
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe self-template synthesis of highly efficient hollow structure Fe/N/C electrocatalysts with Fe–N coordination for the oxygen reduction reaction†
Yue Yua,
Dejian Xiaoa,
Jun Maa,
Changli Chena,
Kai Lia,
Jie Maa,
Yi Liaoa,
Lirong Zheng b and
Xia Zuo*a
b and
Xia Zuo*a
aDepartment of Chemistry, Capital Normal University, Beijing 100048, P. R. China. Fax: +86-10-68903040; Tel: +86-10-68903086
bBeijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 6th July 2018
Abstract
The exploration of highly efficient catalysts to replace noble metal platinum for the oxygen reduction reaction, on which M/N/C catalysts have shed brilliant light, is greatly significant but challenging. This paper presents a strategy for synthesizing highly efficient and stabilized hollow structure Fe/N/C catalysts with iron and nitrogen doped into the carbon layer by the self-template method. The prepared Fe/N/C catalysts with NaCl protection during pyrolysis are characterized by a unique hollow structure, porous morphology and Fe–N coordination as the active sites, all of which significantly endow the materials with excellent properties towards the ORR, including high electrical conductivity, long-term durability and outstanding capacity for methanol tolerance. We employed X-ray absorption fine structure spectrometry to investigate the chemical state and coordination environment of the central iron atoms of the Fe/N/C catalysts, which also clarified the promoting effect of the NaCl protection for Fe–N coordination during pyrolysis. In particular, the Fe/N/C catalysts exhibit positive half-wave potentials (0.84 V vs. RHE) and Tafel slope comparable to 20% commercial Pt/C, possessing four-electron transfer pathway as well as excellent long-term stability and methanol tolerance in alkaline medium.
Introduction
The oxygen reduction reaction (ORR), occurring at cathodes as the rate-limiting step, is of vital importance in electrochemical energy conversion and storage devices such as fuel cells and metal–air batteries.1–5 As the catalyst of ORR, commercial Pt/C has disadvantages including its scarce reserves, high price, poor durability and inferior methanol tolerance.6–8 Therefore, extensive efforts have been devoted to ORR catalysts with the aim of developing superior performance catalysts, which can manifest prominent stability as well as achieve commercial applications on a large scale.9 The non-precious-metal catalysts have grabbed much attention and demonstrated remarkable catalytic activity in the last decades, among which metal- and nitrogen-doped carbon-based catalysts (M/N/C) behave superiorly.10,11 On the one hand, the large-scale reserves of the non-noble metal on the earth and the accessible nitrogen-doped carbon-based materials are the significant advantages of M/N/C catalysts.12,13 On the other hand, the M/N/C catalysts having remarkable poison resistance and superior stability can implement ORR catalytic activity comparable to 20% commercial Pt/C.14–18 Thus, the non-precious metal catalysts M/N/C that can bring the extensive application of the fuel cells into force have promising prospects.As a part of M/N/C catalysts, the carbon materials can strengthen electrical conductivity, assisting mass transport and provide a location for the ORR. Nitrogen doped into graphite can also fabricate abundant defects that can assist reactant (e.g., H+, OH−, O2) adsorption on the carbon materials.19–21 The significant effect of non-noble metals in M/N/C catalysts is another indispensable factor. Even if the precise influence of metal in the materials still remains confusing, the metal can function as a part of the active sites, which is supposed to take a great endeavour to further research.22,23 Moreover, the nanostructure and the specific surface area of the carbon materials are also crucial parameters for the ORR catalyst activity, which determine the electrochemical properties and the possibilities for exposure of active sites.16,24 Whatever is in favour of the removal of the reaction product, e.g. porous carbon materials, can make active sites continually proceed to next reaction.25 Within this context, many efforts have been made to synthesize M/N/C catalysts with controlled nanostructure, such as nanotubes,26 nanosheets,27 hollow spheres,28 core–shell structures29 and other mesoporous constructions of carbon support,30–33 which all demonstrate remarkable ORR activity in both alkaline and acid media.
Herein, we employed a self-template method to prepare a novel kind of Fe/N/C catalyst with hollow nanostructure: Fe and N dual doped carbon layer with the Fe–N coordination was fabricated by a NaCl recrystallization protection strategy during pyrolysis. For the self-template method, the evaporation of melamine-formaldehyde resin (MF) assists the formation of the hollow structure, simultaneously serving as part of the materials during the pyrolyzation. We also exploit the NaCl protection method for facilitating the formation of Fe–N interactions. Significantly, the Fe–N coordination acts as greatly efficient active sites for enhancing the catalytic activities of Fe/N/C, and the incorporation of Fe–N into the carbon frameworks also enhances the synergetic effect with the introduction of the porous structure. As for the ORR performance, the Fe/N/C catalysts reveal superior electrocatalytic activity that is comparable to 20% commercial Pt/C, excellent stability and remarkable methanol tolerance in alkaline solutions.
