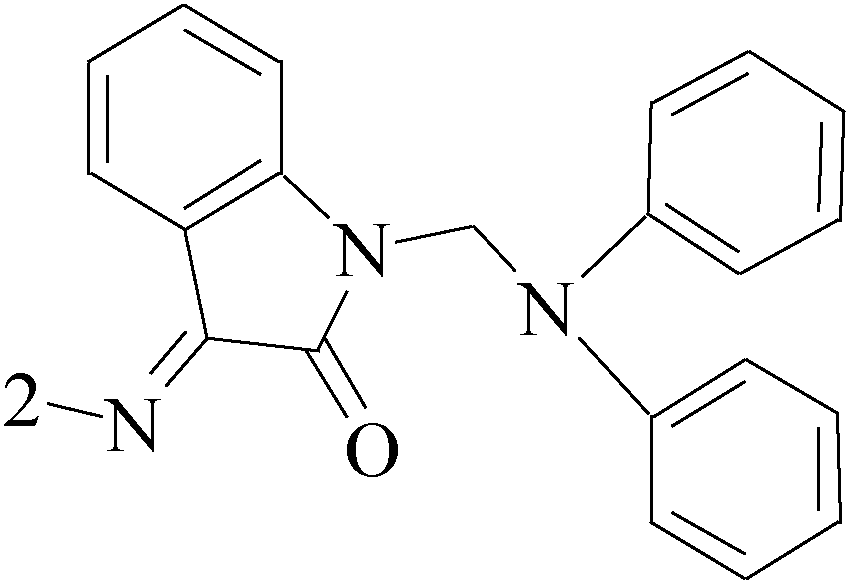
Lai et al.,60 described the identification and characterization of a series of small-molecule, orally bioavailable selective estrogen receptor degraders which are potent antagonists and degraders of ER-α and in which the ER-α degrading properties. In anti-BC screening, candidate 53 (Table 4), the high efficacy degraderst, is currently in clinical trials in women with locally advanced or metastatic ER+ BC.
A series of novel substituted 2-(phenyl)-3H-benzo[d]imidazole-5-carboxylic acids and its methyl esters were synthesized and examined for their antiproliferative effects against three BC cell lines in vitro. Compound 54 (Table 4) bearing 5-fluoro-2-hydroxyphenyl substituent was found to be the most active derivative of the series with GI50 values of 6.23, 4.09 and 0.18 μM against MDA-MB-468, MDA-MB-231 and MCF-7 BC cell lines, respectively.61
A new series of N-sec/tert-butyl 2-arylbenzimidazole derivatives was synthesized and evaluated of antiproliferative activity against MCF-7 and MDA-MB-231. The most potent inhibition against MDA-MB-231 human BC cell line came from compound 55 (Table 4) with IC50 of 29.7 μM.62
A series of new isatin-thiazoline and isatin-benzimidazole derivatives were synthesized and assessed the anti-BC activity. The results showed that compounds 56–58 with IC50 value of 24.95 × 10−3 μM, 26.36 × 10−3 μM and 22.59 × 10−3 μM respectively (Table 4) possessed significant antiproliferative activity against MCF-7 cells.63
7. The tetrazolium functional group
Tetrazole, a heterocyclic compound, contains a carbon atom and four nitrogen atoms in a five-membered ring. Theoretically there are three precursor tetrazole isomers: 1H-tetrazole, 2H-tetrazolium and 5H-tetrazole. Substituted tetrazoles exist as a nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 1H- and 2H-tautomeric forms. Previous studies had indicated that the two positional isomers may be differentiated on the nuclear magnetic resonance (NMR) timescale.64 Tetrazole and its derivatives have a variety of activities, such as anti-tumor, antioxidant, antinociceptive, antibacterial and anti-inflammatory because of their unique structure.65–68
:1 ratio of 1H- and 2H-tautomeric forms. Previous studies had indicated that the two positional isomers may be differentiated on the nuclear magnetic resonance (NMR) timescale.64 Tetrazole and its derivatives have a variety of activities, such as anti-tumor, antioxidant, antinociceptive, antibacterial and anti-inflammatory because of their unique structure.65–68
A series of new 1,2-substituted tetrazole derivatives were synthesized and evaluated on MCF-7 (ER positive), MDA-MB-231 and ZR-75 (ER negative) BC cell lines. Compounds 59–61 showed higher inhibitory effects on MCF-7 cells, while compound 62 exhibited a higher inhibition on MDA-MB-231 cells and ZR-75 cells at a concentration of 10 μM (Fig. 2).69
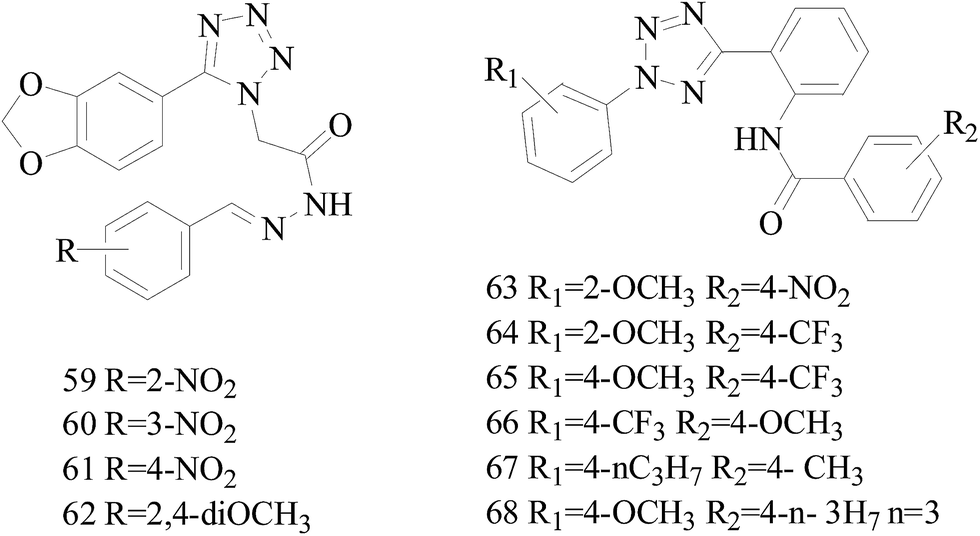 | ||
| Fig. 2 Structure of compounds 59–68. | ||
Köhler et al.,70 synthesized 21 derivatives of the third-generation P-glycoprotein (P-gp, ABCB1) inhibitor HM30181, which is structurally related to tariquidar. The compounds were tested for their inhibitory activities against the BC resistance protein (BCRP, ABCG2) and screened against P-gp and multidrug resistance protein 1 (MRP1, ABCC1) to confirm the selectivity toward BCRP. Compounds 63–65 led to a maximal response comparable to that of the standards the reference inhibitors XR9577 and Ko143. Compounds 66–68 are selective toward BCRP with about 2-fold higher inhibitory activities than the reference Ko143, which is the most potent BCRP inhibitor so far (Fig. 2).
8. The indole or isatin functional groups
Indole ring system as one of the most ubiquitous heterocycles in nature, has been becoming an important structural component in many pharmaceutical agents, such as anticonvulsant, antifungal, antiviral and anti-inflammatory,71–74 particularly in discovery of new antitumor agents.75 A variety of indole derivatives, such as isatin, have also been explored their anticancer activity. Moreover, indoles have promising anti-cancer potential; there has been the emphasis on the synthesis of indole derivatives to overcome problems faced by existing therapeutic agents.76A series of indeno[1,2-b]indole-9,10-dione derivatives were synthesized as human casein kinase II (CK2) inhibitors. Comparison the relative ability of each compound to inhibit CK2, compound 69 (IC50 = 25 × 10−3 μM) (Table 5) has a distinct antiproliferative effect against MCF-7 BC cells and was about orders of magnitude more selective.77
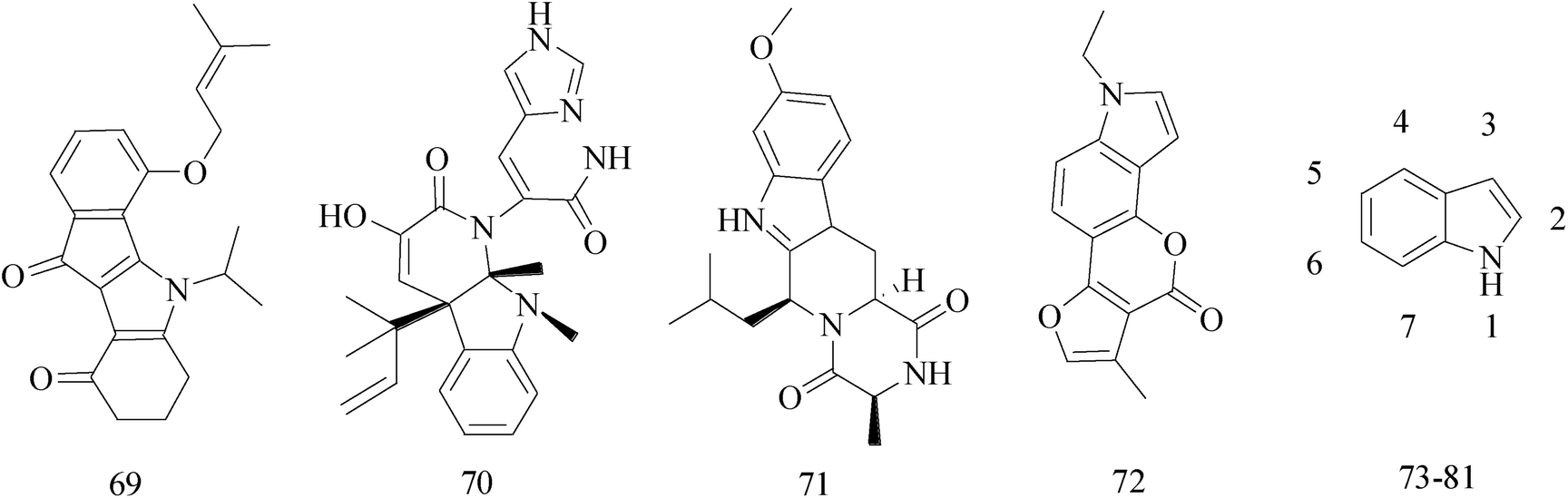
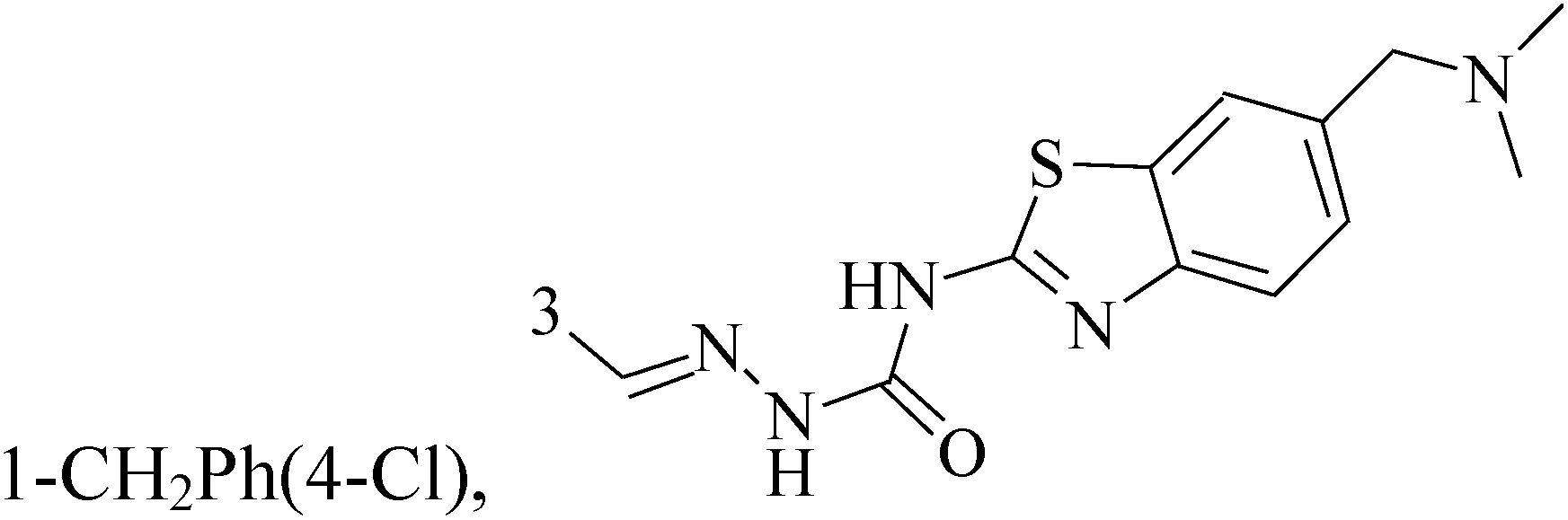
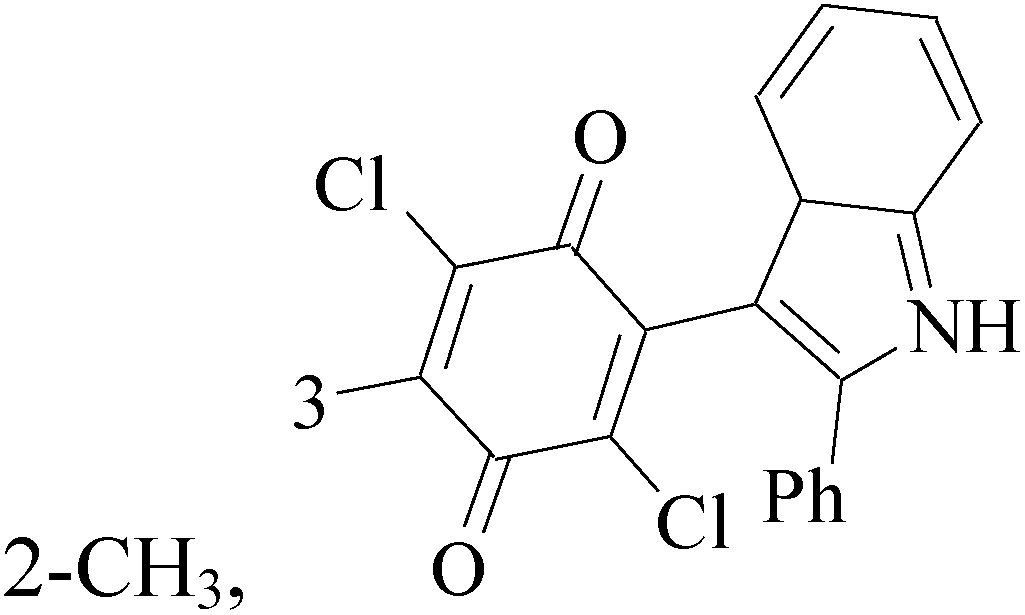
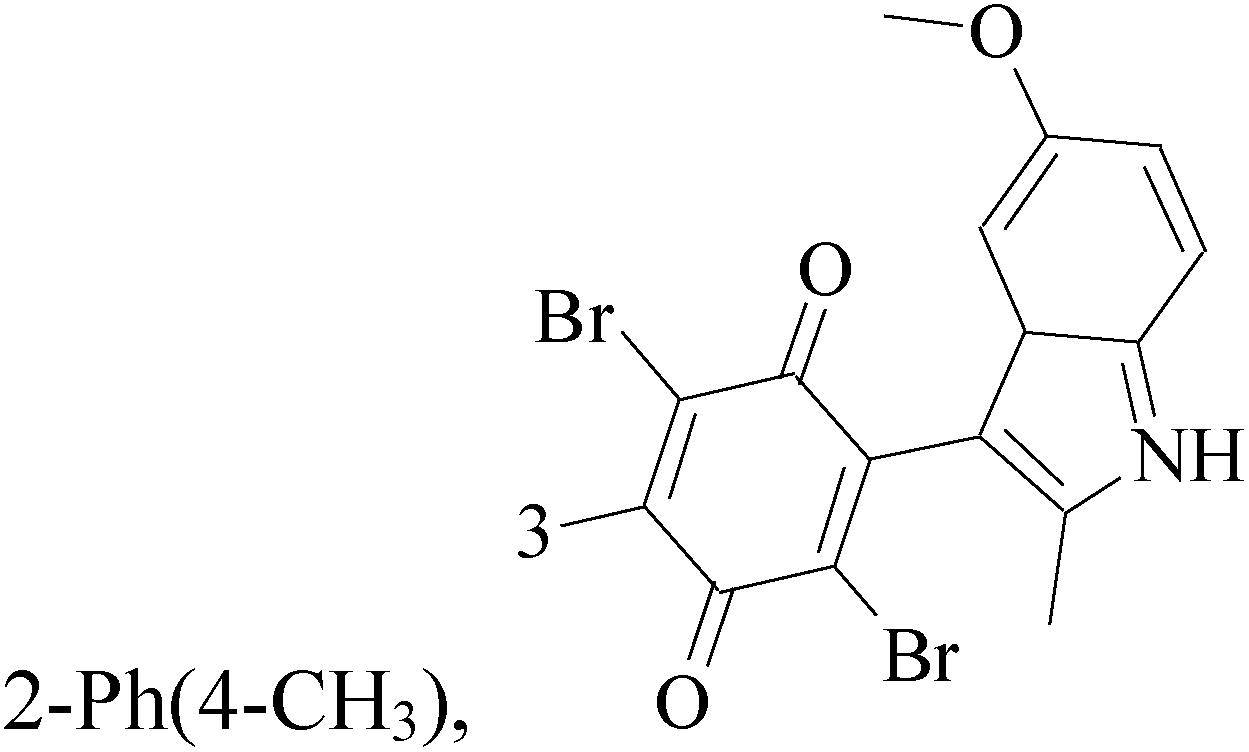
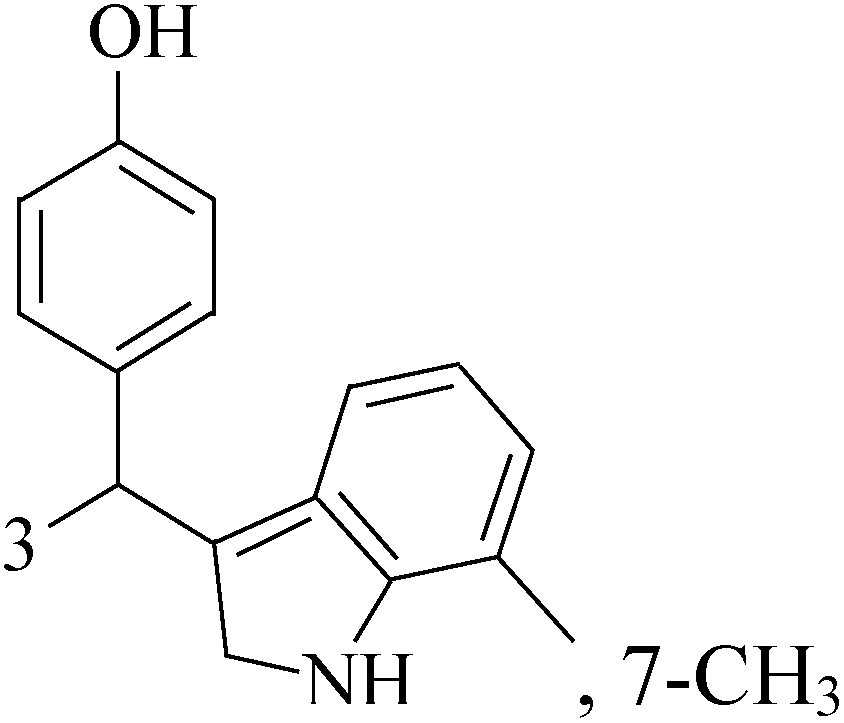
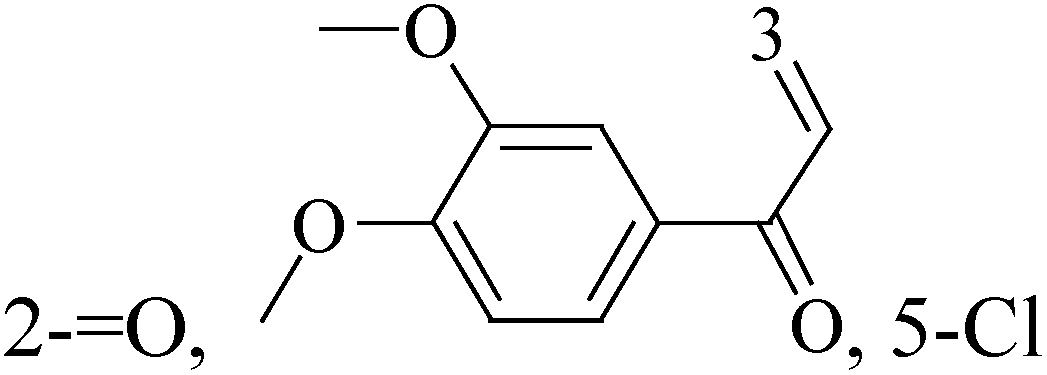
The indole alkaloid meleagrin (70) IC50 value of 1.9, 6.8, 8.9, 2.7 and 2.8 μM respectively for MCF-7, MDA-MB-231, MDA-468, BT-474 and SKBR-3 (Table 5), from the olive tree endophytic fungus Penicillium chrysogenum, was identified as a significant wild and mutant c-Met inhibitor. This was correlated with antiproliferative, antimigratory, and anti-invasive activities against a wide panel of c-Met dependent BC cells including all cell lines but was inactive against the c-Met independent BC cells.78
The design, synthesis, inhibitory potency in vitro, and pharmacokinetic profiles of Ko143 analogs are described. Compared to commonly used Ko143, the new BCRP inhibitor (compound 71) (Table 5) showed the same potency and a significantly improved pharmacokinetic profile in rats.79
A research designed and synthesized novel neo-tanshinlactone analogues in order to inutilize bioisosterism and molecular modification, and to evaluate their bioactivity as anti-BC agents. Compound 72 (Table 5) has been tested for the three non-BC cell lines and the triple negative BC cell lines, but has exhibited no significant cytotoxicity with the average IC50 being greater than 34 μM, compared to neo-tanshinlactone and 4-ethyl neotanshinlactone, and the selectivity towards BC has also diminished.80
A series of novel benzothiazole derivatives bearing indole-based moiety were designed, synthesized and screened for in vitro antitumor activity against four cancer cell lines. Compound 73 (Table 5) exhibited excellent antitumor activity with IC50 values of 0.024, 0.29, 0.84 and 0.88 μM against HT29, H460, A549 and MDA-MB-231, respectively. In addition, the marked pharmacological activity of compound 73 might be ascribed to activation of procaspase-3 and cell cycle arrest.81
A series of novel indolylquinones have been synthesized and evaluated for their antiproliferative activity against human MDA-MB-231 and MCF-7 BC cell lines. Among all these derivatives, 74 (IC50 value = 2.29 μg mL−1 for MCF-7 cells) and 75 (IC50 value = 3.99 μg mL−1 for MDA-MB-231 cells) displayed the most potent antiproliferative activity of the series and inhibited BC cells proliferation by triggering apoptotic cell death (Table 5).82
In the recent study, the synthesis of a novel synthetic aryl methyl ring substituted analog of 3,3′-diindolyl methane, named as phemindole (76) (IC50 = 10.85) was reported as an effective anti-tumor agent against MDAMB-231 cells through inducing apoptosis and restricted the in vitro cell migration through its antimitotic property and the phosphorylation of focal adhesion kinase regulation in MDAMB-231 cells. Furthermore, studies extended to ex ovo and in vivo mice models further validated the efficacy of phemindole.83
