
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon coating and Al-doping to improve the electrochemistry of Li2CoSiO4 polymorphs as cathode materials for lithium-ion batteries†
Hongwei Duab,
Xianhui Zhang *a,
Zhenlian Chena,
Dongyang Wuac,
Zhifeng Zhanga and
Jun Li*a
*a,
Zhenlian Chena,
Dongyang Wuac,
Zhifeng Zhanga and
Jun Li*a
aNano Science and Technology Institute, University of Science and Technology of China, Suzhou 215123, China. E-mail: zhangxianhui@nimte.ac.cn; lijun@nimte.ac.cn; Fax: +86-0574-86685043; Tel: +86-0574-86688074
bNingbo Institute of Material Technology & Engineering, Chinese Academy of Sciences, Ningbo 315201, China
cShanghai University, Shanghai 200444, China
First published on 21st June 2018
Abstract
Li2CoSiO4 has the potential for use as a high safety, high energy-density cathode material for lithium-ion batteries but suffers from bad electrochemical performance. Herein, we demonstrate a profound study on the effects of carbon coating and Al-doping on the electrochemistry of Li2CoSiO4 synthesized by a two-step method. The synthesized 4 at% Al-doped Li2CoSiO4/C allows two lithium removals between 2.5 and 4.6 V, showing a first charge and discharge capacity of 331 and 140 mA h g−1, respectively, and a high capacity retention in cycling with no voltage degradation. The relationship between the improved performance and the supporting structural characteristics was studied by galvanostatic charge/discharge measurements and electrochemical impedance spectroscopy, coupled with material characterizations. This work demonstrates that electrical conductivity plays a central role in controlling the electrochemical performance of the modified Li2CoSiO4. Both the reversibility of delithiation and the irreversible capacity loss are strongly dependent on the electrical condition of the particles, which can be modified by Al-doping and carbon coating. The characteristics of carbon layers are analyzed because of their importance in improving the electrical properties and achieving a solution to the challenges with Li2CoSiO4. We that show Li2CoSiO4 could have unique electrochemical characteristics that satisfy all the requirements of high safety, high energy density, and high compatibility with the current organic electrolytes if appropriately modified.
1. Introduction
The safety of the cathode is a primary concern for the massive application of high energy-density lithium-ion batteries to electric vehicles and smart grids. Most advanced cathode materials, such as layered LiCoO2 oxide, solid solution LiNixMnyCozO2 (NMC) oxides and Li2MnO3-based lithium-rich composites, need to be charged to voltages above 4.4 V in order to obtain a high capacity. Unfortunately, all of these simple metal oxides are unsafe in the deep charged states. A recent work showed that all the compositions in the family of NMC released the lattice oxygen at elevated voltages.1 This oxidizes the electrolyte and is the onset of gas generation, which is one of the critical issues limiting the safety performance and lifetime of lithium-ion batteries. The physical root of lattice oxygen release in simple metal oxides is attributable to the weak oxygen bonding with their ionic framework.The polyoxyanion olivine LiFePO4 is a successful cathode with high safety, which is largely due to its covalent P–O bonding in the tetrahedral PO4 building blocks. Calculations have shown that the P–O bonding is five times stronger than O–O bonding in LiFePO4.2 This should be the key factor in tempering the oxygen bond breaking from the framework, contributing to the high safety of LiFePO4 in the deep charged state. The success of olivine LiMnPO4, operating at 4.1 V (vs. Li+/Li), confirms that the covalent P–O bonding is the mechanism for high safety, regardless of the operation voltage for the olivine family. These rationales make silicates, Li2TSiO4, (T = Fe, Mn, Co), especially Li2CoSiO4, very attractive as highly safe and high-capacity cathode materials. Firstly, the Si–O bond in SiO4 is similar to the P–O bond in PO4; both form strong covalent bonds in tetrahedral building blocks. The average Si–O bond length is 1.63 Å, slightly longer than that of the P–O bond, indicating the strong binding of oxygen in the tetrahedral SiO4 unit. Secondly, the chemical formulas of silicates indicate two lithium extractions, almost doubling the theoretical capacity of the olivine family to above 300 mA h g−1 (e.g., Li2CoSiO4: 325 mA h g−1). Interestingly, our calculations showed that the average Si–O bond length shrinks slightly from 1.657 Å to 1.648 Å when the first lithium is extracted.3 This indicates that strong oxygen bonding may be retained in the charged state of polymorphs. Indeed, the family of silicates has received significant attention as an entirely new class of high-safety and high-capacity lithium intercalation compounds since they were described in 2005.4 Another characteristic that has motivated us to study Li2CoSiO4 is its 4 V redox potential, which is much higher compared to LiFePO4, Li2FeSiO4 and Li2MnSiO4 (all at around 3.0 V).5–7 This makes Li2CoSiO4 (LCSO) very unique, not only as a high safety and high energy-density cathode material but also highly compatible with the full operation window of the 4 V organic electrolytes. However, a clear demonstration of these promising electrochemical characteristics was absent for Li2CoSiO4 in the literature.1–5
Tetrahedrally coordinated cations (XO4) (X = Li, Co, Si) of Li2CoSiO4 form many confusing insulating polymorphs.11,13 Early studies indicated that the βII polymorph, without carbon coating, delivered the first charge and discharge capacity at only 180 and 30 mA h g−1, respectively.11 Several works showed that the discharge capacity decayed quickly to negligible in just a few cycles.11,12 Devaraju and Honma et al. reported the supercritical synthesis of βII Li2CoSiO4 with an initial charge and discharge capacity of 200 and 107 mA h g−1, respectively,3 which was the highest reversible capacity reported in literature from 2007 to 2017.8–12 However, their voltage platform was significantly reduced in the second cycle and degraded to below 3.0 V from the third cycle; such voltage degradation due to inductive polymorph changes was often observed in Fe and Mn silicates.6–8 Several strategies for carbon coating, including multi-walled carbon nanotubes in 2017,8 have been attempted for the improvement of the electrical properties but with marginal effects on the electrochemistry of Li2CoSiO4 in literature.9 Those disappointing results are sharply different from Li2FeSiO4 and Li2MnSiO4, which have demonstrated two complete lithium removals and high reversibility for carbon coated nanostructured particles.9,10
Recently, our group found that 10 at% P substituted Si in Li2CoSiO4/C demonstrates a first charge and discharge capacity of 270 and 144 mA h g−1, respectively, between 2.5–4.6 V.18 It is very interesting that either P-doping or carbon coating cannot achieve improved electrochemistry on their own. Co metal impurities, reduced by the pyrolysis of sucrose, were found to interact in the initial charging process. The new progress has indicated a complicated interplay between carbon coatings and element-doping, which has not yet been studied. While it is well known that P and Al are two representative dopants, donor and acceptor, respectively, for modifying the electrical conductivity of semiconductors, there is no work or theory to predict their new effects in mediating polymorph synthesis, interplaying with carbon coating, and modifying the electrochemical performance of tetrahedral silicates. Al is also an interesting dopant used to improve the cyclability and thermal stability of the layered LiNi0.8Co0.15Al0.05O2 oxide.11,12,20 This work extends to Al-doping to explore new doping effects and serve as a necessary prelude to more complicated solid solutions in the tetrahedral structures, i.e., Li2MSiO4, (M = mixture of multiple elements of redox agent or structural stabilizer). Understanding the underlying working or failure mechanism could lay down the foundation for and open a new avenue to the further improvement of electrochemistry for this and other similar insulating cathode materials.
This work is a profound study on the effects of carbon coating and Al-doping on the electrochemistry of Li2CoSiO4 by systematically varying the Al doping contents. Electrochemical impedance spectroscopic studies have been performed, coupled with other characterizations to provide insight into the improved electrochemistry and to identify potential factors leading to the irreversible loss and cycling decay of capacity. Al-doped Li2CoSiO4/C presents clear and attractive electrochemistry due to combined modifications of bulk and surface properties, which allow two lithium removals between 2.5 and 4.6 V, showing the first charge and discharge capacity of 331 and 140 mA h g−1, respectively, and a high capacity retention in cycling with no voltage degradation. These new improvements confirm the electrochemical characteristics of Li2CoSiO4 as a high-energy cathode material with high redox potential and stable polymorphs in cycling.
