DOI:
10.1039/C8RA02959H
(Paper)
RSC Adv., 2018,
8, 22799-22805
Low-cost CoFe2O4/biomass carbon hybrid from metal-enriched sulfate reducing bacteria as an electrocatalyst for water oxidation†
Received
6th April 2018
, Accepted 28th May 2018
First published on 21st June 2018
Abstract
The development of electrocatalysts for the Oxygen Evolution Reaction (OER) requires extensive and challenging research for the water splitting and fuel cell applications. Herein, we report a low-cost CoFe2O4/biomass carbon (CFO@BC/Zn) hybrid from Co-enriched Sulfate Reducing Bacteria (Co-SRB) as an electrocatalyst for OER. The electrocatalyst exhibits a low potential of 1.53 V at a current density 10 mA cm−2 and Tafel slope of 86 mV dec−1. This method does not require high-cost or long periods of preparation. The density-functional theory (DFT) calculations show a small barrier for oxygen conversion on Fe3+ of CFO (100) surface. The synthesis of CFO@BC/Zn may be a new approach for obtaining low-priced electrocatalysts for OER.
Introduction
The OER is of great significance for the development of water splitting technology due to its ability to produce green and renewable energy in the form of O2 from water.1–3 OER electrocatalysts exhibiting poor performance hinder the reaction process of the Hydrogen Evolution Reaction (HER) at the cathode of a (photo)-electrochemical cell.4,5 Moreover, OER electrocatalysts play a key role in improving the performance of the fuel cells6,7 and metal–air batteries.8 In the field of catalysts, the best catalysts for the OER have been found to be IrO2 and RuO2;1,9,10 however, they are not extensively utilized due to their high costs.4,11,12 Another problem is their poor durability during long-term electrochemical processes because of nanoparticle migration, coalescence, and even detachment from the support materials in both acidic and alkaline electrolytes.13 Effective strategies for lowering the overpotential, such as improving the performance of the catalyst, engineering the surface of the active sites and increasing the surface area of the catalyst, have been reported in the literature. Numerous catalysts have been explored in previous studies, which aimed toward the development of a new catalyst with low over-potential and long-term stability. As OER catalysts, spinel oxides, such as Mg–Co3O4,9 N-CG-CoO14 LiCoO2,15 NiFeOx,16 Co3O4/N-rmGO,17 CFO/NS-rGO8 and Mono-NiTi-MMO,18 were designed or discovered due to their variable valence states.19 Although their low electrical conductivity20 limited the catalytic performance, the proper band gap21 ensured that the catalyst exhibited good catalytic performance. For example, CoMnO@CN,21 a nanoparticle superlattice, exhibits excellent OER performance, with an onset potential of 1.46 V vs. RHE and a high current density of 308 mA cm−2 at 1.65 V vs. RHE. According to the reaction mechanism, the transition metal oxides except IrO2 undergo the process of OOH* formation,22 which is a limited step for the reaction process. The reaction processes are as follows:| | |
OH* + OH− → O* + H2O + e−
| (2) |
| | |
OOH* + OH− → O2 + H2O + e−
| (4) |
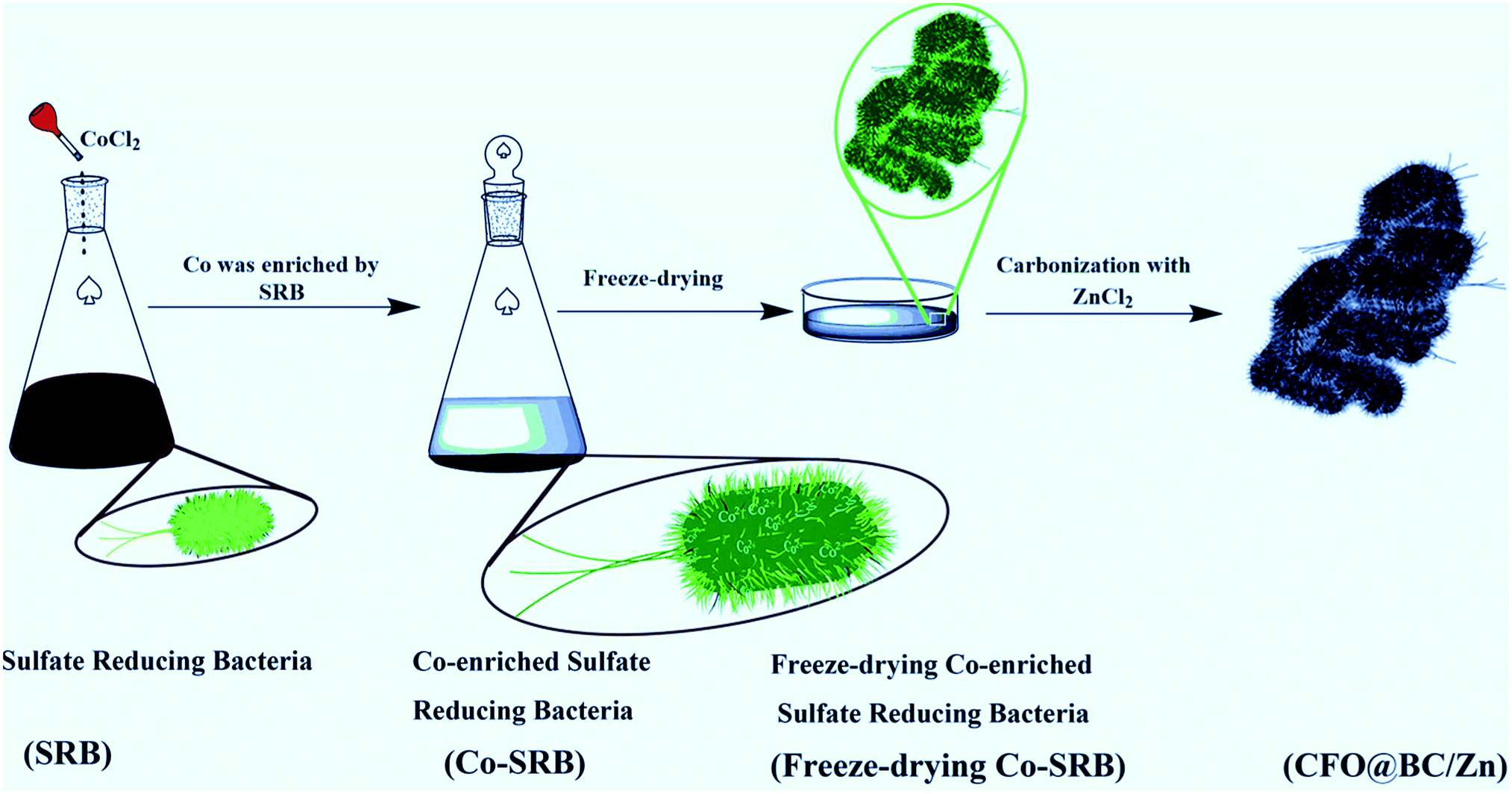
Although most of these spinel oxides above are excellent catalysts, they are prepared by chemical synthesis with complex preparation methods, which require high device costs or long preparation periods. Thus, an electrocatalyst exhibiting superior performance, low cost and a feasible process of industrial production is quite desirable. Therefore, the combination of a spinel transition metal oxide and nitrogen-doped carbon material can be an effective strategy for obtaining efficient water splitting catalysts. To decrease the costs of water splitting catalysts and achieve a