
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew therapeutic strategies based on interference with telomeric DNA synthesis of tumor cells to suppress the growth of tumors
Zhongyan Wang,
Xiuli Zhao,
Yan Liu,
Ting Wang and
Kexin Li *
*
School of Pharmacy, Shenyang Pharmaceutical University, Liaoning Province, China. E-mail: kexinbest1128@163.com; Tel: +8602423986293
First published on 11th July 2018
Abstract
An unusual enzyme called telomerase acts on parts of chromosomes known as telomeres. The enzyme has recently been found in many human tumors and is viewed as a new target for tumor therapy. In this research, we chose the analogue of guanine “2′,3′-dideoxyguanosine” (ddG) as the telomerase inhibitor and prepared the ddG-loaded cationic nanoliposomes (ddG-Clip) to specifically target the tumor tissue and preferentially occupy the telomerase nucleotide binding site. The mean diameter of ddG-Clip is 101.54 ± 2.60 nm and they are cationically charged with a zeta potential of 34.0 ± 9.43 mV; also, the encapsulation efficiency of ddG-Clip is 53.44% ± 2.29%. In vitro cytotoxicity results show that cationic nanoliposomes by themselves are almost non-toxic, but with the increase in ddG concentration, ddG-Clip has the ability to kill S180 tumor cells. The anti-tumor activity study suggests that ddG-Clip could not only suppress the tumor growth, but also inhibit tumor liver metastasis well. In conclusion, reverse transcriptase inhibitor-loaded cationic nanoliposomes could interfere with the synthesis of telomeric DNA and block abnormal proliferation of tumor cells, therefore achieving tumor apoptosis.
Introduction
Telomeres are specialized caps localized at the ends of human chromosomes.1 In normal human cells, telomeres become increasingly shorter with each successive cell division, and upon reaching critical lengths, they elicit DNA-damage responses, thus leading to cell senescence and apoptosis.2 On the contrary, cancer cells, which develop chromosomal aberrations, show activation or re-activation of telomerase upon exposure to a DNA damage signal, bypassing cell cycle checkpoints and leading to uncontrolled growth and proliferation. The basic reason of the difference in DNA division between normal cells and cancer cells is telomerase. The presence of telomerase in various human cancers and its absence in many normal cells indicate that the enzyme might serve as a good target for antitumor drugs.3 The unique reverse transcriptase can maintain cancer cell telomeres at a stable length at all times, ensuring their rapid proliferating potential and immortal capacity.4,5 Hence, agents capable of inhibiting the activity of telomerase might kill tumor cells without disrupting the functioning of many normal cells.6–8 Furthermore, because telomerase occurs in numerous cancers, such agents might work against many tumors.In the process of telomeric DNA catalytic synthesis, single stranded TTAGGG sequences are continuously synthesized and added to the ends of chromosomes to offset the loss. Hence, we chose the analogue of guanine 2′,3′-dideoxyguanosine as telomerase inhibitors, which can replace guanine in telomerase mediated incorporation of newly synthesized DNA chain9–11 and preferentially occupy the telomerase nucleotide binding site, inhibit the synthesis of telomeric DNA and block abnormal clonal proliferation of tumor cells, and consequently achieve the purpose of treating cancer.12–14 Previous research shows that in normal human cells, telomerase activity is tightly regulated, but in addition to tumor cells, telomerase could also be detected in those cells that have to constantly divide, such as hematopoietic cells, stem cells and germ cells.15 Therefore, how to avoid telomerase inhibitors to damage DNA of other normal cells and only target the DNA of tumor cells is the next question we should consider.
Cationic nanoliposomes are a non-viral vector of potential clinical application, having similar cell structure and biological membrane.16–18 When the particle size of cationic nanoliposomes is in the size range of 50–200 nm, they can escape phagocytosis of the reticuloendothelial system (RES) and prolong the circulation time in vivo.19 At the same time, they can also be held up in tumor tissues through the enhanced permeability and retention effect (EPR). Most importantly, the positively charged surface of cationic nanoliposomes and the negatively charged surface of tumor cells can undergo electrostatic attraction. As a result, internalization of carriers will occur by membrane fusion and cell phagocytosis, so as to realize the accumulation of cationic carriers and targeted release of telomerase inhibitors in tumor tissues.20
In this research, we developed ddG-loaded cationic nanoliposomes for treating solid tumors. In addition, we evaluate its inhibitory effects on the tumor growth and preliminary discuss the mechanism of antitumor action.
Experimental
Materials
Phosphatidylcholine (PC) was purchased from Avanti Polar Lipids (Shanghai, China). Cholesterol (CH) was purchased from Tianjin Bodi Chemical (China). Dioleoyl phosphoethanolamine (DOPE) and 3-(N-(N,N-dimethylaminoethane) carbamoyl) cholesterol (DC-chol) were purchased from Sigma(UK). 2′,3′-Deoxyguanosine was obtained from Abcam (Britain). Sepharose 4B was obtained from GE Healthcare. All other reagents purchased were of analytical and molecular biology grades.Preparation and characterization of ddG-Clip
2′,3′-Deoxyguanosine loaded cationic nanoliposomes (ddG-Clip) were prepared by the revised reverse phase evaporation technique.21 First, DOPE and DC-chol with the molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were dissolved in 15 mL diethyl ether.22,23 A uniform lipid film was formed by rotary evaporation at room temperature. Second, the film was redissolved in 15 mL dichloromethane as the oil phase, and 5 mL deionized water containing 5 mg ddG as the aqueous phase was added dropwise to form water-in-oil emulsion. The subsequent vortex oscillation was performed for 5 min in order to increase the uniformity of internal phase distribution. Then, the emulsion was rotary-evaporated under reduced pressure entirely until residual diethyl ether was removed. Lastly, the white residue was thoroughly hydrated for 15 min at 50 °C, well above the Tm for DOPE, and probe-sonicated at 400 W for 120 s to form 5 mL nanoliposomes suspension by deionized water. The preparation method of ddG-Lip is the same as that of ddG-Clip, but DC-chol was changed into cholesterol.
