
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the spontaneous formation of hybrid PdOx/PEDOT films: electroless deposition and oxygen reduction activity†
Julian A. Vigil a,
Michael T. Brumbachb,
Jonathon Duaya and
Timothy N. Lambert*a
a,
Michael T. Brumbachb,
Jonathon Duaya and
Timothy N. Lambert*a
aDepartment of Materials, Devices & Energy Technologies, Sandia National Laboratories, Albuquerque, New Mexico 87185, USA. E-mail: tnlambe@sandia.gov; Tel: +1 505 284 6967
bMaterials Characterization & Performance, Sandia National Laboratories, Albuquerque, New Mexico 87185, USA
First published on 6th July 2018
Abstract
Hybrid palladium oxide/poly(3,4-ethylenedioxythiophene) (PdOx/PEDOT) films were prepared through a spontaneous reaction between aqueous PdCl42− ions and a nanostructured film of electropolymerized PEDOT. Spectroscopic and electrochemical characterization indicate the presence of mixed-valence Pd species as-deposited (19 ± 7 at% Pd0, 64 ± 3 at% Pd2+, and 18 ± 4 at% Pd4+ by X-ray photoelectron spectroscopy) and the formation of stable, electrochemically reversible Pd0/α-PdOx active species in alkaline electrolyte and furthermore in the presence of oxygen. The elucidation of the Pd speciation as-deposited and in solution provides insight into the mechanism of electroless deposition in neutral aqueous conditions and the electrocatalytically active species during oxygen reduction in alkaline electrolyte. The PdOx/PEDOT film catalyses 4e− oxygen reduction (n = 3.97) in alkaline electrolyte at low overpotential (0.98 V vs. RHE, onset potential), with mass- and surface area-based specific activities competitive with, or superior to, commercial 20% Pt/C and state-of-the-art Pd- and PEDOT-based nanostructured catalysts. The high activity of the nanostructured hybrid PdOx/PEDOT film is attributed to effective dispersion of accessible, stable Pd active sites in the PEDOT matrix.
Introduction
Overcoming the kinetic challenges of the electrocatalytic oxygen reduction reaction (ORR) is central to addressing some of the limitations of next-generation energy conversion devices, including the cathodic overpotential in fuel cells.1 Pt-based materials have been the benchmark for electrocatalytic ORR activity, but concerns regarding the abundance, cost, and stability of Pt have motivated the identification of Pt-free electrocatalyst materials.2 Furthermore, the enhanced stability of Pt-free materials and more facile kinetics in alkaline conditions, along with recent advances in anion exchange membranes, have revitalized interest in Anion Exchange Membrane Fuel Cells (AEMFCs).2 Thus, materials based on alternative transition metals, heteroatom-doped carbon, and transition metal oxides that exhibit promising ORR activity and stability in alkaline electrolytes have been developed.2–6As an emerging class of organic materials, conducting polymers (CPs) offer high electrical conductivity and, being polymeric in nature, may serve as a single replacement for both carbon and binders in composite electrode assemblies.7 In addition, catalytic activity discovered from CP-only electrodes such as polyaniline and poly(3,4-ethylenedioxythiophene) (PEDOT) has generated interest in developing CP-based electrocatalysts for the ORR.7–9 Winther-Jensen et al. first reported ORR activity competitive with Pt from a PEDOT air electrode prepared by vapor phase polymerization.9 Since then, studies have notably demonstrated that the ORR pathway on PEDOT-only electrodes is determined by the polymerization method10 and that an oxygen-induced doping mechanism may be responsible for the sustained conductivity of PEDOT at the highly reducing potentials of the ORR.11 PEDOT has also served as a conductive support in composite ORR electrocatalysts with semiconducting metal oxides, including CoMn2O4,12 Fe3O4,13 FeO,14 and MnOx.15,16 Our group has reported two approaches to prepare highly active MnOx/PEDOT composite electrocatalysts for the ORR: anodic co-electrodeposition15 and spontaneous reduction of MnO4− ions to form MnOx on PEDOT.16 The latter approach provided a composite film with improved specific activity and active site utilization for catalysing the ORR.16
In addition to first-row transition metal oxides, Pd-based electrocatalysts are promising alternatives to Pt-based AEMFC cathodes owing to similar electronic properties to Pt (i.e. d-band center) and superior electrochemical activity and stability in alkaline electrolytes.17,18 This may be attributed to reduced dissolution of oxidized Pd (compared to Pt) and suitable reversibility of Pd redox in the ORR potential window.19 Pd is also approximately 50 times more abundant on Earth than Pt.17
Here we report on PdOx/PEDOT films prepared by a spontaneous redox reaction between PdCl42− ions and a reduced PEDOT film in neutral aqueous solution (Fig. 1a), and the electrocatalytic activity of the hybrid films toward the ORR. Previously, spontaneous electroless deposition using Pd2+ ions and PEDOT has been reported exclusively in acidic aqueous conditions,20–22 and the chemical nature (e.g. Pd valence) of the as-deposited Pd species has not been studied. Such information regarding Pd speciation would provide insight across a wide range of conditions as Pd0, PdO, and PdO2 are all thermodynamically stable at pH > 2. For the first time, we elucidate the Pd speciation as-deposited in neutral aqueous conditions and in basic electrolyte to identify the deposition mechanism and active species involved in catalysing the ORR, respectively. The specific ORR activity of the hybrid PdOx/PEDOT film at low overpotential (0.8–0.9 V vs. RHE) is superior to commercial 20% Pt/C and competitive with state-of-the-art Pd/C and PEDOT-based nanostructured electrocatalysts at low Pd loading (∼5 wt%).