A series of ten novel isatin analogs have been synthesized and screened in vitro for their anti-BC activity against MCF-7 cell line. All the tested compounds showed highly potent activity against MCF-7 cell line with especially compounds 77–80 (Table 5) exhibited demonstrative antiproliferative effects on MCF-7 BC cell line compared to reference adramycin (doxorubicin) and GI50 < 0.02 μM.84
A novel series of 3-(2-oxo-2-phenylethylidene)indolin-2-ones incorporating pharmacophoric elements of isatins and chalcones were designed and synthesized and evaluated for anticancer activity against three BC cell lines. In all the promising compound, compound 81 (Table 5) was found to be the most active in the series with GI50 values of 8.54, 4.76 and 3.59 μM against MDA-MB-231, MDA-MB-468 and MCF-7 cells, respectively.85
9. The miscellaneous functional groups of organic compounds
Bhat et al.,86 reported a new series of 2-cyclohexyl-N-[(Z)-(substituted phenyl/furan-2-yl/thiophene-2-yl)methylidene]hydrazinecarbothioamide derivatives. Compound 82 (Fig. 3) markedly inhibited the cell migration and cell adhesion of MDA-MB-231, MDA-MB-468, SKBr-3 and MCF-7 BC cell lines with an IC50 value of 88.89 μM, 46.66 μM, 30.94 μM and 49.90 μM, respectively.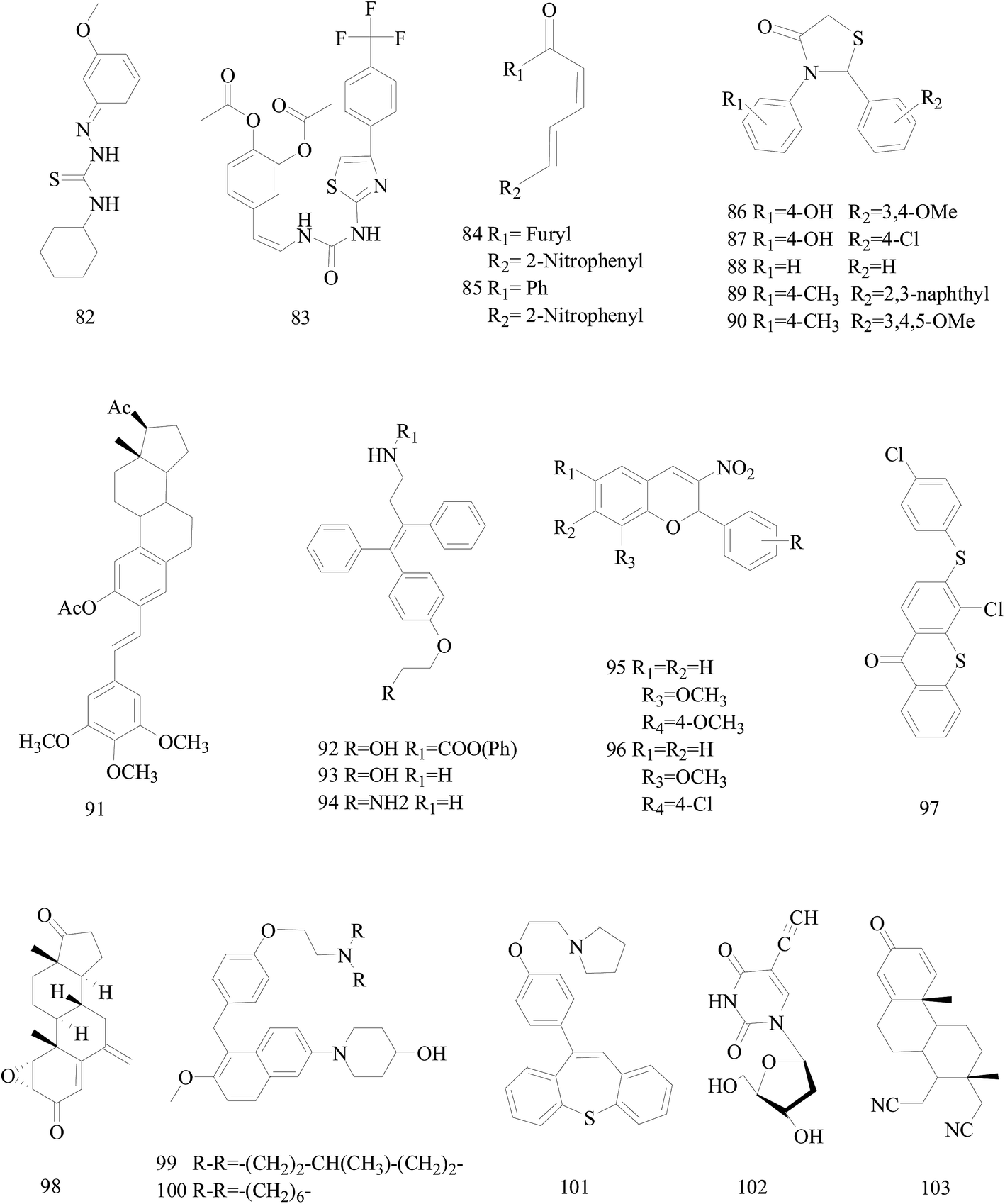 | ||
| Fig. 3 Structure of compounds 82–103. | ||
Mudjupa et al.,87 designed 4-(2-(3-(4-(4-(trifluoromethyl)phenyl)thiazol-2-yl)ureido)vinyl)-1,2-phenylene diacetate (compound 83), a small molecule EGFR inhibitor in silico by using caffeic scaffold as core structure, inhibited the growth of EGFR-overexpressing MDA-MB-468 triple-negative BC cells (TNBC) and wild-type non-small-cell lung cancer H460 cells via programmed cell death and suppressed EGFR expression. Alternatively, compound 83 may be able to decrease hormone receptors, ER and PR expression in the invasive MCF-7 cancer cells resulting in the potent anti-tumor activity (Fig. 3).
Weldon et al.,88 synthesized a series of cinnamylideneacetophenones via a modified Claisen–Schmidt condensation reaction and evaluated for cytotoxicity against BC cells using the Alamar Blue™ assay. Derivatives 84 and 85 (Fig. 3) displayed significantly exhibited cytotoxicity against MDA-MB-468 and MCF-7 cells in the nanomolar to sub-micromolar range yet exhibited substantially less cytotoxicity in non-tumorigenic MCF-10A cells. In fact, the selectivity index for both compounds appears to be superior to that observed with the established chemotherapeutic agent doxorubicin.
Sala et al.,89 synthesized a library of 2,3-thiazolidin-4-one derivatives which showed strong inhibitory effects on BC cell growth. Among all tested compounds 86 and 87 showed cytotoxic activity on MCF-7 cells while in SKBR3 cells compounds 88–90 determined a significant inhibition starting from the lowest dose (Fig. 3).
Parihar et al.,90 synthesized twenty two combretastatin A4 analogues on steroidal framework from gallic acid with a possibility of anti-BC agents. Among these, compound 91 (Fig. 3) was the most active in MCF-7 by inducing apoptosis and MDA-MB-231 cells with IC50 of 7.5 mM and 5.5 mM respectively and showed potent antitubulin effect. Docking experiments also supported strong binding affinity of 91 to microtubulepolymerase and was found to be non-toxic up to 300 mg kg−1 dose in Swiss albino mice in acute oral toxicity.
Kaur et al.,91 described the synthesis of some novel ospemifene derived analogs and their evaluation as anti-BC agents against human BC cell lines. Compound 92 was relatively more cytotoxic to MCF-7 cell lines similar to ospemifene and tamoxifen, while most potent compounds 93 and 94 were equally effective in inhibiting growth of both MCF-7 (ER-positive) and MDA-MB-231 (ER-negative) human BC cell lines (Fig. 3).
A series of 2-aryl-3-nitro-2H-chromenes were synthesized tested against BC cell lines including MCF-7, T-47D and MDA-MB-231. Representatively, compound 95 with IC50 = 0.2 μM against MCF-7 cells, was 36-times more potent than etoposide, and antitumor activity for selected compounds 95 and 96 are identified by apoptosis pathway (Fig. 3).92
A series of 3-substituted-4-chloro-thioxanthones and their corresponding S,S-dioxidethioxanthone derivatives were designed and synthesized and the inhibition activities of the synthesized compounds on cell viability were evaluated. Among the synthesized compounds, compound 97 (with a 4-chlorophenylthio group) was the most-active compound exhibiting potent inhibitory activity on the cell viability of MCF-7 and MDA-MB-468 cell lines with respective IC50 values of 7.2 and 3.9 μM (Fig. 3).93
Varela et al.,94 synthesized some already identified exemestane metabolites. All the studied metabolites reduced cell viability of MCF-7aro cells in a dose and time dependent manner, and metabolite 98 (Fig. 3) in much lower concentrations than the other metabolites and even exemestane dramatically decrease MCF-7aro cells viability. Therefore, their data suggest for the first time that exemestane originates active metabolites after metabolic transformation, which are also able to inhibit aromatase and reduce hormone-dependent BC cells viability.
A study has led to the identification of 1-(5-(4-(2-(dialkylamino)ethoxy)benzyl)-6-methoxynaphthalen-2-yl)piperidin-4-ols as potential novel SERMs. Two of the newly designed compounds 99 and 100 displayed far superior cytotoxicity towards the estrogen-responsive human MCF-7 BC cell line than tamoxifen. Compound 100 also showed significant binding and antagonistic effects against human ERα in an in vitro assay (Fig. 3).95
A targeted library of constrained tricyclic compounds (three prototypes I, II, and III) of substituted dibenzo[b,f]thiepine and dibenzo[b,f]oxepines and structurally analogous to tamoxifen were synthesized as a new class of anti-BC agents. Of all the compounds tested, 101 (Fig. 3) exhibited potent in vitro antiproliferative activity at 1.33 μM and 5 μM concentration in MCF-7 and MDA-MB-231 cell lines.96
Meneni et al.,97 synthesized a series of 5-alkynyl-20-deoxyuridines tested for antiproliferation properties against MCF-7 and MDA-MB-231 human mammary carcinoma cell lines. Compound 102 (Fig. 3), the only nucleoside in the series containing a terminal acetylene, showed the highest potency with IC50 (μM) 0.4 ± 0.3 for MCF-7 and 4.4 ± 0.4 for MDA-MB-231 exceeding cisplatin and 5-fluorouracil.
Nikolić et al.,98 reported the synthesis of steroidal 16,17-seco-16,17a-dinitriles and investigated their antitumor cell properties. Strikingly, 103 (Fig. 3), a 1,4-dien-3-on derivative, displayed selective submicromolar antiproliferative activity against both MCF-7 and MDA-MB-231 BC cells (IC50 0.52, 0.11 μM, respectively).
Dyari et al.,99 reported the synthetic ω-3 epoxides (104–106) of saturated fatty acids as antiproliferative and proapoptotic agents in MDA-MB-231 BC cells. Compounds 104–106 impaired ATP production, enhanced caspase-3 activity, and activated c-jun-N-terminal-kinase signaling, leading to cyclin D1 down-regulation and cell cycle arrest in G1-phase. Fatty acid ω-3 monoepoxides may represent a novel class of antitumor agents (Fig. 4).
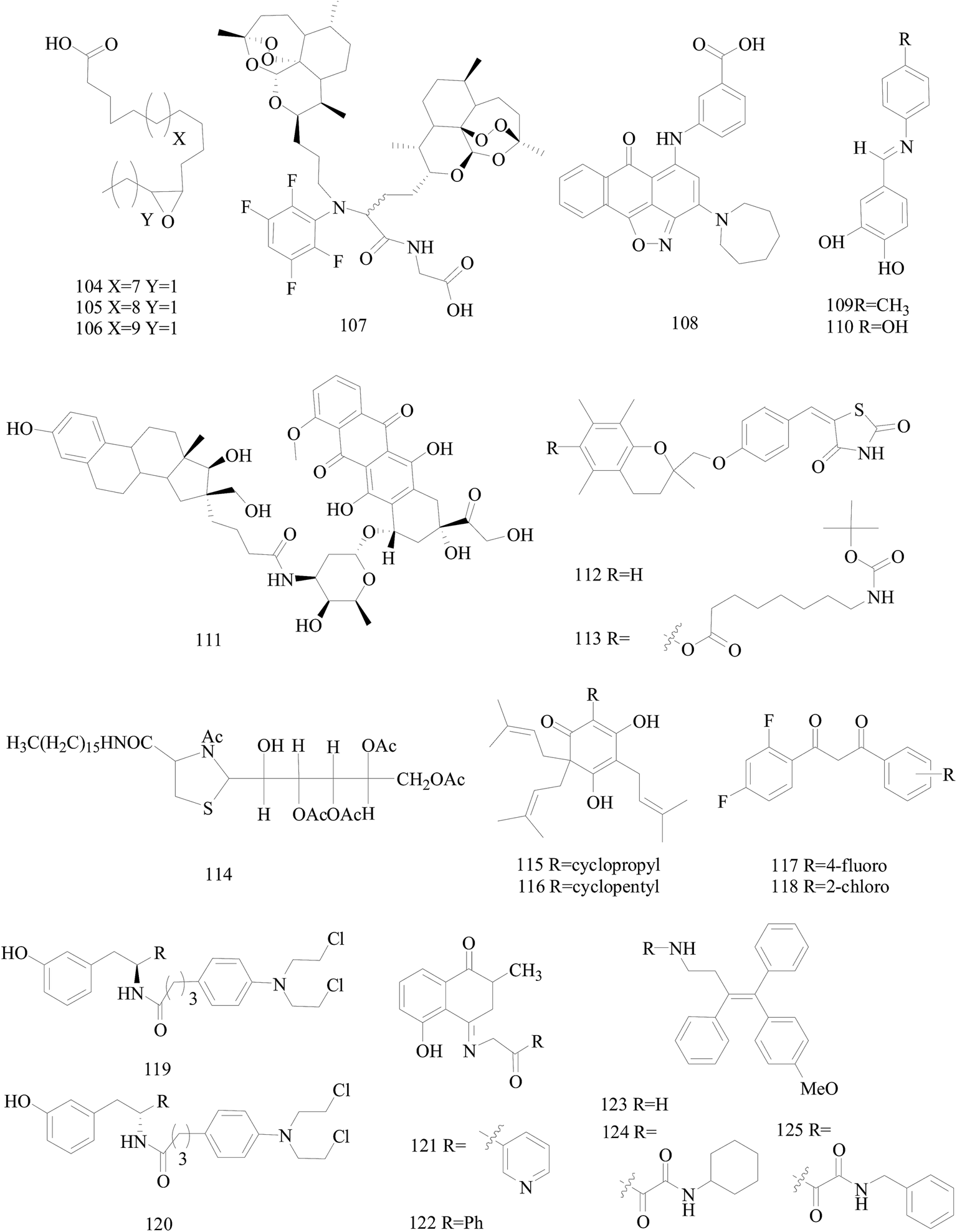 | ||
| Fig. 4 Structure of compounds 104–125. | ||
Wang et al.,100 prepared a series of artemisinin monomers and dimers by using the Ugi reaction, a convenient synthesis. With BT-474 cell lines, all the Ugi dimers showed remarkable activities. Especially, dimer 107 (Fig. 4) showed an IC50 value of 12 nM, approximately more than 600 times more potent than artesunate as a positive control, and a low toxicity when tested on MCF-10A, a nontumorigenic cell line.