:1 were dissolved in 15 mL diethyl ether.22,23 A uniform lipid film was formed by rotary evaporation at room temperature. Second, the film was redissolved in 15 mL dichloromethane as the oil phase, and 5 mL deionized water containing 5 mg ddG as the aqueous phase was added dropwise to form water-in-oil emulsion. The subsequent vortex oscillation was performed for 5 min in order to increase the uniformity of internal phase distribution. Then, the emulsion was rotary-evaporated under reduced pressure entirely until residual diethyl ether was removed. Lastly, the white residue was thoroughly hydrated for 15 min at 50 °C, well above the Tm for DOPE, and probe-sonicated at 400 W for 120 s to form 5 mL nanoliposomes suspension by deionized water. The preparation method of ddG-Lip is the same as that of ddG-Clip, but DC-chol was changed into cholesterol.
Nanoliposomes were characterized by a Dynamic Light Scattering Instrument (LS 320; Beckman, American) and mean size was expressed as the volume diameter. The zeta potential was measured using a zeta potential analyzer (Delsa 440SX; Beckman, American). The measurements were carried out at 25 °C using cationic nanoliposome suspensions diluted in deionized water. Average values were based on measurements using three different suspensions. Dialysis method was used to determine the release behavior of cationic nanoliposomes in vitro. Sephadex G50 column was used to separate the free drug and nanoliposomes by deionized water as eluent in order to reduce the damage of ions on vehicles. Encapsulation efficiency (EE%) was calculated by the following formula (n = 3): EE% = [(total amount of ddG added − amount of free ddG detected)/total amount of ddG added] × 100%.
In vitro cytotoxicity assays
In vitro cytotoxicity test of ddG solution, ddG-Lip and ddG-Clip against S180 was evaluated by the standard MTT assay. S180 was seeded at a density of 5000 cells per well in a 96-well plate and incubated overnight to allow cell attachment for subsequent studies. Then, the cells were treated with ddG solution, ddG-Lip and ddG-Clip at different ddG concentrations of 0.1, 1 and 10 μg per well for 24 h and 48 h, respectively. Furthermore, 20 μL of MTT (5 mg mL−1) was added and further incubated for 4 h, and 150 μL DMSO was added to dissolve the formazan crystals formed in the live cells. The absorbance at 570 nm was recorded using a spectrometer and the cell viability rate (%) was calculated as follows:| Cell viability rate (%) = (Asample − Ablank/Acontrol − Ablank) × 100% |
Tumor model establishment
Healthy female KM mice (weight, 18–22 g) were purchased from animal center of Shenyang Pharmaceutical University in Liaoning province, China. Female BALB/c nude mice (18–22 g) were purchased from Beijing HFK Bioscience Co., Ltd. All animals were housed separately with free access to food and water according to SPF standards. Animals were maintained in accordance with the guidelines of Regulations for the Administration of Affairs Concerning Experimental animals, China, and approved by the Institutional Animal Care and Use Committee (IACUC) of Shenyang Pharmaceutical University. The tumor bearing mice were produced by inoculating 0.2 mL suspension of S180 cells (2 × 107 cells per mL) subcutaneously into the right axillary fossa. When the tumor reached a volume of 100 mm3, the subsequent experiments could be carried out on the tumor bearing mice.Anti-tumor study of ddG-Clip