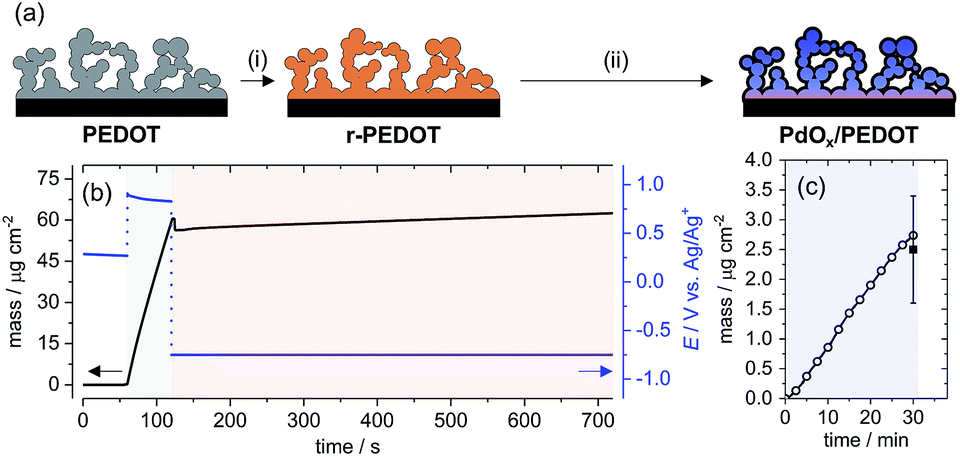 | ||
| Fig. 1 (a) Schematic representation of the preparation of PdOx/PEDOT: (i) reduction of electrodeposited PEDOT in CH3CN, (ii) reaction of r-PEDOT with PdCl42− in neutral aqueous solution; (b) working electrode potential and mass change (measured by QCM) during preparation of the r-PEDOT film: t = 0–60 s, open circuit potential; t = 60–120 s, electropolymerization; t = 120–720 s, PEDOT reduction; (c) mass change of a representative r-PEDOT film during electroless deposition of Pd species (circles) and average mass loading of Pd species after 30 min (square, 3 measurements). | ||
Experimental
Materials
Potassium tetrachloropalladate(II) (K2PdCl4, 99.99%), lithium perchlorate (LiClO4, 99.99%), 3,4-ethylenedioxythiophene (EDOT, 97%), potassium hydroxide (KOH, reagent grade >85%) and Nafion solution (5 wt% in lower alcohols) were obtained from Sigma-Aldrich. Acetonitrile (CH3CN, Certified ACS) and isopropanol (IPA, Certified ACS) was obtained from Fisher Scientific. Ethanol (EtOH, Certified ACS) was obtained from Pharmco-Aaper. Commercial 20% Pt/C (Vulcan XC-72) catalyst powder was obtained from E-TekSM. All materials were used as-received.Preparation of r-PEDOT and PdOx/PEDOT films
Glassy carbon (GC) electrodes were polished with 0.05 μm alumina slurry and rinsed with DI H2O and EtOH. A standard three-electrode cell was used for electropolymerization of PEDOT, including a GC working electrode (pyrolized GC electrodes on SiO2 substrates or commercial rotating ring disk working electrodes), graphite rod counter electrode, and Ag/Ag+ reference electrode (polished Ag wire, 10 mM AgNO3/0.1 M LiClO4, CH3CN). The cell was controlled by a Solartron Analytical 1287 Potentiostat Galvanostat (Ametek, Inc.). Electropolymerization of PEDOT proceeded in a bath of 0.05 M EDOT monomer and 0.1 M LiClO4 in CH3CN at constant anodic current density of 1 mA cm−2 for 60 s. The pristine PEDOT film was electrochemically reduced at constant potential of −0.75 V vs. Ag/Ag+ for ten min. The reduced PEDOT film (r-PEDOT) was transferred to a water bath for five min to allow for wetting and removal of unreacted monomer and supporting electrolyte. For PdOx/PEDOT, the r-PEDOT film was transferred directly from the water bath to a deposition solution of 1 mM K2PdCl4 for 30 min at room temperature. The electrode surface was inverted in the deposition solution to ensure the Pd species deposited on the film originated from reaction with PEDOT, and not instability in the bulk solution resulting in precipitation onto the electrode surface. Finally, the PdOx/PEDOT film was likewise transferred to a water bath for five min.Results and discussion