2. Experimental section
2.1 Two-step synthesis of carbon-coated Li2CoSiO4 nanoparticles
Fig. 1 outlines the synthesis process. The first step involves the synthesis of the pure βII polymorph by a hydrothermal reaction with and without Al-doping. Nanosized SiO2-Cabosil M5 (Cabot Co., USA) and LiOH·H2O (Aldrich, 98%) were mixed in 50 mL deionized water at the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 and processed in an ultrasonic bath. CoCl2·6H2O (Aldrich, 99%) and Al2(SO4)3 (Aldrich, 99%) at the designed doping concentration (x) were added to 25 mL of ethylene glycol and stirred. The two solutions were then mixed with stirring and transferred to a 100 mL Teflon-lined autoclave. The remaining volume was topped up with deionized water. The sealed autoclave was heated at 150 °C for 72 h. The product was filtered and dried at 120 °C for 12 h in the vacuum oven. This procedure produces only pure βII polymorph nanoparticles as reported in our recent work.18
:4 and processed in an ultrasonic bath. CoCl2·6H2O (Aldrich, 99%) and Al2(SO4)3 (Aldrich, 99%) at the designed doping concentration (x) were added to 25 mL of ethylene glycol and stirred. The two solutions were then mixed with stirring and transferred to a 100 mL Teflon-lined autoclave. The remaining volume was topped up with deionized water. The sealed autoclave was heated at 150 °C for 72 h. The product was filtered and dried at 120 °C for 12 h in the vacuum oven. This procedure produces only pure βII polymorph nanoparticles as reported in our recent work.18
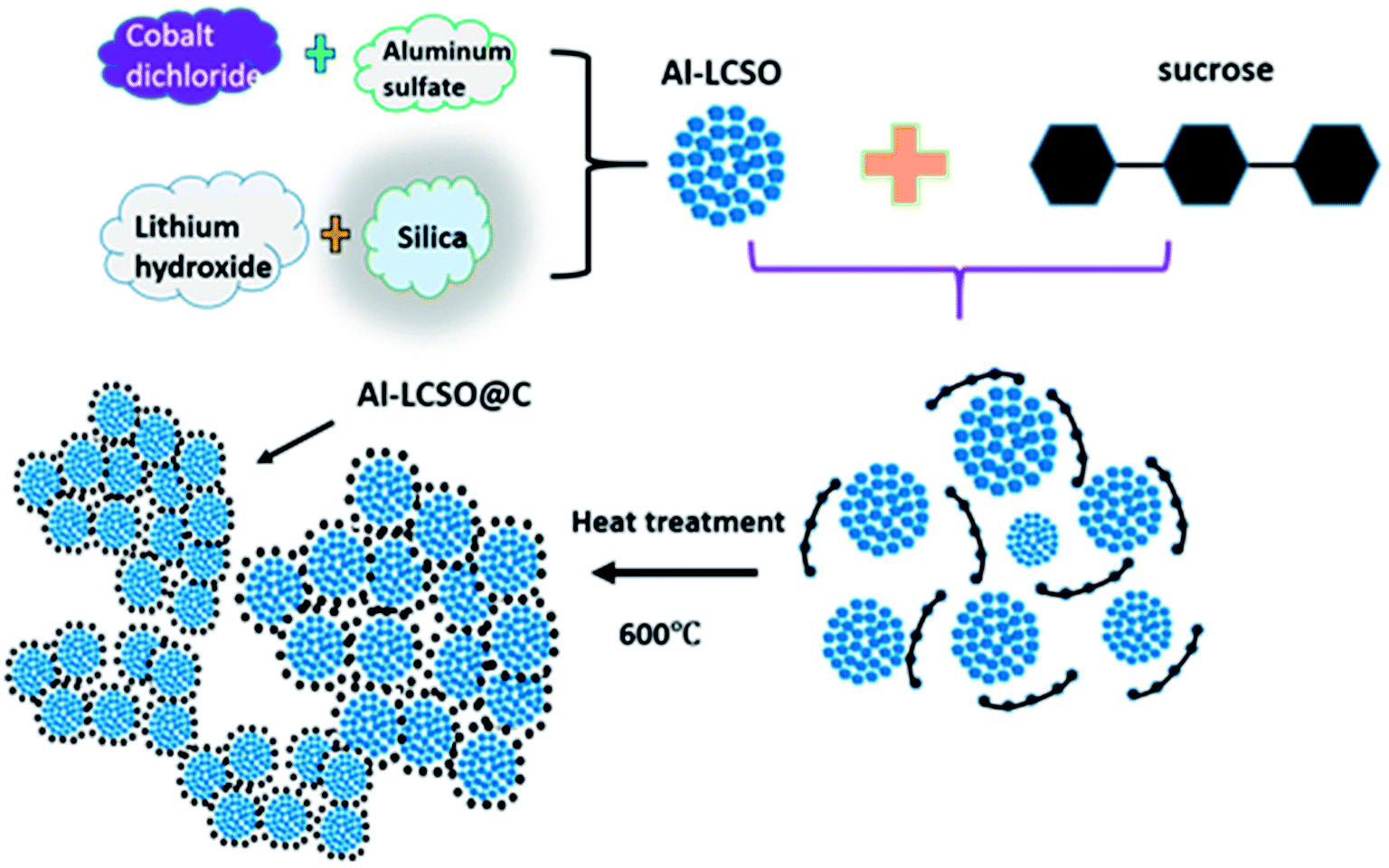 | ||
| Fig. 1 The two-step synthesis of carbon-coated Al–Li2CoSiO4 nanoparticles. | ||
The second step is the pyrolysis of sucrose on βII polymorph nanoparticles. The samples of the as-prepared xAl-Li2CoSiO4 from the first step were mixed with sucrose in the molar ratio of 7:1 (the content of carbon was about 10 wt%). The mixture was ball milled for 12 h and calcined at 600 °C for 1 h in the Ar atmosphere. In this work, the pristine sample, the carbon-coated sample and Al-doped with carbon coated Li2CoSiO4 samples were labelled LCSO, C-LCSO and xAl-LCSO/C, respectively, where x = 0.02, 0.04, 0.05, 0.08, 0.10, 0.12 and 0.15 (interchangeable as 2, 4, 5, 8, 10, 12 and 15 at% in this text).
2.2 Characterization
Crystal structures of the prepared samples were evaluated by X-ray diffraction (XRD, XRD-6100, Shimadzu) using Cu-Kα radiation (λ = 1.5418 Å). Diffraction data were collected over the range of 2θ between 10° and 80°. The morphology of materials was studied by scanning electron microscope (SEM, FEI, QUANTA 250 FEG) coupled with an energy dispersive X-ray detector (EDX). Carbon coating in all samples was characterized by Raman spectrometry (InVia-reflex, Renishaw). The morphology of the coated carbon was observed with a FEI Tecnai G2 F20 transmission electron microscope (TEM). The contents of carbon and Co impurities were determined by thermogravimetric analysis (TGA, SDT Q600). All the measurements were conducted under the same conditions to obtain reproducible results.To better track the bulk phase evolution of the carbon-coated polymorphs a notation was introduced to describe the different polymorphs. The βII polymorph has often been assigned to the space group Pmn21 in literature. However, Rietveld refinement of XRD, neutron powder diffraction, and Li MAS NMR have pointed out the existence of cationic disorder between CoO4 and LiO4 in the βII polymorph,13 which is inconsistent with the symmetric operations on their Wyckoff sites. Our modelling work has identified the key CoO4–LiO4 tetrahedral connection in the βII polymorph, which is associated with an easily recognizable XRD pattern, a double peak (labelled as DP in this work) between 20° and 25° (for radiation source: Cu).21 This characteristic can easily distinguish the βII polymorph from the βI polymorph (space group Pbn21); the latter shows a triple peak (labelled as TP in this work) in the same 2θ region (c.f. Fig. 2). Therefore, this work uses DP and TP phases as interchangeable terms for βII and βI polymorphs, respectively. This helps to identify and track the characteristics of βII and βI polymorphs to show the effects of Al-doping on the cationic ordering in the particles.