Tumor bearing KM mice were randomly divided into four groups with six mice in each group. On days 1, 3, 5, 7 and 9, the tumor-bearing mice were treated with saline, ddG solution, ddG-Lip and ddG-Clip via tail vein injection at a dosage of 5 mg kg−1. Tumor-bearing KM mice were fed normally and we observed the growth status, recorded the weight of mice, and measured the length and width of tumors using vernier calipers every two days. The tumor volume was then calculated using the formula: volume = 0.5 × (length) × (width)2 (mm3). The curves of mice weight to time and tumor volume to time could be plotted and the tumor volume inhibition rate (TIR) was also calculated. TIR = (tumor volume in control group − tumor volume in test group)/tumor volume in control group × 100%. The experiment was terminated at day 21 post-inoculation, and the major organs (liver and kidney) and parts of the tumors were removed and fixed in formalin and cut into 5 mm slices for hematoxylin and eosin (H&E) staining.The imaging of blood vessels in tumor tissue
Large molecule fluorescein FITC-dextran (Mr 500000) was dissolved in saline to form 50 mg mL−1 solution. Then, 0.2 mL of this solution was injected into the tail vein of S180-bearing tumor mice.24 After 5 minutes, the mice were sacrificed by breaking the neck. The tumor tissue was taken out and soaked overnight with PBS solution containing 4% paraformaldehyde to fix the tissue and then, the soaking was continued in 20% sucrose for 2 days to dehydrate the tissue. Then, the tissue was cut into 80 μm slices by Frozen Section Technique. FITC fluorescence of tissue slices was observed by confocal microscopy.Statistical analysis
All of the data are presented as the mean ± standard deviation (SD) and all experiments were performed with at least 3 independent repetitions. Differences between groups were examined using Student's t-test between 2 groups or one-way analysis of variance (ANOVA) among ≥3 groups.Results and discussion
Characterization of cationic nanoliposomes
As shown in Fig. 1, the mean diameter of ddG-Clip is 101.54 ± 2.60 nm. Also, they were positively charged with the zeta potential value of 34.0 ± 9.43 mV and the EE% was 53.44% ± 2.29%. The diameters of ddG-Lip were 96.25 ± 7.11 nm, the zeta potential was −21.7 ± 5.82 mV, and the EE% of was 40.2% ± 6.03%. The lower EE% of ddG-Lip as compared with that of ddG-Clip was due to the lack of positive charges on the surface of the nanoliposomes, which led to less drug adsorption. The observation through the transmission electron microscope showed that the morphology of ddG-Clip was spherical and uniform in size. In addition, they were large single chamber liposomes with visible bilayered structure.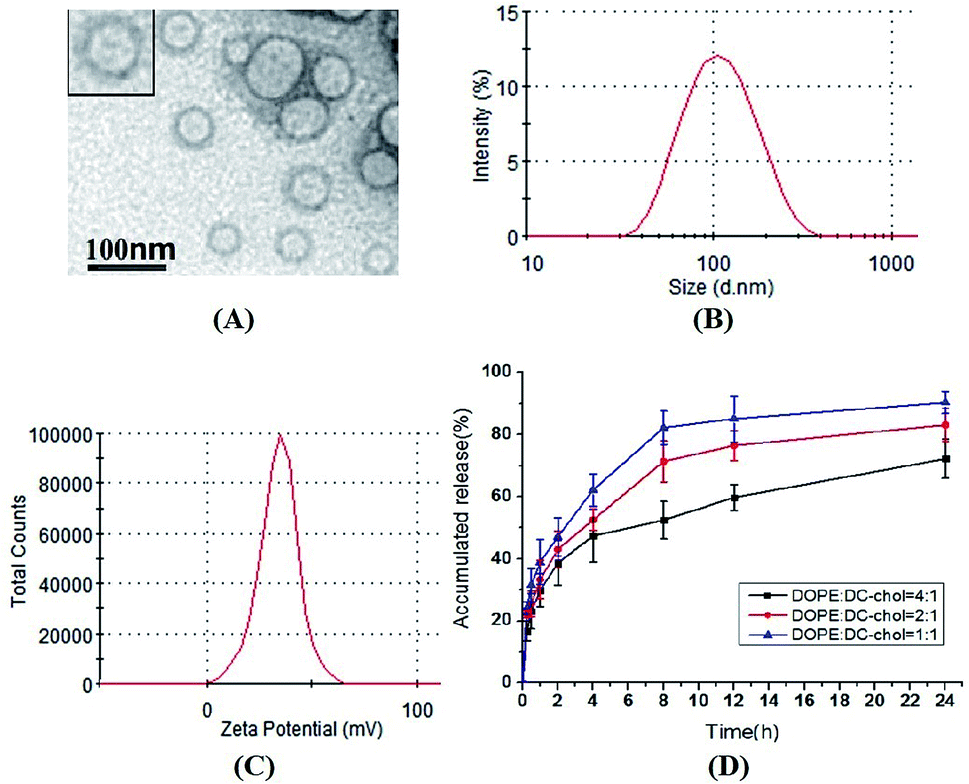 | ||
| Fig. 1 Characterization of 2′,3′-deoxyguanosine loaded cationic nanoliposomes. (A) The transmission electron microscopy images of ddG-Clip. (B) and (C) The particle size distribution and the zeta potential of ddG-Clip. (D) The cumulative release of ddG-Clip with the different DOPE:DC-chol ratios of 4:1, 2:1 and 1:1 in pH 7.4 PBS at 37 °C. | ||
The release curves in vitro indicated that with the increase in ratio of the cationic lipid, electrostatic interactions are also enhanced between the adjacent positively charged tertiary amine structures. Additionally, the broadened gap on the surface of the lipid bilayer membrane accelerated the release rate of ddG and led to significant burst effect.25,26 Probably the free drug could be adsorbed on the surface of the nanoliposomes depending on the electrostatic adsorption, but the binding force was weak. Hence, the release was faster at first, but gradually slowed down at the late release stage.27 At 24 h, the cumulative release of ddG-Clip with the DOPE:DC-chol ratio of 4:1 was 72.23% ± 6.34%, which showed that ddG-Clip had a certain extent of sustained release.