The EDOT monomer was electropolymerized galvanostatically from CH3CN with LiClO4 electrolyte onto a GC electrode (see Experimental section for details).16 The resulting oxidized PEDOT film (doped with perchlorate anions) was electrochemically reduced at constant potential in the CH3CN electrolyte for 10 min, denoted r-PEDOT [Fig. 1a, (i)]. The r-PEDOT film was then used directly as a template for the electroless deposition of Pd species from aqueous K2PdCl4 for 30 min at room temperature [Fig. 1a, (ii)]; denoted PdOx/PEDOT – see discussion below on the as-deposited Pd speciation.The preparation of the hybrid PdOx/PEDOT film was reproduced on Ti/Pt quartz crystal electrodes monitored by a Quartz Crystal Microbalance (QCM) to understand the mass change and doping/de-doping behaviour of the PEDOT film during the electrochemical reduction and Pd deposition steps. Anodic electropolymerization of PEDOT results in a linear increase in film mass at the electrode surface, with an average deposition rate of 0.98 ± 0.03 μg cm−2 s−1 (Fig. 1b). Subsequently, the reduction of PEDOT is accompanied by an initial decrease in film mass, explained by rapid de-doping of the PEDOT film,23 followed by an overall increase in film mass over 10 min. The average mass of the r-PEDOT film was 63 ± 5 μg cm−2.
Mass uptake of Pd species by the r-PEDOT film was approximately linear over the 30 min deposition time, resulting in a mass loading of Pd species of 2.5 ± 0.9 μg cm−2 (Fig. 1c), or 3.9 ± 1.4 wt% relative to the total film mass. Elemental analysis of PdOx/PEDOT films digested in acid was also investigated by inductively coupled plasma-mass spectrometry, indicating 4.8 ± 0.3 wt% Pd composition. Based on the following discussion, we propose that the spontaneous reaction of PdCl42− with r-PEDOT produces a PdOx/PEDOT composite formed as follows: (i) initial e− transfer between r-PEDOT and Pd2+ ions to form PEDOT-bound Pd0; (ii) subsequent growth of a mixed-valence Pd2+/Pd4+ oxide by reactions between PEDOT-bound Pd0, Pd2+ ions, H2O and dissolved or adsorbed O2.24
Scanning electron microscope (SEM) images of the PdOx/PEDOT and r-PEDOT films (Fig. 2a and S1†) demonstrate a porous, interconnected nanostructure. The similar morphology of the r-PEDOT and PdOx/PEDOT films suggests the electroless deposition produces a homogenous coverage of Pd species on and within the polymeric film. This was confirmed by SEM with energy dispersive spectroscopy, where elemental mapping shows uniform distribution of Pd (Fig. S2†). Individual Pd species (particles) were not observed, suggesting they may be extremely small, on the order or smaller than surface features resulting from Pt sputter coating. X-ray diffraction studies on a PdOx/PEDOT film produced no diffraction pattern (not shown), which may also be indicative of small particle size. In contrast, electroless deposition of Pd species in 0.1 M H2SO4 results in the formation of spherical particles with >20 nm diameter within two minutes.22
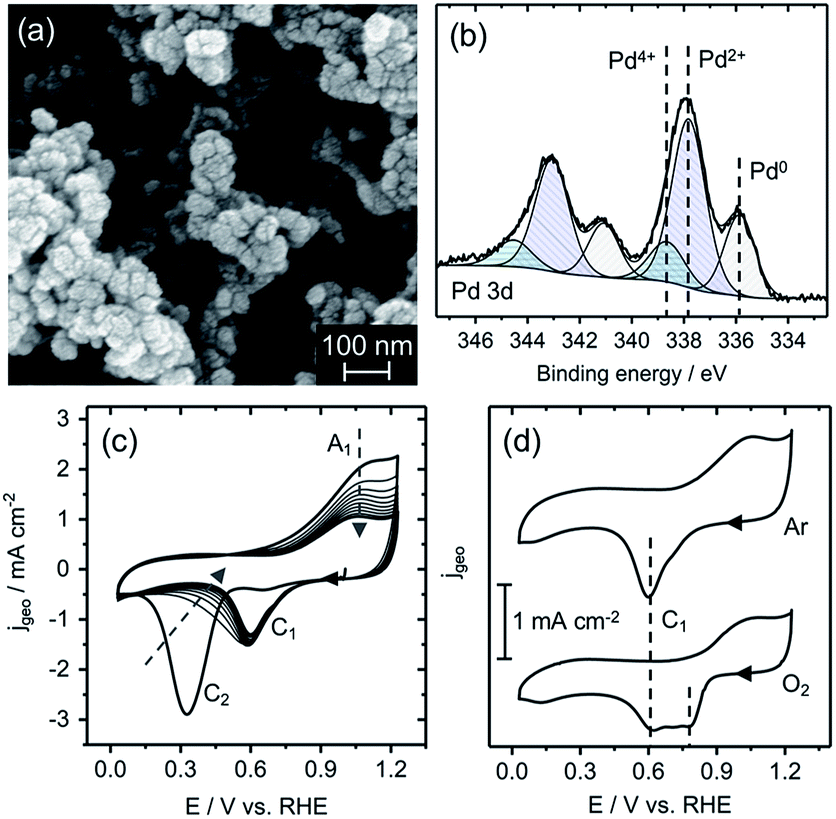 | ||
| Fig. 2 (a) SEM image of a PdOx/PEDOT film as-deposited on glassy carbon; (b) representative XPS Pd 3d spectrum and symmetric core-level fits with valence assignments for a PdOx/PEDOT film as-deposited; (c) CV scans of a PdOx/PEDOT film in Ar-saturated 0.1 M KOH electrolyte, 50 mV s−1; (d) CV scans of a PdOx/PEDOT film sequentially cycled in Ar-saturated and O2-saturated 0.1 M KOH, 50 mV s−1. | ||