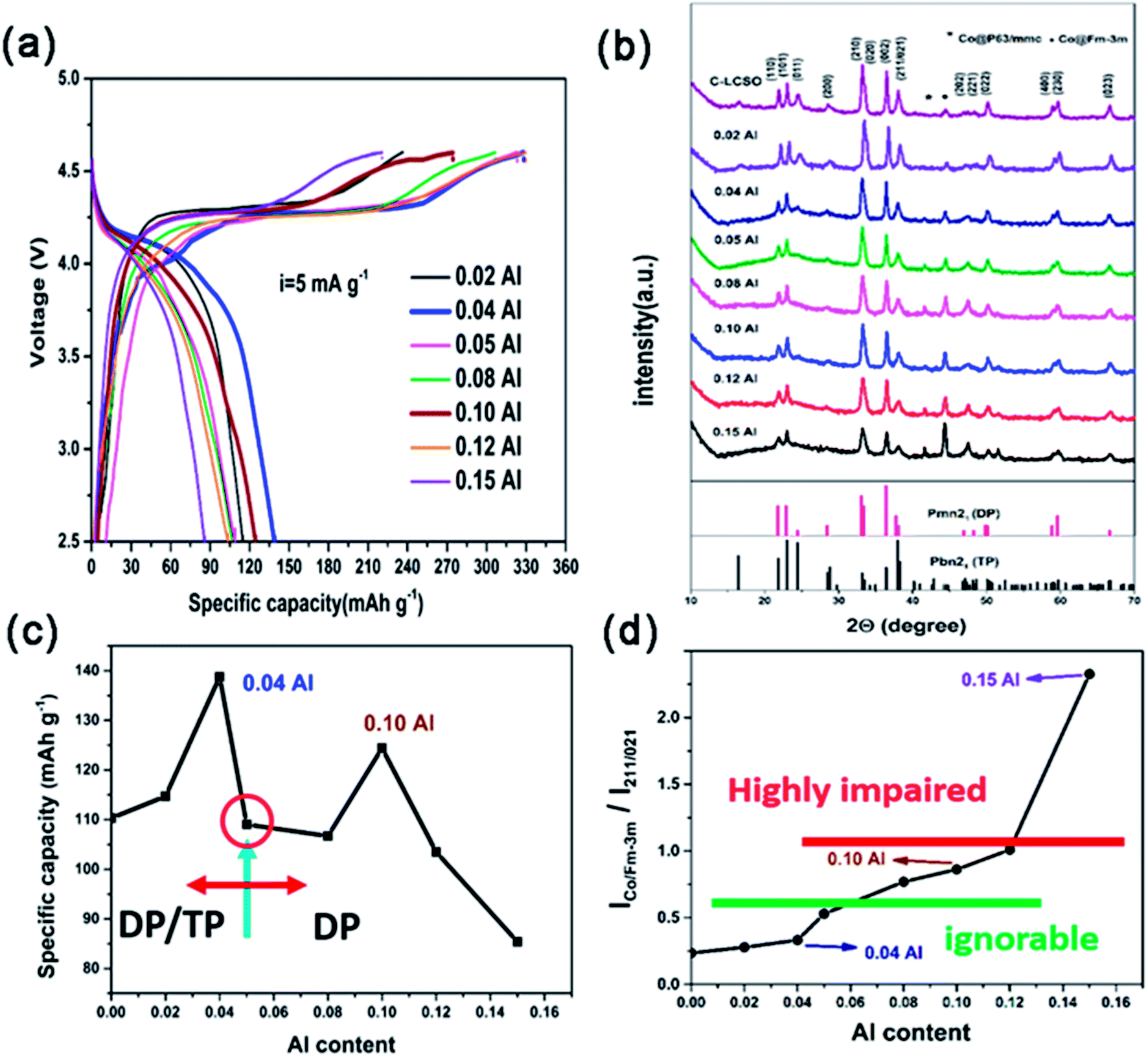 | ||
| Fig. 2 (a) The first charge/discharge curve comparison among xAl–LCSO/C samples; (b) XRD patterns of C–LCSO and xAl–LCSO/C samples, peaks labeled by “*” and “●” indicate two different reduced Co metal impurities; (c) the reversible capacities evolution with Al content; (d) the evolution of the ICo/Fm-3m/I211/021 ratio with respect to the doped-Al content of LCSO. | ||
2.3 Electrochemical measurement
The working electrodes were fabricated by mixing the active material, super P carbon and polyvinylidene fluoride (PVDF) in an 80:10:10 (w/w/w) ratio, using N-methyl-pyrrolidone (NMP) as the solvent. CR2032 coin cells were assembled with the prepared electrode as the cathode, lithium foil as the anode, and a few drops of electrolyte (1 M LiPF6 dissolved in EC/DMC with the volume ratio of 1:1) in an Ar-filled M-Braun glove box. The electrochemical performance of the cells was tested on a Land 2001A battery tester (Wuhan, China) in the voltage range of 2.5–4.6 V (vs. Li+/Li) at the current density of 5 mA g−1 at 25 °C. The electrochemical impedance spectroscopy (EIS) studies of the coin cells were conducted over the frequency range of 0.1 MHz to 0.1 Hz using SI 1470 (Solartron Analytical, Cambridge, UK).
3. Results and discussion
3.1 Electrochemical performance of Al-doping samples
Galvanostatic charge/discharge measurements were carried out over the potential window of 2.5–4.6 V (vs. Li+/Li) at the current density of 5 mA g−1. The first charge/discharge curves of all the Al-doped samples are presented in Fig. 2a; the xAl-LCSO/C samples with x = 0.02, 0.04, 0.05, 0.08, 0.10, 0.12 and 0.15 delivered initial discharge capacities of 114.8, 140, 109, 106.7, 124.5, 103.5 and 85.4 mA h g−1, respectively. Without carbon coating, the initial reversible capacity of LCSO was only 40 mA h g−1. The carbon coating C-LCSO, without Al-doping, delivered a reversible capacity of 110 mA h g−1. The reversibility of delithiation in Al-doped samples, xAl-LCSO/C, presented a non-linear dependence on Al doping contents, which is related to the cationic ordering in the doped samples.Fig. 2b displays the XRD patterns of all the carbon-coated samples. The polymorph retained the dominating DP phase, i.e. βII polymorph, even at pyrolysis temperatures higher than 400 °C. This differs significantly from the polymorph transitions reported by Bruce et al.4,13 and Nazar et al.,2 which showed that the coated βII polymorph transitioned to other silicates, and the Co ion was reduced at a sintering temperature as low as 400 °C. The polymorph mediating effect can be clearly observed in the systematic evolution of XRD characteristics with Al-doping content. With no Al doping, the C-LCSO presented mixed polymorphs of DP and TP, confirming the easy transition from DP to TP; the latter is only 2 meV lower in total energy.14 Note that the third peak (011), which should be a high peak of the pure TP phase (i.e., βI polymorph), diminishes consistently with the increase in Al content.
From the identification of DP and TP signatures, two kinds of particles evolved along with Al-doping contents. For low doping contents, x ≤ 0.05, samples are a mixture of βII and βI polymorphs and have better reversibility than C-LCSO samples, shown in Fig. 2c. In this region, the reversible capacity peaks occurred at x = 0.04 with 140 mA h g−1, which is only slightly lower than our recent 10 at% P-doping with 144 mA h g−1.18 Calculations have indicated that P-doping will smooth the Peierls distortion in the delithiated phase of doped models, therefore promoting the electrochemical performance.18 Here the 0.04 Al-doped sample shows a mixing phase of βII and βI polymorphs, which may have an interface between phases to release Peierls distortion, and can also achieve a similar performance to the purer βII polymorphs of the 10 at% P-doping sample. This finding may provide a new design strategy, analogous to Li2MnO3–LiNixMnyCozO2 composites, for lithium cobalt silicates.