In vitro cytotoxicity evaluation
The cytotoxicity of ddG solution, ddG-Lip and ddG-Clip on S180 cells was determined by MTT assay with different ddG concentrations of 0.1, 1, and 10 μg per well for 24 h and 48 h, as shown in Fig. 2.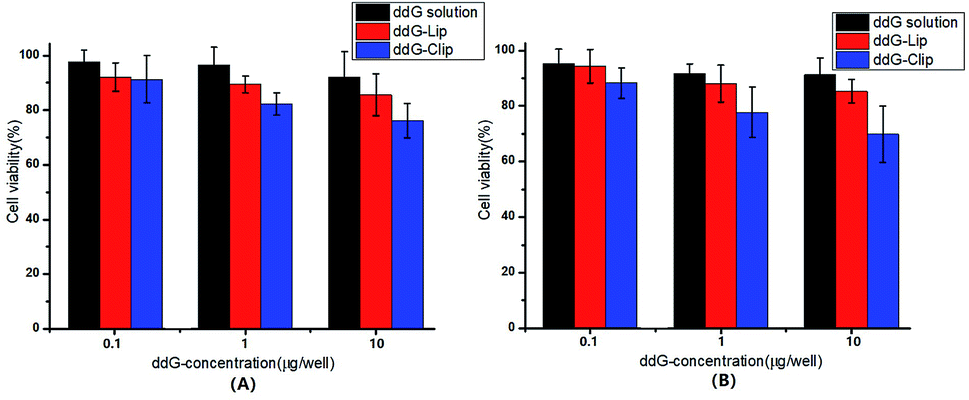 | ||
| Fig. 2 Cell viability of S180 cells after treatment with ddG solution, ddG-Lip and ddG-Clip at different concentrations of 0.1, 1, 10 μg per well for 24 h (A) and 48 h (B). | ||
In case of ddG solution and ddG-Lip groups, no marked growth inhibition effect was found even after treatment with ddG concentration of 10 μg per well, suggesting that ddG and ddG-Lip do not have significant cytotoxicity, and the results therefore proved that ddG containing non-cationic nanoliposomes could not be effectively taken up by the S180 cells. In case of the ddG-Clip group, at a lower concentration of 0.1 μg per well, the cationic nanoliposomes were almost non-toxic, which showed that the materials DOPE and DC-chol used in this study could reduce its toxicity and increase its biocompatibility. Also, with the increase in ddG concentrations from 0.1 to 10 μg per well, the cell viability decreased proportionately both at 24 h and 48 h. At the same time, we could also find that there is no significant difference between the cell viabilities shown in Fig. 2(A) and (B); thus, time had no significant effect on cell viability. We speculated that the reason was related to the release characteristics of ddG-Clip (shown in Fig. 1(D)). The loaded ddG basically released completely within 24 h and later, there was almost no drug release. Hence, even if time was extended, the survival rate still could not change significantly. The above findings illustrate that S180 cells were more susceptible to cationic nanoliposomes and ddG-Clip could successfully be internalized into tumor cells. Between the ddG-Lip and ddG-Clip groups, the root difference in S180 cell viability was that the positive charges on the surface of ddG-Clip could interact with the negatively charged phospholipids, proteoglycans, glycosylation, and membrane proteins such as sialic acid on the newborn endothelial cells of tumor angiogenesis. Then, ddG-Clip would enter the cell through the endocytosis of the cell or fusion of the membrane, and after being transported to lysosomes by endosomes, the degradation would be carried out and 2′,3′-deoxyguanosine would be released near the nucleus, therefore interfering with the synthesis of DNA.28
Antitumor efficacy
Fig. 3 indicates the antitumor efficacy in tumor-bearing mice after treatment with the experimental drug formulations, namely, saline, ddG, ddG-Lip and ddG-Clip.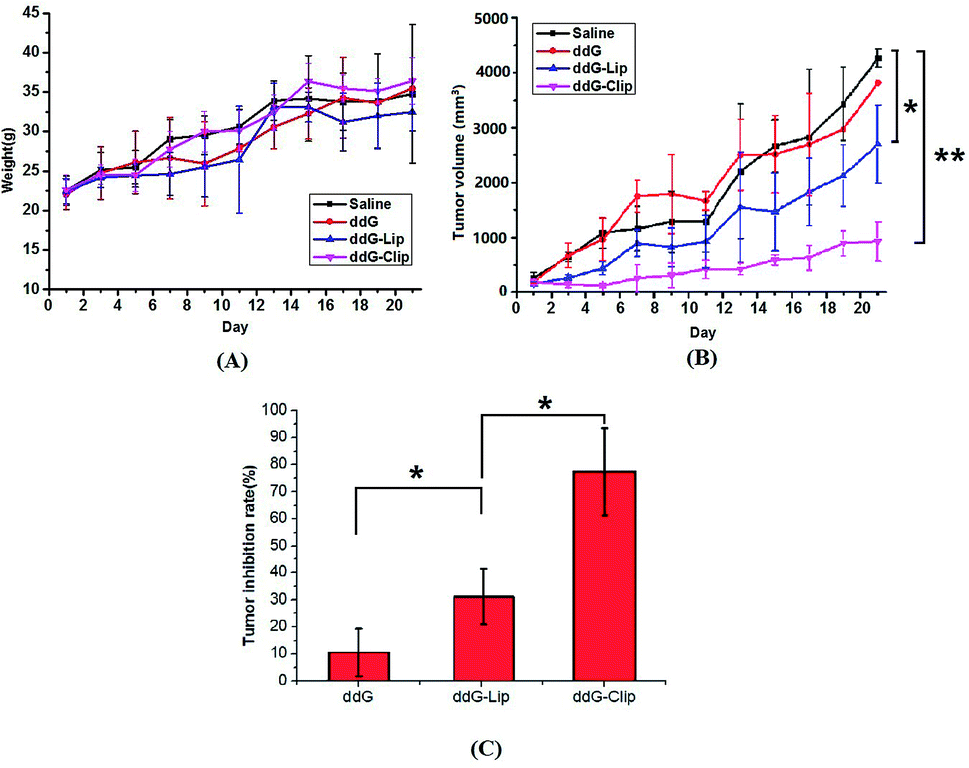 | ||
| Fig. 3 In vivo antitumor activities of different formulations in S180 tumor bearing mice. (A) The mice weight–time curve, (B) the tumor volume–time curve and (C) the tumor inhibition rates of ddG solution, ddG-Lip and ddG-Clip groups as compared with saline group. *p < 0.05 is considered statistically significant, **p < 0.01 is considered extremely significant. The results are representative of replicate experiments and are presented as the mean value with standard deviation (mean ± SD). | ||
Fig. 3(A) indicates that there was no significant difference in weight of mice. The mice in saline group and ddG solution group gradually ate less and became thinner and more inactive, but the tumor increased rapidly. Hence, the weight was still on the rise and the mice in the two groups exhibited ulcerated external skin of tumor site and limited action of the left forelimb because of the larger tumor. The mice weight in ddG-Lip group increased slowly at first but rapidly after day 11, indicating that the growth rate of tumor suddenly increased rapidly. However, in the mice of ddG-Clip group, normal physiological activity was observed, and the mice maintained a stable weight without toxic symptoms.