A representative high-resolution X-ray photoelectron spectroscopy (XPS) spectrum collected on a PdOx/PEDOT film (as-deposited) in the Pd 3d binding energy (Eb) region is shown in Fig. 2b. All PdOx/PEDOT Pd 3d spectra were fit with three symmetric components; average Eb(Pd 3d5/2)/Eb(Pd 3d3/2) positions of 336.0 ± 0.2/341.2 ± 0.1 eV, 337.8 ± 0.1/343.0 ± 0.1 eV, and 338.8 ± 0.1/344.5 ± 0.2 eV correspond with peak positions and spin-splitting values assigned to Pd0, Pd2+, and Pd4+, respectively.25 In addition, the Eb shift of 1.8 eV between the Pd0 and Pd2+ lines is indicative of the PdO stoichiometry, rather than Pd0 with adsorbed oxygen (Pd-Oads), which is characterized by a Eb shift of less than 1 eV.25 Atomic concentrations of Pd0, Pd2+, and Pd4+ calculated from the Pd 3d core levels are 19 ± 7%, 64 ± 3%, and 18 ± 4%, respectively. The presence of Pd in the 0, 2+ and 4+ valence states reinforces the proposed mechanism for PdOx/PEDOT formation, and suggests an equilibrium favouring Pd2+ in what is most likely to be the PdO stoichiometry or a hydrated analogue (e.g. Pd(OH)2, PdO·nH2O). Full XPS quantitative analysis, Eb values, and survey spectra are provided in Fig. S3 and Tables S1 and S2.†
Based on previous work on the redox behaviour of Pd-based films, Pd oxides have been classified by two types (α and β) with characteristic oxide-reduction profiles.19 Thus, the redox potentials and valence states of the PdOx/PEDOT film can also be revealed by examining oxide-reduction profiles by cyclic voltammetry (CV). α-PdOx is characterized by Pd2+ valence in an anhydrous monolayer or dense multilayer structure and exhibits a reduction wave between 0.6 and 0.9 V vs. RHE, representative of the Pd2+/0 couple.19 In contrast, β-PdOx is typically amorphous, hydrated, and porous with valence assigned to Pd2+, Pd4+, or Pd6+, exhibiting a reduction wave negative of 0.6 V vs. RHE.19 This wave has been interpreted as the reduction of both hydrated Pd2+ and Pd4+ oxides to Pd0.26,27
An as-deposited PdOx/PEDOT film was transferred to a three-electrode cell and cycled in Ar-saturated 0.1 M KOH electrolyte between 0.03 and 1.23 V vs. RHE (Fig. 2c). Beginning from the open circuit potential (OCP) of approximately 1.1 V vs. RHE, the first cathodic scan shows a weak reduction wave with peak potential (Ep) of 0.65 V vs. RHE (C1) and an intense reduction wave at 0.33 V vs. RHE (C2), followed by a broad oxidation wave A1 (Ep of 1.0 V vs. RHE) in the anodic scan. C1 is indicative of α-PdOx reduction and the dominant C2 peak is associated with the reduction of the β-PdOx species.19,26,27 A mixed Pd2+/Pd4+ oxide is also suggested by the OCP of the PEDOT/PdOx film, 1.1 V vs. RHE, a mixed-potential between the formal potentials of the Pd2+/0 (0.9 V vs. RHE) and Pd4+/2+ (1.2–1.28 V vs. RHE) couples.19,24 The OCP and reduction profile on the first cathodic scan are also consistent with the average quantitative analysis by XPS on PdOx/PEDOT films as-deposited (see above).