No signal was shown for x > 0.05 of the βI polymorph, indicating only purer βII polymorphs for high Al-doping contents. However, their performances, except 10 at% Al, deteriorated with the increase of Al-doping contents. It is a surprise that 10 at% Al has smaller discharge capacity at 124.5 mA h g−1 than both the 0.04 Al doping and 10 at% P-doping samples. This demonstrates that Al-doping affects not only the thermal stability of DP and TP phases in pyrolysis but also the reactivity of a cationic network of doped polymorphs. Our calculation has shown that there is a small difference in the edge states and gap opening between DP and TP phases during lithium extraction.3 These states may have different reactivity with respect to Co reduction by coated carbon layers, especially in different doping schemes. Experimentally, there is a correlation between Co impurity and DP concentration of polymorphs. The larger the proportion of DP presented in Al-doped samples, the higher the intensity of the Co impurity signal shown in the XRD of Fig. 2b. The suppression of the Co-impurity coincides with the TP ordering in polymorphs. This agrees with the observation that the DP phase is much more inclined to be reduced than the TP phase.2,4 Since Al is not a redox agent, its concentration is not directly convertible to reversible capacity, but is indirectly associated with increased reversibility through modifying the cationic network by releasing Peierls distortion, similar to P doping.18 Thus, the non-linear performance-dopant dependence, especially two peaks at around 0.04 and 0.1 Al-doping, is attributed to the interplay of cationic ordering in doped phases and Co reduction by carbon coating.
As marked in Fig. 2b, there are two types of Co metal impurity corresponding to P63/mmc and Fm-3m space groups, respectively. When there is less than 0.04 Al-doping, Fm-3m Co is the main impurity, but its content is relatively small. For Al content above 0.05, peaks of Fm-3m increase and P63/mmc also becomes visible. Fig. 2d plots the evolution of Co impurity with respect to the Al-doping content in terms of a relative measurement between the integrated intensity ratios of Fm-3m Co to (211/021) reflections of as-prepared samples. The amounts of Co impurity monotonically grow with respect to the Al-doping content, but there are approximately three different regions. When the doping content x > 0.12, the impurity ratio becomes greater than 1 and the reversible capacity becomes much lower than that of C-LCSO. This is a highly impaired region. On the other end, when the impurity ratio is under 0.5, Co impurity has an ignorable effect on the reversibility. Between 0.5 and 1, the Co impurity is the major factor to decrease the discharge capacity of Al-doped samples. This gives an estimation that Co impurity may account for at least a 20 mA h g−1 decrease in the reversible capacity.
To verify the reactivity of Co impurity in carbon-coated polymorphs, thermal gravimetric analyses (TG) were performed at a heating rate of 8 °C min−1 in the temperature range from 30 °C to 800 °C under air atmosphere for the C-LCSO and xAl-LCSO/C samples. Co metal is oxidized into CoO or other oxides in the air when the temperature is above 300 °C, which should result in a weight gain on the TG curve. As shown in Fig. 3, the weight of C-LCSO steadily decreases with the increasing temperature and then undergoes a significant loss between 300 °C and 500 °C, which agrees well with the pattern of “internal” carbon oxidation of Li2CoSiO4 reported by Nazal et al.2 No weight gain is observed for low Al-doping samples (x < 0.05). This confirms that the concentration of reduced Co metal impurity is negligibly small for oxidation in air. When the Al concentration is above 0.05, a weight gain after 300 °C is observed before a loss at around 400 °C, and the proportion of weight gain grows with the increase in the Al content. This confirms the oxidation of Co metal impurity in the air and its positive correlation with the impurity ratios of the XRD signals. We checked the impurity ratio of the 10 at%. P-doping in our previous work,18 which was less than 0.5, falling in the ignorable region. This differs from the Al-doping, confirming the different effects of different dopants on bulk modification and their ensuing interplay with carbon coating. It is therefore important to control the Co impurity ratio under 0.5 for better electrochemistry.
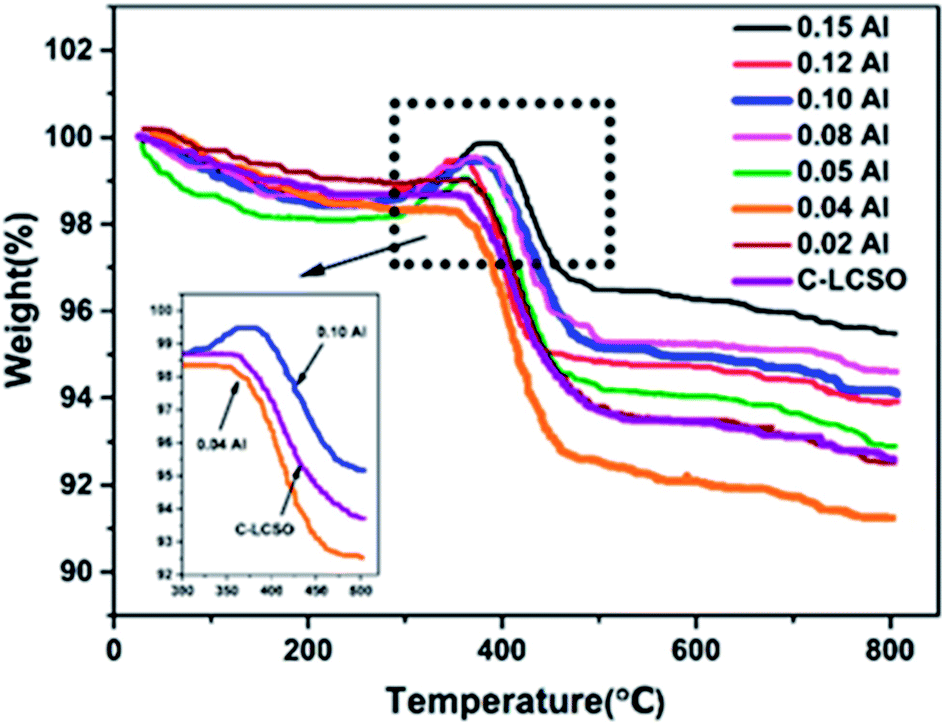 | ||
| Fig. 3 The TG curves of C–LCSO and xAl–LCSO/C under flowing air. | ||
Reversibility has been one of the fatal drawbacks of LCSO, resulting in very low discharge capacity, often below ∼100 mA h g−1, in previous works. By systematically varying the Al doping content in carbon-coated samples, we show that the initial discharge capacity can now be significantly increased in both mixed and pure polymorphs. This may help to address a long-standing question of LCSO, which polymorph would be better for electrochemistry. Several polymorphs, i.e. βII, βI and γ0 polymorphs, have been tested in previous studies.4 From this study, it was concluded that βI helps to suppress Co reduction in pyrolysis and βII shows better overall reversibility. The first peak at 0.04 Al-doping contains contributions from both DP and TP phases, while the second one at 0.1 Al-doping has only decreased the contribution of DP due to Co impurity.