Mani et al.,101 synthesized compound 108 (Fig. 4) which impaired invasion migration and adhesion in breast MDA-MB-231, inhibiting matrix metalloproteinase breakdown of the extracellular matrix and its activity, also impairing metastasis to the lungs with only four of the treated mice showing severe or marked metastasis, in comparison to ten for the untreated mice.
Novel aza-resveratrol analogs were synthesized, structurally characterized and evaluated for cytotoxic activity against BC cell lines, which exhibited superior inhibitory activity than parent resveratrol compound. Compounds 109 and 110 exhibited most potent activity with IC50 value of 21 and 29 μM against MDA-MB-231(with 65–75% cytotoxicity) and 32 and 44 μM against T47D cells (Fig. 4).102
Saha et al.,103 designed, prepared and evaluated in vitro the first estrogen–doxorubicin conjugates at 16 aposition of estradiol termed E–DOXs. The IC50 of E–DOX 111 (Fig. 4) on ER+ MCF-7 and HT-29 human colon carcinoma cells are 14 and 18 μM, respectively whereas it is mainly inactive (>50 μM) on M21 human skin melanoma and ER− MDA-MB-231 cells, showing that the selectivity of 111 is potentially a promising conjugate to target ER+ BC.
Salamone et al.,104 synthesised new derivatives of troglitazone. Compounds 112 and 113 indicated good micromolar activity against hormone-dependent (MCF-7) and hormone-independent (MDA-MB-231) BC cell lines and poor toxicity towards hepatocytes in comparison to troglitazone (Fig. 4).
A set of 2-galactosylthiazolidine-4-carboxylic acid amides was synthesized. For the derivatives tested, the compound 114 (Fig. 4) is the highest activity with an IC50 of 17.0 μM for A375 melanoma and 5.8 μM for MCF-7 cell lines and also showed cytotoxicity against triple negative cancer cell line HCC1806. What's more, the compound showed the activation of apoptotic pathways and also DNA damages with blockage of the cell cycle in the S-phase and appearance of peaks in G0/G1-phase.105
The novel unnatural lupulone derivatives were synthesised found to be more toxic to MDA-MB-231 cell lines than the parent lupulone itself. Interestingly the most potent derivative 115 demonstrated an enhanced PARP cleavage for MDA-MB-231 cells while derivative 116 shows a significantly greater effect upon MCF-7 cells (Fig. 4).106
A series of difluorinated propanediones were synthesized and evaluated for in vitro cytotoxic activity on the 4-cell line panel consisting of MCF-7 (BC), HOP62 (lung cancer), A498 (renal cancer) and MIAPACA2 (pancreatic cancer). Compounds 117 and 118 showed favorable anti-proliferative activity in all of the four tested cancer cell lines (Fig. 4).107
Ortho-, meta- and para-tyrosine–chlorambucil analogs were synthesized in order to generate new anticancer drugs with structural diversity. The m-tyrosine–chlorambucil hybrids (119 and 120) showed greater cytotoxicity compared to the other regioisomers (Fig. 4).108
Novel plumbagin hydrazonates were prepared, structurally characterized and evaluated for anti-proliferative activity against the BC cell lines. Compounds 121 and 122 were especially promising against triple negative MDA-MB-231 (IC50 values 1.9 and 2.1 μM, respectively) and MDA-MB-468 BC cell lines via inhibiting NF-B expression (Fig. 4).109
The synthesis of novel triarylethylene analogs, designed based on well-known Selective Estrogen Receptor Modulators, as potential anti-BC agents is described. Compounds 123 and 124 exhibited remarkable activity against both MCF-7 and MDAMB-231 cell lines, whereas the oxalamide 125 was selectively active against MDA-MB-231 cells (Fig. 4).110
Neo-tanshinlactone derivatives (126–130) (Fig. 5) exerted potent and selective anti-breast cancer activity with IC50 values of 0.3, 0.2, 0.1 and 0.1 μg mL−1, respectively, against the ZR-75-1 cell lines. Importantly, analog 127 had an approximately 12-fold ratio of SKBR-3/MCF-7 selectivity. Analog 128, 23 times more active against ZR-75-1 than MCF-7, exhibited high selectivity. In addition, analog 130 showed potent activity against a ZR-75-1 xenograft model, and high selectivity against breast cancer cell line compared with normal breast tissue-derived cell lines.111
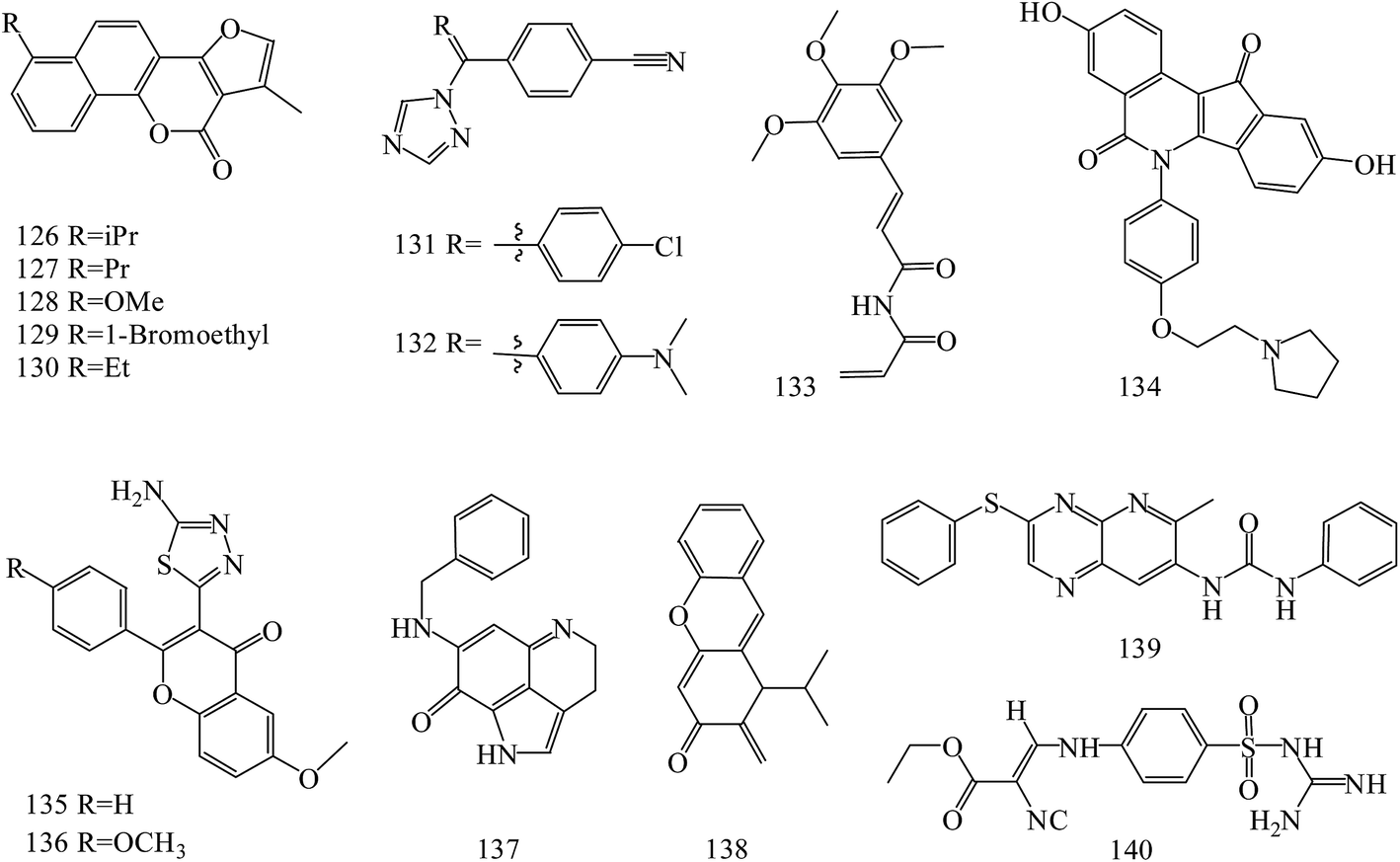 | ||
| Fig. 5 Structure of compounds 126–140. | ||
A novel series of letrozole analogs was designed and synthesized to find new potential anti-breast cancer agents. Cytotoxicity evaluation revealed that compounds 131 and 132 (Fig. 5) were the most potent compounds with comparative activity with etoposide.112
A series of five analogous compounds based on the structure of piperlongumine were designed, synthesized and evaluated in cell migration and cytotoxicity assays. Among these, the analogue designed by molecular simplification (133) (Fig. 5) was the most active of the series, with an EC50 of 1.5 ± 1 μM.113
A series of novel substituted pyridopyrazine derivatives have been rationally designed and evaluated as multi-kinase inhibitors in the PI3K pathway. The most potent compound 134 (Fig. 5) showed low micromolar cytotoxic potency in all BC cell lines and inhibited the growth of a HER2 amplified BC xenograft tumors. Analysis of excised tumors from the treated animals showed a significantly reduced population of Ki-67 positive cells and down-regulated levels of phosphorylated AKT, ERK1/2 and SRC compared to vehicle treated animals. Finally, the specificity of 89 was assessed in a panel of 31 kinases where a mild, but direct, inhibition of the MET receptor tyrosine kinase was observed.114
Novel flavone derivatives were synthesized, characterized and examined for their antitumor activities against breast cancer cell lines. In initial screening, analogs 135 and 136 (Fig. 5) were found to be effective against the estrogen receptor negative cell line (MDA-MB-453).115
A novel synthetic compound 137 (Fig. 5), an analog of a naturally-occurring marine compound, was found to be the most active out of more than 40 related compounds. In a dose dependent manner, it inhibited cell growth and induced apoptosis and cell cycle arrest in human MCF-7 and MDA-MB-468 breast cancer cells in vitro, and showed in vivo efficacy in mice bearing MCF-7 or MDA-MB-468 xenograft tumors. Preclinical data indicated that 137 was a potential therapeutic agent for breast cancer that has multiple hormone-, Her2- and p53-independent mechanisms of action.116
New synthetic α-methylene-δ-lactones ware tested cytotoxicity and anticancer activity in comparison to parthenolide as a positive control. The most potent compound 138 (Fig. 5) reduced the activity of viable MDA-MB-231 and MCF-7 cells (IC50 values of 5.3 μM and 3.54 μM, respectively). It activated the intrinsic pathway of apoptosis, related to the loss of mitochondrial membrane potential and changes in Bax/Bcl-2 ratio and also suppressed the movement of both types of breast cancer cells. Suppression of cell migration and invasion was the result of the decreased secretion of enzymes responsible for the degradation of the extracellular matrix, metalloproteinase-9 and urokinase plasminogen activator.117
A series of 6-aryl-indenoisoquinolone derivatives as dual ERα and vascular endothelial growth factor receptors (VEGFR)-2 inhibitors were synthesized and evaluated. These compounds possessed good ERα binding affinity and ERα antagonistic activity and potent VEGFR-2 inhibitory potency, as well as excellent anti-proliferative activities against MCF-7, MDA-MB-231, Ishikawa and HUVEC cell lines. Among these, compound 139 (Fig. 5) may inhibit the activation of VEGFR-2 and the signaling transduction of Raf-1/MAPK/ERK pathway in MCF-7 cells.118
Novel N-(guanidinyl)benzenesulfonamides were prepared and evaluated for their anticancer activity against human tumor breast cell line (MCF-7). It was found that the most potent compounds was 139 (IC50 values = 49.5 μM) (Fig. 5).119
10. The ferrocenyl functional group
Ferrocene possesses many chemical properties, one of which is robust enough to be functionalized on its cyclopentadienyl rings.120,121 Because of it, as an electron acceptor, disrupting electron transfer in biological systems, ferrocenyl derivatives have been found to possess biological activity, for instant, anti-tumour, antimalarial, antibacterial, and antifungal activity.122–125Eight ferrocenyl derivatives were synthesized and assessed the anti-BC activities. Of the eight compounds, only compounds 141 (IC50 value of 56 and 61 μM respectively for MCF-7 and MDA-MB-231) and 142 (IC50 value of 47 and 87 μM respectively for MCF-7 and MDA-MB-231) showed cytotoxicity in both MCF-7 and MDA-MB-231 cell lines (Table 6).126
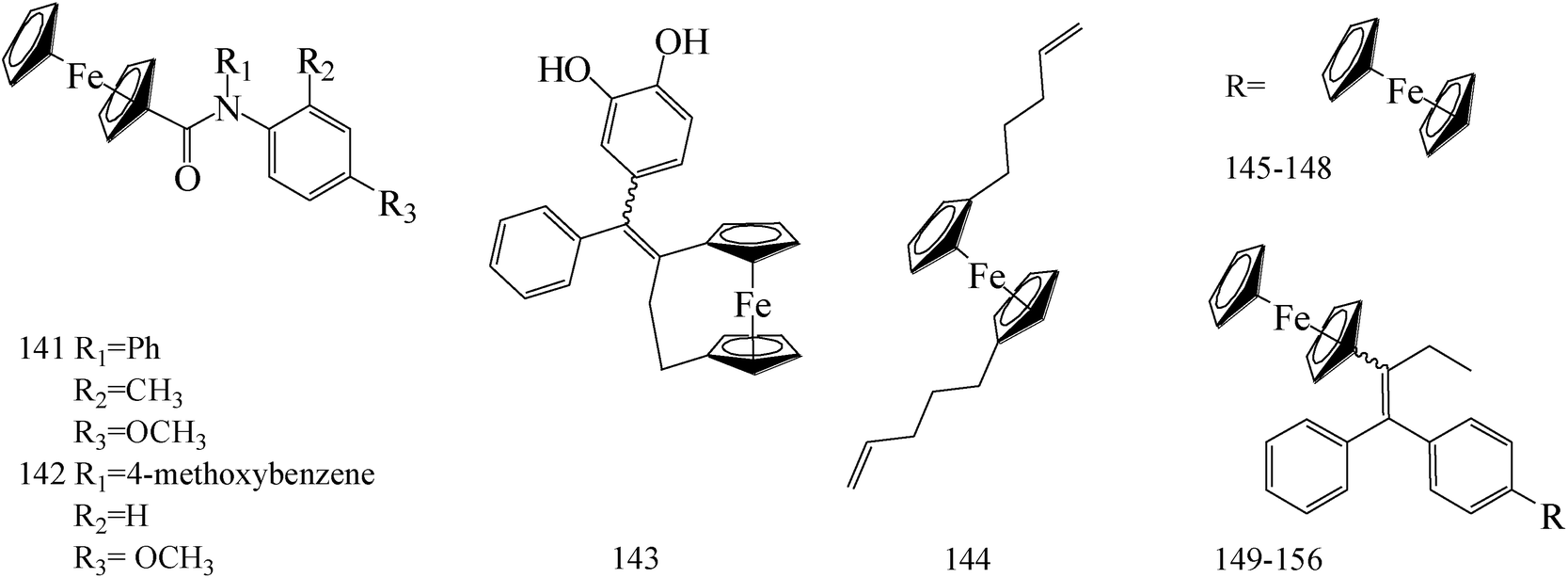
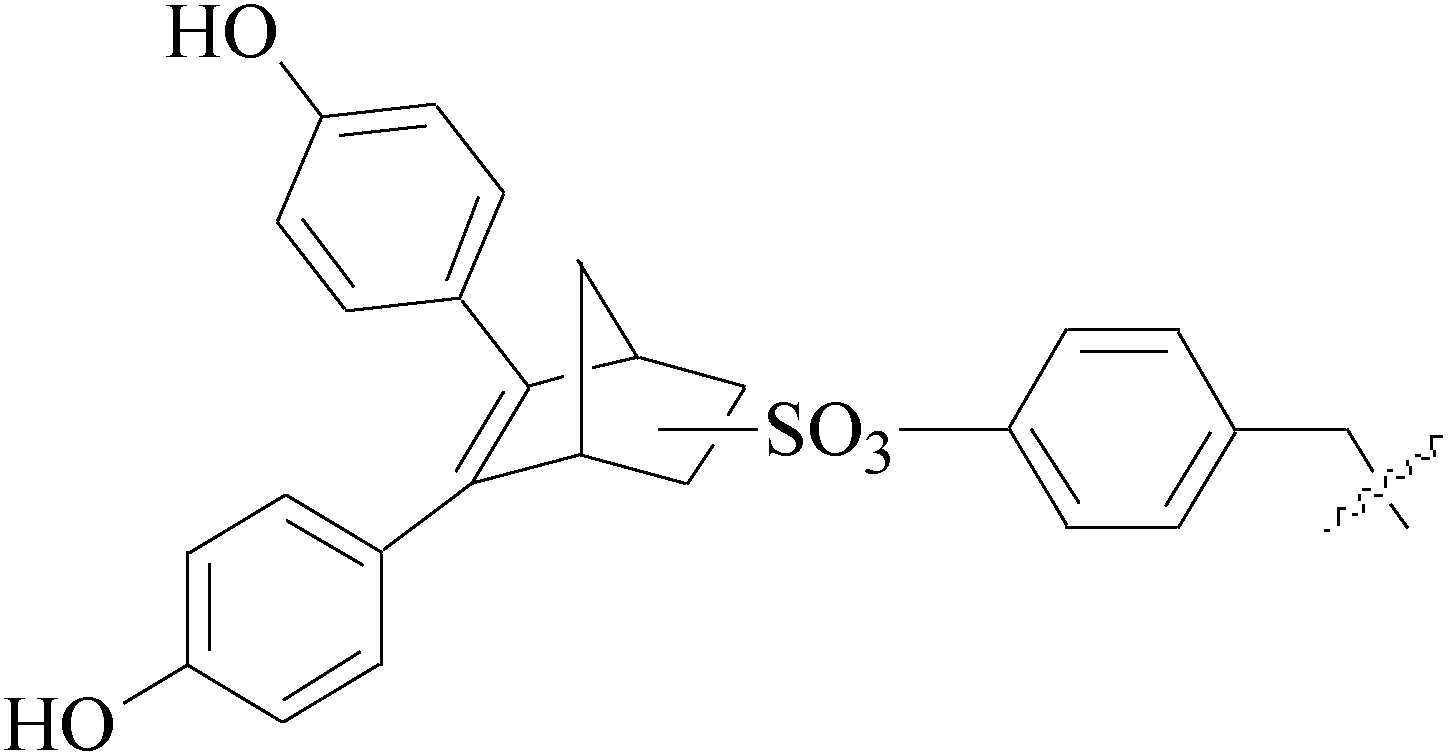
Tan et al.,127 synthesized and assessed anti-tumoral properties of a series of compounds possessing a ferrocenyl group tethered to a catechol through a conjugated system. Compound 143 (IC50 = 0.48 μM for MDA-MB-231) (Table 6) displayed the highest anti-proliferative effect amongst the catechol complexes.