Results and discussion
Physical characterization
The Fe–N doped hollow carbon structure Fe/N/C catalysts were synthesized through the self-template method; the procedure is demonstrated in Fig. 1. The polydopamine (PDA) modified melamine-formaldehyde resin (PDA@MF) was produced by utilizing a dopamine (DA) self-crosslinking reaction in the air without catalysts, which can employ PDA to endow the materials with the ability to absorb metal ions. The PDA@MF and FeCl3 were dispersed in supersaturated NaCl solution for the adsorption of Fe3+, and then the water was evaporated for NaCl re-crystallization around the materials. During the carbonization, the materials were equipped with a metal and nitrogen doped carbon layer and a highly porous hollow structure. The prepared MF sphere (Fig. S1†) can serve as the self-template that can assist the formation of the hollow structure. Distinct from the traditional template method, the self-template method not only plays the role of the supporting framework of the traditional template but also directly participates in the formation processes of the materials.34 The microstructures and morphology were detected by SEM and TEM. The Fe/N/C catalysts with the unique hollow structure and highly porous architecture are shown in Fig. 2, which reveals that each Fe/N/C catalyst has an amorphous carbon layer. The images of PDA@MF-Fe-800 are shown in Fig. S2,† having a similar hollow structure to Fe/N/C. The SEM (Fig. S3†) and TEM images all show the aggregation of Fe/N/C, which could form a three-dimensional carbon framework with porous structure generating an extensive network of pores.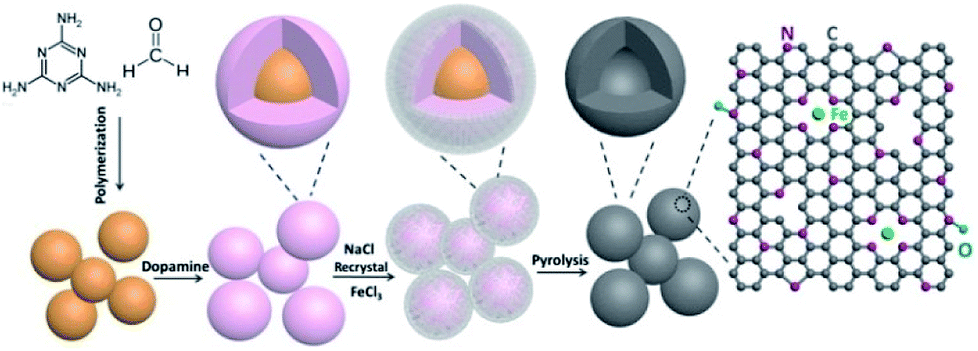 | ||
| Fig. 1 Schematic diagram of the preparation of the Fe/N/C. | ||
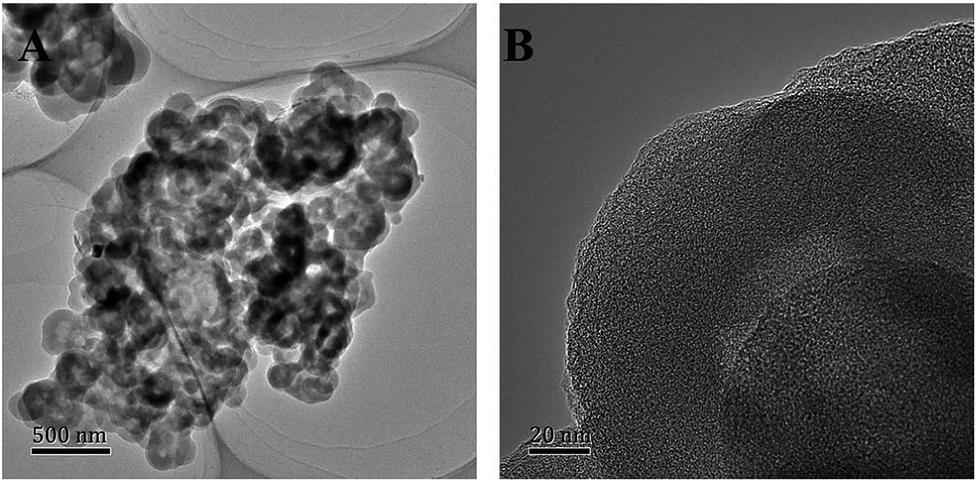 | ||
| Fig. 2 TEM images of Fe/N/C. | ||
We detected the crystal structure of the Fe/N/C by XRD. The intense peaks at around 26.6° and 43.4° could be ascribed to the (002) and (100) planes of carbon, which further confirmed the amorphous character of Fe/N/C. In Fig. 3A, the Fe/N/C and PDA@MF-Fe-800 show much stronger and narrower peaks compared with PDA@MF-800 and MF-800, meaning a higher degree of graphitization;35 this proved that the addition of Fe had an energetic effect on the graphitic structure.36 The previous results indicated that the Fe is an efficient catalyst for the graphitization of carbon during the high-temperature carbonization process; the Fe phase is thermodynamically stable at high temperature, which could increase the dissolved carbon content to form the graphite structure.37,38 It is notable that no other diffraction peaks were observed, meaning that there were no large crystallites of Fe containing species; the superfluous Fe containing species might have been removed by acid leaching. This is in accordance with the TEM data, where no particles were detected, signifying that the rest of the Fe may have been doped into the graphite layer during high-temperature pyrolysis.
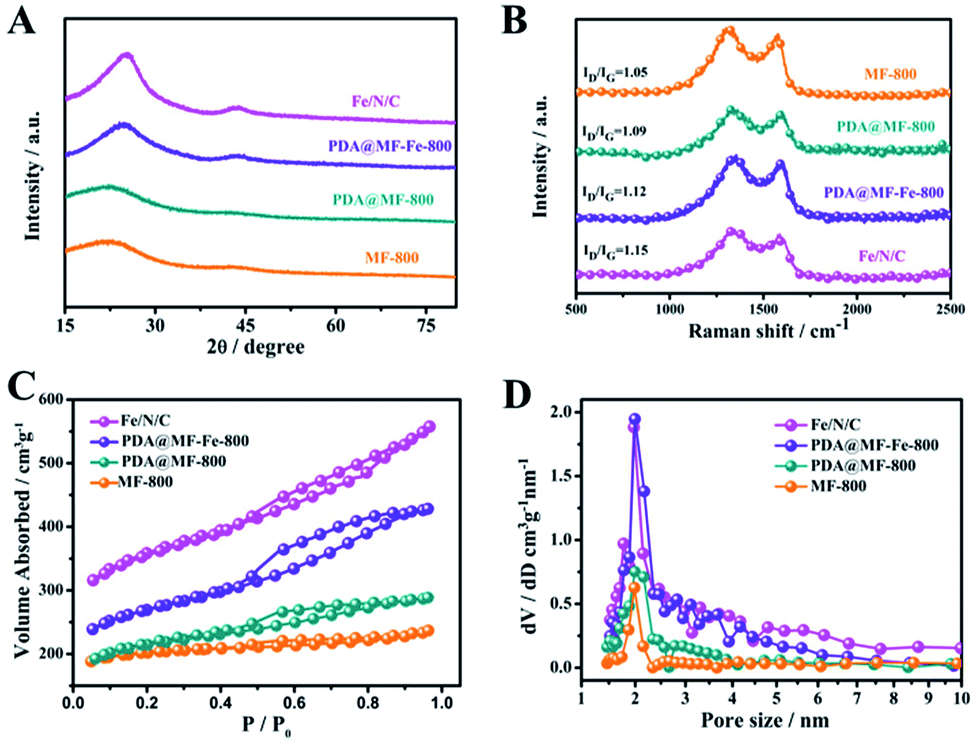 | ||
| Fig. 3 (A) XRD patterns (B) Raman spectra (C) N2 adsorption/desorption isotherms and (D) pore size distribution of Fe/N/C, PDA@MF-Fe-800, PDA@MF-800 and MF-800. | ||
We used Raman spectroscopy to evaluate the degree of graphitization of materials and the level of disorder. As can be seen in Fig. 3B, all spectra of the samples exhibited two characteristic broad peaks for carbonized materials. The G band (at about 1582 cm−1) can confirm the presence of C sp2 atoms within the carbon compounds and the degree of graphitization.39 The D-band (at about 1345 cm−1) can attest to the presence of defects within the hexagonal graphitic structure.40 Therefore, the intensity ratio of the D band to G band (ID/IG) was utilized to ascertain the degree of defects for the carbon-based materials.41–43 The ID/IG value of PDA@MF-800 (1.09) is similar to MF-800 (1.05), meaning almost the same degree of defects, which these catalysts produced without the addition of Fe. The PDA@MF-Fe-800 presents the ID/IG value of 1.12, proving that the addition of Fe species increases the degree of defects. The Fe/N/C samples present the largest ID/IG value of 1.15, which indicates the high density of defective sites in the carbon materials. In order to accurately interpret the graphitization degree of the materials, we added decomposed Raman spectra of the prepared catalysts in Fig. S4.† The spectral region clearly exhibits several overlapping lines, which are respectively located at 1160 cm−1 (polyene-like structures), 1320 cm−1 (a disorder in the nanomaterial), 1500 cm−1 (amorphous sp2 phase) and 1580 cm−1 (in-plane vibration of sp2-bonded carbon atoms).44–47 The proportions of peak areas are as shown in the inset of Fig. S4.† The 1320 cm−1 and 1580 cm−1 of Fe/N/C indicate the disorder in the nanomaterial (70.51%) and the graphitization structure (23.51%), respectively, which are both enhanced at the same time. The increasing proportion of graphite structure is in accordance with the conclusion of XRD.