3.2 Electrochemical stability in cycling
Previous studies have shown voltage changes after initial activation charging for Fe, Mn silicates15,17 and βII LCSO synthesized by a supercritical method.10 Several studies showed that the discharge capacity of LCSO would quickly decay to negligible in cycling.8,9 Fig. 4 compares the first two charge–discharge cycles for C-LCSO, 0.04 and 0.10 Al-doped samples. Fig. 4a indicates that the first two discharging curves of C-LCSO are highly repeatable in the same 4 V discharge platform, with almost no change. The cyclic performances of C-LCSO in Fig. 4b show that the reversible capacity is 60.5 mA h g−1 in the 10th cycle, a significant reversible capacity after 10 cycles compared to previous reports.1–3,5 The two Al-doped samples also show the same cyclic characteristics and retain higher cycling capacity. This is clear evidence of the high stability of these polymorphs in electrochemical cycling. No voltage degradation is observed in all the 10 full cycles. Additional graphical comparison of the charge and discharge curves for the first five cycles of LCSO, C-LCSO, and 0.1 Al-LCSO/C is shown in Fig. S1 of the ESI.† These measurements reveal that the nature of voltage degradation is due to voltage polarization, which will be analyzed by EIS in the next section. Al doping does not change the voltage platform or degrading rate but retains more discharge capacities over cycles.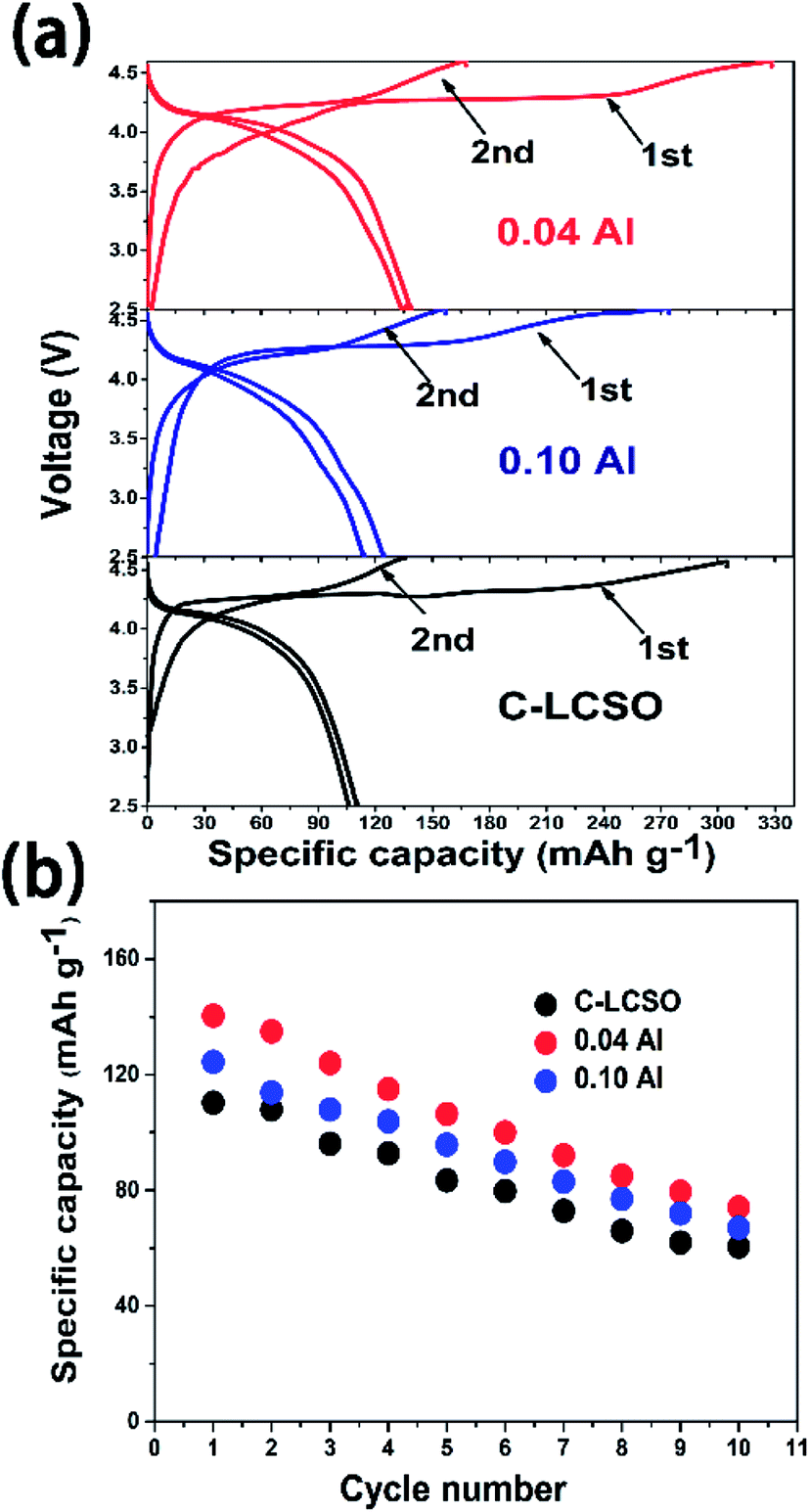 | ||
| Fig. 4 (a) The first two charge/discharge curves; (b) cycling performances up to 10 cycles of C–LCSO, 0.04 Al–LCSO/C and 0.10 Al–LCSO/C. | ||
We analyzed the XRD spectra of LCSO after the first cycle (showed in Fig. S2 of ESI†). No irreversible phase change was identified. Such stability in electrochemical cycling is absent in Fe/Mn silicates.15,16 This confirms the unique electrochemical stability of the delithiated structure in the deep charged state, illustrating the attractive electrochemical characteristics of Li2CoSiO4 as a high-energy cathode candidate, which has stable polymorphs at the 4 V platform, fully compatible with the 4 V voltage window of the state-of-the-art organic electrolytes for battery application.
Both 0.04 and 0.10 Al-LCSO/C have very high charge capacities of 330.6 mA h g−1 and 270.3 mA h g−1, respectively. These values are significantly higher than all the previous reports, c.f. Table 1. The charging-discharging efficiencies in our work are also better than previous reports when comparing the same voltage window. Because the 0.04 Al-LCSO/C contains a lower ratio of Co impurity (inactive in oxidation), its charge capacity of 330.6 mA h g−1, the highest one so far, is strong evidence that Li2CoSiO4 has the potential to extract two Li+ ions with the cut-off voltage at 4.6 V. This conjecture is consistent with previous works, all of which have estimated that more than one lithium may have been extracted according to the first charging capacity at the 4.6 V cut-off voltage. Because the theoretical capacity of LCSO is only 325 mA h g−1, we believe there were side reactions contributing to the charge capacity. We used the inductively coupled plasma (ICP) to estimate the remaining lithium concentration in samples showing charge capacity of 330 mA h g−1. This gave an equivalent formula Li0.38Co1.0Si1.0O4 (charged to 4.8 V for C-LCSO), corresponding to a charge capacity of 260 mA h g−1 from Li deintercalation. This estimation, while showing the incomplete extraction of two lithium ions, is very encouraging, because so far only the family of Li2MnO3-like lithium-rich composites has shown that high capacity in the 4 V voltage window. This also proves that the second delithiation occurs under 4.6 V, disagreeing with the previous calculation that predicted the second lithiation potential of lithium cobalt silicate would be above 5 V,17 and at least a significant portion has been extracted out in this work. Of course, more studies are needed to identify and reduce those side reactions.
| Polymorphs | Potential range | Capacity (mA h g−1) | Efficiency | Ref. | |
|---|---|---|---|---|---|
| Charge | Discharge | ||||
| DP | 2.5–4.6 V | 270.3 | 124.5 | 46% | This work |
| DP/TP | 330.6 | 140 | 42% | ||
| DP | 2.5–4.6 V | 270 | 144 | 54% | 18 |
| TP | 1.5–4.6 V | 200 | 107 | 54% | 10 |
| DP | 2.0–4.6 V | 162 | 33 | 20% | 9 |
| DP | 3.0–4.6 V | 1.4 Li | 0.46 Li | 33% | 12 |
| γ0 | 2.0–4.6 V | 100 | 30 | 30% | 11 |
| DP | 180 | 30 | 17% | ||
| TP | 170 | 60 | 35% | ||
| TP | 2.0–4.7 V | 240 | 56 | 23% | 8 |
3.3 Electrochemical impedance spectroscopic study
To understand the improved electrochemistry, especially to identify the possible origin of the big irreversible capacity loss in the initial performance and the heavy capacity decay in cycling, electrochemical impedance spectroscopic studies were performed.Nyquist plots of C-LCSO and xAl-LCSO/C composites are shown in Fig. 5. The measurements were performed over an applied frequency range of 0.1 Hz to 105 Hz, by using the a.c. amplitude of 10 mV. All plots show one skewed semicircle followed by an inclined straight line; the latter is confined to the low-frequency domain. The high-frequency region where the curve touches the real axis corresponds to the electrolyte and electrodes resistance (Re). The high to medium frequency range is ascribed to a parallel combination of charge transfer resistance (Rct) and electrical double layer capacitance (Cdl), and the straight line originates from the diffusion of charged species through the bulk of the electrode material.