Further cycling demonstrates a decrease in current associated with C2 and corresponding current increase and stabilization of C1 (shifts to Ep of ∼0.6 V vs. RHE), as shown in Fig. 2c. Hence, over time (and with cycling) the Pd oxide species on PEDOT becomes less oxidized and stabilizes in the α-PdOx (Pd2+) form with reversibility upon reduction to Pd0 (C1/A1). It is worth noting that the β-PdOx species can be regenerated and cycled if the anodic switching potential is extended to 1.5 V vs. RHE (Fig. S4†). The stability and electrochemical reversibility of the Pd0/α-PdOx species in the ORR potential window suggests it may act as an ORR-active species. However, the behaviour of the PdOx/PEDOT film in O2-saturated electrolyte using CV and Tafel analysis must also be considered to confirm the active species as Pd oxide films are known to exhibit different speciation and adsorption properties in the presence of O2.28
The electrocatalytic activity of the r-PEDOT and PdOx/PEDOT films toward the ORR was investigated using CV and rotating ring disk electrode (RRDE) linear scanning voltammetry (LSV) methods. A cathodic scan of the PdOx/PEDOT film in O2-saturated 0.1 M KOH produced a catalytic wave with Ep of ∼0.8 V vs. RHE, a 0.25 V reduction in ORR overpotential (ηORR) relative to the r-PEDOT film, Ep = 0.55 V vs. RHE (Fig. 2d and S5†).‡ Furthermore, the reduction of ηORR is evident in RRDE LSVs, where the characteristic onset potential (Eonset) and half-wave potential (E1/2) of PdOx/PEDOT are 0.98 V and 0.86 V vs. RHE, respectively, compared to 0.7 V and 0.49 V vs. RHE for r-PEDOT (Fig. 3a). The PdOx/PEDOT film also notably demonstrates a shift in Eonset and E1/2 to lower ηORR by 30–40 mV relative to the commercial 20% Pt/C catalyst film (Eonset = 0.95 V vs. RHE; E1/2 = 0.82 V vs. RHE).§ Long-term stability was investigated by electrolysis for 3 h at 0.8 V vs. RHE; the PdOx/PEDOT film retained 87% of the initial ORR current, compared to 78% retention for 20% Pt/C (Fig. S6†).
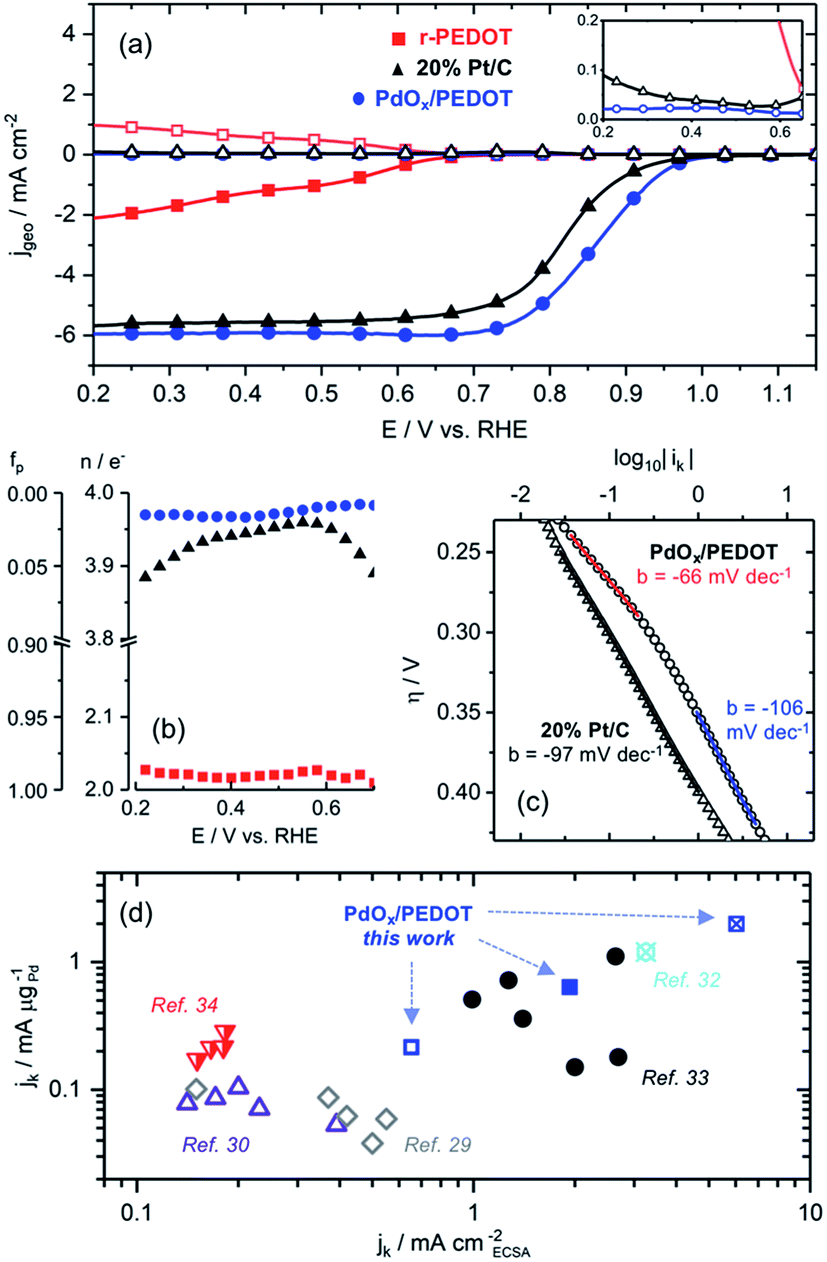 | ||
| Fig. 3 (a) ORR LSVs measured at the disk (closed symbols) and ring (open symbols) electrodes in 0.1 M KOH electrolyte at 2500 RPM, inset: zoom view of ring currents; (b) n and fp parameters calculated from disk and ring current in 0.1 M KOH electrolyte at 2500 RPM; (c) Tafel plots and slopes constructed from the kinetic ORR current in the range of ± 100 mV relative to Eonset; (d) comparison of specific ORR activity on the basis of ECSA and Pd mass loading of the PdOx/PEDOT film and Pd-based electrocatalysts in 0.1 M KOH: 0.8 V vs. RHE (strikethrough symbols), 0.85 V vs. RHE (closed symbols), 0.9 V vs. RHE (open symbols), unspecified potential (half-filled symbols). | ||