feasible production process, we were inspired by the enrichment of metals from Sulfate Reducing Bacteria (SRB). SRB are siderophilic bacteria, and they act as a carbon source rich in C, N, O, S and other trace elements;23 they were selected as a subject for research, particularly because of their unique property – orientated-enrichment of metals. FeS2 was synthesized in the biological process and has been investigated as an electrocatalyst for HER.24 On the other hand, due to the formation of delocalized electrons around oxygen vacancies,25,26 cobalt oxide is also an excellent catalyst for OER. CoCl2 was added in the bacteria to get Co-enriched Sulfate Reducing Bacteria (Co-SRB), and this sample was designed as a precursor for the water splitting catalyst (Scheme 1). Co-SRB was then carbonized with ZnCl2 (denoted as CFO@BC/Zn), and Co-SRB was carbonized in the absence of ZnCl2 (denoted as CFO@BC); the carbonized SRB without CoCl2 was denoted as the blank group (BG). Most importantly, we found that SRB has the potential to be industrialized because 2.36 g of the carbonation hybrid was obtained from 500 mL SRB solution. Moreover, a large number of hybrids could be obtained in a short time because of the simple culture method and unlimited reproduction ability of SRB. Similarly, SRB has the ability to be enriched with other transition metals and thus, it is essential to explore SRB as an electrocatalyst.
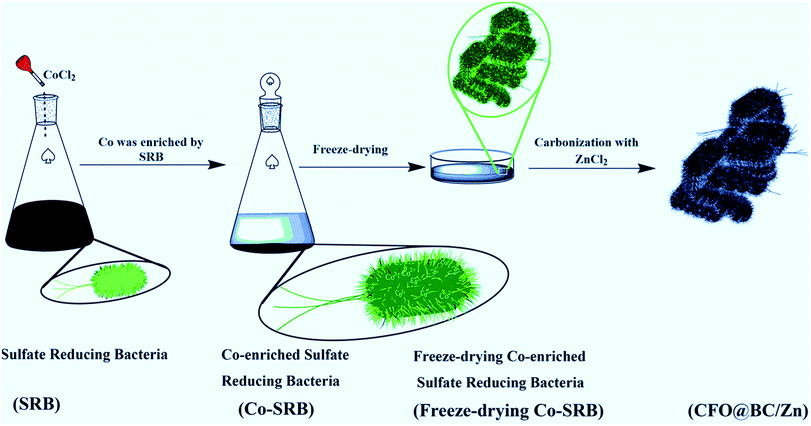 |
| | Scheme 1 Preparation process of CFO@BC/Zn, which shows that Co was enriched with SRB. | |
Experimental
Preparation of CoFe2O4 biomass carbon hybrid and Co3O4/N-rmGO, Co3O4/rmGO
First, 50 mL of CoCl2·6H2O solution was prepared at a concentration of 0.106 mol L−1. This solution was then added to 500 mL SRB culture medium with pH value of 7.8. Then, the resulting mixture was shaken well and set aside under anaerobic conditions for 3 days, which resulted in a high amount of precipitation at room temperature (Rt). Next, Co-SRB was obtained by freeze-drying the precipitated samples. To increase the surface area of biomass carbon materials, a Lewis acid (ZnCl2) was used to admix with freeze-dried bacteria, and the mixture carbonized under high temperature. The freeze-dried bacteria were carbonized at 900 °C for 3 h with ZnCl2 at a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 under protection from Ar atmosphere (100 sccm, 99.999%), and this was followed by cooling to Rt. After carbonization, the blank powder was bathed in H2SO4 solution (0.5 M) for 2 h to remove metal residues (such as Zn, Ca, and K). Afterwards, the powder was washed with ethanol and deionized water several times, dried in a loft drier at 80 °C for 24 h, and denoted as CFO@BC/Zn. The Co-SRB sample that was carbonized in the absence of ZnCl2 was denoted as CFO@BC, and the SRB sample carbonized without CoCl2 was denoted as the blank group (BG). IrO2 was purchased from Aladdin Co. Ltd, and the preparation of Co3O4/N-rmGO and Co3O4/rmGO was conducted according to the procedure reported in ref. 17.
:4 under protection from Ar atmosphere (100 sccm, 99.999%), and this was followed by cooling to Rt. After carbonization, the blank powder was bathed in H2SO4 solution (0.5 M) for 2 h to remove metal residues (such as Zn, Ca, and K). Afterwards, the powder was washed with ethanol and deionized water several times, dried in a loft drier at 80 °C for 24 h, and denoted as CFO@BC/Zn. The Co-SRB sample that was carbonized in the absence of ZnCl2 was denoted as CFO@BC, and the SRB sample carbonized without CoCl2 was denoted as the blank group (BG). IrO2 was purchased from Aladdin Co. Ltd, and the preparation of Co3O4/N-rmGO and Co3O4/rmGO was conducted according to the procedure reported in ref. 17.