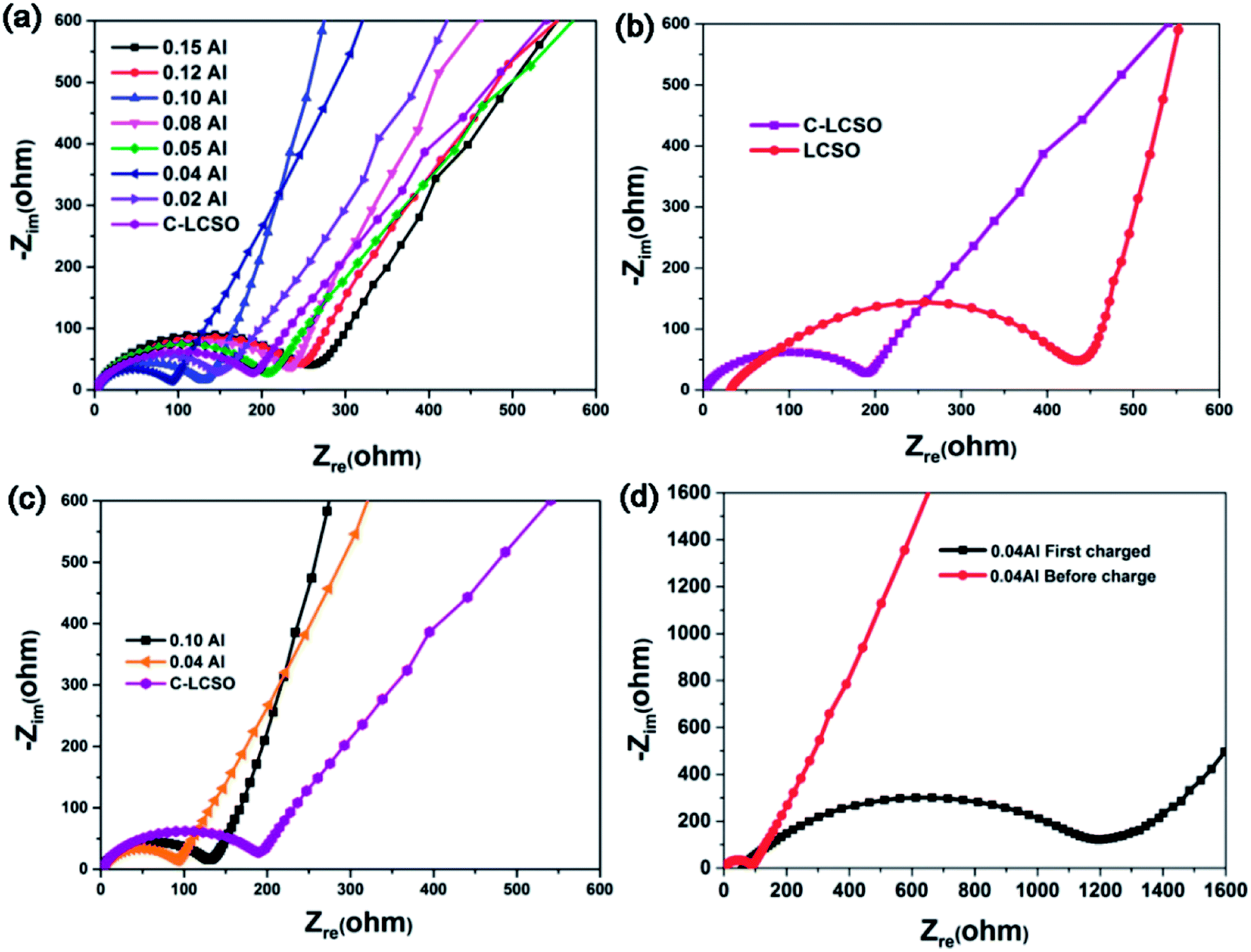 | ||
| Fig. 5 Nyquist plot comparison of (a) the C–LCSO and xAl–LCSO/C samples; (b) the C–LCSO and uncoated LCSO; (c) the C–LCSO, 0.04 Al–LCSO/C, and 0.1 Al–LCSO/C; (d) the 0.04 Al–LCSO/C before and after the first charging. | ||
Fig. 5a compares the results of the C-LCSO and xAl-LCSO/C impedance spectra, which are consistent overall with their reversibility in the first cycling performance shown in Fig. 2c. Fig. 5b compares the impedance spectra of C-LCSO with the undoped and uncoated LCSO. It is very clear that the surface coating has effectively reduced the material's charge transfer resistance (Rct), thus improving the electrochemical reversibility. Furthermore, Fig. 5c shows that the 0.04 and 0.10 Al-LCSO/C samples exhibit significantly smaller charge transfer resistances (Rct) in comparison with LCSO, C-LCSO and other xAl-LCSO/C, confirming that appropriate Al and carbonaceous content collaboratively improve the kinetics of the lithium cobalt silicate materials. The physical cause, why 0.04 Al-doping has more discharge capacity than the 0.10 Al-doping, is also due to its better overall conductivity. Thus, the two capacity peaks at 0.04 and 0.10 Al-doping can be interpreted by the better effective modification of the surface and bulk electrical properties together.
A remaining drawback is the big irreversible capacity loss observable in Fig. 4a. The initial charge capacity of C-LCSO is 312 mA h g−1, giving an efficiency of 35%, which is almost the same as those uncoated LCSO and is lower than 42% of 0.04 Al-LCSO/C and 46% of 0.10 Al-LCSO/C (c.f. Table 1). This indicates that the irreversible capacity loss is hardly separable into the surface or the bulk origin. The doping indeed improves the efficiency a little more. Considering the potential contribution of side reactions to the initial charge capacity, the actual delithiation and re-intercalation efficiency should be higher than the above 35%, 42% and 46%, values.
To address the physical origin of capacity loss or decay in cycling, we examined the impedance behavior of the 0.04 Al-doping sample in the initial charging process. Fig. 5d shows the comparison of the impedance spectra before and after the initial charging of 0.04 Al samples. The Rct at the deep charged state (∼4.6 V) increases almost 10 times, indicating a very bad electrical conductivity of the delithiated state at the end of the initial charging. In one aspect, this interprets the changes in voltage polarizations presented in Fig. 4a and S1 of ESI.† On the other hand, it is a very surprising and big increase, because most of the cathode materials show improved bulk electrical properties in delithiated states. We speculate that there have been some changes on the surfaces of the particles, for example, the formation and breaking of the solid-electrolyte interphase, which has often been a failure mechanism for anode materials so far.18,19 The very low electrical conductivity significantly reduces the re-intercalation of lithium and electrons in the discharging process. This could be the electrical cause accounting for the big irreversible capacity loss. While the exact mechanism needs further study for the charged state, it is clear that the performance of modified LCSO is still very sensitive to any change in electrical properties of either the bulk or the surface. Thus, it is worth examining the characteristics of the coated carbon layers for a better understanding.