The ORR electron transfer number (n) and fraction of peroxide product (fp) were calculated using disk and ring currents measured by RRDE (see ESI† for calculations). These parameters provide insight regarding the ORR pathway, where the limits n = 2 and n = 4 correspond with the 2e− reduction of O2 to peroxide and the 4e− reduction of O2 to OH−, respectively, while n values of 2 < n < 4 suggest a mixed pathway.2 The increase from n ≈ 2 for r-PEDOT to n = 3.97 for PdOx/PEDOT (averaged over 0.2 V to 0.9 V vs. RHE) confirms the Pd species are the active site for catalysing efficient 4e− reduction of O2 to OH− (Fig. 3b). The PdOx/PEDOT composite was also more selective to the 4e− pathway than commercial 20% Pt/C (3.87 < n < 3.97, Fig. 3b). Koutecky–Levich (K–L) analysis was employed to confirm the RRDE results shown in Fig. 3b (see ESI for calculations; Fig. S7†). Average n values calculated using the K–L equation and plots were 3.9 ± 0.1 for PdOx/PEDOT and 3.86 ± 0.09 commercial 20% Pt/C. Furthermore, the calculated kinetic rate constant for the ORR is higher for PdOx/PEDOT (0.12 ± 0.03 cm s−1) than for commercial 20% Pt/C (0.07 ± 0.02 cm s−1).
The relationship between kinetic ORR current (ik) and overpotential is well established for Pd surfaces, where the rate-limiting step is proposed to be the first electron transfer to O2.17,29–31 The Tafel slope (b) for PdOx/PEDOT changes from −66 mV dec−1 at low overpotential to −106 mV dec−1 at higher current densities after Eonset (Fig. 3c). This behaviour is consistent with the literature, where b is expected to change from −60 mV dec−1 to −120 mV dec−1 in the kinetic-limited regime near Eonset, owing to the reduction or protonation of the surface oxides at higher ORR current densities.17,31 Considered together with CV profiles in Ar- and O2-saturated 0.1 M KOH (Fig. 2c and d), we propose the following ORR-active species for PdOx/PEDOT that correspond to distinct potential ranges. In the low current density region positive of Eonset (∼1.23–0.98 V vs. RHE), the ORR is catalysed by the multi-layer α-PdOx identified by CV (Fig. 2c and d) and with b = −66 mV dec−1 (Fig. 3c). In the following high current density regime still under kinetic control (∼0.98–0.85 V vs. RHE), the surface α-PdOx species interacting with O2 and adsorbed oxygen is reduced or protonated, as evidenced by the change in b to −106 mV dec−1 (Fig. 3c). As the potential decreases below 0.85 V vs. RHE, bulk α-PdOx is reduced to Pd0 and diffusion-limited ORR conditions are reached. Ep for the reduction of α-PdOx to Pd0 is ∼0.6 V vs. RHE in the absence and presence of O2, and the film appears to be fully reduced to Pd0 by 0.4 V vs. RHE (Fig. 2d). The reduced bulk Pd0 likely remains covered with adsorbed oxide or hydroxide considering the high pH and constant n value between 0.7 and 0.2 V vs. RHE (Fig. 3b).