Characterization
The blank hybrid powder comprised a variety of elements such as N, P, S and other trace elements. The X-ray photoelectron spectra (XPS) were obtained with the Casa XPS software using a magnesium anode (Al 1486.69 eV) X-ray source. The C 1s line at 284.5 eV was used to correct all XPS spectra with curve fitting and background subtraction. The X-ray diffraction (XRD) patterns of the hybrid was recorded on a X-ray diffractometer (DX-2700) with Cu Kα radiation (λ = 1.540 Å) over the Bragg angles ranging from 10° to 70°. The CFO@BC/Zn hybrid was mapped by a Raman microscope (Lab RAM-HR, France) with an excitation of 532 nm laser. Transmission electron microscopy (TEM) and high resolution transmission electron microscopy (HRTEM) (Zeiss Libra 200FE Germany) characterizations were performed at an accelerating voltage of 200 kV. The morphologies of Co-SRB and CFO@BC/Zn were observed by SEM (JSM-7500F, Japan). Energy dispersive X-ray spectroscopy (EDS) of CFO@BC/Zn was mapped by SEM (JSM-7500F, Japan).
Electrochemical measurement
OER was performed on the CHI 760E electrochemistry workstation with a standard three-electrode system in 1 M NaOH solution saturated with N2 (poured into nitrogen gas for half an hour). Considering that the CFO@BC/Zn hybrid was in a powdered form, a classical method was used in which Nafion was used as a film for attaching the catalyst on a Glass Carbon Electrode (GCE) 3 mm in diameter (0.071 cm−2). The GCE was polished by 30 nm Al2O3 to remove the oxidized material on the surface. Five mg powder was scattered in 1 mL isopropanol/water (volume ratio 1:3) and 35 μL 5 wt% Nafion solution with 2 h sonication to form a homogeneous ink. Then, the 5 μL mixed ink was placed on GCE and dried under room temperature (loading of 0.340 mg cm−2). The Pt sheet (2 × 2 cm2) was used as the counter electrode. To eliminate the impact of oxides present on the surface of Pt, a counter electrode was immersed in 0.5 M H2SO4 for 20 min and then, 5 min sonication was conducted in ethanol/water solution. Afterwards, the material was washed with deionized water several times. An HgO/Hg electrode was used as the reference electrode. Linear sweep voltammetry (LSV) was performed in a three electrode cell under a sweep rate of 5 mV s−1 between 1.0 V and 1.8 V vs. RHE for OER. IrO2 was scattered into ethanol with 5 wt% Nafion and pasted on GCE. An Ni foam (2 × 2 cm2) was used as the substrate to support the catalyst. Twenty mg catalyst and 2 mL polytetrafluoroethylene ethanol dispersion solution (1 mg mL−1) were mixed well by ultrasonicating for 2 h. Then, the uniform suspension was dropped on the Ni foam (hybrid loading of 2.5 mg cm−2 on Ni foam). To standardize the data, an equation was used as a bridge between the HgO/Hg reference electrode and reversible hydrogen electrode without any iR-correction: E (RHE) = E (HgO/Hg) + 0.0591 × pH + 0.098.
Theoretical calculations
In this paper, the Vienna ab initio simulation package (VASP) in the framework of the projector augmented wave (PAW) method was used for all calculations. The cut off for the plane waves set was 520 eV. Hubbard-like, localized term was added to the PBE generalized gradient approximation (GGA) exchange correlation functional, namely, GGA + U. The Ueff (U–J) value of 4.5 eV was applied for both Co3d and Fe3d states. All 2 × 2 × 2 supercells with 4 × 4 × 2 mesh of k-points were used to calculate the electronic structure of CFO. For calculations of adsorption energy, (100) surfaces were modeled using symmetric slabs of one layer with vacuum widths of 10 Å. All atoms were fully relaxed with the energy convergence tolerance of 10−6 eV per atom, and the final force on each atom was <0.01 eV Å−1.
Results and discussion
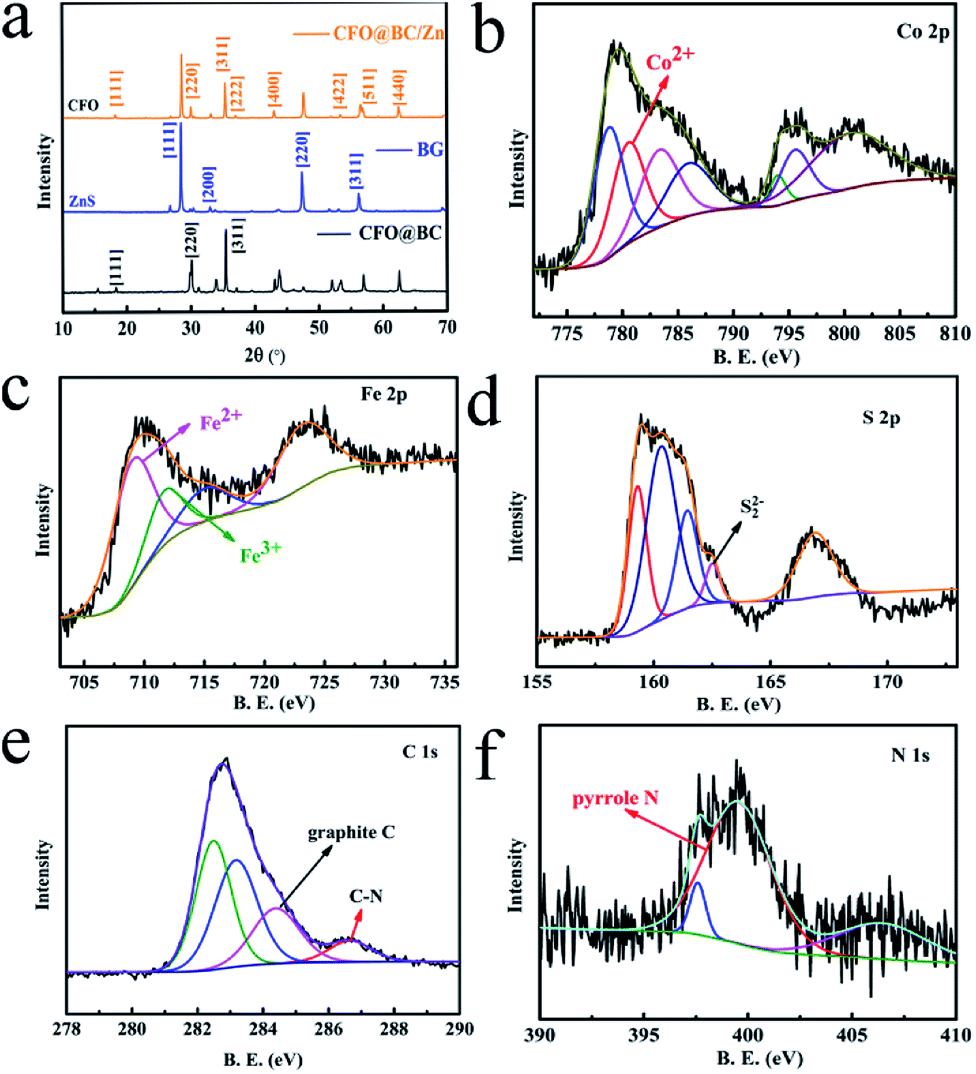