Gao et al.,128 established a synthesis and cytotoxic properties of ferrocenyl ester derivatives, varying the lipophilic character of the pendant groups. Compound 144 (Table 6) showed the best IC50 values, 180 μM for HT-29 and 190 μM for MCF-7 cell lines, with cytotoxicities similar to ferrocene and ferrocenium for comparison.
1-p-(Ferrocenylcarbonylamino-phenyl)-1,2-di(p-hydroxyphenyl)-but-1-ene (145) was synthesized and exhibited a significant antiproliferative activities against MCF-7 and MDA-MB-231 BC cells with 4.53 μM and 1 μM, respectively (Table 6).129
Zheng et al.,130 synthesized a series of novel Selective Estrogen Receptor Modulators (SERMs) bearing a ferrocenyl unit based on a three-dimensional oxabicyclo[2.2.1]heptene core scaffold. The antiproliferative effects of compounds 146–148 (IC50 value of 5.6, 10.4 and 7.5 μM respectively for MCF-7 and IC50 value of 12.2, 126 and 7.8 μM respectively for MDA-MB-231) (Table 6) on MCF-7 cells line does not arise from antiestrogenicity, but rather proceeds through a cytotoxic pathway.
A new class of indeno[1,2-c]isoquinolines containing the ferrocenyl scaffold was synthesized. The most potent compound 149 (Table 6) with IC50 of 0.95 μM displayed high DNA interaction, topoisomerase I and II inhibition, and an in vitro cytotoxicity comparable to etoposide as the reference drug.131
de Jesús Cázares-Marinero et al.,132 synthesized seven new ferrocenyl compounds which showed strong antiproliferative activities against BC cell lines with IC50 values ranging from 0.5 μM to 4.12 μM. Primary amides FcTAMPSA (compound 150) and 151 were the most cytotoxic compounds of all the three series (suberic, adipic and succinic) against MDA-MB-231 cells, with IC50 values of 0.50 μM and 0.54 μM, respectively; while succinimide 152 was the most active compound against hormone dependent MCF-7 BC cells (Table 6).
Heilmann et al.,133 synthesized four new ferrocene compounds and show strong cytotoxic effects against both the hormone-dependent MCF-7 and hormone-independent MDA-MB-231 BC cell lines. Compound 153 showed cytotoxic effects similar in magnitude to those observed with previously described ferrocenyl diphenol 154 (IC50 (153) = 0.5 μM; IC50 (154) = 0.6 μM) (Table 6).
Pigeon et al.,134 investigated the synthesis, cell proliferation effects, and electrochemical behavior of the ferrocenyl aniline 155 and acetanilide 156. Both compounds showed dual estrogenic/cytotoxic activity at concentrations 10 μM on the MCF-7 cell line, whereas antiproliferative with IC50 values of 0.8 μM and 0.65 μM, respectively, on the MDA-MB-231 cell line (Table 6).
11. The titanocene functional group
Several platinum agents, such as cisplatin, which exert antiproliferative activity in BC targeting DNA,135 have produced a strong interest in research of new organometallic compounds as pharmacological anticancer tools. Even though cisplatin and follow-on derivatives are still widely used in the clinic, the onset of toxic side effects and/or chemoresistance represents the principal limitation to their therapeutic efficacy.136,137 Within this context, the antitumor properties of different metal complexes have been evaluated and, amongst them, titanium complexes have received considerable attention because of their cytotoxic activity against solid tumors.138 During the recent years a plethora of modified titanium-based compounds have been synthesized and studied as potential antitumor agents, especially.138–141Sirignano et al.,142 has prepared a series of novel titanocene-complexes and evaluated for their growth regulatory effects in MCF-7 and SkBr-3 BC cells. Among these compounds, that showed moderate to high antitumor activity, the strongest antiproliferative activity against MCF-7 cells was displayed especially by compound 157, whereas 158 elicited relevant repressive effects on SkBr3 cells (Table 7).
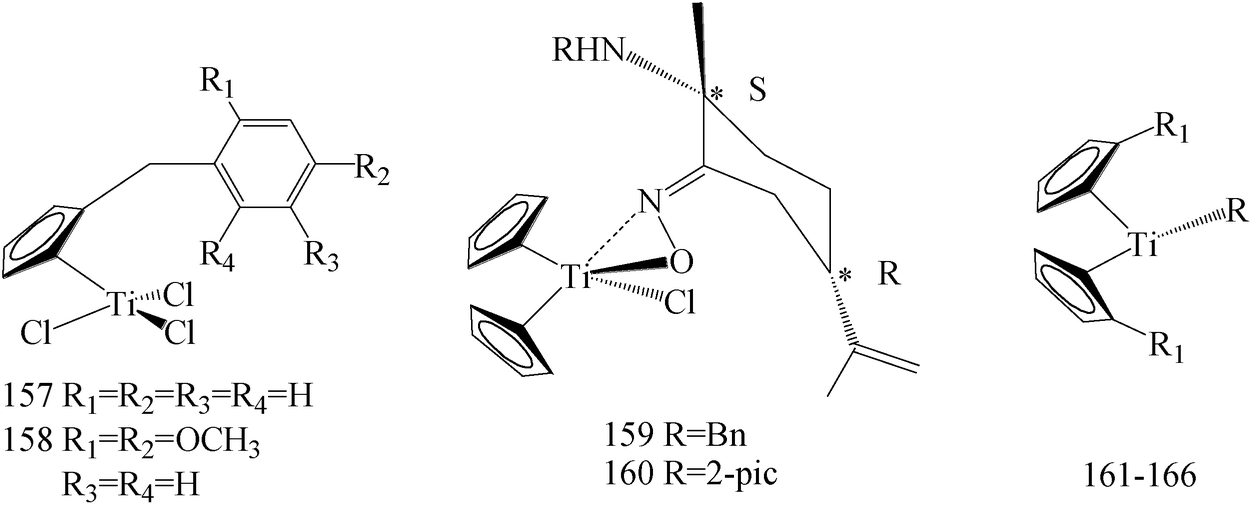
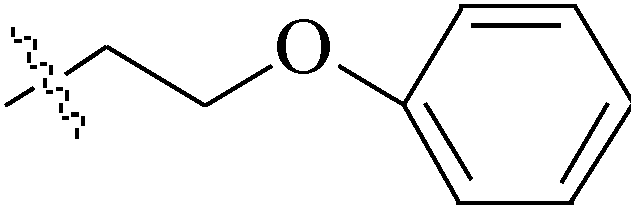
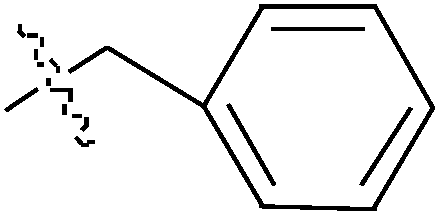
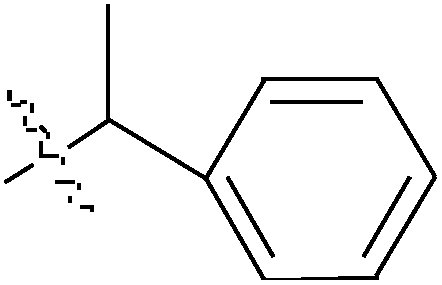
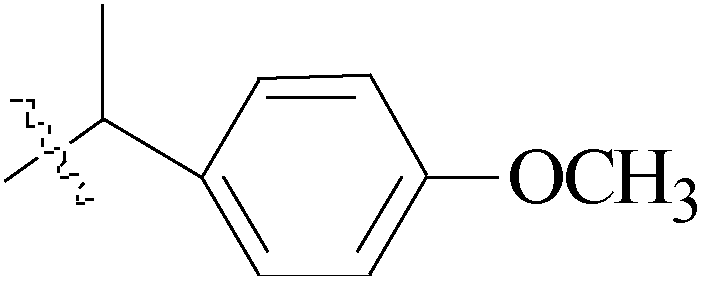
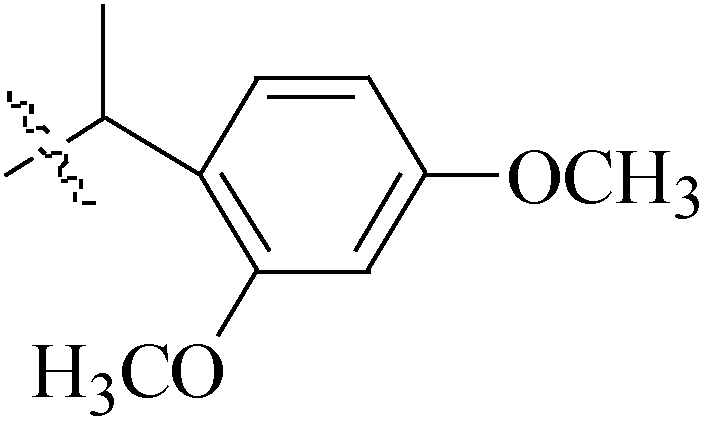
de la Cueva-Alique et al.,143 reported on the synthesis and characterization of a novel family of chiral cyclopentadienyl ammonium- or amino-oximato titanium derivatives. Compounds 159 and 160 (IC50 > 100 μM for MDA-MB-231) (Table 7) are more cytotoxic than additive doses of titanocene dichloride and free oxime proligand, against human renal Caki-1, colon DLD-1 and MDA-MB-231 cell lines.
Saturnino et al.,144 evaluated some synthesized titanocene-complexes, having a ethenyl-phenoxide or a benzyl group as substituents of the cyclopentadienyl rings for their cytotoxic potential against MCF-7 and SkBr-3 human BC cell lines. Compounds 161–163 (IC50 value of 10, 9 and 10 μM respectively for MCF-7 and IC50 value of 10, 6 and 10 μM respectively for SKBR-3) (Table 7) have shown significant anti-proliferative effects, compared to cisplatin.
Chimento et al.,145 reported the synthesis of some new titanocene and half-titanocene compounds having a methyl group on the carbon 6 and a methoxy-naphthyl group as substituent of the cyclopentadienyl. Moreover, the IC50 values of the most active compounds (i.e., 164–166) and of cisplatin have been calculated, evidencing that these new complexes 164–166 (IC50 value of 85.26, 49.16 and 129.8 μM respectively for MCF-7) exerted antiproliferative effect on MCF-7 BC cells, via inhibiting important DNA-metabolizing enzymes, that is, topoisomerase I and II (Table 7).
12. The miscellaneous functional groups of organometallic compounds
Yin Zhang et al.,146 reported the new phosphorescent rhenium(I) polypyridine fructose complex (167, 168). These complexes have been used to image BC cells, where fructose transporters are overexpressed (Fig. 6).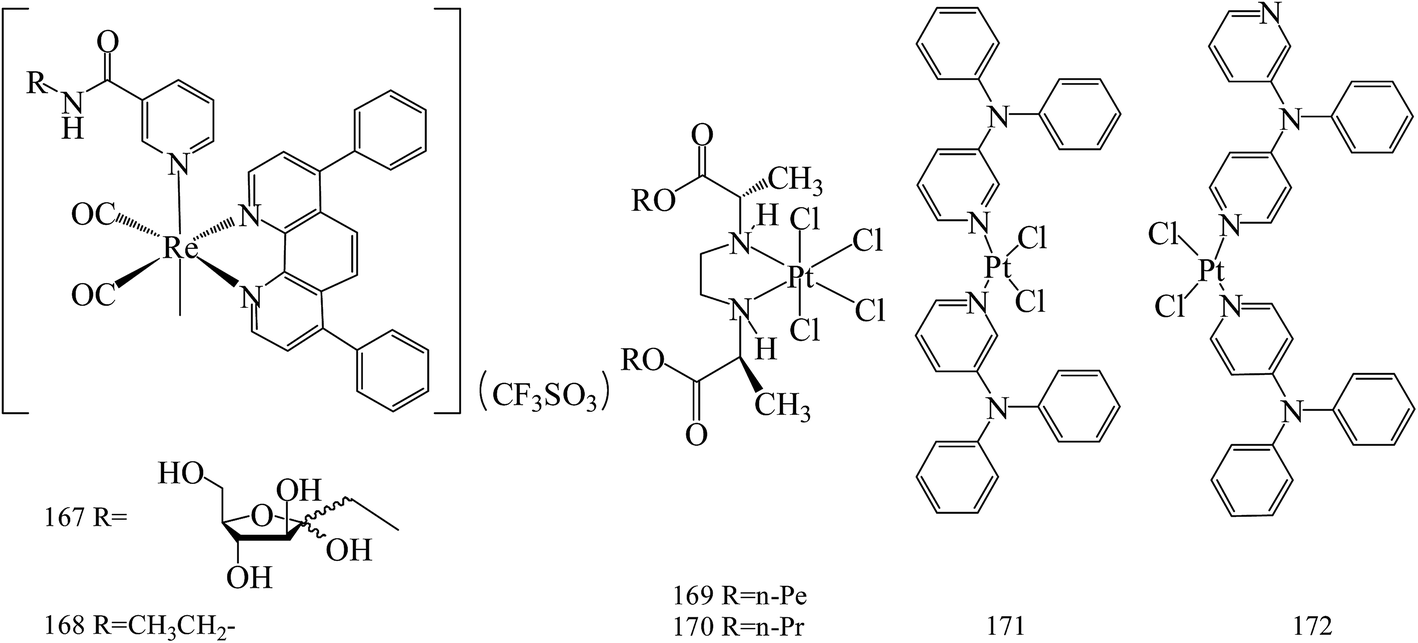 | ||
| Fig. 6 Structure of compounds 167–172. | ||
Stojković et al.,147 reported three novel platinum(IV) complexes and their in vitro antiproliferative activity on tumor cell lines: human colon carcinoma HCT-116 and human breast carcinoma MDA-MB-231. Among these compounds, the highest antiproliferative effects had complex 169 on HCT-116 (IC50 = 64.21 μM), until 170 had the highest effect on MDA-MB-231 (IC50 = 68.023 μM) for 72 h of exposure (Fig. 6).