We investigated the porous structure of the materials with N2 gas adsorption–desorption tests. The MF-800, PDA@MF-800, PDA@MF-Fe-800 and Fe/N/C samples all exhibit type-IV isotherms with obvious hysteresis loops over a relative pressure range of 0.8–0.5 (Fig. 3C), indicating the existence of the hierarchically mesoporous structure of the materials, which is in favour of the mass transport and exposure of more active sites.48 The Brunauer–Emmett–Teller (BET) surfaces of all samples are listed in Table S1.† The specific surface area of Fe/N/C is 1146.75 m2 g−1, which is much larger than other samples due to the formation of the hollow sphere structure with the protection of NaCl during the carbonation process. The PDA@MF-Fe-800 also has a hollow structure with the specific surface area of 858.03 m2 g−1, which is much smaller than that of Fe/N/C. These results indicate that NaCl can protect the porous structure. The NaCl was recrystallized around the materials before pyrolysis. During the pyrolysis, the NaCl melted at about 800 °C around the materials (melting point of NaCl is 800.9 °C). During the formation of the hollow structure process, some gases were released from the samples. The molten NaCl around the materials could be the obstruction of the departure of these gases, which could assist the development of the porous structure. The addition of FeCl3 can also promote the production of the porous structure, which is confirmed by the specific surface area of PDA@MF-800 without the addition of Fe during the production. The pore size distribution curve (Fig. 3D) of Fe/N/C is similar to PDA@MF-Fe-800, which has a major peak centered around 2 nm revealing the main pore diameter, and might have been created by the release of some gases during pyrolysis.
We further analyzed the chemical contents on the surfaces of as-synthesized samples by X-ray photoelectron spectroscopy (XPS). The XPS spectra (Fig. S5†) verified that all the samples consisted of the peaks of C, N, and O; the diminutive Fe peak of Fe/N/C and PDA@MF-Fe-800 were also observed and the atomic surface concentrations of the as-prepared samples are outlined in Table S2.† The high-resolution C 1s (Fig. 4A) spectrum can be deconvoluted into four bonding types, centered at 284.7 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 285.8 eV (CN and C–O), 287.2 eV (C–N and CO), 289.8 eV (O–CO), demonstrating the existence of the heteroatoms, especially O and N in the hybrids. The high-resolution Fe 2p spectra of Fe/N/C (Fig. 4B) have the characteristic peaks of Fe2+ and Fe3+, which can be divided into 738.22 eV (Fe3+ 2p1/2), 724.62 eV (Fe2+ 2p1/2), 717.29 eV (satellite peak), 712.71 eV (Fe3+ 2p3/2), 710.81 eV (Fe2+ 2p3/2), suggesting the existence of Fe–N coordination.49 Furthermore, the N 1s peak of the samples (Fig. 4C and S6†) can be deconvoluted into pyridine-N (398.38 eV), pyrrole-N (399.56 eV), graphite-N (400.78 eV), and oxidized-N (402.84 eV), which were measured to investigate the nitrogen surface groups on the samples. It is worth noting that the nitrogen bound to the metal (Fe–N) and the pyridine-N have almost the same binding energy, so the peak located at 398.38 eV is also ascribed to Fe–N.50 The relative ratios of the deconvoluted peak areas of the N 1s spectra are summarized, and the five types of nitrogen are schematically depicted in Fig. 1. Notably, the merging N dopants in the catalysts could boost conductivity to assist electron transportation during electrochemical processes believed to be responsible for the ORR activity of Fe–N–C catalysts.51 The nitrogen content of Fe/N/C (6.71 at%) is higher than PDA@MF-Fe-800 (6.13 at%), which proves that the addition of NaCl during pyrolyzation can improve the content of nitrogen. The molten NaCl during the pyrolysis process could hinder the departure of nitrogen-containing gases, which could also facilitate nitrogen remaining in the materials, consequently enhancing the N-doping content. The catalysts were dominated by pyridinic-N (Fe–N) and graphite-N. The content of graphite-N increased with the increase in temperature, possibly on account of the greater thermal stability at high temperatures. The pyridinic-N is proposed to be located at the edges and defects of carbon layers, which is specifically connected with the ORR reactivity.
C), 285.8 eV (CN and C–O), 287.2 eV (C–N and CO), 289.8 eV (O–CO), demonstrating the existence of the heteroatoms, especially O and N in the hybrids. The high-resolution Fe 2p spectra of Fe/N/C (Fig. 4B) have the characteristic peaks of Fe2+ and Fe3+, which can be divided into 738.22 eV (Fe3+ 2p1/2), 724.62 eV (Fe2+ 2p1/2), 717.29 eV (satellite peak), 712.71 eV (Fe3+ 2p3/2), 710.81 eV (Fe2+ 2p3/2), suggesting the existence of Fe–N coordination.49 Furthermore, the N 1s peak of the samples (Fig. 4C and S6†) can be deconvoluted into pyridine-N (398.38 eV), pyrrole-N (399.56 eV), graphite-N (400.78 eV), and oxidized-N (402.84 eV), which were measured to investigate the nitrogen surface groups on the samples. It is worth noting that the nitrogen bound to the metal (Fe–N) and the pyridine-N have almost the same binding energy, so the peak located at 398.38 eV is also ascribed to Fe–N.50 The relative ratios of the deconvoluted peak areas of the N 1s spectra are summarized, and the five types of nitrogen are schematically depicted in Fig. 1. Notably, the merging N dopants in the catalysts could boost conductivity to assist electron transportation during electrochemical processes believed to be responsible for the ORR activity of Fe–N–C catalysts.51 The nitrogen content of Fe/N/C (6.71 at%) is higher than PDA@MF-Fe-800 (6.13 at%), which proves that the addition of NaCl during pyrolyzation can improve the content of nitrogen. The molten NaCl during the pyrolysis process could hinder the departure of nitrogen-containing gases, which could also facilitate nitrogen remaining in the materials, consequently enhancing the N-doping content. The catalysts were dominated by pyridinic-N (Fe–N) and graphite-N. The content of graphite-N increased with the increase in temperature, possibly on account of the greater thermal stability at high temperatures. The pyridinic-N is proposed to be located at the edges and defects of carbon layers, which is specifically connected with the ORR reactivity.
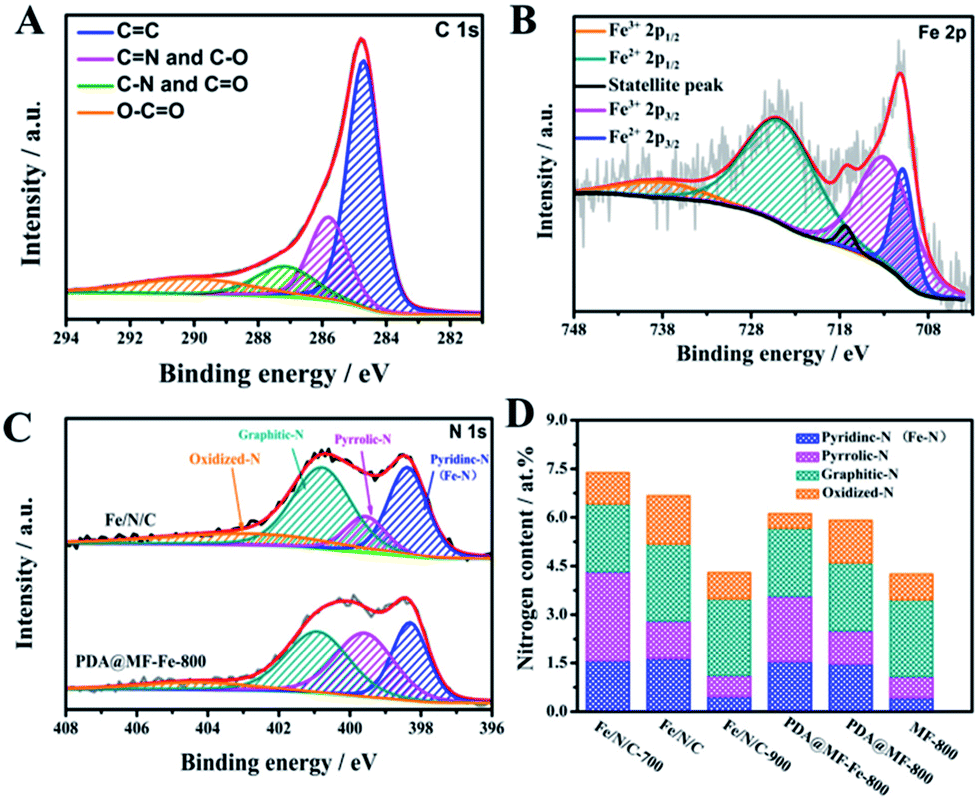 | ||
| Fig. 4 XPS spectra in the (A) C 1s and (B) Fe 2p of Fe/N/C; (C) N 1s spectra of Fe/N/C, PDA@MF-Fe-800, PDA@MF-800 and MF-800; (D) nitrogen bonding configurations of Fe/N/C-700, Fe/N/C, Fe/N/C-900, PDA@MF-Fe-800, PDA@MF-800 and MF-800. | ||
Further detail on the Fe–N coordination was investigated by X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), which are well-established and useful tools for clarifying the coordination environment of the center iron atoms in spite of the low concentration. Fig. 5A shows the Fe K-edge spectra XANES of the Fe/N/C, as well as PDA@MF-Fe-800, Fe foil and iron phthalocyanine (FePc). The commercial FePc has the distinctive feature of the Fe–N4 structure. The Fe/N/C samples similar to that of FePc exhibit a weak pre-edge peak at around 7113.3 eV, assigned as a fingerprint of the Fe–N square-planar configuration, verifying the existence of Fe–N coordination in Fe/N/C samples.52 By comparison, the Fe K-edge XANES spectrum of PDA@MF-Fe-800 shows a weak pre-edge peak at around 7119 eV, which is more similar to that of Fe foil, meaning the presence of iron-based structures. Furthermore, the Fourier transform Fe K edge EXAFS spectra were also investigated. As shown in Fig. 5B, the EXAFS spectra of Fe/N/C had a main signal peak located at 1.48 Å, similar to that of FePc, which was recognized as the Fe–N distance stemming from a nitrogen shell surrounding iron atoms.53 The spectrum of PDA@MF-Fe-800 mainly shows two peaks at 1.62 Å and 2.04 Å, assigned to Fe–N and Fe–Fe interactions. It is noteworthy that the Fe–N peak of PDA@MF-Fe-800 had low intensity compared with Fe/N/C, corroborating the less developed Fe–N environment compared to Fe/N/C. The consequences demonstrate that Fe–N coordination does exist in Fe/N/C and the NaCl recrystallization protection strategy assists the coordination between nitrogen and iron.
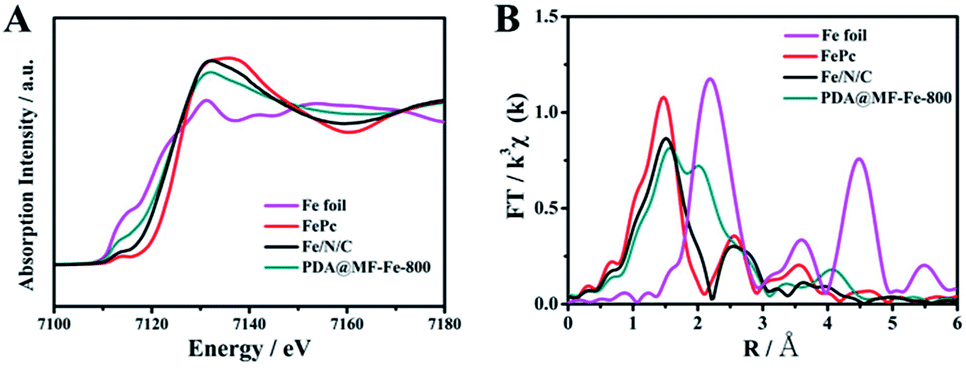 | ||
| Fig. 5 (A) The normalized Fe K-edge XANES spectra (B) Fourier transform Fe K-edge EXAFS spectroscopy of Fe foil, FePc, Fe/N/C and PDA@MF-Fe-800. | ||
Electrochemical evaluation
The electrochemical performance of Fe and N doped carbon layer electrocatalysts with multiple mesoporous hollow structure are measured by rotating disk electrode (RDE) for estimating. The ORR performance of the catalysts was determined by cyclic voltammetric (CV) measurements in N2 and O2-saturated 0.1 M KOH aqueous solutions, with a scan rate of 20 mV s−1. As shown in Fig. 6A, the CV curves of all samples had no obvious redox peaks in N2-saturated 0.1 M KOH aqueous solutions. The cathode reduction current peaks, which are indicative of the oxygen reduction reaction, were found at 0.88 V vs. RHE, 0.86 V vs. RHE, 0.75 V vs. RHE, 0.69 V vs. RHE in O2-saturated 0.1 M KOH aqueous solutions for Fe/N/C, PDA@MF-Fe-800, PDA@MF-800, MF-800, respectively. The Fe/N/C exhibited the most positive peak potential at 0.88 V vs. RHE, suggesting superior ORR catalytic activity. Furthermore, linear sweep voltammograms (LSVs) were carried out to evaluate the ORR activity levels of these catalysts. In Fig. 6B, the Fe/N/C catalysts exhibited the most positive half-wave potential (0.84 V vs. RHE), as compared to MF-800 (0.67 vs. RHE), PDA@MF-800 (0.76 vs. RHE) and PDA@MF-Fe-800 (0.82 vs. RHE). The Fe/N/C showed remarkable ORR activity with the half-wave potential at 0.84 vs. RHE, which was comparable to that of the 20% commercial Pt/C catalysts. Fig. 6C shows the LSV curves of the Fe/N/C catalysts in O2-saturated 0.1 M KOH solution at rotating speeds from 400 to 1600 rpm. To gain insight into the ORR kinetics, the electron transfer number for Fe/N/C calculated from Koutecky–Levich plots (inset Fig. 6C) is 3.96, which indicates the oxygen reduction process, mainly involving a four-electron reaction pathway, is the same as for 20% commercial Pt/C. The Tafel slope was employed to further clarify the kinetics of Fe/N/C catalysts. The Tafel slope of Fe/N/C (91 mV dec−1) is comparable to the 20% commercial Pt/C (87 mV dec−1), which indicates the semblable ORR kinetic process.54 The onset potential (Eonset), the half-wave potential (E1/2), limiting current density, and calculated electron transfer numbers are listed in Table S2,† which are all the significant parameters connected with ORR activity. The results above all indicate that Fe/N/C is indeed an efficient electrocatalyst toward ORR. The ORR performances of similar catalysts in literature have been summarized in Table S4,† among which the Fe/N/C still has remarkable catalytic performance. | ||
| Fig. 6 (A) CV curves of Fe/N/C, PDA@MF-Fe-800, PDA@MF-800 and MF-800 in O2 saturated 0.1 M KOH aqueous solution with a scan rate of 20 mV s−1. (B) LSV curves of Fe/N/C, PDA@MF-Fe-800, PDA@MF-800 and MF-800 in O2 saturated 0.1 M KOH aqueous solution at a rotation rate of 1600 rpm. (C) LSV curves of Fe/N/C at different rotation rates (the inset in (C) shows the fitting K–L plots); (D) Tafel plots. (E) Linear fitting of Δj of Fe/N/C and PDA@MF-Fe-800 vs. scan rates at a given potential. (F) LSV of Fe/N/C in O2-saturated 0.1 M KOH on a rotating speed at 1600 rpm with and without KSCN. | ||
On the basis of the preceding results, the positive impact of the Fe species must be pointed out. In comparison with PDA@MF-800, the addition of Fe in the PDA@MF-Fe-800 results in a material possessing a greater degree of defects and larger specific area. The PDA@MF-Fe-800 has great potential for implementing better ORR activity, so more detailed information on the Fe species was investigated using XAFS, which confirmed the Fe–N coordination in Fe/N/C. The Fe–N coordination is vital for enhancing the electrochemical performance,55–57 and acts as greatly efficient active sites for enhancing the catalytic activities of Fe/N/C. The NaCl protective effect was also measured. The electrical double-layer capacitance (Cdl) at the solid–liquid interface was calculated by a simple CV method (Fig. S8†), which was positively correlated to the electrochemically active surface area.58,59 The capacitances of Fe/N/C and PDA@MF-Fe-800 are 27.6 μF cm−2 and 14.2 μF cm−2, respectively (Fig. 6E). The electrochemically active surface area of Fe/N/C wrapped with NaCl during pyrolyzation is much higher, compared with PDA@MF-Fe-800 without NaCl protection. This consequence indicates that NaCl can protect the construction of the Fe/N/C, preventing aggregation and interior structure collapse during the carbonization process. Together with the results of XAFS, the NaCl can not only endow the catalysts with a much greater electrochemical surface area, but also provide more opportunities for the formation of Fe–N coordination. In order to confirm the crucial role of the Fe species in the ORR process, thiocyanate (SCN−) was employed as a complexing agent for shielding the Fe in Fe/N/C. As shown in Fig. 6F, after the introduction of 5 mM SCN−, the ORR activity of Fe/N/C was severely decreased with the 90 mV negative shift for E1/2 and a serious decrease in the diffusion-limiting current. This demonstrates that the Fe–N has a crucial impact on the electrochemical performance. It is further implied that the Fe–N coordination plays the part of active sites in Fe/N/C catalysts.60
The chronoamperometric tests of Fe/N/C and 20% commercial Pt/C were assessed in O2-saturated 0.1 M KOH to investigate the stability of the Fe/N/C toward ORR. As shown in Fig. 7A, the Fe/N/C presents low attenuation with high current retention of 92.6% for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s, while the relative current of the 20% commercial Pt/C was decreased to 72.3%. This proves that the Fe/N/C has a much better stability than the 20% commercial Pt/C. Moreover, the methanol tolerance of the Fe/N/C was also remarkable, and there was no obvious loss with the addition of methanol, compared to 20% commercial Pt/C (Fig. 7B).
000 s, while the relative current of the 20% commercial Pt/C was decreased to 72.3%. This proves that the Fe/N/C has a much better stability than the 20% commercial Pt/C. Moreover, the methanol tolerance of the Fe/N/C was also remarkable, and there was no obvious loss with the addition of methanol, compared to 20% commercial Pt/C (Fig. 7B).
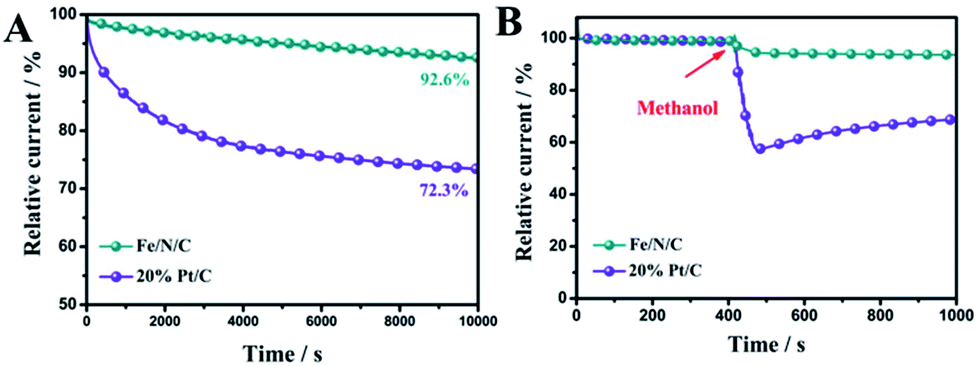 | ||
| Fig. 7 (A) Chronoamperometric response of Fe/N/C and Pt/C in O2-saturated 0.1 M KOH aqueous solution. (B) The durability test of Fe/N/C and Pt/C for methanol. | ||
Conclusions
In summary, iron and nitrogen doped hollow structured carbon materials with Fe–N coordination were readily prepared by a simple self-template method with NaCl protection during pyrolysis. The Fe/N/C catalysts also possess larger electrical double-layer capacitance (27.6 μF cm−2), specific surface area (1146.75 m2 g−1) and nitrogen content (6.71%), compared with the PDA@MF-Fe-800, which means that the NaCl protection strategy can produce an effect on crucial factors for the ORR catalytic activity. XANES and EXAFS further confirmed that the NaCl protection strategy can assist the formation of Fe–N coordination, indicating that the strategy can endow the materials with more active sites. The fabricated Fe/N/C catalysts have demonstrated outstanding electrocatalytic activity for the ORR in alkaline media, including the positive half-wave potential (0.84 V vs. RHE), the predominant four-electron transfer process (n = 3.96), and the superior stability and methanol tolerance compared to 20% commercial Pt/C. This work presents significant progress in the synthesis of highly efficient ORR catalysts with a distinctive structure by the self-template method, confirming the promoting effect of NaCl protection for the formation of the active site, which can provide guidance for the synthesis of non-precious ORR catalysts with controlled morphology and high-density active sites.Experimental
Chemicals and materials
Melamine (Sigma-Aldrich), formaldehyde (37% aqueous solution, Alfa Aesar), sodium dodecyl sulfate (SDS, Alfa Aesar), dopamine (Alfa Aesar), tris(hydroxymethyl)aminomethane (Tris, Alfa Aesar), HCl (Beijing Chemical Reagent Factory), FeCl3 (Alfa Aesar), NaCl (Beijing Chemical Reagent Factory), KOH (Beijing Chemical Reagent Factory), iron phthalocyanine (Alfa Aesar) were used in this experiment.Preparation of melamine-formaldehyde resin (MF) and PDA@MF
In a typical procedure,61 1.2 g of melamine was dissolved in 20 mL deionized water (Millipore, 18.2 MΩ) with the addition of 9.4 g of formaldehyde solution. After vigorous stirring at 60 °C for about 10 minutes, the mixed solution was cooled to room temperature, and 1.2 g of SDS added with stirring at a low speed. The homogeneous precursor solution was then transferred into a 50 mL autoclave at 120 °C for 3 h. After hydrothermal reaction, the obtained white products were collected by centrifuging and washed several times with deionized water and dried at 60 °C for 12 h in a vacuum oven to obtain the MF product.MF (0.2 g) and 0.4 g dopamine were uniformly dispersed in 40 mL Tris–HCl buffer solution (pH = 8.8) by ultrasonication for 10 min, then the mixed solution was stirred for 24 h in order to prepare PDA@MF.62 After centrifuging and washing with deionized water, the resultant was dried at 60 °C for 12 h remove the solvent residuals.
Preparation of Fe/N/C catalyst
The resultant PDA@MF and FeCl3 (mass ratio = 1.2:1) were dispersed in 15 mL saturated NaCl solution with continuous stirring for 8 h and the solvent was allowed to evaporate. The dried mixture was transferred into a quartz boat, which was put into a tubular furnace and calcined under an argon atmosphere at 800 °C for 2 h with a programming rate of 3 °C min−1, then finally cooled to room temperature. The carbonized products were put into 50 mL of 3 M H2SO4 for 8 h in order to remove some unstable components. Ultimately, the Fe/N/C product was obtained by centrifugation and washed with a large amount of deionized water for completely removing NaCl, then dried at 60 °C in a vacuum. The products were denoted as Fe/N/C-700, Fe/N/C and Fe/N/C-900. As a control, the electrocatalyst was also prepared without the presence of NaCl via the same procedure and was denoted as PDA@MF-Fe-800. The 800 °C carbonized MF and PDA@MF were denoted as MF-800 and PDA@MF-800, respectively.
Physical characterization
The microstructures of the synthesized samples were observed by scanning electron microscopy (SEM, Hitachi, Japan), transmission electron microscopy (TEM, Hitachi, Japan), and high-resolution transmission electron microscopy (HR-TEM, FEI Tecnai F20, USA). The crystal structures of the samples were acquired by Powder X-ray diffraction (XRD, D8 Advance, Bruker, Germany) with a Cu Kα radiation source (λ = 1.5406 Å). The chemical states and the surface properties of the catalysts were characterized using X-ray photoelectron spectroscopy (XPS) (ESCALAB 250, UK) with Al Kα radiation as the excitation source. The Raman spectroscopy data were obtained using a confocal Raman microscope (InVia Reflex, Renishaw) with a 633 nm excitation laser. The specific surface area and pore size distribution were characterized by a Quantachrome NOVA 1000e Brunauer–Emmet–Teller (BET) analyzer at 77 K. The pore size distribution was calculated by the Barrett–Joyner–Halenda (BJH) model.The electronic structure and coordination environment of the central Fe atom in the catalysts were confirmed by X-ray absorption fine structure spectroscopy (XAFS). The Fe K-edge spectra were acquired using a double-crystal Si (111) monochromator in the fluorescence mode. X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) were also used, and the Athena software package was used to analyze the data.63
Electrochemical measurement
Electrochemical measurements were executed using a standard three-electrode test system, which was fabricated using platinum wire as the counter electrode, saturated calomel electrode (SCE) as the reference electrode and the catalyst modified glassy carbon electrode (GCE) as the working electrode (ϕ = 4 mm). Potential versus SCE was converted to the RHE scale according to E (vs. RHE) = E (vs. SCE) + 0.0591 × pH + 0.244. The working rotating disk electrodes (RDEs) were prepared as follows: the working RDEs were polished successively by 1 μm, 0.5 μm and 0.05 μm alumina suspensions to a mirror finish before use. Initially, 5 mg of catalyst was dispersed in 490 μL ethanol and 10 μL Nafion solution (5 wt%); the mixture was then agitated via ultrasonication for 30 min. Subsequently, 10 μL of the ink was deposited onto the electrode surface and dried in air at room temperature.The ORR performance was evaluated by cyclic voltammograms (CV) and linear sweep voltammetry (LSV) in O2-saturated 0.1 M KOH at a scan rate of 20 mV s−1 and 5 mV s−1, respectively, which were performed by SP-200 electrochemical workstation (Bio-Logic SA). The slopes of the ORR polarization curves with different rotation rates, 400 rpm, 625 rpm, 900 rpm, 1225 rpm, 1600 rpm, were then evaluated, which were used to calculate the number of electrons transferred (n) by the first-order Koutecky–Levich (K–L) equation:
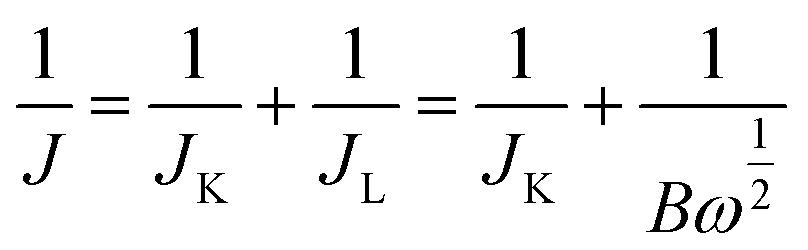 | (1) |
| B = 0.62nFC0(D0)2/3ν−1/6 | (2) |
485 C mol−1), C0 is the bulk concentration of O2 (1.9 × 10−3 mol L−1), D0 is the diffusion coefficient of O2 in the KOH electrolyte (1.1 × 10−5 cm2 s−1), ν is the electrolyte viscosity (0.01 cm2 s−1).
The stability was tested by chronoamperometric measurement (CA) in an O2-saturated 0.1 M KOH electrolyte at −0.2 V (SCE) with a rotation rate of 1600 rpm. The methanol tolerance test was conducted by adding 3 M methanol to the O2-saturated electrolyte solution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support of this study by the National Natural Science Foundation (no. 11179033), the Capacity Building for Sci-Tec Innovation-Fundamental Scientific Research Funds (025185305000/210), the Beijing Natural Science Foundation (no. 2102012 and no. 2182012).Notes and references
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef PubMed.
- R. F. Service, Science, 2002, 296, 1222–1224 CrossRef PubMed.
- I. E. L. Stephens, A. S. Bondarenko, U. Grønbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744–6762 RSC.
- S. Y. Wang and S. P. Jiang, Natl. Sci. Rev., 2017, 4, 163–166 Search PubMed.
- S. Wang, L. I. Yunxiao, D. Yan, Y. Zou, C. Xie, Y. Wang and Y. Zhang, J. Mater. Chem. A, 2017, 5, 25494–25500 RSC.
- J. Wu and H. Yang, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef PubMed.
- M. Shao, Q. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef PubMed.
- T. Chen, Z. Cai, Z. Yang, L. Li, X. Sun, T. Huang, A. Yu, H. G. Kia and H. Peng, Adv. Mater., 2011, 23, 3594–3657 Search PubMed.
- J. Yu, G. Chen, S. Jaka, Y. Zhu, R. Ran, Z. Zhu, W. Zhou and Z. Shao, Adv. Sci., 2016, 3, 1600060 CrossRef PubMed.
- W. Niu, L. Li, X. Liu, N. Wang, J. Liu, W. Zhou, Z. Tang and S. Chen, J. Am. Chem. Soc., 2015, 137, 5555–5562 CrossRef PubMed.
- S. J. Kim, J. Mahmood, C. Kim, G. F. Han, S. W. Kim, S. M. Jung, G. Zhu, J. J. D. Yoreo, G. Kim and J. B. Baek, J. Am. Chem. Soc., 2018, 140, 1737–1742 CrossRef PubMed.
- W. Xia, A. Mahmood, Z. Liang, R. Zou and S. Guo, ChemInform, 2016, 47, 2650–2676 Search PubMed.
- M. A. Molina-García and N. V. Rees, Appl. Catal., A, 2017, 84, 65–70 Search PubMed.
- Y. Wang, J. Li and Z. Wei, ChemElectroChem, 2018, 5, 1–12 CrossRef.
- M. Yang, H. Chen, D. Yang, Y. Gao and H. Li, J. Power Sources, 2016, 307, 152–159 CrossRef.
- P. Cheng, S. Li, R. Li, J. Yan, W. Yu, X. Shao, Z. Hu and D. Yuan, RSC Adv., 2015, 5, 107389–107395 RSC.
- L. Yang, Y. Su, W. Li and X. Kan, J. Phys. Chem. C, 2015, 119, 11311–11319 CrossRef.
- M. Q. Wang, W. H. Yang, H. H. Wang, C. Chen, Z. Y. Zhou and S. G. Sun, ACS Catal., 2014, 4, 3928–3936 CrossRef.
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878–1889 CrossRef PubMed.
- J. Zhu, M. Xiao, C. Liu, J. Ge, J. Stpierre and W. Xing, J. Mater. Chem. A, 2015, 3, 21451–21459 RSC.
- C. Zhu, S. Fu, J. Song, Q. Shi, D. Su, M. H. Engelhard, X. Li, D. Xiao, D. Li and L. Estevez, Small, 2017, 13, 1603407 CrossRef PubMed.
- C. D. Tomas, I. Suarez-Martinez, F. Vallejos-Burgos, M. J. Lopez, K. Kaneko and N. A. Marks, Carbon, 2017, 19, 1–9 CrossRef.
- J. Ma, D. Xiao, C. L. Chen, Q. Luo, Y. Yu, J. Zhou, J. Ma and L. Zheng, J. Power Sources, 2018, 378, 491–498 CrossRef.
- W. Yang, X. Liu, X. Yue, J. Jia and S. Guo, J. Am. Chem. Soc., 2015, 137, 1436–1439 CrossRef PubMed.
- L. Xu, Q. Jiang, Z. Xiao, X. Li, J. Huo, S. Wang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef PubMed.
- G. L. Tian, M. Q. Zhao, D. Yu, X. Y. Kong, J. Q. Huang, Q. Zhang and F. Wei, Small, 2014, 10, 5277–5281 Search PubMed.
- Q. Wang, Z. Y. Zhou, Y. J. Lai, Y. You, J. G. Liu, X. L. Wu, E. Terefe, C. Chen, L. Song and M. Rauf, J. Am. Chem. Soc., 2014, 136, 10882–10885 CrossRef PubMed.
- X. Jin, Y. Jiang, Q. Hu, S. Zhang, Q. Jiang, L. Chen, L. Xu, Y. Xie and J. Huang, RSC Adv., 2017, 7, 56375–56381 RSC.
- Z. Huang, H. Pan, W. Yang, H. Zhou, N. Gao, C. Fu, S. Li, H. Li and Y. Kuang, ACS Nano, 2018, 12, 208–216 CrossRef PubMed.
- Y. Wang, C. Zhu, S. Feng, Q. Shi, S. Fu, D. Dan, Z. Qiang and Y. Lin, ACS Appl. Mater. Interfaces, 2017, 9, 40298–40306 CrossRef PubMed.
- M. Hoque, S. Zhang, M. L. Thomas, Z. Li, S. Suzuki, A. Ando, M. Yanagi, Y. Kobayashi, K. Dokko and M. Watanabe, J. Mater. Chem. A, 2017, 6, 1138–1149 RSC.
- Y. Wang, L. Tao, Z. Xiao, R. Chen, Z. Jiang and S. Wang, Adv. Funct. Mater., 2018, 28, 1705356 CrossRef.
- P. Zhou, Y. Wang, C. Xie, C. Chen, H. Liu, R. Chen, J. Huo and S. Wang, Chem. Commun., 2017, 53, 11778–11781 RSC.
- Y. Lv, L. Gan, M. Liu, W. Xiong, Z. Xu, D. Zhu and D. S. Wright, J. Power Sources, 2012, 209, 152–157 CrossRef.
- A. Mufundirwa, G. F. Harrington, B. Smid, B. V. Cunning, K. Sasaki and S. M. Lyth, J. Power Sources, 2018, 5, 244–254 CrossRef.
- J. Liu, P. Li, L. Wang, H. Zhang, H. Liu, J. Liu, Y. Wang, W. Tian and X. Wang, Carbon, 2018, 126, 1–8 CrossRef.
- W. S. Jung and B. N. Popov, Carbon, 2017, 122, 746–755 CrossRef.
- N. S. Kim, Y. Tack Lee, J. Park, H. Ryu, H. J. Lee, S. Y. Choi and J. Choo, J. Phys. Chem. B, 2002, 106, 9286–9290 CrossRef.
- N. Tachibana, S. Ikeda, Y. Yukawa and M. Kawaguchi, Carbon, 2017, 115, 515–525 CrossRef.
- X. L. Sui, Z. B. Wang, C. Z. Li, J. J. Zhang, L. Zhao and D. M. Gu, Catal. Sci. Technol., 2016, 6, 3767–3775 RSC.
- C. Guo, W. Liao, Z. Li and C. Chen, Carbon, 2015, 85, 279–288 CrossRef.
- H. Yang, J. Zhu, Q. Lv, C. Liu, Q. Li and X. Wei, Electrochim. Acta, 2015, 155, 335–340 CrossRef.
- L. Tao, Q. Wang, S. Dou, Z. Ma, J. Huo, S. Wang and L. Dai, Chem. Commun., 2016, 52, 2764–2767 RSC.
- I. Abidat, C. Morais, S. Pronier, N. Guignard, J. D. Comparot, C. Canaff, T. W. Napporn, A. Habrioux, A. S. Mamede and J. F. Lamonier, Carbon, 2016, 111, 849–858 CrossRef.
- R. Al-Jishi, B. S. Elman and G. Dresselhaus, Carbon, 1982, 20, 127 CrossRef.
- R. E. Shroder, R. J. Nemanich and J. T. Glass, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 3738 CrossRef.
- M. Veres, S. Tóth and M. Koós, Diamond Relat. Mater., 2008, 17, 1692–1696 CrossRef.
- C. Deng, H. Zhong, X. Li, L. Yao and H. Zhang, Nanoscale, 2016, 8, 1580–1587 RSC.
- H. Zhang, T. Wang, J. Wang, H. Liu, T. D. Dao, M. Li, G. Liu, X. Meng, K. Chang and L. Shi, Adv. Mater., 2016, 28, 3703–3710 CrossRef PubMed.
- W. Yan, L. Wang, C. Chen, D. Zhang, A. J. Li, Z. Yao and L. Y. Shi, Electrochim. Acta, 2016, 188, 230–239 CrossRef.
- J. Shui, M. Wang, D. Feng and L. Dai, Sci. Adv., 2015, 1, e1400129 Search PubMed.
- Q. L. Zhu, W. Xia, L. R. Zheng, R. Zou, Z. Liu and Q. Xu, ACS Energy Lett., 2017, 2, 504–511 CrossRef.
- Y. J. Sa, D. J. Seo, J. Woo, J. T. Lim, J. Y. Cheon, S. Y. Yang, J. M. Lee, D. Kang, T. J. Shin and H. S. Shin, J. Am. Chem. Soc., 2016, 138, 15046 CrossRef PubMed.
- Y. Lei, Q. Wang, Z. Chen, N. Wu, Y. Wang, B. Wang and Y. Wang, J. Mater. Chem. A, 2017, 6, 516–526 Search PubMed.
- A. Mufundirwa, G. F. Harrington, B. Smid, B. V. Cunning, K. Sasaki and S. M. Lyth, J. Power Sources, 2017, 375, 244–254 CrossRef.
- M. M. Hossen, K. Artyushkova, P. Atanassov and A. Serov, J. Power Sources, 2017, 375, 214–221 CrossRef.
- A. Zitolo, V. Goellner, V. Armel, M. T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937 CrossRef PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef PubMed.
- X. Fan, Z. Peng, R. Ye, H. Zhou and X. Guo, ACS Nano, 2015, 9, 7407–7418 CrossRef PubMed.
- X. Zhou, Z. Bai, M. Wu, J. Qiao and Z. Chen, J. Mater. Chem. A, 2015, 3, 3343–3350 RSC.
- Y. Wu, Y. Li, L. Qin, F. Yang and D. Wu, J. Mater. Chem. B, 2012, 1, 204–212 RSC.
- R. Wu, S. Chen, Y. Zhang, Y. Wang, Y. Nie, W. Ding, X. Qi and Z. Wei, J. Mater. Chem. A, 2016, 4, 2433–2437 RSC.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra03672a |
| This journal is © The Royal Society of Chemistry 2018 |