The transition metal Co was enriched by SRB, and CoFe2O4 (CFO) was found in CFO@BC/Zn. Analysis of the XRD patterns showed that the peaks of CFO appeared for CFO@BC/Zn and CFO@BC (Fig. 1a), and according to the OER performances between CFO@BC/Zn and BG, we observed that superior catalytic performance was induced by CFO. The X-ray diffraction (XRD) patterns of CFO@BC/Zn, CFO@BC and BG were indexed to the (220) and (311) planes of the orthorhombic to spinel (Fd3m). Comparing the OER activity of CFO@BC/Zn with that of BG, the catalytic activity absolutely came from CFO as shown in Fig. 1a. CFO (JCPDS no. 22-1086) showed a single reflection around 2θ ≈ 20.09°, 35.44° corresponding to CFO. However, there were no characteristic peaks of C in the XRD pattern, which indicated that the contents of graphitic carbon and N-doped carbon were too low to be detected. Fortunately, the Raman spectrum exhibited D band and G band at 1340 cm−1 and 1595 cm−1 (Fig. s1†), respectively, which meant that the carbonation of SRB was successful. Furthermore, the presence of CFO@BC/Zn hybrid was confirmed by XPS (Fig. 1b and s2†). Significant two core-level signals of Co of CFO@BC/Zn hybrid located at ∼780 and 796 eV were assigned to Co2p3/2 and Co2p1/2, respectively. The high-resolution deconvoluted Co2p spectrum indicated that the peak of Co2+ located at 779.9 eV was the characteristic peak of CFO. Analysis of the XPS data of Fe (Fig. 1c) indicated the presence of substantial Fe3+ species, which was confirmed from the CFO peak located at 710.4 eV and the Fe2+ peak located at 709.1 eV. Additionally, the main state of S was found to be S2− in the hybrid, and a small amount of S22− was confirmed by the peak located at 164.6 eV (Fig. 1d). This indicated that ZnS was the main form of S2− in the hybrid. Likewise, this also explained why the XRD pattern did not show clear characteristic peaks of FeS2. The XPS data of C1s and N1s are shown in Fig. 1e and f, respectively. The presence of graphitic carbon, which can increase conductive ability, was also confirmed by the XPS data. Moreover, pyrrolic nitrogen was found in CFO@BC/Zn (Fig. 1f). Previous studies have shown that the pyrrolic nitrogen plays a key role in the catalytic process in the presence of various nitrogen forms.27 It was believed that the incorporation of nitrogen caused an increase in the degree of delocalization of charge such that the adsorption state of O2 was changed.
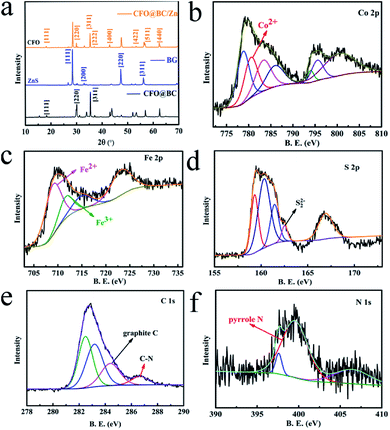 |
| | Fig. 1 (a) XRD patterns of CFO@BC/Zn hybrid and BG; (b), (c), (d), (e) and (f) high-resolution deconvoluted Co2p, Fe2p, S2p, C1s and N1s spectrum, respectively, of XPS data. | |
The activity of the catalyst was affected by porous surface characteristics, long-range structure and controllable material composites.20 Scanning electron microscopy (SEM) images show porous structure and rough surfaces of freeze-dried Co-SRB and CFO@BC/Zn (Fig. 2a and b, respectively). The porous and uniform surface exposed more active sites, which essentially increased the adsorption opportunities between OH−/H2O and the active sites. Energy dispersive X-ray spectroscopy (EDS) elemental mapping by SEM was employed to obtain elemental distribution of Co, Fe, O, S, C and Zn in the hybrid, verifying the uniform dispersion of five elements (Fig. s3†). Likewise, a mesoporous structure was seen on the CFO crystal in TEM images (Fig. 2c and f). There was no doubt that this porous CFO helped oxygen generation and precipitation. To further examine the crystal structure of as-made CFO, HRTEM analysis and Fast Fourier Transformation (FFT) were carried out, and a typical image for the hybrid is shown in Fig. 2d. Crystal lattice fringes could be clearly observed, suggesting the highly crystalline nature. The distances between two fringes in Fig. 2d and h were approximately 0.254 and 0.297 nm, corresponding to distances of the (311) and (220) planes of CFO spinel. Also, the presence of ZnS in the mixture was confirmed by TEM and HRTEM images, as shown in Fig. s4.†
 |
| | Fig. 2 (a) SEM images of the freeze-dried Co-SRB; (b) SEM images of CFO@BC/Zn; (c) and (f) are TEM images, which exhibit a mesoporous structure, as shown on the CFO crystal; (d), (e), (g) and (h) HRTEM and its FFT image of CFO. | |
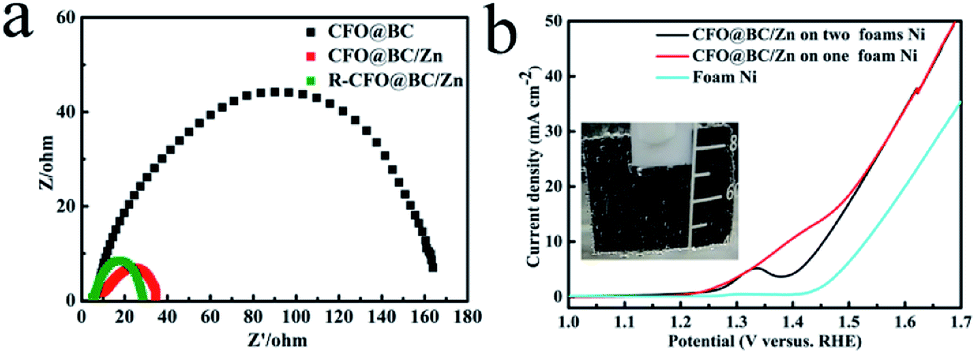