As shown in Fig. 3(B), the tumor volume in the saline group and the ddG solution group increased rapidly, indicating that the saline and ddG solution had no inhibition effect on the growth of the tumor. Compared with the above two groups, the tumor growth rate of mice in ddG-Lip group decreased at first, that is, ddG-Lip could inhibit the tumor growing to some extent. However, the tumor in the later period grew rapidly and the inhibitory effect gradually decreased. In contrast, the treatment with ddG-Clip showed significantly higher antitumor activity than the other three treatments. In case of the mice in ddG-Clip group, during the entire experiment period, the tumor volume increased very slowly, indicating that ddG-Clip had significant inhibiting effect on tumor growth. Fig. 3(C) indicates that the tumor inhibition rates as compared with the saline group at day 21 post-incubation were 10.63% ± 8.36% for ddG solution group, 31.79% ± 10.50% for ddG-Lip group and 77.46% ± 16.50% for ddG-Clip group, which suggested that ddG-Clip had significantly inhibited tumor growth.
In order to further explore the mechanism of ddG-Clip with good antitumor effect, mice in all groups were killed on day 21 after the first injection. Then, the liver, kidney and parts of the tumors were peeled for eosin (H&E) staining.
The tumor histological staining results in Fig. 4 show that in case of the saline and ddG solution groups, a large number of tumor cells accumulated subcutaneously. Due to very rapid tumor growth, the blood supply was not enough, and the tumor exhibited a large hemorrhagic necrotic tissue due to lack of nutrition (see the red area) furthermore, the tumor had invaded the surrounding tissue and skeletal muscle layer. In case of the ddG-Lip group, the tumor also had a necrotic area and invaded the surrounding tissue, but did not invade the muscularis and the structure of tumor cells was intact. However, in case of the ddG-Clip group, we found an interesting phenomenon of aggregation and wrapping of immune cells and fiber cells around the tumor. We suspected that this was part of the reason for preventing further expansion and growth of tumors. But why was there an inflammatory reaction in the tumor tissue? We inferred that this was associated with the aggregation of cationic nanoliposomes at the tumor site. In order to obtain direct and visible evidence, the cationic nanoliposomes labeled with DiR dye were prepared and injected into S180 tumor bearing nude mice through caudal vein injection. An NIR fluorescence imaging system was used within 96 h after administration in order to observe the specific targeting and the retention time of the vehicles to the tumor tissue. Fig. 5 showed that only 5 minutes after the injection of cationic nanoliposomes into the caudal vein, the accumulation in the tumor site that could be seen, even until 96 h, was still very strong. This proved that the carriers could not only rapidly target the tumor site, but also have an evident sustained release effect. Hence, in Fig. 3(B), we could see that even after the last injection at day 9, there was still a significant inhibitory effect on the growth of the tumor in mice of ddG-Clip groups. All these characteristics of tumor targeting and accumulating effectively stimulate the activity of peripheral immune cells and therefore cause the inclusion to the tumor cells. At the same time, ddG-Clip packaged in the tumor tissue could be endocytosed and release ddG near the nucleus, and thus interfere with the synthesis of telomeric DNA. Most importantly, we should make sure that depending on the delivery of cationic nanoliposomes, telomerase inhibitors could only target the tumor cells, effectively avoiding damage to other normal cells. This was very significant for reducing side effects and enhancing safe use of the carriers.