Two cis-isomers of platinum(II) dichloride complexes composed of meta- and para-N,N-diphenyl pyridineamine derivatives (171 and 172) were prepared and found to be effective in blocking the growth and in inducing apoptosis in MCF-7 and MDA-MB-231, similar to cisplatin. In addition, compounds 171 and 172 also inhibited the migration of MDA-MB-231 cells possibly by affecting cytoskeletal organization membrane and vesicle flow, and cell polarity, as well as decreasing the levels of metabolic energy (ATP) (Fig. 6).148
13. Computer-aided synthesis of compounds
The above anticancer compounds are synthesized using traditional computational techniques, such as molecular docking, and in recent years, new approaches like the prediction of drug and drug targets candidates with mathematical and computational techniques was introduced.149–152 Advanced chemoinformatic tools were known as multitasking models for quantitative structure biological effect relationships.153 Such models are able to integrate multiple kinds of chemical and biological data,154–157 where many different kinds of pharmacological activities, pharmacokinetic parameters, and toxicity profiles are simultaneously predicted.158–165A series of pyrazole based VEGFR-2 inhibitors supported by docking and in silico computational studies were designed on the basis of the hybridization approach, synthesized through facile synthetic methods, screened for in vitro antiproliferative activity against the HT-29 (human colon cancer) and MCF-7 (human breast cancer) cell lines and also studied for in vitro inhibitory activity against VEGFR-2 kinase. Among all the tested compounds, compound 173 (Fig. 7) emerged as a potent agent in the antiproliferative study against HT-29 and MCF-7 cells, with IC50 values of 2.36 and 6.59 μM, respectively. Moreover, the same compound exhibited the highest VEGFR-2 inhibitory activity with an IC50 value of 1.89 μM.166
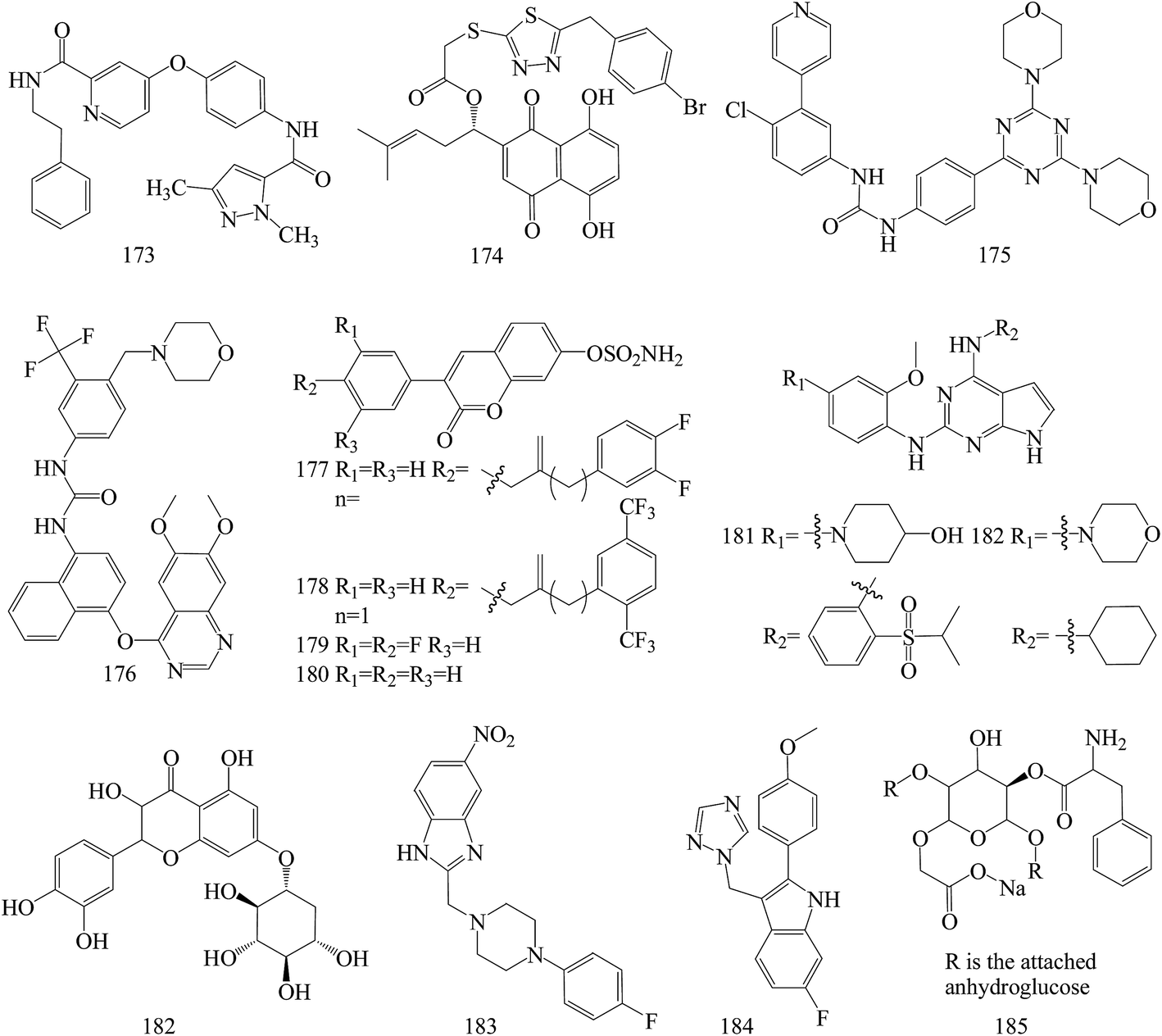 | ||
| Fig. 7 Structure of compounds 173–185. | ||
The in vitro and in vivo results collectively suggest that 174 (Fig. 7), a new type of signal transducer and activator of transcription (STAT)3 inhibitors based on structural modifications on shikonin scaffold, guided by computational modeling, may serve as a promising lead compound for the further development of potential therapeutic anti-neoplastic agents. 174 was found to induce cell apoptosis in MDA-MB-231 cells, associated with the reduction of mitochondrial membrane potential, production of ROS and alteration of the levels of apoptosis-related proteins. Moreover, 174 inhibited constitutive/inducible STAT3 activation, transcriptional activity, nuclear translocation and downstream target genes expression in STAT3-dependent breast cancer cells MDA-MB-231.167
Based on the structures of PI3K inhibitor buparlisib and Hh inhibitor vismodegib, a series of hybrid structures were designed and synthesized utilizing rational drug design and computer-based drug design. Among several compounds, the representative compound 175 (Fig. 7) with the IC50 value of 3.61 μM displayed most excellent antiproliferative activities against MDA-MB-231 cell possibly through inhibiting both PI3K/Akt/mTOR and hedgehog signalings by inhibiting the phosphorylation of S6K and Akt as well as decreasing the SAG elevated expression of Gli1. Compound 175 could also induce apoptosis remarkably in T47D and MDA-MB-231 cells and showed significant inhibition on the migration of MDA-MB-231.168
Computational and experimental studies were applied to the discovery of a series of novel vascular endothelial growth factor receptor 2 (VEGFR-2) inhibitors. Among all compounds, compound 176 (Fig. 7) behaved better than FDA approved drugs, sorafenib and sunitinib, in antiproliferative activity leukemia, non-small cell lung cancer (NSCLC), colon cancer, ovarian cancer and breast cancer cell lines, and it was better or comparable in safety. Compound 176 even demonstrated high potency on one of the drug-resistant cell lines responsible for ovarian cancer and cell lines contributing to prostate cancer, regarded as one of the VEGF/VEGFR pathway drug-resistant tumors.173
Computational docking methods were used to determine the binding modes of 3-(4-aminophenyl)-coumarin-7-O-sulfamate derivatives N-acylated with fluorinated analogs of benzoic or phenylacetic acid as steroid sulfatase (STS) inhibitors and to identify potential interactions between inhibitors and amino acid residues located in the active site of STS. Compounds 177 and 178 demonstrated the highest inhibitory effect in enzymatic STS assays, both with IC50 values of 0.18 μM (the IC50 value of coumarin-7-O-sulfamate is 1.38 μM, used as a reference). Compound 178 exhibited the highest potency against the MCF-7 and T47D cell lines (GI50 values of 15.9 μM and 8.7 μM, respectively), in comparison with tamoxifen as a reference with 6.8 μM and 10.6 μM for MCF-7 and T47D cell lines, respectively. Despite the slightly lower activity of compounds 1 and 2 (both in enzymatic and cell-based experiments) compared to 180, analogues 179 and 180 proved to selectively inhibit the growth of ER- and PR-positive cell lines (Fig. 7).170
Novel small molecules targeting Mps1 were designed by computer assisted docking analyses and synthesized. The lead compounds have strong anti-proliferative potential through Mps1/TTK inhibition in both basal and luminal BC cell lines, exhibiting IC50 values ranging from 0.05 to 1.0 μM. In addition, the lead compounds 181 and 182 inhibit Mps1 kinase enzymatic activity with IC50 values from 0.356 μM to 0.809 μM, and inhibited Mps1-associated cellular functions such as centrosome duplication and the spindle checkpoint in triple negative breast cancer cells. The most promising analog, compound 181, significantly decreased tumor growth in nude mice containing Cal-51 triple negative breast cancer cell xenografts (Fig. 7).171
To identify potential natural allosteric inhibitor for Akt1, a seven-point pharmacophore model was generated and screened it through natural compound library. 182 (Fig. 7) as the best among selected molecules induced dose-dependent inhibition of MDA MB-231 and arrested them in G1 and sub-G phase. This was associated with down-regulation of anti-apoptotic protein Bcl-2, up-regulation of cleaved caspase-3 and PARP. Expression of p-Akt (Ser473) was also down-regulated which might be due to Akt1 inhibition in inactive conformation. the Akt1 and 182 interaction which was observed to have a dissociation constant of 0.246 μM.172
New benzimidazole derivatives were synthesized and assessed for anti-cancer. The computational studies affirmed that almost all of the inspected compounds meet the optimal requirements for good absorption and oral bioavailability. In vitro antitumor testing of these compounds toward liver cancer (HepG2), colon cancer (HCT-116) (IC50 = 0.014 mM) and breast cancer (MCF-7) (IC50 = 0.015 mM) cell lines revealed that compound 183 (Fig. 7) has the highest potency against the three tested cell lines.169
A series of novel non-steroidal molecules containing 2-phenylindole scaffold and moiety of either imidazole or 1,2,4-triazole to enhance their binding capacity with the aromatase were designed and synthesized. Among these molecules, a compound 184 (IC50/aromatase: 14.1 nM; IC50/MCF-7: 325 μM) have the highest inhibitory activity to aromatase and low cytotoxicity. Molecular modelling and simulation techniques were performed to identify the binding modes of letrozole and 184 with the aromatase. Analysis of energy of the two compound–aromatase complexes revealed that the 184 has a low binding energy (strong binding affinity) to the aromatase as compared to letrozole, which was in accordance with the experimental results (Fig. 7).174
The synthesized 185 (Fig. 7) was evaluated as biological and anti-cancer active compound. Among all the tested microorganisms and MCF-7 breast cancer cells, the synthesized nano-cellulose derivative is possible used as safety medicine for microbial infections and cancers. The cytotoxic index (IC50) for MCF-7 breast cancers is 50 μg mL−1. Moreover, the computational study of ADMET (absorption, distribution, metabolism, elimination and toxic) properties, of the molecules showed that, this investigated nanocompound is good oral bioavailability.175
14. Prospect
Due to the rapid social development and people's life pressure, more and more patients were diagnosed with BC.1,2 With the increase in breast cancer patients, early treatment and early diagnosis were promoted. Once diagnosed, to determine chemotherapy is of great significance through differences in the genetic profile of the primary BC.2,176 What's more, participation in an exercise and diet counseling program lead to loss of body fat, improved fitness and quality of life, and increased habitual physical activity in survivors of BC.177,178 And BC patients undergoing breast-conserving therapy had better outcome than modified radical mastectomy.179 Chemical treatment is particularly important, because of its advantages including higher rates of breast-conserving surgery and the possibility of measuring early in vivo response to systemic treatment.180 Thus, the discovery of new compounds is of particular importance, with promising anti-BC properties, which may be proven to be more effective and selective, and possibly free of unwanted side effects. In this review, the compounds designed and synthesized show little difference in the efficacy against breast cancer, whether using traditional computational techniques such as molecular docking or an innovative de novo design approach.15. Conclusion
To sum up, based on the recent references, this review summarized some significant anti-BC compounds which are classified by functional groups. From this review we can see that a lot of people made many attempts to seek anti-BC compounds with more effective, more selective effects and lesser secondary actions. I hope that researchers can have a certain understanding and ideas on design and synthesis of anti-BC compounds through this review.Conflicts of interest
There are no conflicts to declare.References
- R. Siegel, J. Ma, Z. Zou and A. Jemal, Cancer statistics, Ca-Cancer J. Clin., 2014, 64(1), 9–29 CrossRef PubMed.
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Global cancer statistics, Ca-Cancer J. Clin., 2011, 61(2), 69–90 CrossRef PubMed.
- N. N. Su, Q. Wu and J. Cui, Applications and prospects of light environment control technology for vegetable seedling cultivation in factory, China Vegetables, 2013, 4, 006 Search PubMed.
- D. Hanahan and R. A. Weinberg, Hallmarks of cancer: the next generation, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- K. Polyak, Heterogeneity in breast cancer, J. Clin. Invest., 2011, 121, 3786–3788 CrossRef CAS PubMed.
- A. Mullard, 2011 FDA drug approvals, Nat. Rev. Drug Discovery, 2012, 11, 91–94 CrossRef CAS PubMed.
- V. G. Vogel, J. P. Costantino, D. L. Wickerham, W. M. Cronin, R. S. Cecchini, J. N. Atkins, T. B. Bevers, L. Fehrenbacher, E. R. Pajon and J. L. Wade, Update of the national surgical adjuvant breast and bowel project study of tamoxifen and raloxifene (STAR) P-2 trial: preventing breast cancer, Cancer Prev. Res., 2010, 3, 696–706 CrossRef CAS PubMed.
- P. M. O'Neill and S. A. Ward, A Quinoline Carboxamide Antimalarial Drug Candidate Uniquely Targets Plasmodia at Three Stages of the Parasite Life Cycle, Angew. Chem., Int. Ed., 2015, 54, 13504–13506 CrossRef PubMed.
- R. A. Odeh, R. A. Al-Qawasmeh, M. O. Sinnokrot, M. H. Semreen, M. H. Abu-Zarga, B. B. Huthail, H. Tarazi, I. A. Yousef and T. H. Al-Tel, Design, Synthesis and Qualitative Structure Activity Relationship Evaluations of Quinoline-Based Bisarylimidazoles as Antibacterial Motifs, Med. Chem., 2016, 12(6), 563–573 CrossRef.
- S. Cretton, S. Dorsaz, A. Azzollini, Q. Favre-Godal, A. Azzollini, L. Marcourt, J. L. Wolfender, M. Cuendet and P. Christen, Antifungal activity of quinoline alkaloids from Waltheria indica, J. Nat. Prod., 2016, 79, 300–307 CrossRef CAS PubMed.
- X. Q. Deng, M. X. Song, Y. Zheng and Z. S. Quan, Design, synthesis and evaluation of the antidepressant and anticonvulsant activities of triazole-containing quinolinones, Eur. J. Med. Chem., 2014, 73, 217–224 CrossRef CAS PubMed.
- S. K. Gupta and A. Mishra, Synthesis, characterization & screening for anti-inflammatory & analgesic activity of quinoline derivatives bearing azetidinones scaffolds, Antiinflamm. Antiallergy Agents Med. Chem., 2016, 15(1), 31–43 CrossRef CAS PubMed.
- H. Bharathkumar, C. D. Mohan, H. Ananda, J. E. Fuchs, F. Li, S. Rangappa, M. Surender, K. C. Bulusu, K. S. Girish, G. Sethi, A. Bender, Basappa and K. S. Rangappa, Microwave-assisted synthesis, characterization and cytotoxic studies of novel estrogen receptor α ligands towards human breast cancer cells, Bioorg. Med. Chem. Lett., 2015, 25, 1804–1807 CrossRef CAS PubMed.
- A. Zarghi, R. Ghodsi and E. Azizi, Design, Synthesis and Biological Evaluation of 4-(Imidazolylmethyl)-2-(4-Methylsulfonyl Phenyl)-Quinoline Derivatives as Selective COX-2 Inhibitors and In-vitro Anti-breast Cancer Agents, Iran. J. Pharm. Res., 2016, 15(1), 169–177 Search PubMed.
- L. N. Bheemanapalli, A. Kaur, R. Arora, R. R. Akkinepally and N. M. Javali, Synthesis, evaluation of 6,8-dibromo-2-aryl-2,3-dihydroquinolin-4 (1H)-ones in MCF-7 (breast cancer) cell lines and their docking studies, Med. Chem. Res., 2012, 21, 1741–1750 CrossRef CAS.
- E. Leung, J. M. Hung, D. Barker and J. Reynisson, The effect of a thieno[2,3-b]pyridine PLC-γ inhibitor on the proliferation, morphology, migration and cell cycle of breast cancer cells, MedChemComm, 2014, 5, 99–106 RSC.
- L. Yin, Q. Hu and R. W. Hartmann, Tetrahydropyrroloquinolinone type dual inhibitors of aromatase/aldosterone synthase as a novel strategy for breast cancer patients with elevated cardiovascular risks, J. Med. Chem., 2013, 56, 460–470 CrossRef CAS PubMed.
- Q. Hu, L. Yin and R. W. Hartmann, Selective dual inhibitors of CYP19 and CYP11B2: targeting cardiovascular diseases hiding in the shadow of breast cancer, J. Med. Chem., 2012, 55, 7080–7089 CrossRef CAS PubMed.
- N. Mohammadhosseini, M. Pordeli, M. Safavi, L. Firoozpour, F. Amin, S. K. Ardestani, N. Edraki, A. Shafiee and A. Foroumadi, Novel N-2-(Furyl)-2-(Chlorobenzyloxyimino) Ethyl Piperazinyl Quinolones: Synthesis, Cytotoxic Evaluation and Structure–Activity Relationship, Iran. J. Pharm. Res., 2015, 14, 1095–1103 CAS.
- V. G. Ugale and S. B. Bari, Quinazolines: New horizons in anticonvulsant therapy, Eur. J. Med. Chem., 2014, 80, 447–501 CrossRef CAS PubMed.
- V. Nenkep, K. Yun and B. W. Son, Oxysporizoline, an antibacterial polycyclic quinazoline alkaloid from the marine-mudflat-derived fungus Fusarium oxysporum, J. Antibiot., 2016, 69(9), 709–711 CrossRef CAS PubMed.
- M. Chen, P. Li, D. Hu, S. Zeng, T. Li, L. Jin, W. Xue and B. Song, Synthesis, antiviral activity, 3D-QSAR, and interaction mechanisms study of novel malonate derivatives containing quinazolin-4 (3H)-one moiety, Bioorg. Med. Chem. Lett., 2016, 26, 168–173 CrossRef CAS PubMed.
- L. M. Antypenko, S. I. Kovalenko, O. V. Karpenko, A. M. Katsev, V. P. Novikov and N. S. Fedyunina, 1-R-2-([1,2,4]Triazolo[1,5-c]quinazoline-2-ylthio)etanon(ol)s: Synthesis, Bioluminescence Inhibition, Molecular Docking Studies, Antibacterial and Antifungal Activities, Curr. Comput.-Aided Drug Des., 2016, 12(1), 29–41 CrossRef CAS PubMed.
- M. N. Noolvi and H. M. Patel, Synthesis, method optimization, anticancer activity of 2,3,7-trisubstituted quinazoline derivatives and targeting EGFR-tyrosine kinase by rational approach: 1st cancer update, Arabian J. Chem., 2013, 6, 35–48 CrossRef CAS.
- M. S. Mohamed, M. M. Kamel, E. M. Kassem, N. Abotaleb, M. Khedr and M. F. Ahmed, Synthesis, biological evaluation and molecular docking of quinazoline-4(1H)-one derivatives as anti-inflammatory and analgesic agents, Acta Pol. Pharm., 2011, 68, 665–675 CAS.
- M. F. Ahmed, A. Belal and M. Youns, Design, synthesis, molecular modeling and anti-breast cancer activity of novel quinazolin-4-one derivatives linked to thiazolidinone, oxadiazole or pyrazole moieties, Med. Chem. Res., 2015, 24, 2993–3007 CrossRef CAS.
- M. F. Ahmed and A. A. Hashim, Design, synthesis of novel quinazolin-4-one derivatives and biological evaluation against human MCF-7 breast cancer cell line, Res. Chem. Intermed., 2016, 42, 1777–1789 CrossRef CAS.
- P. Rizza, M. Pellegrino, A. Caruso, D. Iacopetta, M. S. Sinicropi, S. Rault, J. C. Lancelot, H. El-Kashef, A. Lesnard, C. Rochais, P. Dallemagne, C. Saturnino, F. Giordano, S. Catalano and S. Andò, 3-(Dipropylamino)-5-hydroxybenzofuro[2,3-f]quinazolin-1(2H)-one (DPA-HBFQ-1) plays an inhibitory role on breast cancer cell growth and progression, Eur. J. Med. Chem., 2016, 107, 275–287 CrossRef CAS PubMed.
- F. L. Faraj, M. Zahedifard, M. Paydar, C. Y. Looi, N. Abdul Majid, H. M. Ali, N. Ahmad, N. S. Gwaram and M. A. Abdulla, Synthesis, Characterization, and Anticancer Activity of New Quinazoline Derivatives against MCF-7 Cells, Sci. World J., 2014, 2014, 212096 Search PubMed.
- S. Yin, C. Tang, B. Wang, Y. Zhang, L. Zhou, L. Xue and C. Zhang, Design, synthesis and biological evaluation of novel EGFR/HER2 dual inhibitors bearing a oxazolo[4,5-g]quinazolin-2(1H)-one scaffold, Eur. J. Med. Chem., 2016, 120, 26–36 CrossRef CAS PubMed.