3.4 Effects of carbon coating on electrochemistry
Because of the “internal” carbon oxidation,9 carbon coating has been a long and difficult task for LCSO. Thin and uniform carbon layers with a high degree of graphitization and nanostructured particles with reduced diffusion distances inside the active material are the basic structure forms widely used to increase the electrical and ionic conductivities of insulting polyanionic cathodes such as LiFePO4. We have shown so far that Al-doping, carbon coating and their interplay are critical to the improvement of the electrochemistry of LCSO. Because of bad performance, there is not much detailed work on carbon coating LCSO. In this section, we examine the characteristics of coated carbon layers.The EDX mapping of 0.02 Al-LCSO/C samples in Fig. 6a confirms the uniformly distributed Si, Co and Al elements throughout the polymorph. The presence of Al element in the carbon coated nanoparticles with Al molar ratio as low as 0.02 confirms the successful introduction of Al in the two-step synthesis. XPS analyses of C-LCSO and xAl-LCSO/C are used to investigate the oxidation state of active elements in samples, and the related spectra are shown in Fig. 6b. The Co 2p spectrum of C-LCSO presents a very characteristic Co2+ signal with two main peaks at 797.06 eV and 781.21 eV for the 2p1/2 and 2p3/2, respectively. After doping, a small shift with the doping content was observed in the Co 2p peak positions, indicating the successful introduction of Al into LCSOs. Fig. S3 (shown in ESI†) provides a systematic comparison of high-resolution XPS spectra of 0.1 Al-LCSO, LCSO and aluminium sulphate (as a reference of Al3+). Fig. S3(a)† shows the Al 2s, Al 2p and O 1s high-resolution XPS spectra of 0.1 Al-LCSO, LCSO and aluminium sulphate. In general, the XPS peaks of Al 2p, O 1s, and S 2p of the Al3+ salt are at 75.6 eV, 532.6 eV, and 169.3 eV, respectively, which is consistent with the standard XPS spectra of aluminium sulphate.20 From Fig. S3(a),† it is clear that 0.1 Al-LCSO and aluminium sulphate present distinct Al 2p and Al 2s peaks, which are absent in LCSO. In addition, compared to the aluminium sulphate peaks, the Al 2p and Al 2s peaks of 0.1 Al-LCSO shifted toward lower binding energies. Also, the O 1s peak of 0.1 Al-LCSO exhibited a higher binding energy than that of LCSO, which indicates that Al3+ doping was successfully achieved, and there is also electron transfer between Al3+ and O2−, forming an ionic bonding. It can be verified from Fig. S3(b) and (c)† that the XPS peaks of Co2p and 3p, Si2p and 2s of 0.1Al-LCSO and LCSO have no obvious difference, indicating that the chemical states of Co and Si do not change much before and after Al doping. The SEM and TEM analyses in Fig. 6c and d indicate that both the undoped and doped samples exhibit the same degree of particle aggregation and similar average particle sizes (30–50 nm), but the size distribution is quite wide. Thin and uniform carbon layers are also clearly presented on the surfaces of C-LCSO and 0.10 Al-LCSO/C particles shown in Fig. 6d. A conductive carbon layer on a nano-network of ∼50 nm-sized particles confirmed the improved electrochemical performance as shown in Fig. 5b.
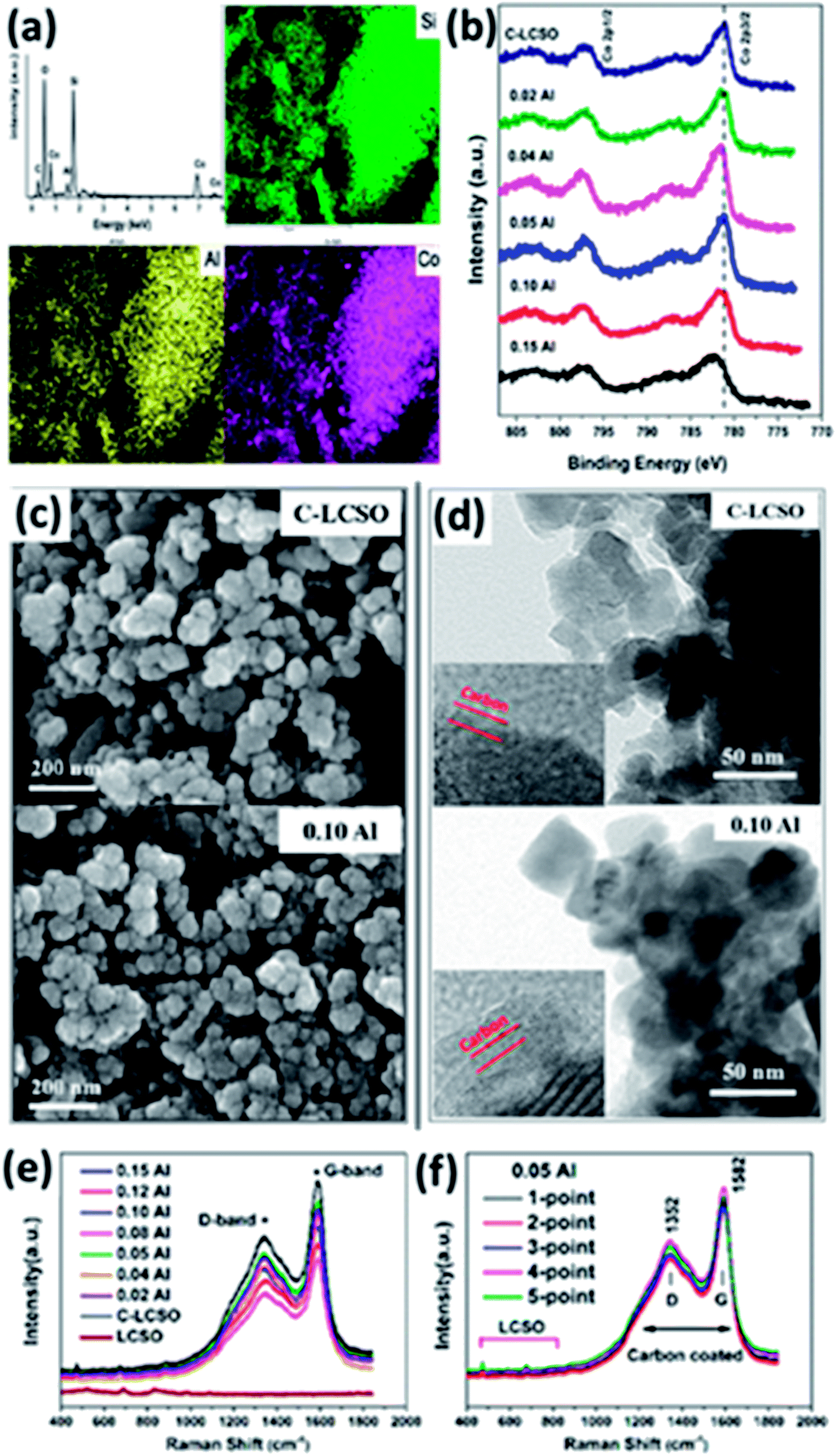 | ||
| Fig. 6 (a) EDX mapping of 0.02 Al–LCSO/C; (b) XPS spectra of the undoped and doped samples C–LCSO and xAl–LCSO/C; (c) SEM and (d) TEM images of C–LCSO and 0.10 Al–LCSO/C. The inset images in (d) indicate the carbon coating layers. Raman spectra of (e) LCSO, C–LCSO and xAl–LCSO/C; (f) 0.05 Al–LCSO/C for five different points on the tested samples. | ||
The percentage of carbon present in all the as-prepared samples was confirmed to be ∼5.9 wt% by infrared spectroscopy. To verify the quality of the carbon layer, Raman spectroscopy was performed. Fig. 6e and f compare the Raman spectra of LCSO, C-LCSO and xAl-LCSO/C. The peaks between 400 and 1000 cm−1 for all presented samples attest to LCSO, confirming the same bulk characteristics. All carbon coated samples exhibit two peaks around 1352 and 1582 cm−1 corresponding to the typical D (sp3 type) and G (sp2 type) bands of carbon, respectively, indicating the success of our direct carbon coating method for lithium cobalt silicates.