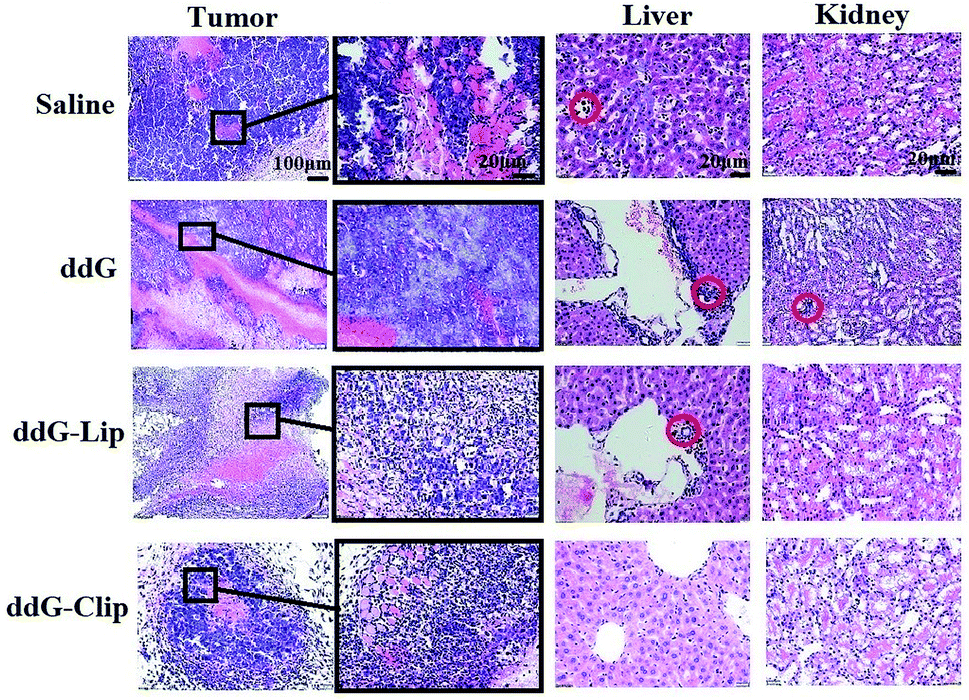 | ||
| Fig. 4 Histological staining of tumors, livers and kidneys on 21 day from mice treated with saline, ddG solution, ddG-Lip and ddG-Clip. Black box area is further enlarged to observe the internal structure of the tumors. Red circles indicate that tumor cells have been transferred to the livers and kidneys. | ||
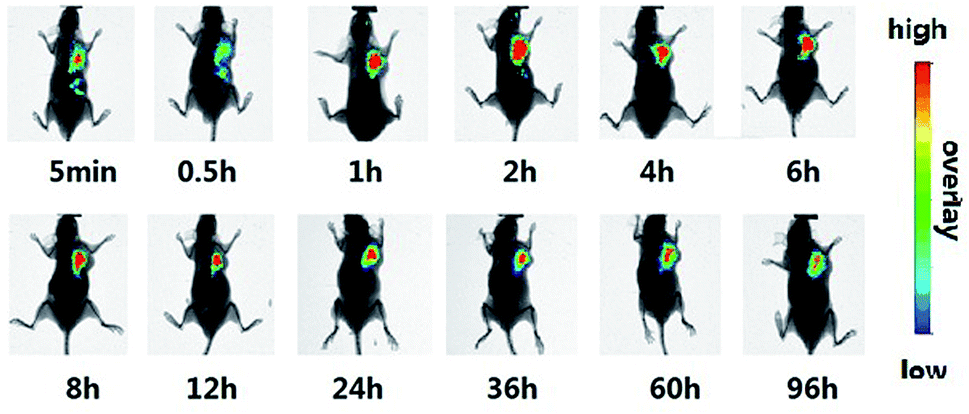 | ||
| Fig. 5 The specific targeting of cationic nanoliposomes to the tumor tissue was evaluated. Cationic nanoliposomes was labeled with DiR dye and injected by caudal vein of female BALB/c nude mice. | ||
In the livers in saline groups, there was tumor cell infiltration around hepatic sinus and central vein, and a few hepatic cells showed degeneration. In case of the solution and ddG-Lip groups, a small amount of tumor invasion occurred in the portal area and hepatic sinus area and the bile canaliculi was also partially invaded. It is well known that tumor metastasis is more likely to occur in vascular rich areas, so the above three groups all showed different degrees of liver metastasis. In sharp contrast, in case of the ddG-Clip group, there was no tumor cell aggregation in the portal area, which demonstrated that the tumor did not transfer to the liver. This indicated that ddG-Clip could significantly reduce the spread of tumor cells in the blood and simultaneously control tumor growth and metastasis effectively. This is of great significance in the clinical treatment of the tumor.
The kidneys of the mice in each group showed different levels of protein tubular denaturation in renal tubular epithelial cells, which was caused by S180 osteosarcoma in vivo, but the denaturation was reversible. In case of the solution group, because the ddG solution showed lack of targeting, it could be distributed in the tissues and organs of the body. Hence, the liver and kidney functions were significantly damaged. The cortex area of the kidney showed a few tumor cells invasion. Protein tubular degeneration was the most significant, but the extent of tumor invasion alleviated was much less as compared to that in the liver, which proved that the main metastasis area of S180 osteosarcoma was the liver and not the kidney. However, in case of the ddG-Clip group, only a few protein tubes were found and the degree of injury was also minimal.
All the above studies suggested that the ddG loaded cationic nanoliposomes not only could effectively suppress the growth of tumor, but also prevent the liver metastasis and eliminate renal damage, therefore showing that ddG-Clip would have good therapeutic effect on tumors.