- H. Yao, M. Ji, Z. Zhu, J. Zhou, R. Cao, X. Chen and B. Xu, Discovery of 1-substituted benzyl-quinazoline-2,4(1H,3H)-dione derivatives as novel poly (ADP-ribose) polymerase-1 inhibitors, Bioorg. Med. Chem., 2015, 23, 681–693 CrossRef CAS PubMed.
- S. Y. Jang, S. Lee, M. Kim, Y. J. Ham, M. Y. Cha, K. H. Suh, Y. H. Kim, A. Kwon, H. J. Kang, K. H. Min and K.-O. Lee, Synthesis of Quinazoline Analogues with Differential Activity for HER2-overexpressing Breast Cancer Cells, Bull. Korean Chem. Soc., 2015, 36, 1933–1935 CrossRef CAS.
- K. V. Sashidhara, J. N. Rosaiah, M. Kumar, R. K. Gara, L. V. Nayak, K. Srivastava, H. K. Bid, R. Konwar and R. Konwar, Neo-tanshinlactone inspired synthesis, in vitro evaluation of novel substituted benzocoumarin derivatives as potent anti-breast cancer agents, Bioorg. Med. Chem. Lett., 2010, 20, 7127–7131 CrossRef CAS PubMed.
- Y. Chen, C. Sun, X. Wen and W. Zhang, Design, synthesis, insecticidal evaluation and molecular docking studies of cis-nitenpyram analogues bearing diglycine esters, Sci. China: Chem., 2013, 56, 159–168 CrossRef CAS.
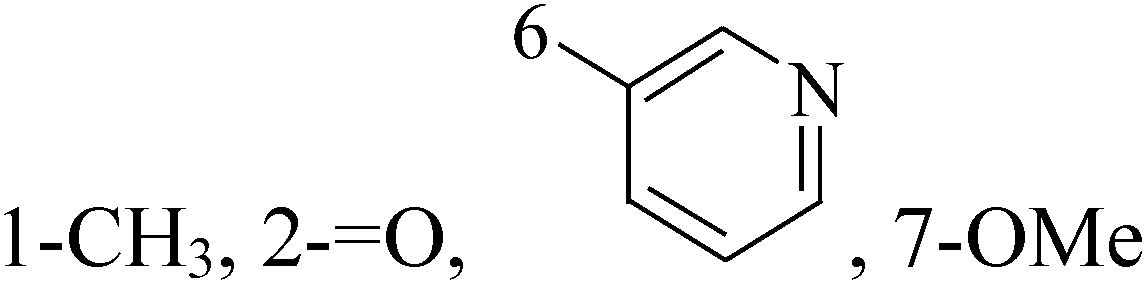
- S. F. Mohamed, H. M. Hosni, A. E. Amr and M. M. Abdalla, Synthesis of novel substituted pyridines from 1-(3-aminophenyl)-3-(1H-indol-3-yl)prop-2-en-1-one and their anticancer activity, Russ. J. Gen. Chem., 2016, 86, 672–680 CrossRef CAS.
- S. Naeem, S. Akhtar, N. Mushtaq, A. Kamil, I. Mahmood, S. Zafar, M. Arif and Z. S. Saify, Analgesic and anxiolytic evaluation of N′-substituted arylsulphonyl and benzoyl derivatives of 4-pyridine carbohydrazide, FUUAST J. Biol., 2014, 4, 123 Search PubMed.
- J. Li, S. Kovackova, S. Pu, J. Rozenski, S. De Jonghe, S. Einav and P. Herdewijn, Isothiazolo[4,3-b]pyridines as inhibitors of cyclin G associated kinase: synthesis, structure–activity relationship studies and antiviral activity, MedChemComm, 2015, 6, 1666–1672 RSC.
- S. Marhadour, P. Marchand, F. Pagniez, M. A. Bazin, C. Picot, O. Lozach, S. Ruchaud, M. Antoine, L. Meijer, N. Rachidi and P. Le Pape, Synthesis and biological evaluation of 2,3-diarylimidazo[1,2-a]pyridines as antileishmanial agents, Eur. J. Med. Chem., 2012, 58, 543–556 CrossRef CAS PubMed.
- N. N. El-Sayed, M. A. Abdelaziz, W. W. Wardakhan and R. M. Mohareb, The knoevenagel reaction of cyanoacetylhydrazine with pregnenolone: synthesis of thiophene, thieno[2,3-d]pyrimidine, 1,2,4-triazole, pyran and pyridine derivatives with anti-inflammatory and anti-ulcer activities, Steroids, 2016, 107, 98–111 CrossRef CAS PubMed.
- S. Naeem, S. Akhtar, N. Mushtaq, A. Kamil, S. Zafar, S. Anwar and M. Arif, Synthesis of novel derivatives of 4-pyridine carboxylic acid hydrazide and their activity on central nervous system, Pak. J. Pharm. Sci., 2014, 27, 1401–1408 CAS.
- C. Pang, C. Sun, J. Wang, D. Xiao, L. Ding and H. Bu, Novel 2H-pyrazolo[4,3-c]hexahydropyridine derivatives: synthesis, crystal structure, fluorescence properties and cytotoxicity evaluation against human breast cancer cells, Sci. China: Chem., 2013, 56, 702–715 CrossRef CAS.
- C. D. Mohan, V. Srinivasa, S. Rangappa, L. Mervin, S. Mohan, S. Paricharak, S. Baday, F. Li, M. K. Shanmugam, A. Chinnathambi, A. Chinnathambi, M. E. Zayed, S. A. Alharbi, A. Bender, G. Sethi, Basappa and K. S. Rangappa, Trisubstituted-Imidazoles Induce Apoptosis in Human Breast Cancer Cells by Targeting the Oncogenic PI3K/Akt/mTOR Signaling Pathway, PLoS One, 2016, 11, e0153155 Search PubMed.
- S. S. Prasad, K. S. Kumar, S. H. Jayaprakash, B. S. Krishna, C. S. Sundar, P. V. Rao, T. M. Babu, W. Rajendra and C. S. Reddy, Design, Synthesis, Antioxidant, and Anti-Breast Cancer Activities of Novel Diethyl (alkyl/aryl/heteroarylamino)(4-(pyridine-2-yl)phenyl)methylphosphonates, Arch. Pharm., 2013, 346, 380–391 CrossRef CAS PubMed.
- S. Radi, S. Tighadouini, O. Feron, O. Riant, M. Bouakka, R. Benabbes and Y. N. Mabkhot, Synthesis of novel keto enol derivatives tethered pyrazole, pyridine and furan as new potential antifungal and anti-breast cancer agents, Molecules, 2015, 20, 20186–20194 CrossRef CAS PubMed.
- R. Kaur, P. Kaur, S. Sharma, G. Singh, S. Mehndiratta, P. M. Bedi and K. Nepali, Anti-cancer pyrimidines in diverse scaffolds: a review of patent literature, Recent Pat. Anti-Cancer Drug Discovery, 2015, 10, 23–71 CrossRef CAS PubMed.
- Z. M. Nofal, H. H. Fahmy, E. S. Zarea and W. El-Eraky, Synthesis of new pyrimidine derivatives with evaluation of their anti-inflammatory and analgesic activities, Acta Pol. Pharm., 2011, 68, 507–517 CAS.
- S. A. Rahaman, Y. Rajendra Pasad, P. Kumar and B. Kumar, Synthesis and anti-histaminic activity of some novel pyrimidines, Saudi Pharm. J., 2009, 17, 255–258 CrossRef PubMed.
- A. K. El-Ansary, A. M. Kamal and M. A. H. Al-Ghorafi, Synthesis and evaluation of 4-anilinoquinazoline bioisosteres as potential anti-breast cancer agents, Eur. J. Med. Chem., 2014, 86, 202–210 CrossRef CAS PubMed.
- C. H. Zhang, M. W. Zheng, Y. P. Li, X. D. Lin, M. Huang, L. Zhong, G. B. Li, R. J. Zhang, W. T. Lin, Y. Jiao, R. Xiang, L.-J. Chen, Y.-L. Zhao, W. Cheng, Y.-Q. Wei and S.-Y. Yang, Design, synthesis, and structure–activity relationship studies of 3-(phenylethynyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine derivatives as a new class of Src inhibitors with potent activities in models of triple negative breast cancer, J. Med. Chem., 2015, 58, 3957–3974 CrossRef CAS PubMed.
- N. K. Yellapu, N. Atluri, K. Kandlapalli, R. B. Kilaru, J. R. Vangavaragu, H. Osuru, N. Chamarthi, P. Sarma and B. Matcha, Design, synthesis, in silico, and in vitro evaluation of novel pyrimidine phosphonates with cytotoxicity against breast cancer cells, Med. Chem. Res., 2014, 23, 317–328 CrossRef CAS.
- A. A. Patravale, A. H. Gore, D. R. Patil, G. B. Kolekar, M. B. Deshmukh and P. V. Anbhule, Trouble-free multicomponent method for combinatorial synthesis of 2-amino-4-phenyl-5-H-indeno[1,2-d]pyrimidine-5-one and their screening against cancer cell lines, Ind. Eng. Chem. Res., 2014, 53, 16568–16578 CrossRef CAS.
- H. Lin, M. J. Schulz, R. Xie, J. Zeng, J. I. Luengo, M. D. Squire, R. Tedesco, J. Qu, K. Erhard, J. F. Mack, K. Raha, R. Plant, C. M. Rominger, J. L. Ariazi, C. S. Sherk, M. D. Schaber, J. McSurdy-Freed, M. D. Spengler, C. B. Davis, M. A. Hardwicke and R. A. Rivero, Rational design, synthesis, and SAR of a novel thiazolopyrimidinone series of selective PI3K-beta inhibitors, ACS Med. Chem. Lett., 2012, 3, 524–529 CrossRef PubMed.
- R. M. Kumbhare, T. L. Dadmal, M. J. Ramaiah, K. S. Kishore, S. N. Pushpa Valli, S. K. Tiwari, K. Appalanaidu, Y. K. Rao and M. P. Bhadra, Synthesis and anticancer evaluation of novel triazole linked N-(pyrimidin-2-yl)benzo[d]thiazol-2-amine derivatives as inhibitors of cell survival proteins and inducers of apoptosis in MCF-7 breast cancer cells, Bioorg. Med. Chem. Lett., 2015, 25, 654–658 CrossRef CAS PubMed.
- M. R. Reddy, B. Akula, S. C. Cosenza, S. Athuluridivakar, M. R. Mallireddigari, V. R. Pallela, V. K. Billa, D. V. Subbaiah, E. V. Bharathi, R.-D. V. Carpio, A. Padgaonkar, S. J. Baker and E. P. Reddy, Discovery of 8-cyclopentyl-2-[4-(4-methyl-piperazin-1-yl)-phenylamino]-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidine-6-carbonitrile (7x) as a potent inhibitor of cyclin-dependent kinase 4 (CDK4) and AMPK-related kinase 5 (ARK5), J. Med. Chem., 2014, 57, 578–599 CrossRef CAS PubMed.
- C. R. Ross, K. W. Temburnikar, G. M. Wilson and K. L. Seley-Radtke, Mitotic arrest of breast cancer MDA-MB-231 cells by a halogenated thieno[3,2-d]pyrimidine, Bioorg. Med. Chem. Lett., 2015, 25, 1715–1717 CrossRef CAS PubMed.
- D. Schemeth, C. Kappacher, M. Rainer, R. Thalinger and G. K. Bonn, Comprehensive evaluation of imidazole-based polymers for the enrichment of selected non-steroidal anti-inflammatory drugs, Talanta, 2016, 153, 177–185 CrossRef CAS PubMed.
- I. I. Abu Hashim, N. F. Ghazy and M. H. El-Shabouri, Potential use of phospholipids in combination with hydrophilic carriers for enhancement of the dissolution and oral bioavailability of imidazole antifungal class II drugs, Pharmazie, 2015, 70, 706–715 CAS.
- R. R. Sonawane and C. Magdum, Synthesis, Anticonvulsant Screening of Some Novel 1,5-Disubstituted-4-Chloro-1H-Imidazole Derivatives, Asian J. Biomed. Pharm. Sci., 2015, 5, 1 Search PubMed.
- I. K. Kostakis, N. Pouli, P. Marakos, O. C. Kousidou, A. Roussidis, G. N. Tzanakakis and N. K. Karamanos, Design, synthesis and cell growth inhibitory activity of a series of novel aminosubstituted xantheno[1,2-d]imidazoles in breast cancer cells, Bioorg. Med. Chem., 2008, 16, 3445–3455 CrossRef CAS PubMed.
- A. Lai, M. Kahraman, S. Govek, J. Nagasawa, C. Bonnefous, J. Julien, K. Douglas, J. Sensintaffar, N. Lu, K. J. Lee, A. Aparicio, J. Kaufman, J. Qian, G. Shao, R. Prudente, M. J. Moon, J. D. Joseph, B. Darimont, D. Brigham, K. Grillot, R. Heyman, P. J. Rix, J. H. Hager and N. D. Smith, Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts, J. Med. Chem., 2015, 58, 4888–4904 CrossRef CAS PubMed.
- C. Karthikeyan, V. R. Solomon, H. Lee and P. Trivedi, Synthesis and biological evaluation of 2-(phenyl)-3H-benzo[d]imidazole-5-carboxylic acids and its methyl esters as potent anti-breast cancer agents, Arabian J. Chem., 2013, 10, S1788–S1794 CrossRef.
- A. S. Abdul Rahim, S. Muhamad Salhimi, N. Arumugam, L. Chung Pin, N. Shy Yee, N. N. Muttiah, W. B. Keat, S. A. Hamid, H. Osman and I. B. Mat, Microwave-assisted synthesis of sec/tert-butyl 2-arylbenzimidazoles and their unexpected antiproliferative activity towards ER negative breast cancer cells, J. Enzyme Inhib. Med. Chem., 2013, 28, 1255–1260 CrossRef CAS PubMed.
- A. T. Taher, N. A. Khalil and E. M. Ahmed, Synthesis of novel isatin-thiazoline and isatin-benzimidazole conjugates as anti-breast cancer agents, Arch. Pharmacal Res., 2011, 34, 1615–1621 CrossRef CAS PubMed.
- J. H. Toney, P. M. Fitzgerald, N. Grover-Sharma, S. H. Olson, W. J. May, J. G. Sundelof, D. E. Vanderwall, K. A. Cleary, S. K. Grant, J. K. Wu, J. W. Kozarich, D. L. Pompliano and G. G. Hammond, Antibiotic sensitization using biphenyl tetrazoles as potent inhibitors of bacteroides fragilis metallo-beta-lactamase, Chem. Biol., 1998, 5, 185–196 CrossRef CAS PubMed.
- M. Alam, M. J. Alam, S. A. Nami, D. U. Lee, M. Azam and S. Ahmad, Computational and anti-tumor studies of 7a-aza-B-homostigmast-5-eno[7a,7-d]tetrazole-3β-yl chloride, J. Mol. Struct., 2016, 1108, 411–426 CrossRef CAS.
- S. R. K. Reddy, S. M. Surya, M. Shaik and P. R. Kanuparthy, Copper complexes of pyridyl–tetrazole ligands with pendant amide and hydrazide arms: synthesis, characterization, DNA-binding and antioxidant properties, Transition Met. Chem., 2016, 41, 517 CrossRef.
- S. Gajanan Khanage, A. Raju, P. Baban Mohite and R. Bhanudas Pandhare, Analgesic activity of some 1,2,4-triazole heterocycles clubbed with pyrazole, tetrazole, isoxazole and pyrimidine, Adv. Pharm. Bull., 2013, 3, 13–18 Search PubMed.
- M. Maria Dorathi Anu, M. Jayanthi, S. Damodar Kumar, S. Raja and S. V. Thirunavukkarasu, Synthesis, characterization, antibacterial & anti-inflammatory effects of substituted tetrazole derivatives based on different types of carbazone and benzaldehyde, Int. J. ChemTech Res., 2013, 5, 1982–1990 CAS.
- M. Arshad, A. R. Bhat, S. Pokharel, J. E. Kim, E. J. Lee, F. Athar and I. Choi, Synthesis, characterization and anticancer screening of some novel piperonyl–tetrazole derivatives, Eur. J. Med. Chem., 2014, 71, 229–236 CrossRef CAS PubMed.
- S. C. Köhler and M. Wiese, HM30181 derivatives as novel potent and selective inhibitors of the breast cancer resistance protein (BCRP/ABCG2), J. Med. Chem., 2015, 58, 3910–3921 CrossRef PubMed.
- P. Ahuja and N. Siddiqui, Anticonvulsant evaluation of clubbed indole-1,2,4-triazine derivatives: a synthetic approach, Eur. J. Med. Chem., 2014, 80, 509–522 CrossRef CAS PubMed.
- M. Z. Zhang, N. Mulholland, D. Beattie, D. Irwin, Y. C. Gu, Q. Chen, G.-F. Yang and J. Clough, Synthesis and antifungal activity of 3-(1,3,4-oxadiazol-5-yl)-indoles and 3-(1,3,4-oxadiazol-5-yl)methyl-indoles, Eur. J. Med. Chem., 2013, 63, 22–32 CrossRef CAS PubMed.
- M. Z. Zhang, Q. Chen and G. F. Yang, A review on recent developments of indole-containing antiviral agents, Eur. J. Med. Chem., 2015, 89, 421–441 CrossRef CAS PubMed.
- N. P. Singh, U. P. Singh, M. Rouse, J. Zhang, S. Chatterjee, P. S. Nagarkatti and M. Nagarkatti, Dietary Indoles Suppress Delayed-Type Hypersensitivity by Inducing a Switch from Proinflammatory Th17 Cells to Anti-Inflammatory Regulatory T Cells through Regulation of MicroRNA, J. Immunol., 2016, 196, 1108–1122 CrossRef CAS PubMed.
- S. R. Pedada, N. S. Yarla, P. J. Tambade, B. L. Dhananjaya, A. Bishayee, K. M. Arunasree, G. H. Philip, G. Dharmapuri, G. Aliev, S. Putta and G. Rangaiah, Synthesis of new secretory phospholipase A2-inhibitory indole containing isoxazole derivatives as anti-inflammatory and anticancer agents, Eur. J. Med. Chem., 2016, 112, 289–297 CrossRef CAS PubMed.
- J. Singh Sidhu, R. Singla and V. Jaitak, Indole Derivatives as Anticancer Agents for Breast Cancer Therapy: A Review, Anti-Cancer Agents Med. Chem., 2016, 16, 160–173 CrossRef.