Finally, ik was normalized on the basis of electrochemical surface area (ECSA) and Pd mass loading to provide measures of specific activity. The ECSA of the PdOx/PEDOT film based on Pd oxide reduction as determined by CV was 1.05 cm2 (see ESI† for calculations). The specific kinetic current density (jk) of the hybrid PdOx/PEDOT film based on ECSA (mA cmECSA−2) and Pd loading (mA μgPd−1) between 0.8 and 0.9 V vs. RHE is shown in Fig. 3d. For comparison, Fig. 3d and Table S3† include reported jk values for state-of-the-art Pd-based electrocatalysts in 0.1 M KOH electrolyte.29,30,32–34 The combined mass- and ECSA-based specific activity of the hybrid PdOx/PEDOT film is among the highest reported to date; for example, at 0.9 V vs. RHE (0.65 mA cmECSA−2, 0.22 mA μgPd−1), PdOx/PEDOT surpasses the activity of size-controlled Pd nanocubes29 and nanoparticles30 supported on conductive carbon. In addition, the mass-specific activity of PdOx/PEDOT is superior to commercial 20% Pt/C at 0.9 V vs. RHE (0.14 mA μgPt−1).
Table 1 provides a comparison of the most active PEDOT-based ORR electrocatalysts reported in 0.1 M KOH.16,32 The Eonset values for the Pd-based composites are markedly positive of that for the MnOx composite (by ∼100 mV), suggesting significantly higher intrinsic activities at low ηORR on Pd. Despite this, E1/2 values for both PdOx/PEDOT and the P-MnOx-20 film16 are >0.8 V vs. RHE and within 30 mV, indicating fast kinetics for both systems in the kinetic-limited regime. The specific activities of PdOx/PEDOT represent a >150% improvement in those reported for the Pd/PEDOT/rGO catalyst32 on the basis of both ECSA and Pd mass (also shown in Fig. 3d). This is notable considering the present work eliminates the need for additional conductive carbon (e.g. rGO), demonstrating the promise of PEDOT to act as both a conductive support and polymeric binder. Analogous to the highly active P-MnOx-20 catalyst films,16 we attribute the high ORR activity of the PdOx/PEDOT films to the electroless deposition method, which affords the following: (i) a porous nanostructure that improves surface area and mass transfer for catalysis; (ii) deposition of a stable, electrochemically reversible ORR-active Pd0/α-PdOx species; (iii) high dispersion and low-mass loading of exposed Pd species maximizes active site utilization and specific activity.
Conclusions
Nanostructured hybrid PdOx/PEDOT films were prepared by a spontaneous reaction between aqueous PdCl42− ions and reduced PEDOT films in neutral aqueous solution. The Pd species as-deposited on PEDOT was characterized for the first time by XPS and CV, revealing Pd in the 0, 2+ and 4+ valence states. Thus, we propose the spontaneous redox reactions proceed to form PEDOT-bound Pd0 with a mixed-valence Pd2+/Pd4+ oxide. Cycling the hybrid PdOx/PEDOT films in the absence (and presence) of O2 in alkaline electrolyte reveals a stable Pd0/α-PdOx species, which acts as the active site in electrocatalysis of the ORR. The PdOx/PEDOT films display high activity and selectivity toward efficient 4e− ORR, competitive with or superior to the activity of state-of-the-art Pd/C and PEDOT-based composites and commercial 20% Pt/C, demonstrating the promise of the dual utility of PEDOT as a conductive substrate and polymeric binder.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DE-NA0003525. The views expressed in the article do not necessarily represent the views of the U.S. Department of Energy or the United States Government.Notes and references
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2016, 16, 57–69 CrossRef PubMed.
- Q. He and E. J. Cairns, J. Electrochem. Soc., 2015, 162, F1504–F1539 CrossRef.
- T. N. Lambert, J. A. Vigil, S. E. White, D. J. Davis, S. J. Limmer, P. D. Burton, E. N. Coker, T. E. Beechem and M. T. Brumbach, Chem. Commun., 2015, 51, 9511–9514 RSC.
- T. N. Lambert, J. A. Vigil, S. E. White, C. J. Delker, D. J. Davis, M. Kelly, M. T. Brumbach, M. A. Rodriguez and B. S. Swartzentruber, J. Phys. Chem. C, 2017, 121, 2789–2797 CrossRef.
- J. A. Vigil, T. N. Lambert, J. Duay, C. J. Delker, T. E. Beechem and B. S. Swartzentruber, ACS Appl. Mater. Interfaces, 2018, 10, 2040–2050 CrossRef PubMed.
- D. J. Davis, A.-R. O. Raji, T. N. Lambert, J. A. Vigil, L. Li, K. Nan and J. M. Tour, Electroanalysis, 2014, 26, 164–170 CrossRef.
- E. Antolini and E. R. Gonzalez, Appl. Catal., A, 2009, 365, 1–19 CrossRef.
- M. C. Lefebvre, Z. Qi and P. G. Pickup, J. Electrochem. Soc., 1999, 146, 2054–2058 CrossRef.
- B. Winther-Jensen, O. Winther-Jensen, M. Forsyth and D. R. MacFarlane, Science, 2008, 321, 671 CrossRef PubMed.