Blood vessels imaging in tumor tissues
We used FITC-dextran to mark new blood vessels, providing a basis for further study of tumor vasculature in vivo. Since FITC-dextran (Mr 500000) is a macromolecule, it cannot to pass through the endothelial gap and cannot be absorbed by endothelial cells in a short time after intravenous injection in vivo.29 Therefore, it could be used to label the process of angiogenesis in vivo.In Fig. 6(A) and (B), we could see that in the saline and ddG solution groups, the mice tumor grew rapidly and the blood supply was not enough, so vascular structure was destroyed and necrosis occurred in the tumors. From frozen slices, we could see large areas of strong green fluorescence and there was no complete vascular structure. Fig. 6(C) shows that the edge of tumor in the ddG-Lip group was expanding rapidly to the surrounding tissue and blood vessels, and rapid growth of the tumor stimulated a large number of vascular proliferations. Therefore angiogenesis was particularly active, and we could clearly see the regeneration of the vascular network. However, there was less neovascularization in the center of the tumor and the fluorescence intensity was relatively weak. In Fig. 6(D), we could clearly observe the characteristics of intratumoral vessels and the vascular network of the tumor was disordered and distributed unevenly. In addition, the new vessels in tumors were more twisted and uneven in thickness and sometimes, a node grew many branches of the blood vessel. These results suggested that in case of the ddG-Clip group, the vascular structure of tumor was very clear because the tumor growth rate was relatively slow in the entire process, and this was consistent with the histological slice of the tumor tissues.
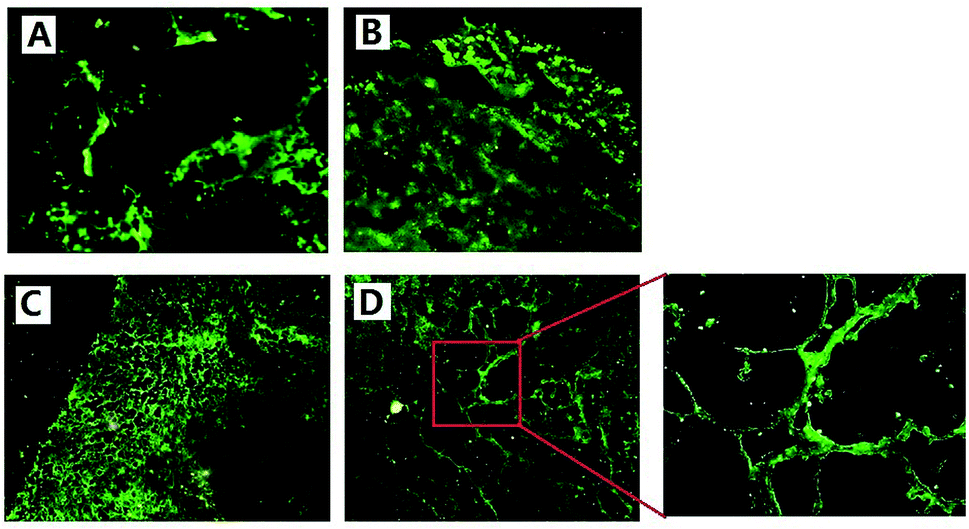 | ||
| Fig. 6 Frozen slice of tumor tissue in saline (A), ddG solution (B), ddG-Lip (C) and ddG-Clip (D) groups treated with the large molecule fluorescein FITC-dextran. Red region is further enlarged to observe the branching structure of the tumor vasculature. | ||
Conclusions
In this study, ddG-Clip with ideal physicochemical characteristics was able to specifically target the tumor cells and had significant inhibitory effects to tumor growth. Action mechanism for the enhanced antitumor efficacy could be explained by the following aspects (Fig. 7). (i) After intravenous injection, the positive charge of ddG-Clip could form the stronger adsorption water film, shield the hydrophobic binding site, reduce the recognition and uptake of nanoliposomes by RES, significantly prolong the circulation time in vivo and increase the accumulation into tumor sites via the EPR effect.30 (ii) The high zeta potential value on the surface of ddG-Clip also could facilitate the cellular uptake and internalization of the carriers; hence, the loaded ddG could be released into the cytoplasm and transported to the nucleus. (iii) Bases were added using RNA as template and by preferentially occupying the binding site of telomerase, ddG could replace guanine and interfere with the synthesis of telomere DNA. (iv) Eventually, the telomeres were shortened and tumor cell division was stopped, and the inhibiting effect of tumor growth was achieved. In other words, cationic nanoliposomes loading telomerase inhibitor could significantly suppress the growth and metastasis of tumor. This must open a new chapter in the field of tumor treatment.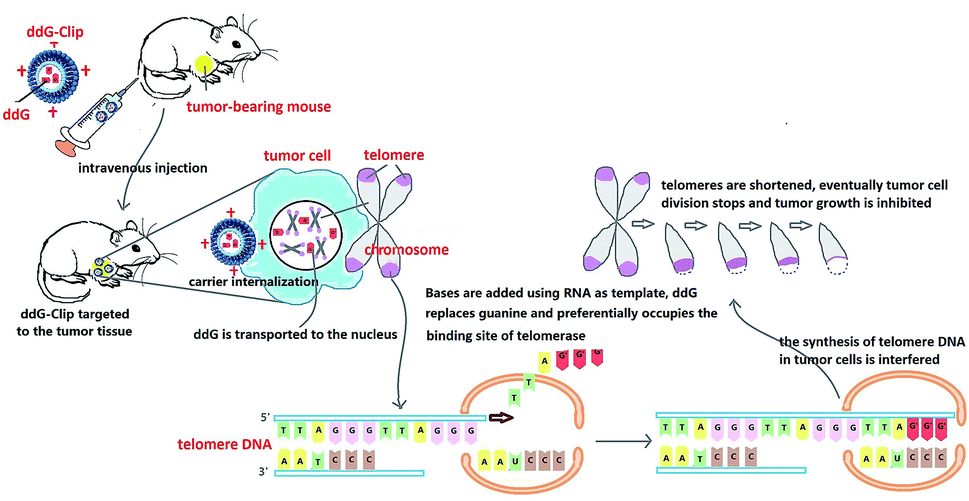 | ||
| Fig. 7 Schematic of ddG loaded cationic nanoliposomes as a tumor therapeutic carrier based on interfering DNA synthesis of telomere in tumor cells. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to financial support by Natural Science Foundation of Liaoning Province (No.201602696) and the National Natural Science Foundation of China (Grant No.81302721, 81202483, 81773668).References
- S. E. Artandi and R. A. DePinho, Carcinogenesis, 2010, 31, 9–18 CrossRef PubMed.