- G. Jabor Gozzi, Z. Bouaziz, E. Winter, N. Daflon-Yunes, D. Aichele, A. Nacereddine, C. Marminon, G. Valdameri, W. Zeinyeh, A. Bollacke, J. Guillon, A. Lacoudre, N. Pinaud, S. M. Cadena, J. Jose, M. Le Borgne and A. Di Pietro, Converting potent indeno[1,2-b]indole inhibitors of protein kinase CK2 into selective inhibitors of the breast cancer resistance protein ABCG2, J. Med. Chem., 2014, 58, 265–277 CrossRef PubMed.
- M. S. Mady, M. M. Mohyeldin, H. Y. Ebrahim, H. E. Elsayed, W. E. Houssen, E. G. Haggag, R. F. Soliman and K. A. El Sayed, The indole alkaloid meleagrin, from the olive tree endophytic fungus Penicillium chrysogenum, as a novel lead for the control of c-Met-dependent breast cancer proliferation, migration and invasion, Bioorg. Med. Chem., 2016, 24, 113–122 CrossRef CAS PubMed.
- Y. Li, J. Woo, J. Chmielecki, C. Q. Xia, M. Liao, B. C. Chuang, J. J. Yang, M. Y. Guan, M. Plesescu and S. R. Prakash, Synthesis of a new inhibitor of breast cancer resistance protein with significantly improved pharmacokinetic profiles, Bioorg. Med. Chem. Lett., 2016, 26, 551–555 CrossRef CAS PubMed.
- J. D. Brattlie, Progress towards the design and synthesis of novel anti-tumor neo-tanshinlactone analogues, The University of North Carolina at Chapel Hill, 2014 Search PubMed.
- J. Ma, G. Bao, L. Wang, W. Li, B. Xu, B. Du, J. Lv, X. Zhai and P. Gong, Design, synthesis, biological evaluation and preliminary mechanism study of novel benzothiazole derivatives bearing indole-based moiety as potent antitumor agents, Eur. J. Med. Chem., 2015, 96, 173–186 CrossRef CAS PubMed.
- X. Li, S. L. Zheng, X. Li, J. L. Li, O. Qiang, R. Liu and L. He, Synthesis and anti-breast cancer activity of new indolylquinone derivatives, Eur. J. Med. Chem., 2012, 54, 42–48 CrossRef CAS PubMed.
- S. Chakraborty, S. Ghosh, B. Banerjee, A. Santra, A. Adhikary, A. K. Misra and P. C. Sen, Phemindole, a synthetic di-indole derivative maneuvers the store operated calcium entry (SOCE) to induce potent anti-carcinogenic activity in human triple negative breast cancer cells, Front. Pharmacol., 2016, 7, 114 Search PubMed.
- B. Debnath and S. Ganguly, Synthesis, biological evaluation, in silico docking, and virtual ADME studies of 2-[2-oxo-3-(arylimino)indolin-1-yl]-N-arylacetamides as potent anti-breast cancer agents, Monatsh. Chem., 2016, 147, 1–10 CrossRef.
- C. Karthikeyan, V. R. Solomon, H. Lee and P. Trivedi, Design, synthesis and biological evaluation of some isatin-linked chalcones as novel anti-breast cancer agents: a molecular hybridization approach, Biomedicine & Preventive Nutrition, 2013, 3, 325–330 Search PubMed.
- M. A. Bhat, A. Al-Dhfyan, A. A. Khan, N. Al-Harbi, P. S. Manogaran, A. M. Alanazi, H.-K. Fun and M. A. Al-Omar, Targeting HER-2 over expressed breast cancer cells with 2-cyclohexyl-N-[(Z)-(substituted phenyl/furan-2-yl/thiophene-2-yl)methylidene]hydrazine-carbothioamide, Bioorg. Med. Chem. Lett., 2015, 25, 83–87 CrossRef CAS PubMed.
- C. Mudjupa, S. Abdelhamed, A. Refaat, S. Yokoyama, I. Saiki and O. Vajragupta, Lead compound bearing caffeic scaffold induces EGFR suppression in solid tumor cancer cells, J. Appl. Biomed., 2015, 13, 305–317 CrossRef.
- D. J. Weldon, M. D. Saulsbury, J. Goh, L. Rowland, P. Campbell, L. Robinson, C. Miller, J. Christian, L. Amis, N. Taylor, C. Dill, W. Davis Jr, S. L. Evans and E. Brantley, One-pot synthesis of cinnamylideneacetophenones and their in vitro cytotoxicity in breast cancer cells, Bioorg. Med. Chem. Lett., 2014, 24, 3381–3384 CrossRef CAS PubMed.
- M. Sala, A. Chimento, C. Saturnino, I. M. Gomez-Monterrey, S. Musella, C. Milite, M. S. Sinicropi, A. Caruso, R. Sirianni, P. Tortorella, E. Novellino, P. Campiglia and V. Pezzi, Synthesis and cytotoxic activity evaluation of 2,3-thiazolidin-4-one derivatives on human breast cancer cell lines, Bioorg. Med. Chem. Lett., 2013, 23, 4990–4995 CrossRef CAS PubMed.
- S. Parihar, A. Kumar, A. K. Chaturvedi, N. K. Sachan, S. Luqman, B. Changkija, M. Manohar, O. Prakash, D. Chanda, F. Khan, C. S. Chanotiya, K. Shanker, A. Dwivedi, R. Konwar and A. S. Negi, Synthesis of combretastatin A4 analogues on steroidal framework and their anti-breast cancer activity, J. Steroid Biochem. Mol. Biol., 2013, 137, 332–344 CrossRef CAS PubMed.
- G. Kaur, M. P. Mahajan, M. K. Pandey, P. Singh, S. R. Ramisetti and A. K. Sharma, Design, synthesis and evaluation of ospemifene analogs as anti-breast cancer agents, Eur. J. Med. Chem., 2014, 86, 211–218 CrossRef CAS PubMed.
- S. Rahmani-Nezhad and M. Safavi, Synthesis, in vitro cytotoxicity and apoptosis inducing study of 2-aryl-3-nitro-2H-chromene derivatives as potent anti-breast cancer agents, Eur. J. Med. Chem., 2014, 86, 562–569 CrossRef CAS PubMed.
- C. L. Chen, T. C. Chen, C. C. Lee, L. C. Shih, C. Y. Lin, Y. Y. Hsieh, A. A. A. Ali and H. S. Huang, Synthesis and evaluation of new 3-substituted-4-chloro-thioxanthone derivatives as potent anti-breast cancer agents, Arabian J. Chem., 2015, 1–14 Search PubMed.
- C. L. Varela, C. Amaral, E. T. da Silva, A. Lopes, G. Correia-da-Silva, R. A. Carvalho, S. C. Costa, F. M. Roleira and N. Teixeira, Exemestane metabolites: synthesis, stereochemical elucidation, biochemical activity and anti-proliferative effects in a hormone-dependent breast cancer cell line, Eur. J. Med. Chem., 2014, 87, 336–345 CrossRef CAS PubMed.
- A. Jha, Y. Yadav, A. B. Naidu, V. K. Rao, A. Kumar, V. S. Parmar, W. J. MacDonald, C. K. Too, J. Balzarini, C. J. Barden and T. S. Cameron, Design, synthesis and bioevaluation of novel 6-(4-hydroxypiperidino)naphthalen-2-ol-based potential selective estrogen receptor modulators for breast cancer, Eur. J. Med. Chem., 2015, 92, 103–114 CrossRef CAS PubMed.
- M. I. Ansari, M. K. Hussain, A. Arun, B. Chakravarti, R. Konwar and K. Hajela, Synthesis of targeted dibenzo[b,f]thiepines and dibenzo[b,f]oxepines as potential lead molecules with promising anti-breast cancer activity, Eur. J. Med. Chem., 2015, 99, 113–124 CrossRef CAS PubMed.
- S. Meneni, I. Ott, C. D. Sergeant, A. Sniady, R. Gust and R. Dembinski, 5-Alkynyl-2′-deoxyuridines: chromatography-free synthesis and cytotoxicity evaluation against human breast cancer cells, Bioorg. Med. Chem., 2007, 15, 3082–3088 CrossRef CAS PubMed.
- A. R. Nikolić, E. T. Petri, O. R. Klisurić, A. S. Ćelić, D. S. Jakimov, E. A. Djurendić, K. M. Penov Gaši and M. N. Sakač, Synthesis and anticancer cell potential of steroidal 16,17-seco-16,17a-dinitriles: identification of a selective inhibitor of hormone-independent breast cancer cells, Bioorg. Med. Chem., 2015, 23, 703–711 CrossRef PubMed.
- H. R. E. Dyari, T. Rawling, K. Bourget and M. Murray, Synthetic ω-3 Epoxyfatty Acids as Antiproliferative and Pro-apoptotic Agents in Human Breast Cancer Cells, J. Med. Chem., 2014, 57, 7459–7464 CrossRef CAS PubMed.
- S. Wang and T. Sasaki, Synthesis of artemisinin dimers using the Ugi reaction and their in vitro efficacy on breast cancer cells, Bioorg. Med. Chem. Lett., 2013, 23, 4424–4427 CrossRef CAS PubMed.
- T. Mani, F. Wang, W. E. Knabe, A. L. Sinn, M. Khanna, I. Jo, G. E. Sandusky, G. W. J. Sledge, D. R. Jones, R. Khanna, K. E. Pollok and S. O. Meroueh, Small-molecule inhibition of the uPAR·uPA interaction: synthesis, biochemical, cellular, in vivo pharmacokinetics and efficacy studies in breast cancer metastasis, Bioorg. Med. Chem., 2013, 21, 2145–2155 CrossRef CAS PubMed.
- A. Siddiqui, P. Dandawate, R. Rub, S. Padhye, S. Aphale, A. Moghe, A. Jagyasi, K. Venkateswara Swamy, B. Singh, A. Chatterjee, A. Ronghe and H. K. Bhat, Novel aza-resveratrol analogs: synthesis, characterization and anticancer activity against breast cancer cell lines, Bioorg. Med. Chem. Lett., 2013, 23, 635–640 CrossRef CAS PubMed.
- P. Saha, S. Fortin, V. Leblanc, S. Parent, É. Asselin and G. Bérubé, Design, synthesis, cytocidal activity and estrogen receptor α affinity of doxorubicin conjugates at 16α-position of estrogen for site-specific treatment of estrogen receptor positive breast cancer, Steroids, 2012, 77, 1113–1122 CrossRef CAS PubMed.
- S. Salamone, C. Colin, I. Grillier-Vuissoz, S. Kuntz, S. Mazerbourg, S. Flament, H. Martin, L. Richert, Y. Chapleur and M. Boisbrun, Synthesis of new troglitazone derivatives: anti-proliferative activity in breast cancer cell lines and preliminary toxicological study, Eur. J. Med. Chem., 2012, 51, 206–215 CrossRef CAS PubMed.
- A. C. Serra, A. M. A. R. Gonsalves, M. Laranjo, A. M. Abrantes, A. C. Gonçalves, A. B. Sarmento-Ribeiro and M. F. Botelho, Synthesis of new 2-galactosylthiazolidine-4-carboxylic acid amides. Antitumor evaluation against melanoma and breast cancer cells, Eur. J. Med. Chem., 2012, 53, 398–402 CrossRef CAS PubMed.
- E. Tyrrell, R. Archer, M. Tucknott, K. Colston, G. Pirianov, D. Ramanthan, R. Dhillon, A. Sinclair and G. A. Skinner, The synthesis and anticancer effects of a range of natural and unnatural hop β-acids on breast cancer cells, Phytochem. Lett., 2012, 5, 144–149 CrossRef CAS.
- T. Pathan, S. Ingale, A. Sharma, R. Mohan and C. S. Ramaa, Synthesis and preliminary evaluation of difluorinated 1,3-propanediones as potential agents in the treatment of breast cancer, Med. Chem. Res., 2012, 21, 584–589 CrossRef CAS.
- C. Descôteaux, K. Brasseur, V. Leblanc, S. Parent, E. Asselin and G. Bérubé, SAR study of tyrosine-chlorambucil hybrid regioisomers; synthesis and biological evaluation against breast cancer cell lines, Amino Acids, 2012, 43, 923–935 CrossRef PubMed.
- P. Dandawate, E. Khan, S. Padhye, H. Gaba, S. Sinha, J. Deshpande, K. Venkateswara Swamy, M. Khetmalas, A. Ahmad and F. H. Sarkar, Synthesis, characterization, molecular docking and cytotoxic activity of novel plumbagin hydrazones against breast cancer cells, Bioorg. Med. Chem. Lett., 2012, 22, 3104–3108 CrossRef CAS PubMed.
- G. Kaur, M. P. Mahajan, M. K. Pandey, P. Singh, S. R. Ramisetti and A. K. Sharma, Design, synthesis, and anti-breast cancer evaluation of new triarylethylene analogs bearing short alkyl- and polar amino-amido-ethyl chains, Bioorg. Med. Chem. Lett., 2016, 26, 1963–1969 CrossRef CAS PubMed.
- Y. Z. Dong, Q. Shi, H. C. Pai, C. Y. Peng, S. L. Pan, C. M. Teng, N. Nakagawa-Goto, D. L. Yu, Y. N. Liu, P. C. Wu, K. F. Bastow, S. L. Morris-Natschke, A. Brossi, J. Y. Lang, J. Hsu, M. C. Hung, E. Y. H. P. Lee and K. H. Lee, Antitumor Agents. 272. Structure–Activity Relationships and In Vivo Selective Anti-Breast Cancer Activity of Novel Neotanshinlactone Analogs, J. Med. Chem., 2010, 53, 2299–2308 CrossRef CAS PubMed.
- M. Vosooghi, L. Firoozpour, A. Rodaki, M. Pordeli, M. Safavi, S. K. Ardestani, A. Dadgar, A. Asadipour, M. H. Moshafi and A. Foroumadi, Design, synthesis, docking study and cytotoxic activity evaluation of some novel letrozole analogs, Daru, J. Pharm. Sci., 2014, 22, 83 CrossRef PubMed.
- M. Valli, W. Altei, R. N. Santos, E. C. Lucca, M. A. Dessoy, R. M. Pioli, F. Cotinguiba, X. Cachet, S. Michel, M. Furlan, L. C. Dias, A. D. Andricopulob and V. S. Bolzani, Synthetic Analogue of the Natural Product Piperlongumine as a Potent Inhibitor of Breast Cancer Cell Line Migration, J. Braz. Chem. Soc., 2017, 28, 475–484 CrossRef CAS.
- O. Argyros, N. Lougiakis, E. Kouvari, A. Papafotika, C. P. Raptopoulou, V. Psycharis, S. Christoforidis, N. Pouli, P. Marakos and C. Tamvakopoulos, Design and synthesis of novel 7-aminosubstituted pyrido[2,3-b]pyrazines exhibiting anti-breast cancer activity, Eur. J. Med. Chem., 2017, 126, 954–968 CrossRef CAS PubMed.
- V. Thakor, M. Poddar, S. Dey, S. N. Manjula, S. V. Madhunapantula, R. Pawara, H. M. Patel and M. N. Noolvi, Exploring the anti-breast cancer potential of flavonoid analogs, RSC Adv., 2016, 6, 79166–79179 RSC.
- W. Wang, E. R. Rayburn, S. E. Velu, D. Chen, D. H. Nadkarni, S. Murugesan, D. Chen and R. Zhang, A novel synthetic iminoquinone, BA-TPQ, as an anti-breast cancer agent: in vitro and in vivo activity and mechanisms of action, Breast Cancer Res. Treat., 2010, 123, 321–331 CrossRef CAS PubMed.
- A. Wyrezbska, K. Gach, U. Lewandowska, K. Szewczyk, E. Hrabec, J. Modranka, R. Jakubowski, T. Janecki, J. Szymański and A. Janecka, Anticancer Activity of New Synthetic α-Methylene-δ-Lactones on Two Breast Cancer Cell Lines, Basic Clin. Pharmacol. Toxicol., 2013, 113, 391–400 CrossRef PubMed.
- Z. Tang, C. Wu, T. Wang, K. Lao, Y. Wang, L. Liu, M. Muyaba, P. Xu, C. He, G. Luo, Z. Qian, S. Niu, L. Wang, Y. Wang, H. Xiao, Q. You and H. Xiang, Design, synthesis and evaluation of 6-aryl-indenoisoquinolone derivatives dual targeting ERalpha and VEGFR-2 as anti-breast cancer agents, Eur. J. Med. Chem., 2016, 118, 328–339 CrossRef CAS PubMed.
- M. M. Ghorab, M. G. El-Gazzar and M. S. Alsaid, Synthesis and anti-breast cancer evaluation of novel N-(guanidinyl)benzenesulfonamides, Int. J. Mol. Sci., 2014, 15, 5582–5595 CrossRef CAS PubMed.
- L. Liu, A. A. Zhang, R. J. Zhao, F. Li, T. J. Meng, N. Ishida, M. Murakami and W. X. Zhao, Asymmetric Synthesis of Planar Chiral Ferrocenes by Enantioselective Intramolecular C–H Arylation of N-(2-Haloaryl) Ferrocenecarboxamides, Org. Lett., 2014, 16, 5336–5338 CrossRef CAS PubMed.
- A. Esparza-Ruiz, C. Herrmann, J. Chen, B. O. Patrick, E. Polishchuk and C. Orvig, Synthesis and in vitro anticancer activity of ferrocenyl-aminoquinoline-carboxamide conjugates, Inorg. Chim. Acta, 2012, 393, 276–283 CrossRef CAS.
- J. Skiba, K. Kowalski, A. Prochnicka, I. Ott, J. Solecka, A. Rajnisz and B. Therrien, Metallocene-uracil conjugates: synthesis and biological evaluation of novel mono-, di- and tri-nuclear systems, J. Organomet. Chem., 2015, 782, 52–61 CrossRef CAS.
- F. Dubar, C. Slomianny, J. Khalife, D. Dive, H. Kalamou, Y. Guérardel, P. Grellier and C. Biot, The ferroquine antimalarial conundrum: redox activation and reinvasion inhibition, Angew. Chem., Int. Ed., 2013, 52, 7690–7693 CrossRef CAS PubMed.
- K. Kowalski, A. Koceva-Chyła, Ł. Szczupak, P. Hikisz, J. Bernasińska, A. Rajnisz, J. Solecka and B. Therrien, Ferrocenylvinyl-flavones: synthesis, structure, anticancer and antibacterial activity studies, J. Organomet. Chem., 2013, 741, 153–161 CrossRef.
- R. Rubbiani, O. Blacque and G. Gasser, Sedaxicenes: potential new antifungal ferrocene-based agents?, Dalton Trans., 2016, 45, 6619–6626 RSC.
- C. Lu, X. Wang, Y. Yang and X. Liu, Ferrocenyl compounds derived from the reaction of phenylamines with ferrocenecarbonyl chloride: synthesis, characterisation and their biological activity, Inorg. Chim. Acta, 2016, 447, 121–126 CrossRef CAS.