As the electrocatalyst obtained from SRB, CFO@BC/Zn hybrid was first evaluated by linear sweep voltammetry (LSV) on the CHI 760E electrochemistry workstation with a standard three-electrode in 1 M NaOH solution saturated with N2. As expected, BG did not exhibit OER performance. CFO@BC/Zn showed excellent performance with a low potential of 1.53 V when it was supported on GC (0.340 mg cm−2), and a low potential of 1.45 V was obtained at a current density 10 mA cm−2 when it was supported on the Ni foam (Fig. 4b); the small specific surface areas of CFO@BC/Zn (3.51 m2 g−1) are shown in Fig. s5.† This performance was superior to those of the currently used Co3O4/N-rmGO and Co3O4/rmGO (Fig. s6c†). The cyclic voltammetry (CV) and time dependence of the current density under a constant potential were used to check the stability of the CFO@BC/Zn hybrid (Fig. 3d and s6b†). The long-term stability of CFO@BC/Zn was reflected in the time dependence of the current density under a constant potential. In theory, as the O2p states pass through Fermi level, the central O p-band moves closer to the Fermi level, which was believed to be the reason of this inverse spinel CFO for the decrease.28 The decline of system may be due to electrochemical deposition. However, the activity of the CFO@BC/Zn hybrid was retained after performing OER with oxygen for 24 hours, and it was better than that of commercial IrO2 (Fig. s6a†). To increase the activity of CFO@BC/Zn hybrid, it was activated by cathodic treatment13,29 at an overpotential of −500 mV vs. RHE for 24 h (R-CFO@BC/Zn). The potential was reduced to 1.47 V at a current density of 10 mA cm−2, which was smaller than 1.57 V for IrO2, and the potential was found to be 1.63 V when the current density was 100 mA cm−2 (Fig. 3b). To the best of our knowledge, this potential was excellent compared to those of the other OER catalysts reported previously (Table s1†). Importantly, an outstanding electrocatalyst possesses not only a low overpotential, but also a small Tafel slope.30,31 The CFO@BC/Zn hybrid exhibited a small Tafel slope of 86 mV dec−1 (Fig. 3c), which was smaller than 92 mV dec−1 of IrO2 and close to the reported value of 95 mV dec−1 in alkaline medium.32 After cathodic treatment, the Tafel slope of R–CFO@BC/Zn deceased to 47 mV dec−1. The reason for this phenomenon was the increase in the contents of reduced metals such as Co2+ and Fe2+.13 These reduced metals degraded electrochemical impedance in the reaction process. As is shown in Fig. 4a, the impedance of R-CFO@BC/Zn decreased to 28 Ω (CFO@BC/Zn: 34 Ω). The excellent electrochemical activities of R-CFO@BC/Zn, CFO@BC/Zn and CFO@BC can thus be understood in terms of the significantly decreased charge-transfer resistance (Fig. 4a). Due to small resistance, the catalysts exhibited a low initial potential. After further evaluation of these catalysts, we found that CFO@BC/Zn exhibited a low initial potential 1.47 V (0.5 mA cm−2); the initial potentials of CFO@BC and R-CFO@BC/Zn were 1.52 V and 1.45 V, respectively.
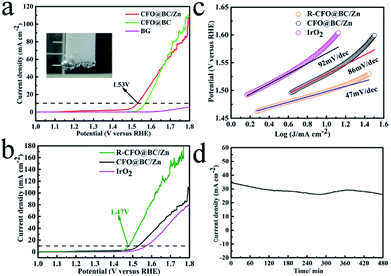 |
| | Fig. 3 (a) LSV curves of CFO@BC/Zn, CFO@BC and BG; (b) LSV curves of R-CFO@BC/Zn, CFO@BC/Zn and IrO2; (c) Tafel slopes of R-CFO@BC/Zn, CFO@BC and IrO2; (d) time dependence of the current density under a constant potential 1.53 V of CFO@BC/Zn hybrid. | |
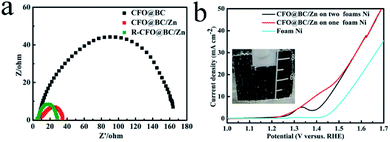 |
| | Fig. 4 (a) Electrochemical impedance spectra for R-CFO@BC/Zn, CFO@BC/Zn and CFO@BC at 1.55 V vs. RHE between 0.01 Hz to 100000 Hz (b) LSV curves of CFO@BC/Zn on the Ni foam. | |
The catalytic ability of CFO@BC/Zn hybrid on Ni foam was evaluated in 1 M NaOH solution by LSV at potential between 0 V and 0.8 V vs. HgO/Hg for water splitting. Under small loading on Ni foam, the CFO@BC/Zn hybrid still exhibited an excellent OER performance with an overpotential of 220 mV at a current density 10 mA cm−2, which was superior to that of R-CoOx@CN13 under approximate test conditions (Fig. 4b). Fig. 4b shows OER of the supported catalyst with Ni foam at 1.48 V. The current density did not change much for the high-current oxygen precipitation process on the Ni foam.