The intensity ratio between D and G bands (ID/IG) generally provides a useful indication of the quality of carbon layers to improve electrical conductivity. A lower ratio of ID/IG corresponds to more sp2 type carbon in the sample and means better electrical conductivity. Table 2 presents the calculated intensity ratios ID/IG of C-LCSO and xAl-LCSO/C. They were found to be in the range of 1.425–1.500, which is within the error range. The similar ID/IG ratio of all the xAl-LCSO/C samples implies that the amorphous degrees of the coated carbons are consistent in the two-step process. Moreover, the ID/IG ratio, less than 1.500, is even smaller than some Li2FeSiO4/C and Li2MnSiO4/C nanocomposites,6,8,21 which are well-known for the remarkable improvement of electrical conductivity by carbon coating. We further examined the Raman spectra of 0.05 Al-LCSO/C on five random points of the tested sample. It is clearly shown that the five curves almost coincide with each other, reflecting the uniform carbon layer on the silicate sample, which agrees with the TEM images. These Raman data perfectly validate the consistent surface modification of carbon coating in different samples and in different areas of the same sample.
| ID/IG | C-LCSO | 0.02 Al | 0.04 Al | 0.05 Al |
| 1.488 | 1.497 | 1.444 | 1.456 | |
| ID/IG | 0.08 Al | 0.10 Al | 0.12 Al | 0.15 Al |
| 1.425 | 1.429 | 1.452 | 1.50 |
It can be concluded that the carbon layers coating the Al-doped samples are almost the same and the doped Al is well distributed in all the samples with negligible effects on the morphologies of the particles as well as the oxidation states of Co element. This is the physical root for the improved electrochemistry reported in this work.
Our carbon coating improves the initial performance and cycling stability considerably, compared to previous works on LCSO (c.f. Table 1), but it is still far from satisfactory with respect to those improvements in carbon coating in advanced LiFePO4. One possible cause is the low intrinsic electrical conductivity of LCSO. The work conducted in 2017 on functionalized multi-walled carbon nanotubes (MWCNTs) in LCSO showed an improvement of the diffusion coefficient of lithium only from 1 × 10−14 to 8 × 10−14 cm2 s−1,8 which was still far below the intrinsic conductivity of LiFePO4 (∼10−9 S cm−1). The characterization of carbon coating in this work was performed before electrochemical testing. The condition of carbon coating may be changed in the cycling; such a change may also account for the heavy capacity decay in just 10 cycles. This possibility is evidenced by the 10 times increase of Rct and its effect on the initial capacity loss (c.f. Fig. 5d). Even though the performances of the modified LCSO reported in this work are not comparable to the advanced cathode materials of NMC or LiFePO4, it is clear that LCSO has the potential to become a unique high-energy cathode material, which has all the requirements of high safety, high capacity and high voltage. The future improvements should continue to focus on the strategies to combine both bulk and surface modifications in a better synthetic way. Limitations exist in our analysis and examination due to limited facilities used so far; analytical characterizations, especially in operando, should be the means toward comprehending the underlying working and degradation mechanisms in subsequent studies.
4. Conclusion
In summary, under the combined effects of carbon coating and Al-doping, this work presents the intrinsic electrochemical properties of Li2CoSiO4 that likely allow two lithium removals at the 4 V redox potential in stable cycling, which is an attractive characteristic of a high energy cathode candidate for next generation lithium-ion batteries. From this study, carbon coating, the phase of polymorphs and Co impurity are three main factors that impact the electrochemical performance of Li2CoSiO4. A conductive carbon nano-network is a key factor to improve the electrochemical performance of Li2CoSiO4, which shows significant effects on the electrochemistry as evidenced in the electrochemical impedance spectroscopy. The pristine carbon coated Li2CoSiO4 exhibits a reversible capacity of 110.3 mA h g−1 above 2.5 V. Al-doping can enhance electrochemical performance by further improving ionic conductivity. The mixture of polymorphs, 0.04 Al-Li2CoSiO4/C, exhibits the best electrochemical performance and cycling stability, showing a first charge and discharge capacity of 331 and 140 mA h g−1, respectively. This may be related to a phase interface, releasing Peierls distortion in a similar way to element dopants. Furthermore, the irreversible capacity loss and heavy decay of cycled capacities have been attributed to the significant change in the electrical conductivity, which may, in turn, be related to the change in the carbon coating layers or the formation of solid-electrolyte interphases.19Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge programs supported by National Research program of China (2018YFB0704302, 2018YFB0704300), Zhejiang Province Key Science and Technology Innovation Team (2013PT16), Zhejiang Natural Science Foundation (LY16B030007), and Ningbo Municipal Science and Technology Innovative Research Team (2016B10005).References
- S. Thayumanasundaram, V. S. Rangasamy, J. W. Seo and J.-P. Locquet, Ionics, 2017, 1–9 Search PubMed.
- G. He, G. Popov and L. F. Nazar, Chem. Mater., 2013, 25, 1024–1031 CrossRef.
- M. K. Devaraju, Q. D. Truong and I. Honma, RSC Adv., 2013, 3, 20633 RSC.
- C. Lyness, B. Delobel, A. R. Armstrong and P. G. Bruce, Chem. Commun., 2007, 4890–4892, 10.1039/b711552k.
- Z. L. Gong, Y. X. Li and Y. Yang, J. Power Sources, 2007, 174, 524–527 CrossRef.
- J. Billaud, C. Eames, N. Tapia-Ruiz, M. R. Roberts, A. J. Naylor, A. R. Armstrong, M. S. Islam and P. G. Bruce, Adv. Energy Mater., 2017, 7, 1601043 CrossRef.
- Q. Cheng, W. He, X. Zhang, M. Li and L. Wang, J. Mater. Chem. A, 2017, 5, 10772–10797 RSC.
- J. Yang, J. Zheng, X. Kang, G. Teng, L. Hu, R. Tan, K. Wang, X. Song, M. Xu, S. Mu and F. Pan, Nano Energy, 2016, 20, 117–125 CrossRef.
- Z. Chen, S. Qiu, Y. Cao, J. Qian, X. Ai, K. Xie, X. Hong and H. Yang, J. Mater. Chem. A, 2013, 1, 4988–4992 RSC.
- D. Rangappa, K. D. Murukanahally, T. Tomai, A. Unemoto and I. Honma, Nano Lett., 2012, 12, 1146–1151 CrossRef PubMed.
- A. Grenier, H. Liu, K. M. Wiaderek, Z. W. Lebens-Higgins, O. J. Borkiewicz, L. F. J. Piper, P. J. Chupas and K. W. Chapman, Chem. Mater., 2017, 29, 7345–7352 CrossRef.
- S. H. Ju, H. C. Jang and Y. C. Kang, Electrochim. Acta, 2007, 52, 7286–7292 CrossRef.
- A. R. Armstrong, C. Lyness, M. Ménétrier and P. G. Bruce, Chem. Mater., 2010, 22, 1892–1900 CrossRef.
- C. Zhang, Z. Chen and J. Li, Chem. Phys. Lett., 2013, 580, 115–119 CrossRef.
- Y.-X. Li, Z.-L. Gong and Y. Yang, J. Power Sources, 2007, 174, 528–532 CrossRef.
- X. Lu, H. Wei, H.-C. Chiu, R. Gauvin, P. Hovington, A. Guerfi, K. Zaghib and G. P. Demopoulos, Sci. Rep., 2015, 5, 8599–8607 CrossRef PubMed.
- Y. L. Guohua Zhong, P. Yan, Z. Liu, M. Xie and H. Lin, J. Phys. Chem. C, 2010, 114, 3693–3700 CrossRef.
- A. Bhaskar, M. Deepa, T. N. Rao and U. V. Varadaraju, J. Electrochem. Soc., 2012, 159, A1954–A1960 CrossRef.
- S. S. Zhang, K. Xu and T. R. Jow, J. Electrochem. Soc., 2002, 149, A1521 CrossRef.
- L. V. Duong, B. J. Wood and J. T. Kloprogge, Mater. Lett., 2005, 59, 1932–1936 CrossRef.
- Q. Cheng, W. He, X. Zhang, M. Li and L. Wang, J. Mater. Chem. A, 2017, 5, 10772–10797 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02555j |
| This journal is © The Royal Society of Chemistry 2018 |