- R. Kerr, C. Pozo-Gonzalo, M. Forsyth and B. Winther-Jensen, ECS Electrochem. Lett., 2013, 2, F29–F31 CrossRef.
- E. Mitraka, M. J. Jafari, M. Vagin, X. Liu, M. Fahlman, T. Ederth, M. Berggren, M. P. Jonsson and X. Crispin, J. Mater. Chem. A, 2017, 5, 4404–4412 RSC.
- A. D. Chowdhury, N. Agnihotri, P. Sen and A. De, Electrochim. Acta, 2014, 118, 81–87 CrossRef.
- G. Gnana kumar, C. Joseph Kirubaharan, D. J. Yoo and A. R. Kim, Int. J. Hydrogen Energy, 2016, 41, 13208–13219 CrossRef.
- R. Kerr, C. Pozo-Gonzalo, M. Forsyth and B. Winther-Jensen, Electrochim. Acta, 2015, 154, 142–148 CrossRef.
- J. A. Vigil, T. N. Lambert and K. Eldred, ACS Appl. Mater. Interfaces, 2015, 7, 22745–22750 CrossRef PubMed.
- J. A. Vigil, T. N. Lambert, M. Kelly and R. Aidun, Mater. Chem. Front., 2017, 1, 1668–1675 RSC.
- D. Diabaté, T. W. Napporn, K. Servat, A. Habrioux, S. Arrii-Clacens, A. Trokourey and K. B. Kokoh, J. Electrochem. Soc., 2013, 160, H302–H308 CrossRef.
- F. H. B. Lima, J. Zhang, M. H. Shao, K. Sasaki, M. B. Vukmirovic, E. A. Ticianelli and R. R. Adzic, J. Phys. Chem. C, 2007, 111, 404–410 CrossRef.
- M. Grdeń, M. Łukaszewski, G. Jerkiewicz and A. Czerwiński, Electrochim. Acta, 2008, 53, 7583–7598 CrossRef.
- S. N. Eliseeva, V. V. Malev and V. V. Kondraťev, Russ. J. Electrochem., 2009, 45, 1045 CrossRef.
- S. N. Eliseeva, E. V. Ubyivovk, A. S. Bondarenko, O. F. Vyvenko and V. V. Kondratiev, Russ. J. Gen. Chem., 2010, 80, 1143–1148 CrossRef.
- V. V. Kondratiev, T. A. Babkova and S. N. Eliseeva, Russ. J. Electrochem., 2012, 48, 205–211 CrossRef.
- A. Bund and S. Neudeck, J. Phys. Chem. B, 2004, 108, 17845–17850 CrossRef.
- J. P. Hoare, J. Electrochem. Soc., 1964, 111, 610–615 CrossRef.
- K. S. Kim, A. F. Gossmann and N. Winograd, Anal. Chem., 1974, 46, 197–200 CrossRef.
- V. I. Birss, M. Chan, T. Phan, P. Vanysek and A. Zhang, J. Chem. Soc. Faraday Trans., 1996, 92, 4041–4047 RSC.
- A. E. Bolzán and A. J. Arvia, J. Electroanal. Chem., 1992, 322, 247–265 CrossRef.
- L. M. Vracar, D. B. Sepa and A. Damjanovic, J. Electrochem. Soc., 1989, 136, 1973–1977 CrossRef.
- M. Lüsi, H. Erikson, A. Sarapuu, K. Tammeveski, J. Solla-Gullón and J. M. Feliu, Electrochem. Commun., 2016, 64, 9–13 CrossRef.
- L. Jiang, A. Hsu, D. Chu and R. Chen, J. Electrochem. Soc., 2009, 156, B643–B649 CrossRef.
- L. M. Vracar, D. B. Sepa and A. Damjanovic, J. Electrochem. Soc., 1986, 133, 1835–1839 CrossRef.
- J. E. Choe, M. S. Ahmed and S. Jeon, J. Power Sources, 2015, 281, 211–218 CrossRef.
- Q. Wu, Z. Rao, L. Yuan, L. Jiang, G. Sun, J. Ruan, Z. Zhou and S. Sang, Electrochim. Acta, 2014, 150, 157–166 CrossRef.
- W. Yan, Z. Tang, L. Li, L. Wang, H. Yang, Q. Wang, W. Wu and S. Chen, ChemElectroChem, 2017, 4, 1349–1355 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization methods and calculations; additional characterization and electrocatalytic data, literature comparison for alkaline Pd/C ORR electrocatalysts. See DOI: 10.1039/c8ra03505a |
| ‡ For a comparison to the ORR activity of an oxidized PEDOT film under the conditions reported here, refer to ref. 16. |
| § The ORR activity for the commercial 20% Pt/C electrocatalyst is the highest obtained in our hands. The activity is consistent with a range of reported values, where alkaline electrolytes are known to inhibit the ORR activity on Pt surfaces compared to acid (see ref. 2 and 18 for further discussion). |
| This journal is © The Royal Society of Chemistry 2018 |