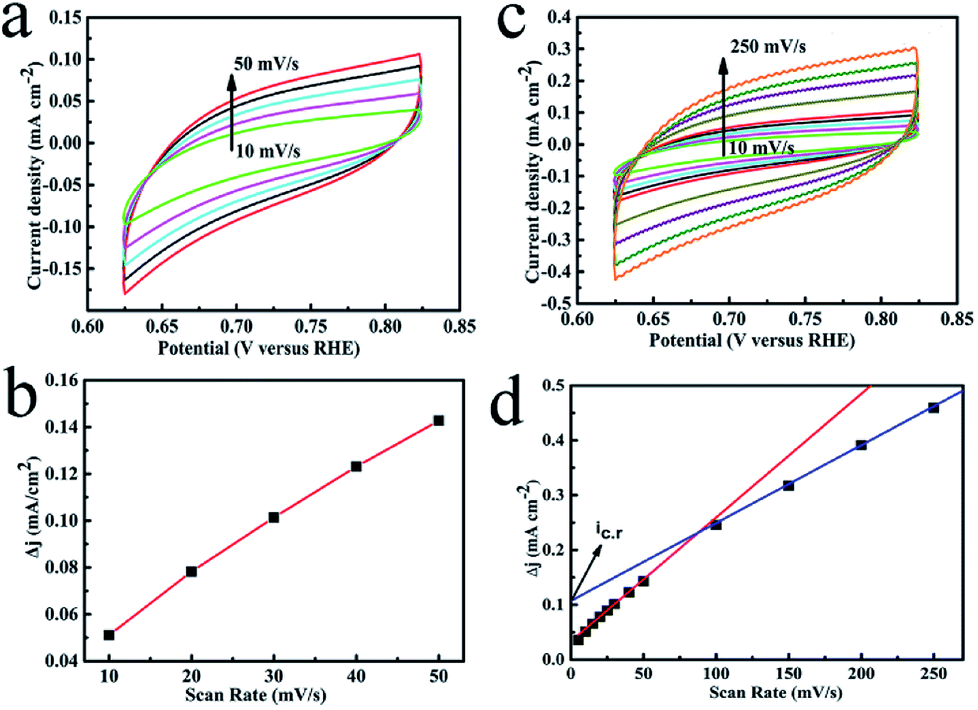
It is generally believed that N-doped thin carbon or high specific surface biomass carbon obtained from Lewis acid (ZnCl2) are susceptible to electron permeation and transfer.33 Electrochemical active surface area (ECSA) was used to study the size of surface areas per mass of materials, which participate in the electrolysis for OER. Thus, cyclic voltammetry measurements (CV) were conducted by sweeping the potential across the non-faradic region to rationally evaluate the hybrid. The double layer capacitance of the catalyst was enhanced due to ZnCl2: 1.25 mF cm−2 of CFO@BC/Zn and 0.63 mF cm−2 of CFO@BC (Fig. 5b and s7†). However, the ECSA did not reflect the effect of Zn on electrocatalytic activity. In accordance with the electrochemical double layer theory, hydrophilic groups of biomass carbon were assembled on the surface of CFO (inner layer) in the double layer capacitance because of vacant 3d orbitals from Fe or Co. However, Zn has no vacant 3d orbital, and doped Zn or ZnS will decrease the assembly of hydrophilic groups on the surface of CFO. The reason for this phenomenon is that the surface charging of conductive metallic oxide is much more complex, and it also depends on the oxide through a proton injection/ejection process involving the solid-state surface redox transition.34 Since we could not obtain the value of the theoretical double-layer capacitance of the CFO@BC/Zn hybrid, morphology factor (φ), a more accurate value, was used to characterize the active sites for metallic oxide. In short, the penetration of electrons in the double capacitor was assumed to be ideal so that the differential capacity for the external capacitor (Ce) and the internal capacitor (Ci) could be taken into account respectively (details in ESI†). The roughness factor was 0.391 for CFO@BC (Fig. 5d) and 0.475 for CFO@BC/Zn (Fig. S6†). This showed that the effect was a decrease in Ci on ECSA by Zn. Other improvement in icr (scan rate → 0) that represented limited diffusion current was enhanced from 0.035 mA cm−2 (CFO@BC) to 0.111 mA cm−2 (CFO@BC/Zn). This indicated an easy diffusion of ions on the CFO@BC/Zn surfaces. Thus, the three advantages due to the presence of Zn were enlarged ECSA, diminished internal capacity and enhanced limited diffusion current; thus, it was easy for the electrons to enter the electrochemical double layer and move to OH−. To check the effects of Zn, we added an appropriate amount of sodium dodecylsulfate (SDS) to reduce the surface tension, and we produced O2 easily through hydrophobic groupings of Ce. The potential was also reduced to 1.42 V at 10 mA cm−2 (Fig. s8†). This electrocatalyst derived from biological bacteria provided a new way for oxygen evolution reaction, and the research obstacles can hopefully be solved by simple preparation conditions, low costs and high activity.
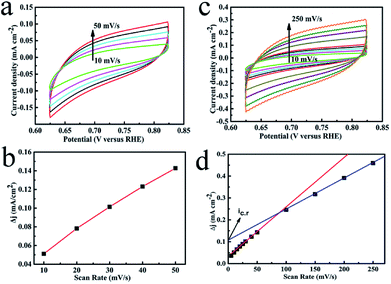 |
| | Fig. 5 (a) CV curves of CFO@BC/Zn from 10 to 50 mV s−1; (b) charging current density differences (Δj = ja − jc) at 0.724 V vs. RHE; (c) CV curves of CFO@BC/Zn from 10 to 250 mV s−1; (d) dependence of the capacitive voltammetric current (Δj = ja − jc) at 0.724 V vs. RHE. | |
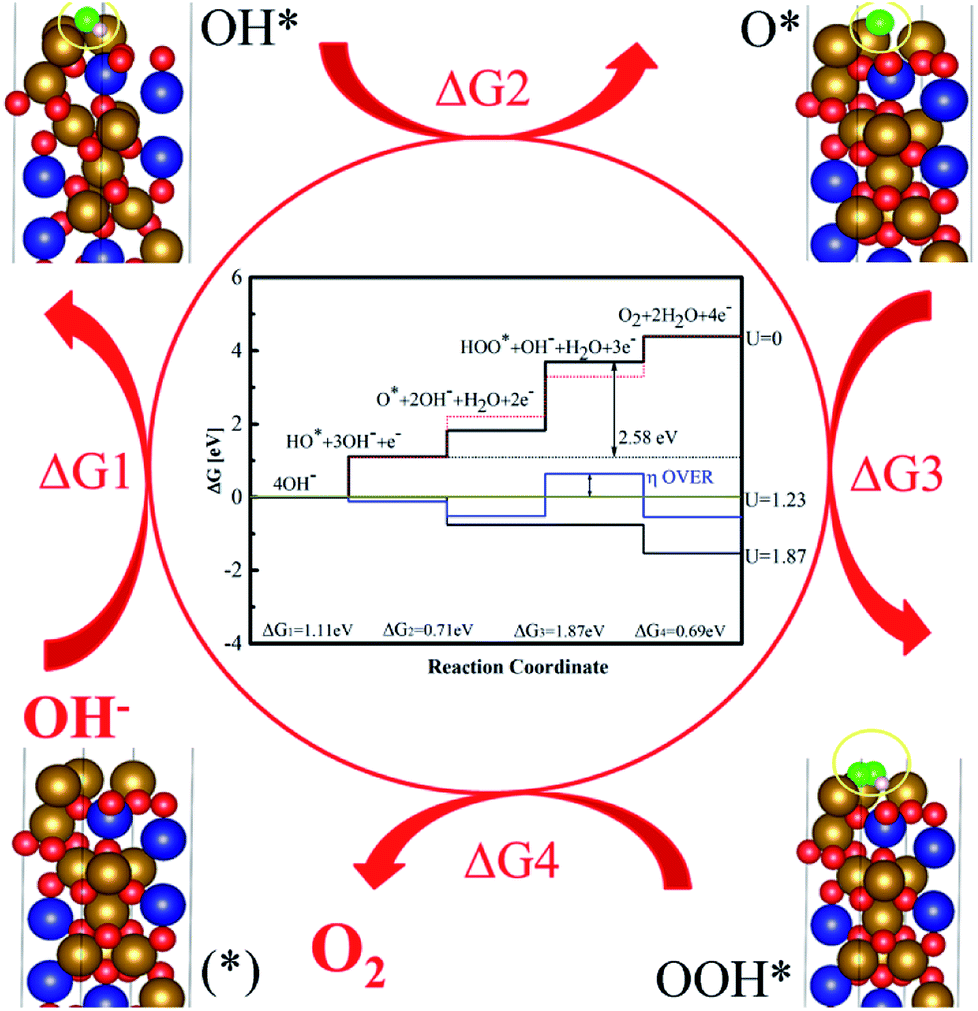
To gain better understanding of the superior catalytic performance from the quantum chemical properties of CFO, density-functional theory (DFT) calculations were performed using model structures in supercells. After the system was optimized, we found that the energy of the (100) surface was lower than those of the (110) and (111) surfaces, which indicated that the (100) surface was the optimum surface for reaction. On the (100) surface, we found that OH− ions move to Fe3+ ions with 1.96 Å Fe–O bond, though 2.08 Å hydrogen bond emerged before optimization (Fig. 6). It was indicated that Fe3+ was the active site of the water oxidation because of valence states. The adsorption energies of OH− and O2 of CFO on the (100) surface were 1.291 eV (chemical adsorption) and 0.479 eV (physical adsorption), respectively, which suggested that OH− was likely to be adsorbed and O2 was easily desorbed on the (100) surface of CFO (Fig. s10†). Correspondingly, the distances of O2 and OH− to the crystal plane (100) were 2.15 Å and 2.04 Å, respectively. (details in EIS†).
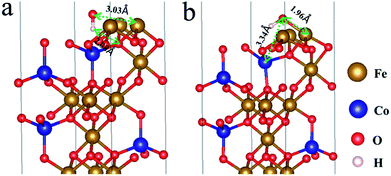 |
| | Fig. 6 (a) Adsorption of OH− on the (100) surface of CFO before the system was optimized; (b) adsorption of OH− on the (100) surface of CFO after the system was optimized. | |
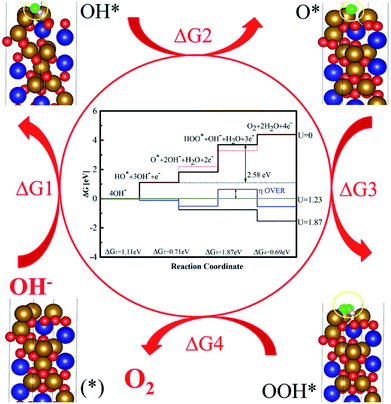 |
| | Fig. 7 Free energy diagram for the dissociative mechanism of oxygen evolution reaction on the Fe3+ of CFO (100) surface. | |
Reaction mechanism and density of states are important ways to reveal a reaction pathway. The total densities of states (DOSs), Co3d, Fe3d and O2p of CFO were plotted (Fig. s9†). As for CFO, there was a narrow bandgap of 1.79 eV. This narrow bandgap led to the enhanced excitation of charge carriers to the conduction band. A coordination to the formation of OOH* was limited step in four steps of the reaction mechanism and the DFT overpotential of CFO on crystal plane (100) was 0.64 V, which was higher than the experimental value. It was believed that the formation of OOH* in the reaction was the limited step, which had a coordination energy of 1.87 eV (Fig. 7). The theoretical enhancement in the experimental value was observed due to the increased conductivity from the biomass carbon and other experiments from CNTs, GO and graphite carbon. Moreover, on the catalytic sites of Fe3+, the oxygen molecule was adsorbed in a bridge position between Co or Co and Fe atoms with an activated oxygen–oxygen bond of 1.34 Å of OOH*, which indicated a preference of the CFO phase for dissociative 4e− mechanism of OER with minimal production of H2O2.4
Conclusions
In this study, the hybrid catalyst CFO@BC/Zn has been prepared successfully from sulfate reducing bacteria by enrichment of Co, and it is an excellent catalyst for water oxidation. The calculated results of the adsorption energies of OH− and O2 for the OER active sites of CFO show a small barrier for oxygen conversion. Thus, the hybrid catalyst exhibits excellent performance. Most importantly, the simple preparation conditions and low cost makes SRB promising as a new source for the development of clean energy technology and industrialized development to further promote water splitting technology and fuel cells. The necessary efforts should be made to understand the catalytic mechanism and the mechanism of the biological preparation in future studies, as much is unknown regarding the biological preparation of the catalyst.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (2017GZ0268). We especially thank the culture technology of SRB for supporting this work by Senior Engineer Fude Li.
Notes and references
- G. Yelena and F. J. Thomas, J. Am. Chem. Soc., 2010, 132, 13712 Search PubMed
 .
. - M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu and J. Zhou, J. Am. Chem. Soc., 2013, 135, 8452 CrossRef PubMed .
- M. Favaro, W. S. Drisdell, M. A. Marcus, J. M. Gregoire, E. J. Crumlin and J. A. Haber, ACS Catal., 2017, 7, 1248 CrossRef .
- K. Fan, H. Chen, Y. F. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. Li, Y. Luo and L. C. Sun, Nat. Commun., 2016, 7, 11981 CrossRef PubMed .
- Y. F. Li and A. Selloni, ACS Catal., 2014, 4, 1148 CrossRef .
- A. Serov, N. I. Andersen, A. J. Roy, I. Matanovic, K. Artyushkova and P. Atanassov, Journal of the Electrochemical Society, 2015, 162, F449 CrossRef .
- J. T. Zhang, Z. H Zhao, Z. H Xia and L. M. Dai, Nat. Nanotechnol., 2015, 10, 1038 Search PubMed .
- W. Yan, X. Cao, J. Tian, C. Jin, K. Ke and R. Yang, Carbon, 2016, 99, 195 CrossRef .
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399 CrossRef PubMed .
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef PubMed .
- J. Rosen, G. S. Hutchings and F. Jiao, J. Am. Chem. Soc., 2013, 135, 4516 CrossRef PubMed .
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed. Engl., 2015, 54, 6251 CrossRef PubMed .
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688 CrossRef PubMed .
- S. Mao, Z. Wen, T. Huang, Y. Hou and J. Chen, Energy Environ. Sci., 2014, 7, 609 RSC .
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3949 CrossRef PubMed .
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef PubMed .
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang and T. Regier, Nat. Mater., 2011, 10, 780 CrossRef PubMed .
- Y. F. Zhao, X. D. Jia, G. B. Chen, L. Shang, G. I. N. Waterhouse, Li-Z. Wu, C.-Ho. Tung, D. O. ' Hare and T. Zhang, J. Am. Chem. Soc., 2016, 138, 6517 CrossRef PubMed .
- J. Bao, X. D. Zhang, B. Fan, J. J. Zhang, M. Zhou, W. L. Yang, X. Hu, H. Wang, B. C. Pan and Y. Xie, Angew. Chem., 2015, 127, 7507 CrossRef .
- Z. Chen, M. Waje, W. Li and Y. Yan, Angew. Chem., 2007, 46, 4060 CrossRef PubMed .
- J. Li, Y. Wang, T. Zhou, H. Zhang, X. Sun and J. Tang, J. Am. Chem. Soc., 2015, 137, 14305 CrossRef PubMed .
- M. Bajdich, M. García-Mota, A. Vojvodic, J. K. Nørskov and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 13521 CrossRef PubMed .
- R. Eugene, Nature, 2003, 421, 456 CrossRef PubMed .
- J. P. Gramp, J. M. Bigham, F. S. Jones and O. H. Tuovinen, J. Hazard. Mater., 2010, 175, 1062 CrossRef PubMed .
- J. Bao, X. D. Zhang, B. Fan, J. J. Zhang, M. Zhou, W. L. Yang, X. Hu, H. Wang, B. C. Pan and Y. Xie, Angew. Chem., 2015, 127, 7507 CrossRef .
- Y. F. Sun, S. Gao, F. C. Lei, J. W. Liu, L. Liang and Y. Xie, Chem. Sci., 2014, 5, 3976 RSC .
- K. Gong, F. Du, Z. Xia, D. Michael and D. Liming, Science, 2009, 323, 760 CrossRef PubMed .
- A. Grimaud, K. J. May, C. E. Carlton, Y. L. Lee, M. Risch and W. T. Hong, Nat. Commun., 2013, 4, 2439 CrossRef PubMed .
- J. Duan, S. Chen, J. Mietek and S. Qiao, ACS Nano, 2015, 9, 931 CrossRef PubMed .
- X. Jia, Y. Zhao, G. Chen, L. Shang, R. Shi and X. Kang, Adv. Energy Mater., 2016, 6, 1502585 CrossRef .
- Q. Jiang, L. Xu, N. Chen, H. Zhang, L. Dai and S. Wang, Angew. Chem., 2016, 55, 13849 CrossRef PubMed .
- Y. Tong, J. Xu, H. Jiang, F. Gao and Q. Lu, Chem. Eng. J., 2017, 316, 225 CrossRef .
- L. Wei, H. E. Karahan, K. Goh, W. C. Jiang, D. S. Yu, Ö. Birer, R. R. Jiang and Y. Chen, J. Mater. Chem. A, 2015, 3, 7210 RSC .
- L. M. D. Silva, L. A. D. Faria and J. F. C. Boodts, Electrochim. Acta, 2001, 47, 395 CrossRef .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra02959h |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Yongyi Yaob
*a and
Yongyi Yaob