- M. C. Bibby, Mol. Biotechnol., 2003, 24, 295–301 CrossRef PubMed.
- M. Daniel, G. W. Peek and T. O. Tollefsbol, Gene, 2012, 498, 135–146 CrossRef PubMed.
- Y. N. Chen and Y. M. Zhang, Pharmacol. Ther., 2016, 163, 24–47 CrossRef PubMed.
- Y. J. Park and E. K. Kim, Cancer Lett., 2016, 370, 222–231 CrossRef PubMed.
- Y. J. Park, E. K. Kim and S. Moon, Anticancer Res., 2014, 34, 6389–6395 Search PubMed.
- M. Ruden and N. Puri, Novel anticancer therapeutics targeting telomerase, Cancer Treat. Rev., 2013, 39, 444–456 CrossRef PubMed.
- J. W. Shay and W. N. Keith, Br. J. Cancer, 2008, 98, 677–683 CrossRef PubMed.
- A. V. Schepdael, N. Ossembe, L. Jie and P. Herdewijn, et al., Int. J. Pharm., 1991, 73, 105–110 CrossRef.
- P. J. Pockros, Clin. Liver Dis., 2013, 17, 105–110 CrossRef PubMed.
- A. P. Cunningham, W. K. Love and R. W. Zhang, et al., Curr. Med. Chem., 2006, 13, 2875–2888 CrossRef PubMed.
- H. Chen, Y. Li and T. O. Tollefsbol, Mol. Biotechnol., 2009, 41, 194–199 CrossRef PubMed.
- A. W. Philip, R. J. Mark and V. Gouverneur, Eur. J. Med. Chem., 2017, 125, 117–129 CrossRef PubMed.
- C. O. Marian, W. E. Wright and J. W. Shay, Int. J. Cancer, 2010, 127, 321–331 Search PubMed.
- S. T. Yu, L. Chen and H. J. Wang, Int. J. Oncol., 2009, 35, 329–336 CrossRef PubMed.
- G. Shim, M. G. Kim and J. Y. Park, Asian J. Pharm. Sci., 2013, 8, 72–80 CrossRef.
- N. f. Sun, Z. A. Liu and W. B. Huang, Crit. Rev. Oncol. Hematol., 2014, 89, 352–357 CrossRef PubMed.
- L. George, H. Elizabeth and L. George, Neuroimmune Biol., 2005, 5, 95–101 Search PubMed.
- K. Greish, Drug Discovery Today: Technol., 2012, 9, 161–166 CrossRef PubMed.
- A. K. Iyer, G. Khaled and J. Fang, Drug Discovery Today, 2006, 11, 812–818 CrossRef PubMed.
- S. Y. Yang, J. Y. Chen and D. Zhao, Int. J. Pharm., 2012, 434, 155–160 CrossRef PubMed.
- J. Li, X. L. Wang and T. Zhang, et al., Asian J. Pharm. Sci., 2015, 10, 81–98 CrossRef.
- Y. Zhang, H. M. Li, J. Sun and J. Gao, et al., Int. J. Pharm., 2010, 390, 198–207 CrossRef PubMed.
- D. Yoshida, H. Todo and T. Hasegawa, Int. J. Pharm., 2008, 356, 181–186 CrossRef PubMed.
- Y. Maitani, S. Igarashi and M. Sato, et al., Int. J. Pharm., 2007, 342, 33–39 CrossRef PubMed.
- S. Y. Yang, Y. Zheng and J. Y. Chen, et al., Colloids Surf., B, 2013, 101, 6–13 CrossRef PubMed.
- Y. Yoshizaki, E. Yuba and N. Sakaguchi, Biomaterials, 2014, 35, 8186–8196 CrossRef PubMed.
- P. P. Qi, M. Cao and L. J. Song, et al., Environ. Toxicol. Pharmacol., 2016, 47, 159–164 CrossRef PubMed.
- N. Gupta, A. A. Patel and R. Nassar, et al., Colloids Surf., A, 2004, 245, 137–142 CrossRef.
- A. A. Natfji, D. Ravishankar and H. M. I. Osborn, et al., Journal of Pharmaceutical Sciences., 2017, 106, 3179–3187 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |