
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
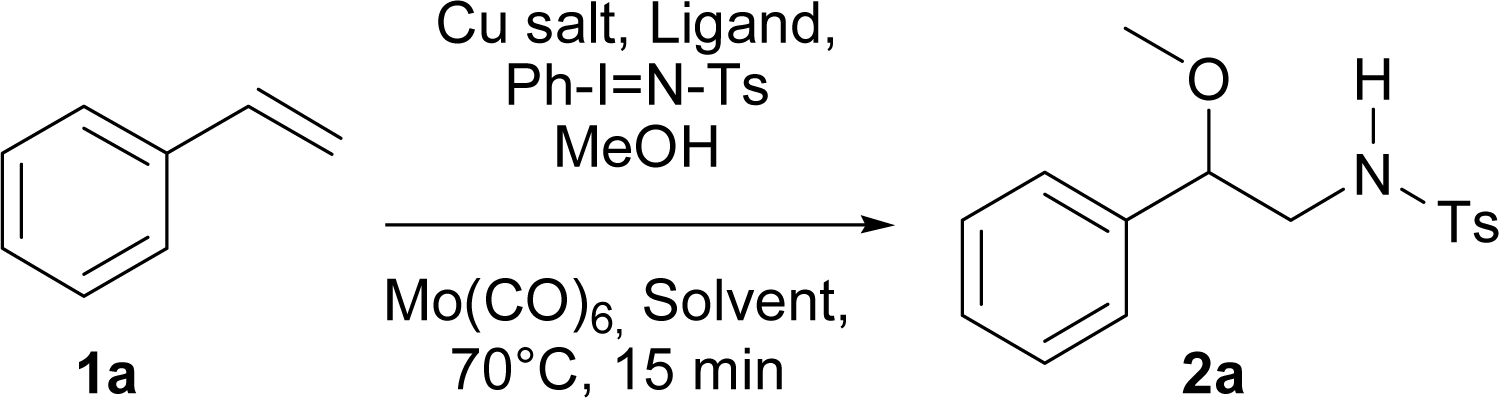
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of β-alkoxy-N-protected phenethylamines via one-pot copper-catalyzed aziridination and ring opening†
Jorge Saavedra-Olavarríaa,
Matías Madrid-Rojasb,
Iriux Almodovarb,
Patricio Hermosilla-Ibáñezb and
Edwin G. Pérez *a
*a
aDepartment of Organic Chemistry, Faculty of Chemistry, Pontificia Universidad Católica de Chile, Av. Vicuña Mackenna 4860, Casilla 306, Correo 22, Santiago, Chile. E-mail: eperezh@uc.cl
bFacultad de Química y Biología, Universidad de Santiago de Chile, USACh, Santiago, Chile
First published on 6th August 2018
Abstract
A regioselective, copper-catalyzed, one-pot aminoalkoxylation of styrenes using primary and secondary alcohols and three different iminoiodanes as alkoxy and nitrogen sources respectively, is reported. The β-alkoxy-N-protected phenethylamines obtained were used to synthesise β-alkoxy-N-benzylphenethylamines which are interesting new compounds that could act as possible neuronal ligands.
Aziridines are an important class of nitrogen-containing heterocycles that can be found in a number of biologically active compounds1 and have also been synthesised by several routes.2 Aziridines are suitable synthetic scaffolds or intermediates for the synthesis of many kinds of organic compounds through their ring opening by different nucleophiles including cyanide, aromatic and olefinic compounds, hydride, alcohols, thiols, amines, and halogens, affording various 1,2-difunctionalised compounds.3 Among these 1,2-difunctionalised products, vicinal amino ethers have been obtained using different methodologies including inorganic protic or Lewis acids such as BF3·Et2O, Sn(OTf)2, (NH4)2Ce(NO3)6, ionic liquids, [Ag(COD)2]PF6, and also by an aprotic imidazolium zwitterion, an N,N′-dioxide–Mg(OTf)2 complex, sulphated zirconia, Ag(I), Au(I), phosphomolybdic acid supported on silica gel, montmorillonites and ceric ammonium nitrate, but always using previously isolated aziridines.4 In addition, very recently, the aziridination of alkenes with subsequent ring opening using alcohols under continuous flow was reported.5 As far as we know, only a small number of one-pot methodologies has been reported using rhodium, iron(II) phthalocyanine and ruthenium as the catalysts and different nucleophiles to generate N-protected difunctionalised alkenes. However, the number of N-protected β-amino ethers is limited.6
N-Protected β-aminophenyl ethers are useful intermediates in the synthesis of substituted indolines,7 and the corresponding N-deprotected β-alkoxyphenethylamines have shown some interesting biological activities.8 For these reasons, we decided to look for a one-pot aziridination-ring opening process but using inexpensive copper catalysts and combining different styrenes, alcohols and phenyl iminoiodinanes.
Results and discussion
Initially, and based on a recent methodology reported by us, we decided to use styrene (1a) as a model substrate, Cu(MeCN)4BF4 and neocuproine as the catalyst and ligand respectively, with Ph-I![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-Ts acting as the nitrene source, plus methanol.9 In addition, we chose Mo(CO)6 as an additive to improve the yield.10 Using the conditions mentioned in Table 1, entry 1, N-tosyl-β-methoxyphenethylamine (2a) was obtained in 48% yield, while none of this desired product was formed when the copper salt was omitted under similar conditions (entry 2). Replacing Cu(MeCN)4BF4 with Cu(BF4)2·H2O the yield increased slightly (entry 3). Conversely, when Cu(BF4)2·H2O and nitromethane were used as the catalyst and solvent, the isolated yield increased to 74% (Table 1, entry 4). Additional trials using other solvents did not further improve the yield (entries 5–9). 2,2′-Bipyridine and 1,10-phenanthroline were tested as ligands, but the yields dropped to 57 and 39% respectively (entries 10 and 11). Decreasing the catalyst loading to 5% resulted in a dramatic detriment of the yield (entry 12). When less equivalents of methanol were used (entries 15 and 16), the yields were similar to that found in entry 4. Finally, decreasing Mo(CO)6 to 0.1 or increasing neocuproine to 0.2 equivalents were found to be less efficient in promoting 2a formation (entries 17 and 18).
N-Ts acting as the nitrene source, plus methanol.9 In addition, we chose Mo(CO)6 as an additive to improve the yield.10 Using the conditions mentioned in Table 1, entry 1, N-tosyl-β-methoxyphenethylamine (2a) was obtained in 48% yield, while none of this desired product was formed when the copper salt was omitted under similar conditions (entry 2). Replacing Cu(MeCN)4BF4 with Cu(BF4)2·H2O the yield increased slightly (entry 3). Conversely, when Cu(BF4)2·H2O and nitromethane were used as the catalyst and solvent, the isolated yield increased to 74% (Table 1, entry 4). Additional trials using other solvents did not further improve the yield (entries 5–9). 2,2′-Bipyridine and 1,10-phenanthroline were tested as ligands, but the yields dropped to 57 and 39% respectively (entries 10 and 11). Decreasing the catalyst loading to 5% resulted in a dramatic detriment of the yield (entry 12). When less equivalents of methanol were used (entries 15 and 16), the yields were similar to that found in entry 4. Finally, decreasing Mo(CO)6 to 0.1 or increasing neocuproine to 0.2 equivalents were found to be less efficient in promoting 2a formation (entries 17 and 18).
|
||||
|---|---|---|---|---|
| Entry | Copper salt 10% | Ligand 10% | Solvent | Yieldb,c[%] |
| a Reactions were carried out with 1a (1.0 mmol) in 3.0 mL of solvent, Ph-IN-Ts (1.5 eq.), MeOH (10 eq.), Mo(CO)6 (0.25 eq) in an open tube, unless otherwise noted.b NMR-determined yields in a 0.20 mmol scale reaction of 1a using 1,3,5-trimethoxybenzene as internal standard are shown in parentheses.c Isolated yields.d Catalyst 5%.e 30 min.f MeOH (5.0 eq.).g MeOH (2.0 eq).h Mo(CO)6 (0.1 eq.).i Neocuproine (20%). |
||||
| 1 | Cu(MeCN)4BF4 | Neocuproine | DCE | (48) |
| 2 | — | Neocuproine | DCE | — |
| 3 | Cu(BF4)2·H2O | Neocuproine | DCE | 53(54) |
| 4 | Cu(BF4)2·H2O | Neocuproine | MeNO2 | 74(75) |
| 5 | Cu(BF4)2·H2O | Neocuproine | CH3CN | (45) |
| 6 | Cu(BF4)2·H2O | Neocuproine | Toluene | (53) |
| 7 | Cu(BF4)2·H2O | Neocuproine | Ph-Cl | (66) |
| 8 | Cu(BF4)2·H2O | Neocuproine | EtOAc | (27) |
| 9 | Cu(BF4)2·H2O | Neocuproine | MeOH | (66) |
| 10 | Cu(BF4)2·H2O | 2,2′-Bipyridine | MeNO2 | (57) |
| 11 | Cu(BF4)2·H2O | 1,10-Phenanthroline | MeNO2 | (39) |
| 12d | Cu(BF4)2·H2O | Neocuproine | MeNO2 | (36) |
| 13e | Cu(BF4)2·H2O | Neocuproine | MeNO2 | (72) |
| 15f | Cu(BF4)2·H2O | Neocuproine | MeNO2 | (73) |
| 16g | Cu(BF4)2·H2O | Neocuproine | MeNO2 | (69) |
| 17h | Cu(BF4)2·H2O | Neocuproine | MeNO2 | (57) |
| 18i | Cu(BF4)2·H2O | Neocuproine | MeNO2 | (36) |
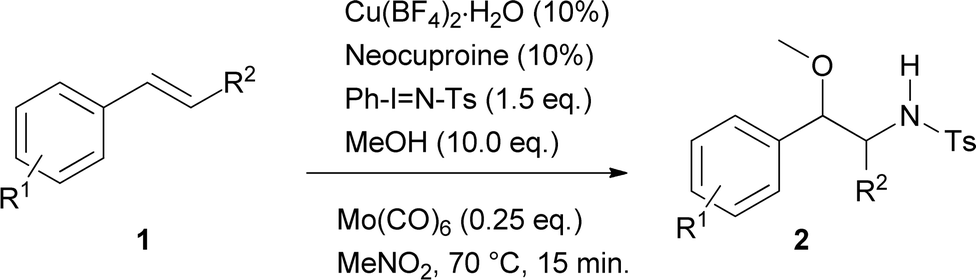
Using these optimised conditions, the scope of the reaction was investigated. Substrates were tested including changes in the scaffold of the styrene, as well as that of the double bond, the alcohol and the oxidant. The results showed that this is a robust methodology since it tolerates a wide assortment of modifications. Besides, complete regioselectivity was observed considering that only 2-alkoxy-N-protected-phenethylamines were detected.
Table 2 displays the outcomes of the reactions using different styrenes under the previously selected conditions, giving rise to products 2a–2p in yields varying from 15 to 92%. Aryl substituents (R1) of the styrene used differ not only in electronic nature, but also in their position on the aromatic ring. R1 structure included both electron-donor and electron-acceptor groups. Unmodified styrene gave 2a in 75% yield. The products from alkyl-substituted styrenes, p-methylstyrene (2b), p-t-butylstyrene (2g) and m-methylstyrene (2h) were obtained in 71, 78 and 63% yield respectively. Compound 2k was produced from m-methoxystyrene in 53% yield. Halogenated substrates tested including p-chloro, p-fluoro, and p-bromostyrene, providing 2c (64%), 2e (69%), 2d (72%). Furthermore m-chloro, m-fluoro, and o-fluorostyrene produced 2i (47%), 2o (56%) and 2j (50%). An acetoxy substituent at the para-position was also tested, giving 2f in 92% yield. A strong electron-acceptor group like trifluoromethane at the ortho-position gave 2p in 15% yield. Other substitutions at the ortho-position such as bromo, methyl and phenyl gave the products 2q, 2r and 2s in 43, 30 and 65% yield respectively. Interestingly, no steric hindrance was observed for the latter compound.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: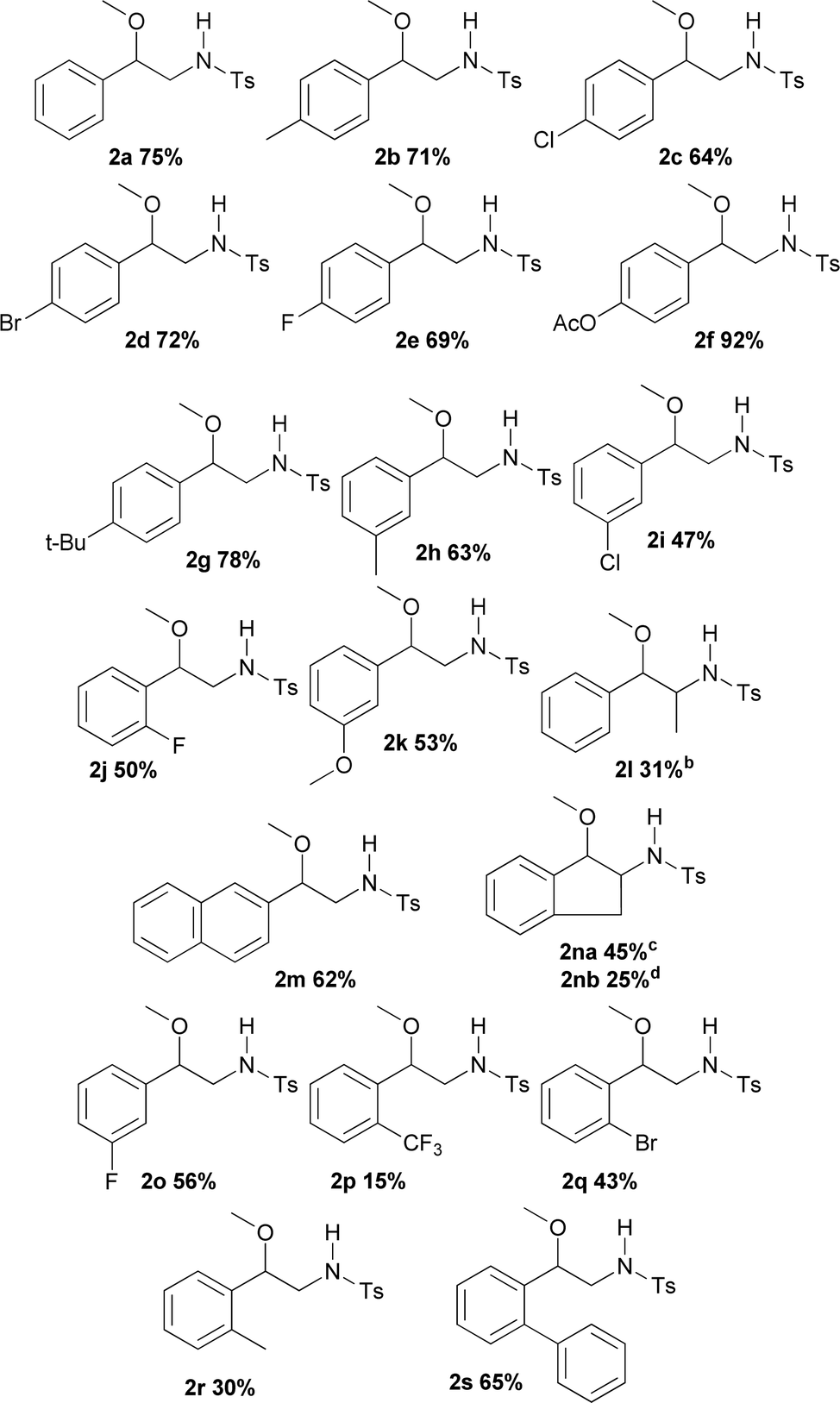
When changes in the scaffold of the double bond were made, β-methylstyrene, an internal alkene, furnished 2l in 31% yield. On the other hand, 1H-indene, as an example containing an endocyclic double bond, generated both the (2na) and (2nb) products in 45 and 25% yield respectively, showing a slight, but not uninteresting diastereoselectivity in this addition reaction. Unfortunately, with ortho or para-methoxystyrene and unactivated olefins (including 1-hexene, 1-octene and cyclohexene) the desired products were not obtained.
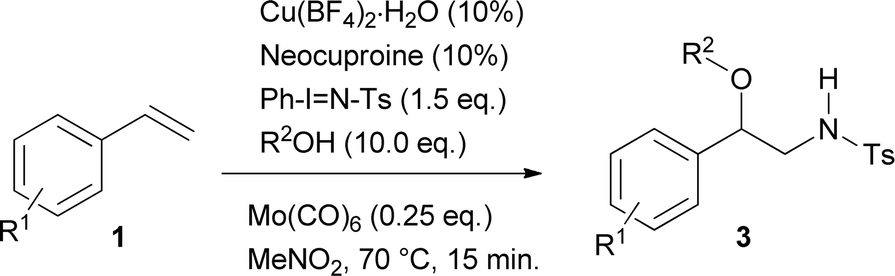
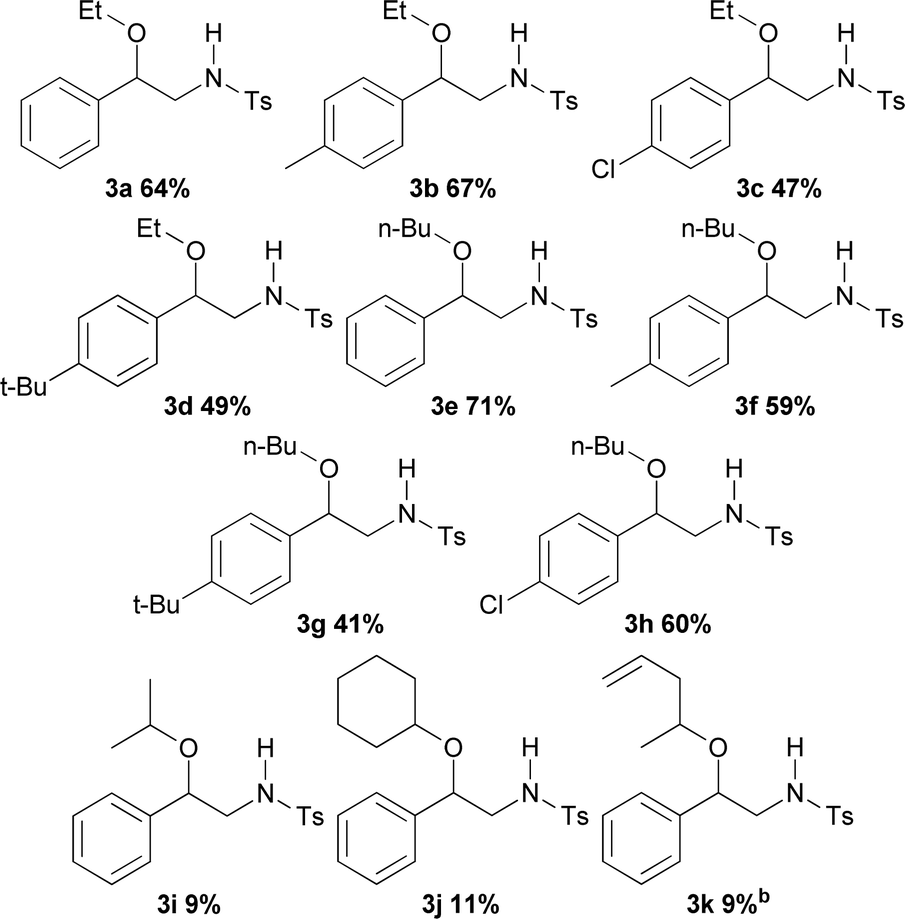
Reactions with alcohols other than methanol supplied compounds 3a–3h in yields going from 71 to 41% (Table 3). With ethanol and n-butanol as reactants and styrene, p-methylstyrene, p-t-butylstyrene and p-chlorostyrene as substrates, the results showed no apparent influence of either the structure of the alcohol or that of the styrene on the reaction yields. Thus, unsubstituted styrene afforded the ethoxy derivative 3a in 64% and the butoxy derivative 3e in 71% yield; p-methylstyrene generated the aminoethoxylated and aminobutoxylated products 3b (67%) and 3f (59%) respectively. Nevertheless, the reaction of styrene bearing the bulker t-butyl substituent showed slightly decreased yields for both the ethoxy (3d) and butoxy (3g) derivatives, 49 and 41% respectively. On the other hand, aminoalkoxylation of p-chlorostyrene with ethanol provided product 3c in 47% yield while the reaction with n-butanol gave 60% of the butoxy derivative 3h. Introduction of steric hindrance by using isopropyl alcohol, cyclohexanol and allylmethylcarbinol as reactants, caused a significant decrease in the reaction yields with styrene leading to 3i (9%), 3j (11%) and 3k (9%) respectively. The structure of 3d was unequivocally assigned by X-ray diffraction analysis (see ESI† for more details).11
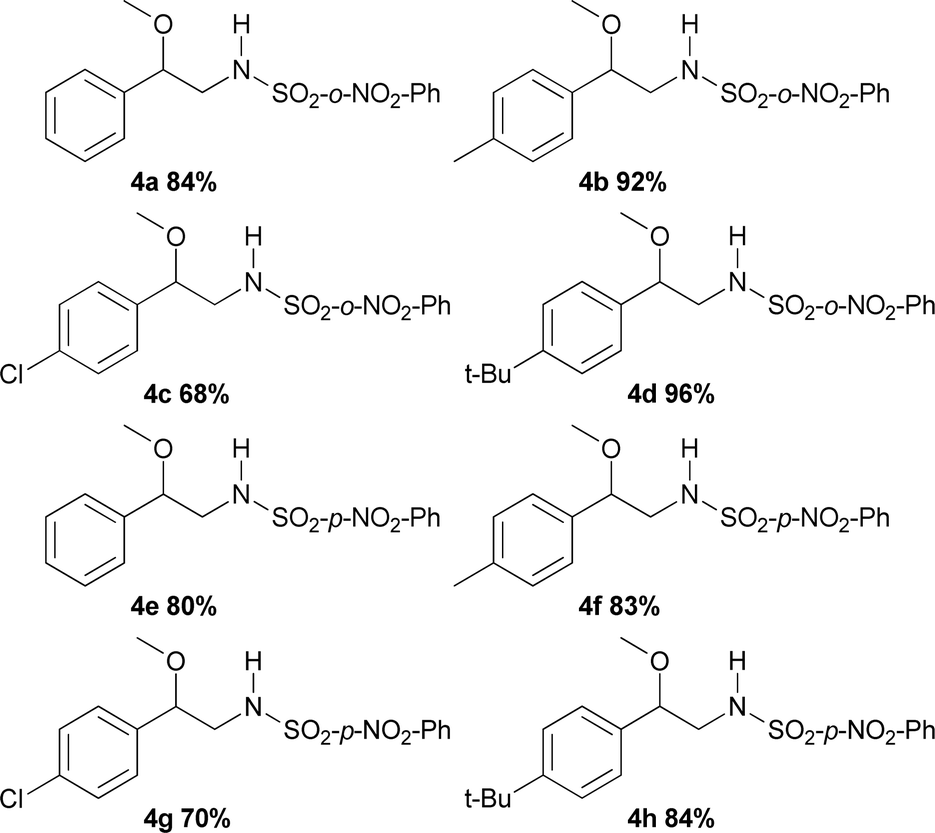
The substrate scope of phenyliodinanes was then investigated. As shown in Table 4, (N-(o- and p-nitro phenylsulphonyl)imino)phenyliodinanes were used as oxidants in these reactions, affording compounds 4a to 4h. Moderately increased yields were observed when the N-(sulphonyl)imino substituent was changed from Ts (Table 2) to p-NO2Ph to o-NO2Ph, for the substrates under study, except for the chloro compound. The products from the reactions of styrenes and o-NO2Ph-, (H-), (Me-), (Cl-) and (t-Bu-)phenyliodinanes,4a, 4b, 4c and 4d, were obtained in 84, 92, 68 and 96% yield, respectively. On the other hand, the use of p-NO2Ph-substituted phenyliodinane as nitrogen source with different styrenes gave 4e (H-), 4f (Me), 4g (Cl-) and 4h (t-Bu-) in 80, 83, 70 and 84% yield respectively.
|
|---|
| a Yields of isolated products after column chromatography of reaction mixtures on a 1.0 mmol scale. |
 |
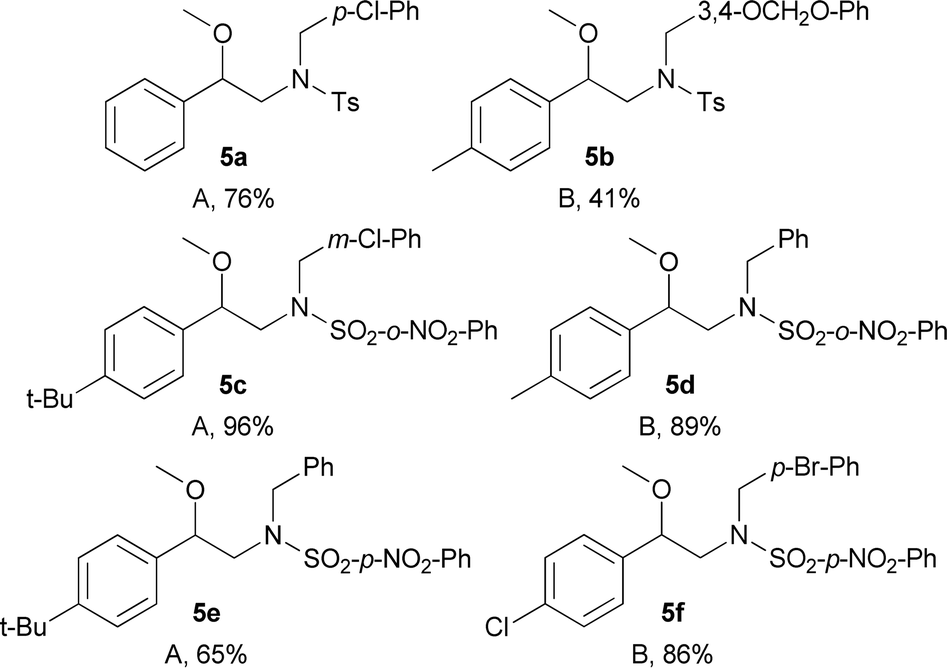
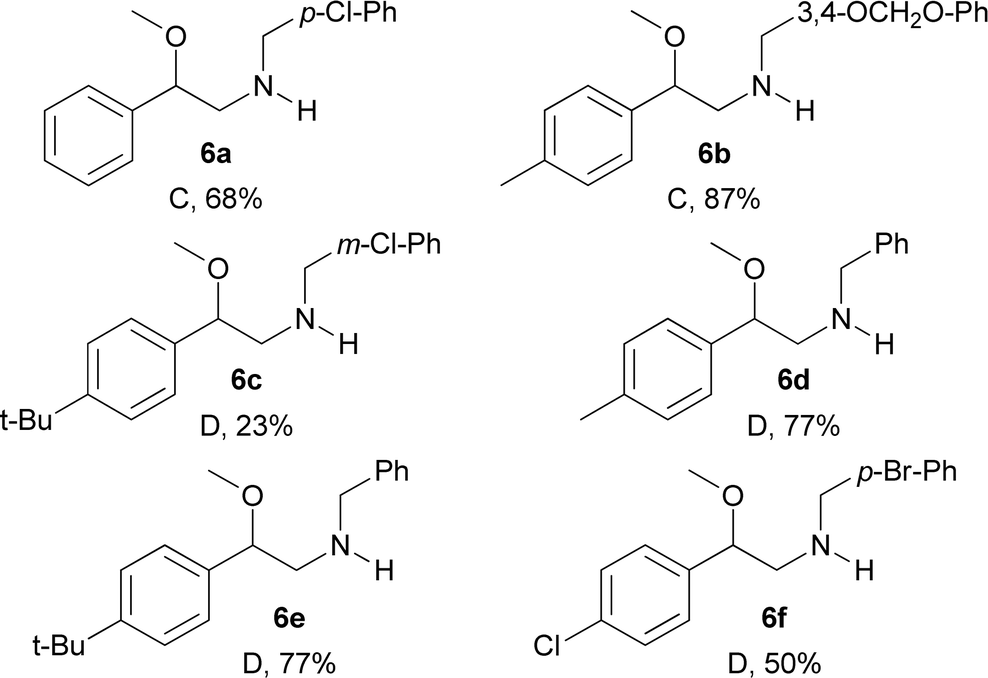
To show the usefulness of the new compounds as building blocks and continuing with our interest in the synthesis of substances with possible activity on neuronal targets, we decided to synthesise the N-benzylated derivatives 6a–6f.12 To achieve this goal, we initially took advantage of the acidity of the sulphonamide N–H bond present in compounds 2a, 2b, 4d, 4b, 4h and 4g which were N-benzylated using basic alkylation conditions (A) or Mitsunobu conditions (B) furnishing the corresponding N-protected-N-benzylated-β-methoxyphenethylamines 5a–5f in yields in the 41–96% range (Table 5).
|
|---|
| a Yields of isolated products after column chromatography of reactions carried out on a 0.5 mmol scale. Conditions: (A) β-methoxysulphonamide (1.0 eq.), NaH (1.1 eq.), DMF, benzyl halide (1.1 eq.), 0 °C to r.t., 12 h. (B) β-Methoxysulphonamide (1.0 eq.), triphenylphosphine (1.4 eq.), benzyl alcohol (1.4 eq.), DIAD (2.0 eq.), THF, 0 °C to r.t., 12 h. |
 |

In addition, compounds 5a–5f were subsequently deprotected using Mg in methanol with ultrasound activation (condition C) or basic 2-mercaptoethanol (condition D) for Ts or Ns groups, respectively.13,14 Thus, the desired N-benzylated β-methoxyphenethylamines 6a–6f were obtained in yields in the 23–87% range (Table 6).
|
|---|
| a Yields of isolated products after column chromatography of reactions on a 0.3 mmol scale. Conditions: (C) Mg, MeOH, ultrasound, 1 h. (D) 2-Mercaptoethanol, DBU, DMF, r.t., 12 h. |
 |

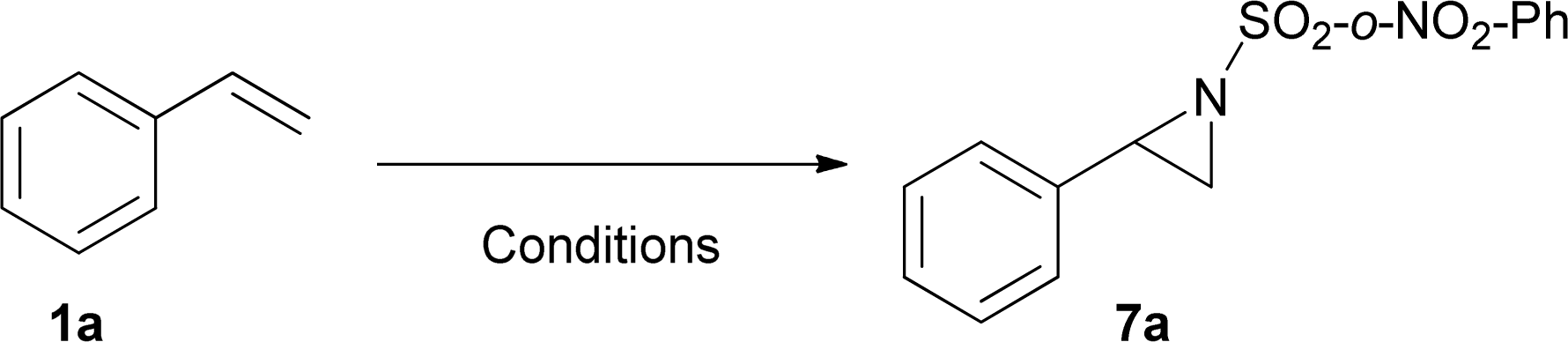
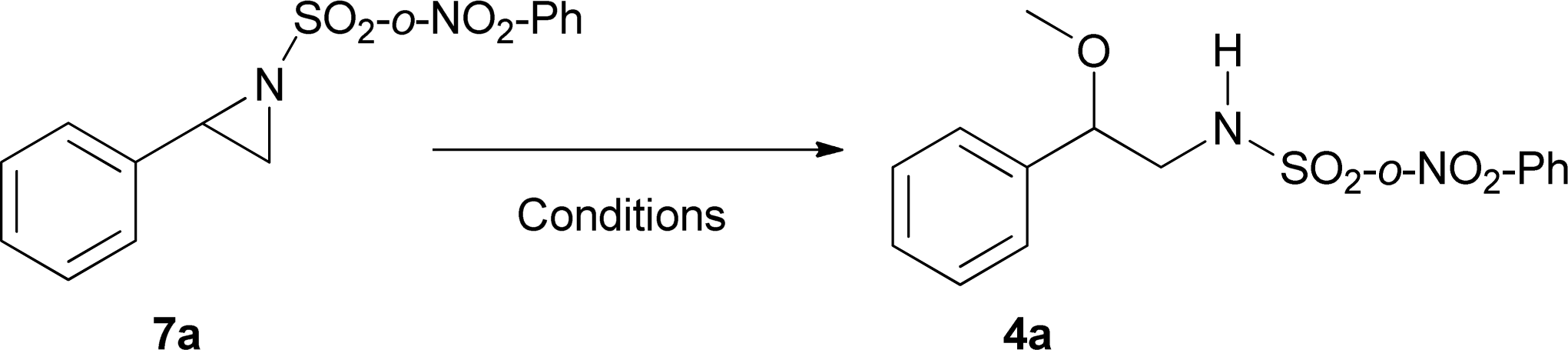
Finally, we carried out some preliminary experiments to elucidate the role of Mo(CO)6 on the reaction pathway. Tables 7 and 8 show the influence of Mo(CO)6 on the nitrene transfer and aziridine ring opening respectively.
|
|||
|---|---|---|---|
| Entry | Cu(BF4)2·H2O | Mo(CO)6 | Yieldb [%] |
| a Conditions: styrene (0.2 mmol), Cu(BF4)2·H2O (0.1 eq.); neocuproine (0.1 eq.); Ph-IN-SO2-o-NO2-Ph (1.5 eq.); Mo(CO)6 (0.25 eq.); MeNO2 (0.6 mL).b Yields were determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard. |
|||
| 1 | + | + | 64 |
| 2 | + | − | 6 |
| 3 | − | + | — |
| 4 | − | − | — |
|
|||
|---|---|---|---|
| Entry | Cu(BF4)2·H2O | Mo(CO)6 | Yieldb [%] |
| a Conditions: aziridine (0.2 mmol), Cu(BF4)2·H2O (0.1 eq.); neocuproine (0.1 eq.); Ph-IN-SO2-o-NO2-Ph (0.5 eq.); MeOH (10 eq.); Mo(CO)6 (0.25 eq.); MeNO2 (0.6 mL).b Yields were determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard. |
|||
| 1 | + | + | 23 |
| 2 | + | − | 23 |
| 3 | − | + | — |
| 4 | − | − | — |
These results suggest that the aziridination reaction requires only the copper salt, but the obtained yield is low (6%). However, when both copper and Mo(CO)6 are present, the yield rose to 64%, suggesting that the Mo(CO)6 or some of its derivatives is a key to improve the yield under the reaction conditions (Table 7).
In contrast, for the ring opening process, it is clear that the presence of the copper salt is essential but this is not the case for Mo(CO)6 (Table 8). In this sense, and to elucidate how the Mo(CO)6 or its derivative(s) improve the yield of the reaction, other mechanistic studies, including synthesis and characterisation of new organometallic complexes are under way in our laboratory.
Interestingly, based on the abovementioned results, a one-pot methodology affords better yields than separated procedures under these reaction conditions.
Conclusions
In summary, we have devised a new one-pot copper-catalysed regioselective aminoalkoxylation of styrenes. The reaction takes place through a nitrene transfer, with subsequent opening of the aziridine ring, and proceeds under mild conditions using Cu(BF4)2·H2O–neocuproine and Mo(CO)6 as the catalytic system. Three different iminoiodanes were used as nitrogen sources as well as primary and secondary alcohols as alkoxy sources. The products obtained should be of interest for the development of β-alkoxyphenethylamines as new compounds possibly acting on neuronal targets.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We are grateful for financial support from the Fondo Nacional para el Desarrollo Científico y Tecnológico, Chile (FONDECYT grant 1171391) and FONDEQUIP (grant EQM120021). I. Almodovar thanks the USACH Vicerrectoría de Investigación, Desarrollo e Innovación (grant DICYT-021641AF). P. H.-I. thanks ACT-1404 (IPMaG).Notes and references
- P. A. S. Lowden, in Aziridine Natural Products, ed. A. K. Yudin, Wiley-VCH, Weinheim, 2006, pp. 399–442 Search PubMed.
- (a) D. Tanner, Angew. Chem., Int. Ed. Engl., 1994, 33, 599 CrossRef; (b) Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199 CrossRef PubMed; (c) C. J. Thibodeaux, W.-C. Chang and H.-W. Liu, Chem. Rev., 2012, 112, 1681 CrossRef PubMed; (d) M. Nonn, A. M. Remete, F. Fülöp and L. Kiss, Tetrahedron, 2017, 73, 5461 CrossRef; (e) L. Degennaro, P. Trichera and R. Luisi, Chem. Rev., 2014, 114, 788 CrossRef PubMed; (f) W. McCoull and F. A. Davis, Synthesis, 2000, 1347 CrossRef.
- (a) S. H. Krake and S. C. Bergmeier, Tetrahedron, 2010, 66, 7337 CrossRef; (b) J. A. Kalow and A. G. Doyle, Tetrahedron, 2013, 69, 5702 CrossRef; (c) X. E. Hu, Tetrahedron, 2004, 60, 2701 CrossRef; (d) P. Lu, Tetrahedron, 2010, 66, 2549 CrossRef; (e) Y. Zhao, G. Wang, S. Zhou, Z. Li and X. Meng, Org. Biomol. Chem., 2014, 12, 3362 RSC; (f) T. Cytlak, M. Saweliew, M. Kubicki and H. Koroniak, Org. Biomol. Chem., 2015, 13, 10050 RSC; (g) L. Ma and J. Xu, Prog. Chem., 2004, 16, 220 Search PubMed.
- (a) N. C. Ghosal, S. Santra, S. Das, A. Hajra, G. Zyryanov and A. Majee, Green Chem., 2016, 18, 565 RSC; (b) B. A. Bhanu Prasad, G. Sekar and V. K. Singh, Tetrahedron Lett., 2000, 41, 4677 CrossRef; (c) S. Chandrasekhar, C. Narsihmulu and S. S. Sultana, Tetrahedron Lett., 2002, 43, 7361 CrossRef; (d) H. Stamm and D. Speth, Arch. Pharm., 1989, 332, 277 CrossRef; (e) B. A. Bhanu Prasad, R. Sanghi and V. K. Singh, Tetrahedron, 2002, 58, 7355 CrossRef; (f) Y. Li, D. Gu, X. Xu and S. Ji, Chin. J. Chem., 2009, 27, 1558 CrossRef; (g) M. Bera, S. Pratihar and S. J. Roy, J. Org. Chem., 2011, 76, 1475 CrossRef PubMed; (h) J. Li, Y. Liao, Y. Zhang, X. Liu, L. Lin and X. Feng, Chem. Commun., 2014, 50, 6672 RSC; (i) J. Llaveria, A. Espinoza, G. Negrón, M. I. Matheu and S. Castillón, Tetrahedron Lett., 2012, 53, 2525 CrossRef; (j) S. Zhang, C. Shan, S. Zhang, L. Yuan, J. Wang, C.-O. Tung, L.-B. Xing and Z. Xu, Org. Biomol. Chem., 2016, 14, 10973 RSC; (k) G. D. K. Kumar and S. Baskaran, Synlett, 2004, 1719 Search PubMed.
- (a) N. Hsueh, G. J. Clarkson and M. Shipman, Org. Lett., 2015, 17, 3632 CrossRef PubMed; (b) N. Hsueh, G. J. Clarkson and M. Shipman, Org. Lett., 2016, 18, 4908 CrossRef PubMed; (c) J. Zakrzewski, A. P. Smalley, M. A. Kabeshov, M. J. Gaunt and A. A. lapkin, Angew. Chem., Int. Ed., 2016, 55, 8878 CrossRef PubMed; (d) M. K. Jackl, L. Legnani, B. Morandi and J. W. Bode, Org. Lett., 2016, 19, 4696 CrossRef PubMed.
- (a) J. Ciesielki, G. Dequirez, P. Retailleau, V. Gandon and P. Dauban, Chem.–Eur. J., 2016, 22, 9338 CrossRef PubMed; (b) H. Sun, C. Yang, R. Lin and W. Xia, Adv. Synth. Catal., 2014, 356, 2775 CrossRef; (c) L. Legnani and B. Morandi, Angew. Chem., Int. Ed., 2016, 55, 2248 CrossRef PubMed.
- A. Mal, G. Goswami, I. A. Wani and M. K. Ghorai, Chem. Commun., 2017, 53, 10263 RSC.
- (a) M. A. Torres, B. Cassels and M. C. Rezende, Synth. Commun., 1995, 25, 1239 CrossRef; (b) M. Osorio-Olivares, M. C. Rezende, S. Sepúlveda-Boza, B. K. Cassels and A. Fierro, Bioorg. Med. Chem., 2004, 12, 4055 CrossRef PubMed; (c) M. A. Torres, M. C. Rezende and B. K. Cassels, Gen. Pharmacol., 1998, 31, 51 CrossRef PubMed.
- (a) J. Saavedra-Olavarría, G. C. Arteaga, J. J. López and E. G. Pérez, Chem. Commun., 2015, 51, 3379 RSC; (b) C. Herrera-Leyton, M. Madrid-Rojas, J. J. López, A. Cañete, P. Hermosilla-Ibáñez and E. G. Pérez, ChemCatChem, 2016, 8, 2015 CrossRef.
- Currently we do not know what the role of Mo(CO)6 may be, but several experiments using different molybdenum salts, in different oxidation states are underway in our laboratories.
- CCDC 1835571.†.
- (a) J. J. López, E. G. Pérez and J. García-Colunga, Neurosci. Lett., 2015, 607, 35 CrossRef PubMed; (b) H. R. Arias, J. J. López, D. Feuerbach, A. Fierro, M. O. Ortells and E. G. Pérez, Int. J. Biochem. Cell Biol., 2013, 45, 2420 CrossRef PubMed; (c) E. G. Pérez, C. Ocampo, D. Feuerbach, J. J. López, G. L. Morelo, R. A. Tapia and H. R. Arias, Med. Chem. Commun., 2013, 4, 1166 RSC; (d) E. G. Pérez, B. K. Cassels, C. Eibl and D. Gündisch, Bioorg. Med. Chem., 2012, 20, 3719 CrossRef PubMed.
- (a) B. Nyasse, L. Grehn and U. Ragnarsson, Chem. Commun., 1997, 1017 RSC; (b) D. A. Alonso and P. G. Andersson, J. Org. Chem., 1998, 63, 9455 CrossRef.
- T. Kan and T. Fukuyama, Chem. Commun., 2004, 353 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1835571. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra03815e |
| This journal is © The Royal Society of Chemistry 2018 |