- Y. L. Tan, P. Pigeon, S. Top, E. Labbé, O. Buriez, E. A. Hillard, A. Vessières, C. Amatore, W. K. Leong and G. Jaouen, Ferrocenyl catechols: synthesis, oxidation chemistry and anti-proliferative effects on MDA-MB-231 breast cancer cells, Dalton Trans., 2012, 41, 7537–7549 RSC.
- L. M. Gao, R. Hernández, J. Matta and E. Meléndez, Synthesis, structure, electrochemistry, and cytotoxic properties of ferrocenyl ester derivatives, Met.-Based Drugs, 2009, 2009, 420784 Search PubMed.
- T. Dallagi, M. Saidi, A. Vessières, M. Huché, G. Jaouen and S. Top, Synthesis and antiproliferative evaluation of ferrocenyl and cymantrenyl triaryl butene on breast cancer cells. Biodistribution study of the corresponding technetium-99m tamoxifen conjugate, J. Organomet. Chem., 2013, 734, 69–77 CrossRef CAS.
- Y. Zheng, C. Wang, C. Li, J. Qiao, F. Zhang, M. Huang, W. Ren, C. Dong, J. Huang and H. B. Zhou, Discovery of novel SERMs with a ferrocenyl entity based on the oxabicyclo[2.2.1]heptene scaffold and evaluation of their antiproliferative effects in breast cancer cells, Org. Biomol. Chem., 2012, 10, 9689–9699 CAS.
- N. Wambang, N. Schifano-Faux, A. Aillerie, B. Baldeyrou, C. Jacquet, C. Bal-Mahieu, T. Bousquet, S. Pellegrini, P. T. Ndifon, S. Meignan, J. F. Goossens, A. Lansiaux and L. Pélinski, Synthesis and biological activity of ferrocenyl indeno[1,2-c]isoquinolines as topoisomerase II inhibitors, Bioorg. Med. Chem., 2016, 15, 651–660 CrossRef PubMed.
- J. de Jesús Cázares-Marinero, S. Top and G. Jaouen, Synthesis and characterization of new ferrocenyl compounds with different alkyl chain lengths and functional groups to target breast cancer cells, J. Organomet. Chem., 2014, 751, 610–619 CrossRef.
- J. B. Heilmann, E. A. Hillard, M. A. Plamont, P. Pigeon, M. Bolte, G. Jaouen and A. Vessières, Ferrocenyl compounds possessing protected phenol and thiophenol groups: synthesis, X-ray structure, and in vitro biological effects against breast cancer, J. Organomet. Chem., 2008, 693, 1716–1722 CrossRef CAS.
- P. Pigeon, S. Top, O. Zekri, E. A. Hillard, A. Vessières, M.-A. Plamont, O. Burieet, E. Labbé, M. Huché, S. Boutamine, C. Amatoreb and G. Jaouen, The replacement of a phenol group by an aniline or acetanilide group enhances the cytotoxicity of 2-ferrocenyl-1,1-diphenyl-but-l-ene compounds against breast cancer cells, J. Organomet. Chem., 2009, 694, 895–901 CrossRef CAS.
- F. Petrelli, A. Coinu, K. Borgonovo, M. Cabiddu, M. Ghilardi, V. Lonati and S. Barni, The value of platinum agents as neoadjuvant chemotherapy in triple-negative breast cancers: a systematic review and meta-analysis, Breast Cancer Res. Treat., 2014, 144, 223–232 CrossRef CAS PubMed.
- L. Kater, J. Claffey, M. Hogan, P. Jesse, B. Kater, S. Strauss, M. Tacke and A. Prokop, The role of the intrinsic FAS pathway in titanocene Y apoptosis: the mechanism of overcoming multiple drug resistance in malignant leukemia cells, Toxicol. In Vitro, 2012, 26, 119–124 CrossRef CAS PubMed.
- M. Hanif, M. V. Babak and C. G. Hartinger, Development of anticancer agents: wizardry with osmium, Drug Discovery Today, 2014, 19, 1640–1648 CrossRef CAS PubMed.
- M. M. Harding and G. Mokdsi, Antitumour metallocenes: structure–activity studies and interactions with biomolecules, Curr. Med. Chem., 2000, 7, 1289–1303 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, Organometallic anticancer compounds, J. Med. Chem., 2010, 54, 3–25 CrossRef PubMed.
- N. Muhammad and Z. Guo, Metal-based anticancer chemotherapeutic agents, Curr. Opin. Chem. Biol., 2014, 19, 144–153 CrossRef CAS PubMed.
- K. M. Buettner and A. M. Valentine, Bioinorganic chemistry of titanium, Chem. Rev., 2011, 112, 1863–1881 CrossRef PubMed.
- E. Sirignano, C. Saturnino, A. Botta, M. S. Sinicropi, A. Caruso, A. Pisano, R. Lappano, M. Maggiolini and P. Longo, Synthesis, characterization and cytotoxic activity on breast cancer cells of new half-titanocene derivatives, Bioorg. Med. Chem. Lett., 2013, 23, 3458–3462 CrossRef CAS PubMed.
- I. de la Cueva-Alique, L. Muñoz-Moreno, Y. Benabdelouahab, B. T. Elie, M. A. El Amrani, M. E. Mosquera, M. Contel, A. M. Bajo, T. Cuenca and E. Royo, Novel enantiopure cyclopentadienyl Ti(IV) oximato compounds as potential anticancer agents, J. Inorg. Biochem., 2016, 156, 22–34 CrossRef CAS PubMed.
- C. Saturnino, E. Sirignano, A. Botta, M. S. Sinicropi, A. Caruso, A. Pisano, R. Lappano, M. Maggiolini and P. Longo, New titanocene derivatives with high antiproliferative activity against breast cancer cells, Bioorg. Med. Chem. Lett., 2014, 24, 136–140 CrossRef CAS PubMed.
- A. Chimento, C. Saturnino, D. Iacopetta, R. Mazzotta, A. Caruso, M. R. Plutino, A. Mariconda, A. Ramunno, M. S. Sinicropi, V. Pezzi and P. Longo, Inhibition of human topoisomerase I and II and anti-proliferative effects on MCF-7 cells by new titanocene complexes, Bioorg. Med. Chem., 2015, 23, 7302–7312 CrossRef CAS PubMed.
- K. Yin Zhang, T. K. Ka-Shun, M. W. Louie, H.-W. Liu and K. K.-W. Lo, A Phosphorescent Rhenium(I) Tricarbonyl Polypyridine Complex Appended with a Fructose Pendant that Exhibits Photocytotoxicity and Enhanced Uptake by Breast Cancer Cells, Organometallics, 2013, 32, 5098–5102 CrossRef.
- D. L. Stojković, V. V. Jevtić, G. P. Radić, D. S. Đačić, M. G. Ćurčić, S. D. Marković, V. M. Ðinović, V. P. Petrović and S. R. Trifunović, Stereospecific ligands and their complexes. Part XII. Synthesis, characterization and in vitro antiproliferative activity of platinum(IV) complexes with some O,O′-dialkyl esters of (S,S)-ethylenediamine-N,N′-di-2-propanoic acid against colon cancer (HCT-116) and breast cancer (MDA-MB-231) cell lines, J. Mol. Struct., 2014, 1062, 21–28 CrossRef.
- J. G. Varela, A. De Chatterjee, P. Guevara, V. Ramirez, A. J. Metta-Magaña, D. Villagrán, A. Varela-Ramirez, S. Das and J. E. Nuñez, Synthesis, characterization, and evaluation of cis-diphenyl pyridineamine platinum(II) complexes as potential anti-breast cancer agents, JBIC, J. Biol. Inorg. Chem., 2014, 19, 967–979 CrossRef PubMed.
- N. Alonso, O. Caamano, F. J. Romero-Duran, F. Luan, M. N. D. S. Cordeiro, M. Yanez, H. Gonzalez-Diaz and X. Garcia-Mera, Model for high-throughput screening of multitarget drugs in chemical neurosciences: synthesis, assay, and theoretic study of rasagiline carbamates, ACS Chem. Neurosci., 2013, 4, 1393–1403 CrossRef CAS PubMed.
- F. J. Romero-Duran, N. Alonso, M. Yanez, O. Caamano, X. Garcia-Mera and H. Gonzalez-Diaz, Brain-inspired cheminformatics of drug-target brain interactome, synthesis, and assay of TVP1022 derivatives, Neuropharmacology, 2016, 103, 270–278 CrossRef CAS PubMed.
- F. J. Romero Duran, N. Alonso, O. Caamano, X. Garcia-Mera, M. Yanez, F. J. Prado-Prado and H. Gonzalez-Diaz, Prediction of multitarget networks of neuroprotective compounds with entropy indices and synthesis, assay, and theoretical study of new asymmetric 1,2-rasagiline carbamates, Int. J. Mol. Sci., 2014, 15, 17035–17064 CrossRef PubMed.
- A. Speck-Planche, V. V. Kleandrova, F. Luan and M. N. D. S. Cordeiro, Unified multi-target approach for the rational in silico design of anti-bladder cancer agents, Anti-Cancer Agents Med. Chem., 2013, 13, 791–800 CrossRef CAS PubMed.
- E. Tenorio-Borroto, F. R. Ramirez, A. Speck-Planche, M. N. Cordeiro, F. Luan and H. Gonzalez-Diaz, QSPR and flow cytometry analysis (QSPR-FCA): review and new findings on parallel study of multiple interactions of chemical compounds with immune cellular and molecular targets, Curr. Drug Metab., 2014, 15, 414–428 CrossRef CAS PubMed.
- F. Luan, M. N. D. S. Cordeiro, N. Alonso, X. Garcia-Mera, O. Caamano, F. J. Romero-Duran, M. Yanez and H. Gonzalez-Diaz, TOPSMODE model of multiplexing neuroprotective effects of drugs and experimental-theoretic study of new 1,3-rasagiline derivatives potentially useful in neurodegenerative diseases, Bioorg. Med. Chem., 2013, 21, 1870–1879 CrossRef CAS PubMed.
- A. Speck-Planche, V. V. Kleandrova and M. N. D. S. Cordeiro, New insights toward the discovery of antibacterial agents: multitasking QSBER model for the simultaneous prediction of antituberculosis activity and toxicological profiles of drugs, Eur. J. Pharm. Sci., 2013a, 48, 812–818 Search PubMed.
- E. Tenorio-Borroto, C. G. Penuelas-Rivas, J. C. Vasquez-Chagoyan, N. Castanedo, F. J. Prado-Prado, X. Garcia-Mera and H. Gonzalez-Diaz, Model for high-throughput screening of drug immunotoxicity – study of the anti-microbial G1 over peritoneal macrophages using flow cytometry, Eur. J. Med. Chem., 2014, 72, 206–220 CrossRef CAS PubMed.
- E. Tenorio-Borroto, C. G. Penuelas Rivas, J. C. Vasquez Chagoyan, N. Castanedo, F. J. Prado-Prado, X. Garcia-Mera and H. Gonzalez-Diaz, ANN multiplexing model of drugs effect on macrophages; theoretical and flow cytometry study on the cytotoxicity of the anti-microbial drug G1 in spleen, Bioorg. Med. Chem., 2012, 20, 6181–6194 CrossRef CAS PubMed.
- V. V. Kleandrova, F. Luan, A. Speck-Planche and M. N. Cordeiro, In silico assessment of the acute toxicity of chemicals: recent advances and new model for multitasking prediction of toxic effect, Mini-Rev. Med. Chem., 2015, 15, 677–686 CrossRef CAS PubMed.
- A. Speck-Planche and M. N. D. S. Cordeiro, Simultaneous modeling of antimycobacterial activities and ADMET profiles: a chemoinformatic approach to medicinal chemistry, Curr. Top. Med. Chem., 2013, 13, 1656–1665 CrossRef CAS PubMed.
- A. Speck-Planche and M. N. D. S. Cordeiro, Chemoinformatics for medicinal chemistry: in silico model to enable the discovery of potent and safer anti-cocci agents, Future Med. Chem., 2014a, 6, 2013–2028 Search PubMed.
- A. Speck-Planche and M. N. D. S. Cordeiro, Simultaneous virtual prediction of anti-Escherichia coli activities and ADMET profiles: a chemoinformatic complementary approach for high-throughput screening, ACS Comb. Sci., 2014b, 16, 78–84 Search PubMed.
- A. Speck-Planche and M. N. D. S. Cordeiro, De novo computational design of compounds virtually displaying potent antibacterial activity and desirable in vitro ADMET profiles, Med. Chem. Res., 2017, 26, 2345–2356 CrossRef CAS.
- F. Luan, M. N. Cordeiro, N. Alonso, X. García-Mera, O. Caamaño, F. J. Romero-Duran, M. Yañez and H. González-Díaz, TOPS-MODE model of multiplexing neuroprotective effects of drugs and experimental-theoretic study of new 1,3-rasagiline derivatives potentially useful in neurodegenerative diseases, Bioorg. Med. Chem., 2013, 21, 1870–1879 CrossRef CAS PubMed.
- H. Gonzalez-Diaz, S. Arrasate, A. G. Juan, N. Sotomayor, E. Lete, A. Speck-Planche, J. M. Ruso, F. Luan and M. N. Cordeiro, Matrix trace operators: from spectral moments of molecular graphs and complex networks to perturbations in synthetic reactions, micelle nanoparticles, and drug ADME processes, Curr. Drug Metab., 2014, 15, 470–488 CrossRef CAS PubMed.
- H. Zheng, J. Hou, M. D. Zimmerman, A. Wlodawer and W. Minor, The future of crystallography in drug discovery, Expert Opin. Drug Discovery, 2014, 9, 125e137 Search PubMed.
- P. Ravula, H. B. Vamaraju, M. Paturi and J. N. G. N. Sharath Chandra, Design, synthesis, in silico and antiproliferative evaluation of novel pyrazole derivatives as VEGFR-2 inhibitors, Arch. Pharm., 2017, 351 DOI:10.1002/ardp.201700234.
- H. Y. Qiu, J. Y. Fu, M. K. Yang, H. W. Han, P. F. Wang, Y. H. Zhang, H. Y. Lin, C. Y. Tang, J. L. Qi, R. W. Yang, X. M. Wang, H. L. Zhu and Y. H. Yang, Identification of new shikonin derivatives as STAT3 inhibitors, Biochem. Pharmacol., 2017, 146, 74–86 CrossRef CAS PubMed.
- W. Li, Q. Sun, L. Song, C. Gao, F. Liu, Y. Chen and Y. Jiang, Discovery of 1-(3-aryl-4-chlorophenyl)-3-(p-aryl)urea derivatives against breast cancer by inhibiting PI3K/Akt/mTOR and hedgehog signalings, Eur. J. Med. Chem., 2017, 141, 721–733 CrossRef CAS PubMed.
- Y. Zhang, Y. Chen, D. Zhang, L. Wang, T. Lu and Y. Jiao, Discovery of Novel Potent VEGFR-2 Inhibitors Exerting Significant Antiproliferative Activity against Cancer Cell Lines, J. Med. Chem., 2017, 61, 140–157 CrossRef PubMed.
- M. Daśko, M. Przybyłowska, J. Rachon, M. Masłyk, K. Kubiński, M. Misiak, A. Składanowski and S. Demkowicz, Synthesis and biological evaluation of fluorinated N-benzoyl and N-phenylacetoyl derivatives of 3-(4-aminophenyl)-coumarin-7-O-sulfamate as steroid sulfatase inhibitors, Eur. J. Med. Chem., 2017, 128, 79–87 CrossRef PubMed.
- Y. Sugimoto, D. B. Sawant, H. A. Fisk, L. Mao, C. Li, S. Chettiar, P. K. Li, M. V. Darby and R. W. Brueggemeier, Novel pyrrolopyrimidines as Mps1/TTK kinase inhibitors for breast cancer, Bioorg. Med. Chem., 2017, 25, 2156–2166 CrossRef CAS PubMed.
- T. Pragna Lakshmi, A. Kumar, V. Vijaykumar, S. Natarajan and R. Krishna, Identification of natural allosteric inhibitor for Akt1 protein through computational approaches and in vitro evaluation, Int. J. Biol. Macromol., 2017, 96, 200–213 CrossRef CAS PubMed.
- N. S. El-Gohary and M. I. Shaaban, Synthesis and biological evaluation of a new series of benzimidazole derivatives as antimicrobial, antiquorum-sensing and antitumor agents, Eur. J. Med. Chem., 2017, 131, 255–262 CrossRef CAS PubMed.
- H. Kang, X. Xiao, C. Huang, Y. Yuan, D. Tang, X. Dai and X. Zeng, Potent aromatase inhibitors and molecular mechanism of inhibitory action, Eur. J. Med. Chem., 2018, 143, 426–437 CrossRef CAS PubMed.
- A. H. Basta, H. El-Saied, M. M. El-Deftar, A. A. El-Henawy, H. H. El-Sheikh, E. H. Abdel-Shakour and M. S. Hasanin, Properties of modified carboxymethyl cellulose and its use as bioactive compound, Carbohydr. Polym., 2016, 153, 641–651 CrossRef CAS PubMed.
- N. Hayashi, M. Iwase, T. Ochi, A. Seki, N. Matsuda and H. Yamauchi, The Role of Preoperative Chemotherapy Depending on Breast Cancer Subtype, Gan to Kagaku Ryoho, 2016, 43, 1149–1156 Search PubMed.
- L. Q. Rogers, K. S. Courneya, P. M. Anton, P. Hopkins-Price, S. Verhulst, S. K. Vicari, R. S. Robbs, R. Mocharnuk and E. McAuley, Effects of the BEAT cancer physical activity behavior change intervention on physical activity, aerobic fitness, and quality of life in breast cancer survivors: a multicenter randomized controlled trial, Breast Cancer Res. Treat., 2015, 149, 109–119 CrossRef PubMed.
- A. K. Swisher, J. Abraham, D. Bonner, D. Gilleland, G. Hobbs, S. Kurian, M. A. Yanosik and L. Vona-Davis, Exercise and dietary advice intervention for survivors of triple-negative breast cancer: effects on body fat, physical function, quality of life, and adipokine profile, Supportive Care Cancer, 2015, 23, 2995–3003 CrossRef PubMed.
- B. S. Abdulkarim, J. Cuartero, J. Hanson, J. Deschênes, D. Lesniak and S. Sabri, Increased risk of locoregional recurrence for women with T1-2N0 triple-negative breast cancer treated with modified radical mastectomy without adjuvant radiation therapy compared with breast-conserving therapy, J. Clin. Oncol., 2011, 29, 2852–2858 CrossRef PubMed.
- M. Untch, G. E. Konecny, S. Paepke and G. von Minckwitz, Current and future role of neoadjuvant therapy for breast cancer, Breast, 2014, 23, 526–537 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |