
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment and characterisation of self-assembled graphene hydrogel-based anodes for bioelectrochemical systems
Mariela I. Lescanoa,
Aurelien Gasnier b,
Maria L. Pedanocd,
Mauricio P. Sicaad,
Daniel M. Pasquevicha and
Maria B. Prados*ad
b,
Maria L. Pedanocd,
Mauricio P. Sicaad,
Daniel M. Pasquevicha and
Maria B. Prados*ad
aInstituto de Energia y Desarrollo Sustentable, Centro Atomico Bariloche, Comision Nacional de Energia Atomica, Av. E. Bustillo 9500, 8400 S. C. de Bariloche, Rio Negro, Argentina. E-mail: mbprados@cab.cnea.gov.ar; Tel: +54 294 444 5107
bGerencia de Investigacion Aplicada, Centro Atomico Bariloche, Comision Nacional de Energia Atomica, CONICET, Av. E. Bustillo 9500, 8400 S. C. de Bariloche, Rio Negro, Argentina
cLab. de Fotonica y Optoelectronica, Centro Atomico Bariloche, Comision Nacional de Energia Atomica, CONICET, Av. E. Bustillo 9500, 8400 S. C. de Bariloche, Rio Negro, Argentina
dInstituto Balseiro, Universidad Nacional de Cuyo, Centro Atomico Bariloche, Av. E. Bustillo 9500, 8400 S. C. de Bariloche, Rio Negro, Argentina
First published on 26th July 2018
Abstract
In this work, we report a simple and scalable method to produce high efficiency 3D graphene-based electrodes (GH) for bioelectrochemical systems. GH were obtained by self-assembly of graphene oxide, through slow reduction with ascorbic acid over conductive mesh-works (carbon cloth and stainless-steel). The GH structure and composition were characterised by electron microscopy (SEM) and spectroscopy (FTIR and Raman), whereas the electrodes' performance was tested by chronoamperometry and cyclic voltammetry in a microbial electrolysis cell (MEC) inoculated with a pure culture of G. sulfurreducens. The hydrogel had a broad pore size distribution (>1 μm), which allowed bacterial colonisation within the framework. The macro-porous structure and chemical properties of the hydrogel rendered a higher bacterial loading capacity and substrate oxidation rate than other carbonaceous materials, including different reported graphene electrodes, which significantly increased MEC performance.
1 Introduction
Bioelectrochemical systems (BES) permit the extraction of energy from microbial anaerobic respiration. These systems are based on electrogenic microorganisms, such as bacteria of the genus Geobacter, that are capable of transferring electrons extra-cellularly during respiration.1,2 Basically, these bacteria oxidise organic compounds, such as acetate, in the cytosol and obtain energy catalysing the transference of the electrons through a respiratory chain to a solid final acceptor outside the cell, such as Fe(III) oxides. This final step is accomplished through extra-cellular electron transfer mechanisms (EET), which include direct-contact between the bacterial cell and the electron acceptor through outer-membrane proteins (cytochromes), diffusion of soluble electron shuttles (e.g. flavins, quinones, phenazines) and electron transport through the extra-cellular biofilm matrix (organic compounds synthesised by the microorganisms). These EET have been described in detail and extensively reviewed.3,4Notably, it has been demonstrated that electrogenic bacteria can use these EET to transfer electrons to an electrode.5 The current thus generated can produce useful work in a microbial fuel cell (MFC)6 or can be used for the production of valuable products in the cathode. For example, microbial electrolysis cells (MEC) are bio-catalysed electrolysis reactors employed in the production of hydrogen.7 Currently, BES represent a promising sustainable green technology to turn organic pollutants present in wastewater into a renewable source of energy.8–10
Additionally, a new generation of BES emerged, applied to desalination,11 electrosynthesis12 and remediation of complexly contaminated sediments.13,14 Interestingly, there is growing evidence of the efficiency of BES in the treatment of refractory pollutants, such as pyridine (commonly found in several industrial wastewaters),15 chloramphenicol16 and azo dye.17
However, the development of BES technology has been hampered by different factors, such as a low rate of charge transfer to the electrodes.
In most BES, like MFC and MEC, the anode potential influence the cellular respiration similarly to the concentration of a soluble electron acceptor. Therefore, respiration will occur only at a certain potential, where the anode accepts electrons from the microorganisms and determines their respiration rate.18 In order to achieve a high energy output with minimal potential losses, electrogenic microorganisms should produce a high current density at a low anode potential (the closest possible to the redox potential of the substrate being oxidised). Improving BES performance requires the structural and material optimisation of the electrode. The optimised structure should increase the active surface area available for bacterial colonisation, while the electrode composition should improve the bacteria-electrode affinity interactions and the catalysis of the electron transfer process.
Recently, graphene has been proposed as a suitable material for electrode applications in MFC.19 Graphene is a single-atom-thick two-dimensional (2D) carbon nano-structured material, where the sp2 hybridisation of all bonds across the sheet gives rise to interesting and unique properties, such as high mechanical strength, flexibility, thermal and electrical conductivity, large specific surface area and stability.20,21 Currently, there are many methods to prepare graphene, such as chemical vapour deposition (CVD), liquid phase exfoliation, mechanical exfoliation, molecular assembly and epitaxial growth.22 The graphene product obtained by each of these methods has different qualities, differing in dimension, shape, type and amount of defects, which strongly affect the properties of the material. In addition, the scale of graphene production also depends on the method of synthesis employed. For the preparation of graphene based electrodes for MFC application, different strategies have been applied. Thin-films of graphene sheets used as anodes of MFC, operated with different bacterial species, demonstrated high bio-compatibility and a good power performance.23,24 However, 2D structures have limited bacterial loading capacity. In addition, the stacking between individual sheets into multi-layered graphene, caused by van der Waals attractions from the basal planes, largely reduces the intrinsic specific area, the conductivity and the mechanical strength of individual graphene sheets.
An effective approach to overcome these problems is the synthesis of three-dimensional (3D) graphene architectures, such as hydrogels, foams and sponges.25 Yong et al.26 have synthesised macro-porous free-standing graphene foam by CVD, employing a nickel foam as substrate, to build anodes for MFC. This architecture substantially increases the electrode surface. However, due to the scarcity of polar organic groups on the surface of the high-quality graphene obtained by CVD, this electrode did not provide anchoring points for bacteria attachment and therefore, it had to be functionalized to provide a significant bacterial loading capacity.
In this work, we present a simple and scalable wet method to prepare graphene hydrogel (GH) based electrodes for BES applications. The assembly of the electrodes involved the chemical preparation of graphene oxide flakes (GO) according to the Tour's method,27 followed by a slow reduction with ascorbic acid over different conductive mesh-works, such as carbon cloth (GH-CC) and stainless-steel (GH-SS). These electrodes were tested in single chamber MECs based on Geobacter sulfurreducens, a model electrogenic bacteria. With no functionalization step, our three-dimensional macro-porous design promoted a high BES performance due to bacterial colonisation of the whole electrode framework and a faster substrate oxidation rate.
2 Materials and methods
2.1 Media and bacteria growth conditions
All the bioelectrochemical assays were performed using Geobacter sulfurreducens. G. sulfurreducens growth medium contained (per litre): 0.1 g of KCl, 1.5 g of NH4Cl, 0.6 g of NaH2PO4, 2.5 g of NaHCO3, 10 ml of vitamin mix and 10 ml of trace mineral mix (Sigma-Aldrich).28 The medium was adjusted to pH 6.8, sterilised and flushed with N2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO2 (80:20) to remove oxygen. Sodium acetate (30 mM) served as the electron donor and as the carbon source and sodium fumarate as the electron acceptor (40 mM). Cells were cultured at 30 °C. Steady-state cells were anaerobically transferred to the electrochemical cell (10% inoculum) at the moment of inoculation.
:CO2 (80:20) to remove oxygen. Sodium acetate (30 mM) served as the electron donor and as the carbon source and sodium fumarate as the electron acceptor (40 mM). Cells were cultured at 30 °C. Steady-state cells were anaerobically transferred to the electrochemical cell (10% inoculum) at the moment of inoculation.
2.2 Hydrogel synthesis
Graphene hydrogels (GHs) were produced by self-assembly of GO through chemical reduction of the aqueous GO sheets over different conductive mesh-works: plain carbon cloth (CC) or a 304 stainless steel mesh (SS, mesh 50). The GO flakes were prepared according to the Tour's method.27 A 9:1 H2SO4/H3PO4 mixture was prepared. In an Erlenmeyer flask 1.0 g of graphite flakes (Sigma-Aldrich) and 6.0 g KMnO4 (Cicarelli) were mixed together. Then 133 ml of the H2SO4/H3PO4 mixture were poured over the solids. The reaction mixture was then heated to 50 °C with mechanical agitation for 12 h. The reaction mixture quickly took a greenish colour and after several hours it turned brown and very thick. Then it was allowed to cool to room temperature and was poured slowly over a mixture of crushed ice (120 g) and 30% H2O2 (3 ml). The obtained yellow suspension was filtered, then the filter cake was washed and filtered successively with 100 ml distilled water, 100 ml 10% HCl (Cicarelli), 2 times 100 ml EtOH (Cicarelli) and finally the solid was coagulated with EtO2 (Cicarelli), filtered, allowed to dry in the hood overnight and placed in the oven at 45 °C for 1 hour. The GO suspension was prepared at concentrations of 6.0 mg ml−1 in deionised sterile water with 1 h of ultrasonication. The biggest aggregates (a very little amount) were separated by 3 cycles of 60 min centrifugation at 4000 rpm, with collection of the supernatant and the solid residues trashed away.
2.3 Preparation of electrodes
The GH electrodes were prepared as follow: a 0.6 cm diameter, 8 cm long glass tube was tapped on the bottom side with a rubber cap. A 6 cm-long mesh (a 0.6 cm-wide band in the case of CC or a 0.6 cm-wide tube in the case of SS) was introduced in the tube, then 6 ml of an ascorbic acid (AA) and GO solution (AA–GO solution, prepared by dissolving 5 mass equivalent of ascorbic acid in aqueous GO at 6 mg ml−1) was poured into the tube (approximately 5 cm of the mesh was submerged in the solution). Then the top of the tube was sealed with a rubber cap and placed in oven at 40 °C for 60 h. During this time, the brownish GO colloid turned to a black rGO hydrogel. The rubber cap was removed and the glass tube containing the gel was placed in a beaker with excess distilled water for washing out over 72 h, replacing the water 3 times every 24 h. Lastly the electrode was pushed away from the tube and the upper part of the mesh not submerged into the GO was electrically connected by a crocodile clip to the electrochemical circuit. The resulting materials were used as anodes in MECs. For comparison purposes, pristine CC or SS meshes of comparable size without GH, and a solid graphite rod (constructed from bars of 0.4 cm in diameter and 10 cm in length), were also used as anodes in control experiments. The graphite rods were polished with sandpaper (P1000 grit), thoroughly rinsed with distilled water and sonicated in a bath (1 minute, 3 times). Additional control experiments in the absence of bacteria were performed. Three independent experiments were performed for each electrode type.2.4 MEC design and operation
The MECs consisted of sterile anaerobic glass single-chamber cells (200 ml) with three electrodes, where the prepared electrodes served as working electrode, a coiled platinum wire was used as counter electrode and a Ag/AgCl electrode was used as reference electrode. The cells were filled with culture media, flushed continuously with N2:CO2 (80:20) and operated in an incubator at constant temperature (30 °C) and stirring. The working electrode was poised at +240 mV vs. Ag/AgCl (PGSTAT101 potentiostat, Metrohm AG) and the MECs were inoculated with 10% stationary-phase cultures of G. sulfurreducens. Sodium acetate (30 mM) was provided as the electron donor, and no electron acceptor other than the electrode was present in the MEC. The MECs were changed from batch to continuous mode 24 h after inoculation, with a hydraulic retention time of 24 h. The current intensity was recorded for 10 days.
2.5 Electrochemical measurements
Cyclic voltammetry (CV) was performed scanning the potential between +240 mV and −550 mV starting from 240 mV (PGSTAT101 potentiostat, Metrohm AG). The scan rate was 1 mV s−1. CVs were performed without stirring and without gas flushing. CVs were run before each inoculation. CVs were also performed over mature biofilms before the end of the experiment, in turnover (in the presence of acetate) and non-turnover conditions (in the absence of acetate). Thus, for non-turnover conditions G. sulfurreducens mature biofilms were starved (the electrolyte was replaced with a medium that lacked acetate) until catalytic features were significantly reduced (less than 10% of maximum current density remained).2.6 Scanning electron microscopy
Electrodes were fixed in glutaraldehyde 2.5% over-night, dehydrated by immersion in a series of ethanol solutions (40, 60, 80, and 100% ethanol in ultra-pure water), critical point dried, and sputtered with gold for the observation by SEM. Samples were observed in a FEI-NOVA Nano SEM at an accelerating voltage of 5 kV.2.7 Raman spectroscopy
Raman spectra of samples of GH (excluding the supporting meshes) were acquired with a Lab Ram HR Evolution, Jobin Yvon, Horiba Scientific Raman Spectrometer, using a 633 nm Laser, 250 hole, 600 grooves per mm grid. 100× objective, and a 800 cm−1 to 3200 cm−1 spectral range, except stated otherwise. The laser power at the source was set to 2 watts (or 30 A current) and internal filter were used to attenuate the out coming power. Except stated otherwise, usual spectra acquisition conditions were: 4 accumulations of 60 s each, at 1% transmitted laser power, with other setting as previously stated. Other studied conditions were 1%, 5%, 10% laser power; 2 or 4 accumulations.2.8 Fourier transformed infrared spectroscopy
Samples of GH were collected after critical point dried and grinded with KBr, and then compressed into thin pellets. The FTIR spectra were obtained on a Fourier Transform Infrared Spectrometer (Spectrum 400-Perkin-Elmer).3 Results and discussion
3.1 Performance of GH anodes in MEC
The performance of the electrodes was analysed in terms of the material stability and current generation during MEC operation.The mechanical stability of the bio-electrodes is very important for its long-term operation. Manipulation and stirring may cause irreversible damage or detachment of substantial portions of the electrode, shortening its durability. Comparing our two models, GH-SS and GH-CC, the former showed a higher durability since the core of the hydrogel is protected wrapped behind the mesh. On the other hand, since in GH-CC the hydrogel is laid on the carbon cloth, it was more prone to detachment during normal operation. For this reason, although our two electrodes showed similar performances in terms of current generation, GH-SS was preferred for long-term MEC operation.
No current generation was appreciated after 240 h of operation of the MECs operated with the pristine meshes, CC and SS, in the presence of bacteria, as well as in any of the MECs operated in the absence of bacteria (data not shown). Therefore, the performance of the GH electrode, in terms of current production, was compared to that of a graphite rod (gr), one of the most employed electrode materials in BES.
The results of current generation, in the presence of bacteria, are shown in Fig. 1 as the anodic (oxidation) current density, considering the geometric area of the portion of the electrodes submerged in the electrolyte. This calculation does not take into account neither the porosity of the materials nor the electro-active surface area. However, considering that the oxidation reaction is carried out inside the microorganisms, these parameters do not necessarily represent proper markers of available surface for catalysis. For example, since the size of a typical bacterium is 0.5–3.0 μm, increasing the specific surface area of the electrode does not imply a proportional increase in the surface area available for bacterial colonisation.
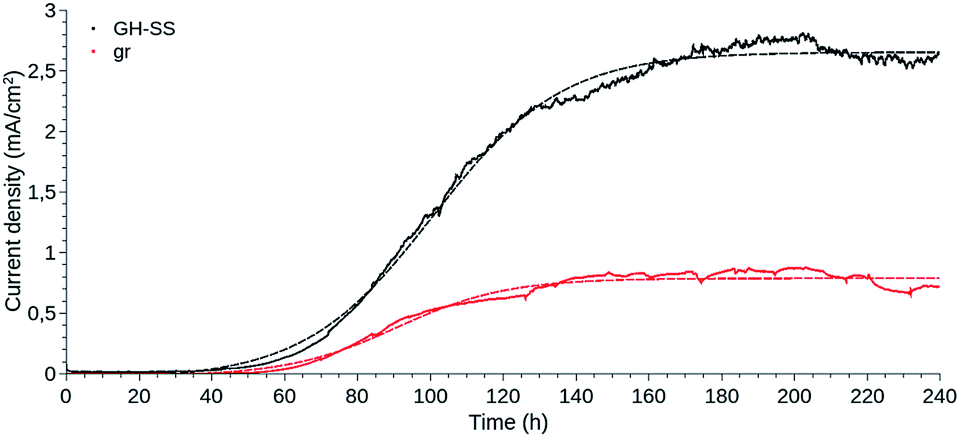 | ||
| Fig. 1 Amperometric response of graphite rod (gr) and graphene hydrogel-based (GH-SS) working electrodes as a function of time, during G. sulfurreducens biofilm growth, under +240 mV vs. Ag/AgCl applied potential. The plotted data are the mean of three independent experiments performed for each electrode type (—). The Boltzmann fit was performed with QtiPlot software and is shown for each electrode (- - -). Current density was calculated considering the geometric area of the portion of the electrodes submerged in the electrolyte. | ||
GH-SS displayed a better performance than gr, reaching a current density approximately three times higher.
The current density evolved likewise for GH-SS and gr, in a manner resembling bacterial growth curves: a lag phase for cell attachment and initial colonisation, followed by a fast exponential growth phase, which indicated that the electrogenic bacteria were transferring electrons to the anode as a result of intra-cellular acetate oxidation, and a stationary phase where the current stabilised and thus the biofilms were considered mature. The lag phase of GH-SS as well as gr lasted ≈ 40 h, during this period cells go through an adaptation process from using a soluble (fumarate) to a solid electron acceptor and begin the biofilm development by secreting exo-polymeric substances. The exponential phase was longer for the GH-SS than the gr, suggesting that GH is capable of sustaining the growth of a higher number of bacterial cells. Accordingly, 1.5 vs. 0.8 mg of protein per cm2 were extracted in alkali for GH-SS and gr respectively (determined by Bradford method29).
To further understand the impact of the material on the biofilm behaviour, we fitted two different models to the amperometries, shown in Fig. 1.
First, a logistic model (eqn (1)) was applied on the assumption that the lag, exponential and plateau phases observed for the curves of current density vs. time parallel similar phases of the biofilm growth.30,31
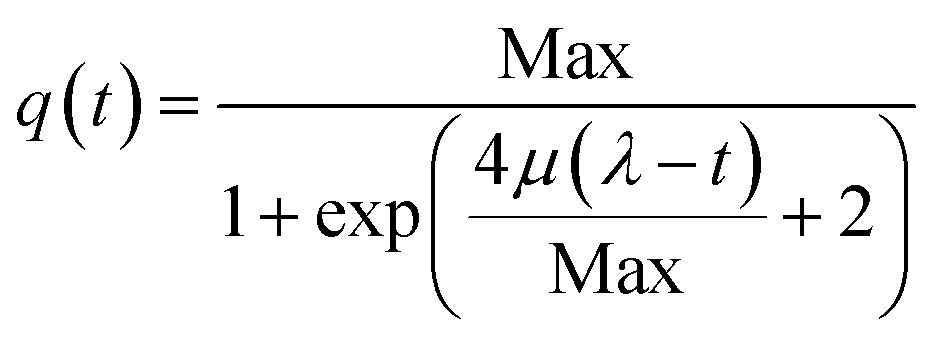 | (1) |
Thus, μ (the tangent in the inflection point) reflects the bacterial maximum growth rate; λ (the x-axis intercept of this tangent) is the lag time and Max (the asymptote) represents the carrying capacity, the maximum population size that the environment can sustain. The μ and Max values are significantly higher for GH-SS (μ = 3.8 × 10−2 mA cm−2 h−1 and Max = 2.7 mA cm−2) than gr (μ = 1.3 × 10−2 mA cm−2 h−1 and Max = 0.8 mA cm−2), while λ are very similar (62.3 h and 59.5 h for GH-SS and gr, respectively). Considering that all the electrochemical cells were operated at the same conditions (inoculum size, temperature, culture media, HRT), a possible explanation of these results is a promoting effect of GH-SS on the biofilm metabolism that affects the growth or substrate oxidation rate. As discussed later, Fig. 5 shows that in our electrodes GH exposes a considerable amount of hydrophilic groups which could enhance its wettability26 and proton transport, and thus improve bacteria-electrode connection and metabolic rates.
An alternative explanation is that GH-SS exposes a larger active surface capable of being colonised by the microorganisms. It is worth mentioning that the effective electrode area depends on the geometry of pores and bacteria, the number of actual contacts per microorganism, which in turn may be affected by chemical characteristics of the material. Thus, the current physical methods employed to estimate solvent exposed areas are of little avail in our case. Therefore, as stated before, the geometric area of the electrode was employed to calculate the current density in the amperometries. But, as shown later by SEM images Fig. 4, bacteria are able to reach intimately the porous structure of GH, consequently the actual active surface could be much greater for GH-SS and the current density overestimated.
To address these hypothesis, we also fitted a Boltzmann equation (eqn (2)) to the amperometries.32
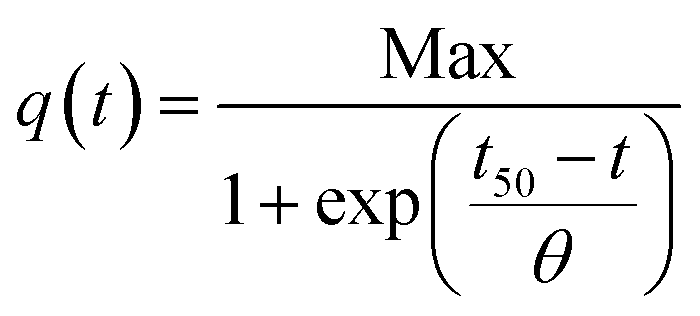 | (2) |
In this equation, Max is the highest current achieved and the slope (θ) at the inflection point (t50) is independent of the number of bacteria, and reflects an intrinsic growth rate. Applying this model, similar slopes were expected if the only difference between GH and gr were the amount of active surface and the biofilms were growing at the same rate. From the θ values obtained (17.7 and 15.0 mA cm−2 h−1 for GH and gr, respectively), GH shows a higher slope suggesting that both, physicochemical and active surface contributions, could explain its increased bacterial loading capacity and better performance.
Finally, we estimated the rate of acetate consumption and supply under continuous operation in our MECs. Considering the following equation for acetate metabolic oxidation, eqn (3),
| 1/8CH3COO− + 3/8H2O → 1/8CO2 + 1/8HCO3− + H+ + e− | (3) |
Interestingly, considering the electrodes bacterial loading previously mentioned (1.5 vs. 0.8 mg of protein per cm2 for GH-SS and gr, respectively), we calculated that the acetate consumption rate per mg of protein is 1.6 times higher for GH-SS than gr (7.8 vs. 4.7 μmol h−1 per mg of protein, respectively). An equal acetate consumption rate per mg of protein would have been expected if the only difference between GH-SS and gr was the increased active surface, which allowed a higher bacterial loading capacity. Instead, this finding supports the hypothesis that the GH chemical properties favours the acetate oxidation reaction.
3.2 Macro-porous structure of GH anodes and electrogenic biofilm establishment
The presence of G. sulfurreducens biofilms in GH anodes was confirmed at day 4 of MEC operation as a characteristic red coating over the electrode surface (Fig. 2). | ||
| Fig. 2 Graphene hydrogel-based working electrode (GH-SS, stainless steel) at the end of the MEC operation. The red coating of the electrode is the G. sulfurreducens mature biofilm. | ||
SEM images of the electrodes surface at the end of the experiments showed that dense biofilms grew over GH displaying heterogeneous pillar structures, similarly to graphite rod (gr) control electrodes (Fig. 3A and B). Only a few bacteria were adhered to the SS and CC pristine meshes (Fig. 3C and D), explaining the absence of a significant current production for these electrodes.
 | ||
| Fig. 3 SEM images of the electrodes surfaces after MEC operation in the presence of a pure culture of G. sulfurreducens: in (A) GH-SS and (B) gr: a dense biofilm was observed over the electrode surface; while in (C) SS and (D) CC only a few cells were found adhered to the electrodes surface. | ||
Cross-sectional SEM images of GH without bacteria (Fig. 4A and B) revealed a hierarchical pore structure, with a broad size distribution of interconnected porous, typical of freeze-dried graphene hydrogels.33 This structure is the result of the partial overlapping of the flexible graphene sheets during the hydrogel formation. Cross-sectional SEM images of GH taken at the end of the experiments revealed a thick layer of cells (>50 μm) on the graphene facing the electrolyte (Fig. 4C). As observed in Fig. 4D, graphene sheets seem to be intercalated between the firsts layers of cells of the biofilm. Bacteria were found along the entire GH framework (Fig. 4E). However, the core of the electrode was not clogged with microorganisms, probably because the pore sizes of the carbon material range in the order of bacteria dimensions, which might prevent a denser bacterial growth. This situation might improve substrate diffusion and ion transport within the electrode and therefore bacterial catalysis, as well as bacteria-electrode direct interaction and charge transfer. Pili-like filaments were present along the thick biofilm (Fig. 4F). Pili are required for optimal biofilm development and for the establishment of an electrical connection with extra-cellular electron acceptors, promoting long-range electrical transfer.34–36
 | ||
| Fig. 4 Cross-sectional SEM images of graphene hydrogel-based electrode before (A and B) and after (C–F) MEC operation in the presence of a pure culture of G. sulfurreducens. (A) Graphene hydrogel cross-sections at (A) low and (B) high magnifications, where the macro-porous structure and pore sizes of the material can be appreciated. (C) Graphene face in contact with the bulk electrolyte after MEC operation, where a thick layer of cells can be observed. (D–F) Higher magnification of (C): in (D) graphene sheets can be distinguished between bacterial cells; in (E) bacterial cells were present in the entire GH framework; in (F) pili-like filaments were observed along the thick biofilm (some of them are pointed by arrows). | ||
Therefore, GH has an enhanced active area per electrode volume capable of increasing the bacterial loading capacity. GH electrodes were able to sustain the development of dense and thick electrogenic biofilms over its surface and inside its framework, favouring an enhanced current production as observed in Fig. 1.
3.3 Chemical characteristics of GH anodes and bacterial interaction
Despite the chemical reduction step, a common feature of rGO is the presence of oxygen-containing functional groups, such as hydroxy, carboxy or epoxy groups.37 Some of these oxygen-containing functional groups are present in GH, as determined by FTIR and Raman spectroscopy (Fig. 5). The FTIR spectrum of GH before MEC inoculation presented an absorption band at 3200–3600 cm−1, characteristic of –OH groups; an absorption band at ≈ 1690–1760 cm−1, attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond; and C–O vibrations at ≈900–1200 cm−1. Finally, the skeletal vibration of CC is observed at ≈ 1620–1680 cm−1. In Raman spectroscopy, the G band (at ≈1582 cm−1) is associated with bond stretching of the sp2 carbon pairs, while the D band (at ≈1350 cm−1, absent in high quality graphene) is observed in all sp2 disordered carbon materials, and thus high D/G ratios indicate higher ordered in the material.38 Interestingly, in our experiments, GH was further reduced during the MEC operation as observed in the FTIR (Fig. 5A) as well as Raman spectra (Fig. 5B, D/G = 1.70 vs. 2.05 before and after operating the MEC respectively). This reduction of GH can be attributed to bacterial activity since the irreversible electrochemical reduction of GO occurs at −1039 mV vs. Ag/AgCl.39,40 Additionally, after MEC operation, a band at around 2900 cm−1 appeared in the FTIR spectrum, consistent with the presence of alkyl groups abundant in cellular membranes.41,42
O bond; and C–O vibrations at ≈900–1200 cm−1. Finally, the skeletal vibration of CC is observed at ≈ 1620–1680 cm−1. In Raman spectroscopy, the G band (at ≈1582 cm−1) is associated with bond stretching of the sp2 carbon pairs, while the D band (at ≈1350 cm−1, absent in high quality graphene) is observed in all sp2 disordered carbon materials, and thus high D/G ratios indicate higher ordered in the material.38 Interestingly, in our experiments, GH was further reduced during the MEC operation as observed in the FTIR (Fig. 5A) as well as Raman spectra (Fig. 5B, D/G = 1.70 vs. 2.05 before and after operating the MEC respectively). This reduction of GH can be attributed to bacterial activity since the irreversible electrochemical reduction of GO occurs at −1039 mV vs. Ag/AgCl.39,40 Additionally, after MEC operation, a band at around 2900 cm−1 appeared in the FTIR spectrum, consistent with the presence of alkyl groups abundant in cellular membranes.41,42
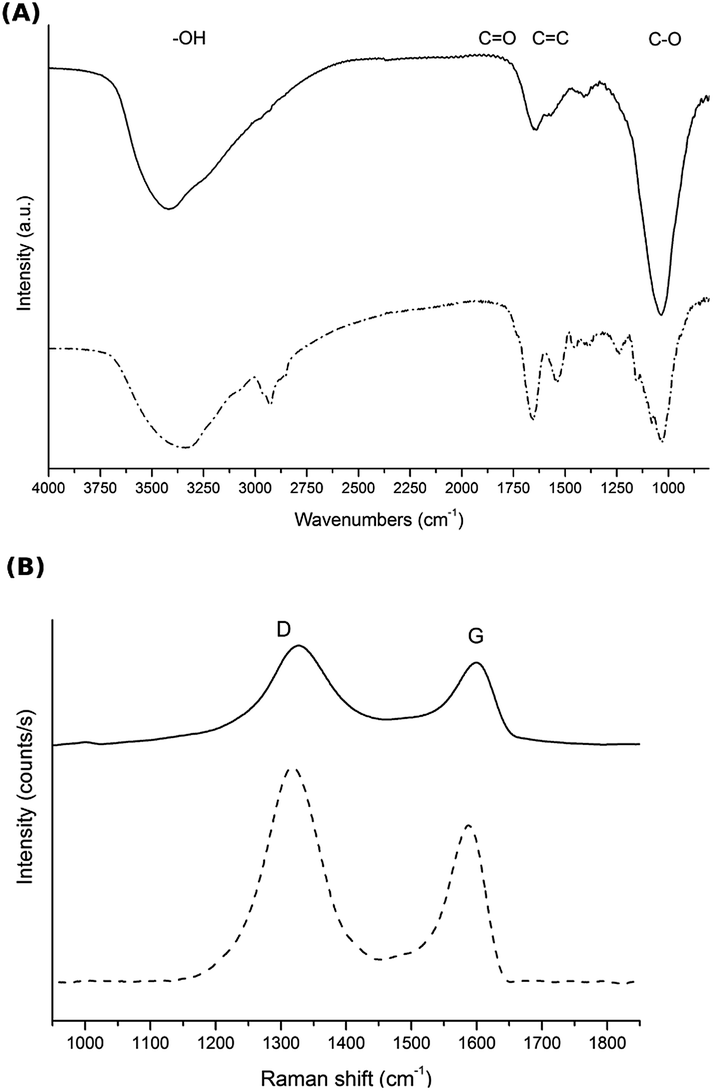 | ||
| Fig. 5 (A) FTIR and (B) Raman spectroscopy of GH before (—) and after (- - -) MEC operation in the presence of a pure culture of G. sulfurreducens. | ||
The presence of hydrophilic functional groups in GH may favour water adsorption onto the graphene sheets, preventing them from re-stacking and stabilising a porous structure with higher surface area. In addition, this feature, considered a defect in graphene, might be beneficial for bacterial-electrode interaction.43 On the one hand, the formation of a biofilm is affected by multiple factors such as the surface topography, charge and wettability, the amount of exo-polymer production by the cells, and cell–cell signalling. It was suggested that wettability enhances the connection between the bacteria and the electrode by altering local polarity at their interface, and extreme hydrophobicity makes high quality graphene unfavourable for bacterial adhesion.26 On the other hand, it was reported that the maximum current densities produced by electrogenic biofilms are limited by respiration inhibition due to proton accumulation inside the biofilm during the substrate oxidation.44 It was demonstrated that in GO, hydrophilic sites help to proton diffusion through hydrogen-bonding networks along the adsorbed water film, turning GO into a highly proton-conductive material.45 Therefore, an enhanced proton conduction in GH containing some degree of oxygenated groups, might enable a faster substrate oxidation, resulting in a faster current production compared to gr, explaining the differences in the slopes observed in the exponential phase of the amperometries.
3.4 Biofilm electron transfer process in GH anodes
Electron transfer from the electrogenic microorganisms towards the anode is coupled to energy conservation, where the electrode functions as a solid electron acceptor during respiration. A great variety of electron transport chains are found among microorganisms.46–48 It was observed that changes in environmental conditions can trigger the expression of a different EET in a bacteria.49 Most studies suggest that G. sulfurreducens biofilms grow on carbon-based anodes by means of direct electron transfer employing two combined strategies: the secretion of non-diffusing mediators (cytochromes) into the biofilm matrix and the production of conductive pili or nano-wires that enable the connection between the cells and the electrode.3In order to gain insight into bacterial EET to the electrode surface on GH-SS anodes, cyclic voltammetry (CV) was carried out under different conditions. First, CV was performed at a low scan rate (1 mV s−1) in the presence of acetate, so that the enzymes that catalyse slow reactions can undergo multiple turnovers at each sampled potential. The voltammograms of both GH-SS and gr electrodes (Fig. 6A), indicate that G. sulfurreducens mature biofilms behave as attached bio-catalysts with heterogeneous electron transfer to the electrode, as previously reported.50,51 Also, both voltammograms show the anodic scan crossing under the cathodic scan at potentials higher than 0 mV vs. Ag/AgCl. This phenomenon has already been observed for full-grown G. sulfurreducens biofilms on a graphite cloth anode.52 The catalytic process started in both electrodes at ≈−490 mV. However, it was limited at −325 mV in gr and at −100 mV in GH-SS. This effect may be related to the hierarchical pore architecture of the GH that allows a better substrate diffusion inside the framework.
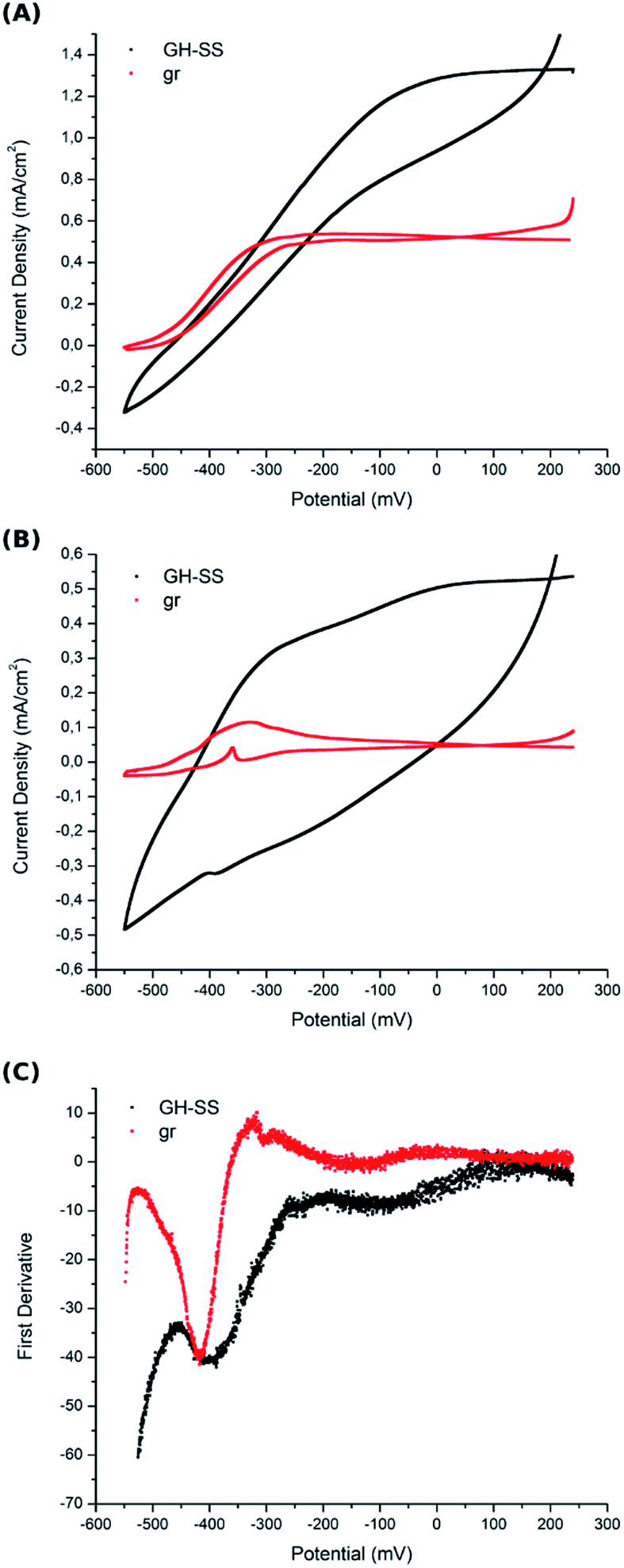 | ||
| Fig. 6 Cyclic voltammetry of graphite rod (gr) and graphene hydrogel-based (GH-SS) working electrodes recorded at a scan rate of 1 mV s−1 in (A) turnover conditions (in the presence of acetate) and (B) non-turnover conditions (in the absence of acetate). (C) First derivative analysis of anodic scan from non-turnover conditions voltammogram (obtained with the potentiostat controlling software, Nova-Metrohm). CVs were performed at 240 h of MEC operation over mature biofilms, where the maximum current density has been reached. The plotted data are the mean of three independent experiments performed for each electrode type. | ||
Under turnover conditions the high catalytic current recorded by CV may obscure individual redox species that could account for EET identification. Thus, CV was also performed in non-turnover conditions (in the absence of acetate): the G. sulfurreducens mature biofilms were starved (the electrolyte was replaced with a medium that lacked acetate) until catalytic features were significantly reduced (less than 10% of maximum current density remained); then CV was performed at a low scan rate (1 mV s−1). Fig. 6B shows voltamogramms of GH-SS and gr, which display peaks associated with more than one redox couple. Examination of the first derivative of anodic scan from Fig. 6B voltammogram, reveals that the potential of the oxidation peaks is similar in both electrode materials (Fig. 6C) and is consistent with cytochromes redox potentials.53 This result suggests that the biofilms in GH-SS and gr might employ the same EET to transfer electrons to the electrode surface. We are currently performing more experiments to confirm this.
4 Conclusions
Graphene hydrogel-based electrodes (GH) have been successfully prepared by self-assembly of graphene oxide through chemical reduction over conductive meshes. The GH proved to be suitable to develop dense and conductive biofilms of Geobacter sulfurreducens, and produces an outstanding current density compared to commonly used gr as well as other reported graphene-based electrodes.54 This result is attributed to the large surface area and pore sizes, provided by the 3D macro-porous structure of the GH, as well as the material chemical properties, that renders a high bacterial loading capacity, and an enhanced mass transport and proton conduction, avoiding thus saturation in the bacteria-mediated acetate oxidation reaction. In conclusion, by coupling the electrode morphology with composition modulation, the GH-SS design presented here upgrades both the long-term stability and output performance of MEC. Noteworthy, this performance was obtained in a cell operated at continuous mode, which resembles a real wastewater treatment coupled to energy production application. Considering that the graphene production method employed in this work allows the preparation of large quantities of graphene and that the anode bio-process studied is shared by many of the BES applications, the presented results turn GH into a promising material for the development of BES technology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Comision Nacional de Energia Atomica (CNEA) and Consejo Nacional de Investigaciones Cientificas y Tecnicas (CONICET), Argentina. Partial support by Fundacion YPF (project: PIO CONICET-YPF 13320130100203CO). Authors acknowledge Tec. P. Troyon and Tec. M. Corte for SEM images acquisition; Lic. P. Sessin for Raman spectra acquisition and processing; Dr L. Fernandez Albanesi for FTIR acquisition-Centro Atomico Bariloche-CNEA; and Dr J. P. Busalmen for his advices and kindly providing G. sulfurreducens cultures, INTEMA-Univ. Nac. Mar del Plata, Argentina.Notes and references
- D. R. Bond, D. E. Holmes, L. M. Tender and D. R. Lovley, Science, 2002, 295, 483–485 CrossRef PubMed.
- D. R. Lovley, Nat. Rev. Microbiol., 2006, 4, 497–508 CrossRef PubMed.
- F. Kracke, I. Vassilev and J. O. Kromer, Front. Microbiol., 2015, 6, 1–18 Search PubMed.
- D. R. Lovley, Annu. Rev. Microbiol., 2012, 66, 391–409 CrossRef PubMed.
- D. R. Bond and D. R. Lovley, Appl. Environ. Microbiol., 2003, 69, 1548–1555 CrossRef PubMed.
- B. E. Logan, B. Hamelers, R. Rozendal, U. Schröder, J. Keller, S. Freguia, P. Aelterman, W. Verstraete and K. Rabaey, Environ. Sci. Technol., 2006, 40(17), 5181–5192 CrossRef PubMed.
- B. E. Logan, D. Call, S. Cheng, H. V. M. Hamelers, T. H. J. A. Sleutels, A. W. Jeremiasse and R. A. Rozendal, Environ. Sci. Technol., 2008, 42, 8630–8640 CrossRef PubMed.
- J. B. a. Arends and W. Verstraete, Microb. Biotechnol., 2012, 5, 333–346 CrossRef PubMed.
- A. Escapa, R. Mateos, E. J. Martínez and J. Blanes, Renewable Sustainable Energy Rev., 2016, 55, 942–956 CrossRef.
- B. E. Logan and K. Rabaey, Science, 2012, 337, 686–690 CrossRef PubMed.
- P. Liang, K. Xiao, Y. Zhou, X. Zhang and B. E. Logan, Environ. Sci. Technol, 2009, 43, 7148–7152 CrossRef.
- K. P. Nevin, S. A. Hensley, A. E. Franks, Z. M. Summers, J. Ou, T. L. Woodard, O. L. Snoeyenbos-west and D. R. Lovley, Appl. Environ. Microbiol., 2011, 77, 2882–2886 CrossRef PubMed.
- H. Wang, H. Luo, P. H. Fallgren, S. Jin and Z. J. Ren, Biotechnol. Adv., 2015, 33, 317–334 CrossRef PubMed.
- Y. Zhao, Z. Li, J. Ma, H. Yun, M. Qi, X. Ma, H. Wang, A. Wang and B. Liang, J. Hazard. Mater., 2018, 349, 168–176 CrossRef PubMed.
- X. Jiang, J. Shen, K. Xu, D. Chen, Y. Mu, X. Sun, W. Han, J. Li and L. Wang, Water Research, 2018, 130, 291–299 CrossRef PubMed.
- F. Sun, H. Liu, B. Liang, R. Song, Q. Yan and A. Wang, Bioresour. Technol., 2013, 143, 699–702 CrossRef PubMed.
- Y. Liu, Y. Zhang, X. Quan, J. Zhang, H. Zhao and S. Chen, Bioresour. Technol., 2011, 102, 2578–2584 CrossRef PubMed.
- C. I. Torres, A. K. Marcus, H. S. Lee, P. Parameswaran, R. Krajmalnik-Brown and B. E. Rittmann, FEMS Microbiol. Rev., 2010, 34, 3–17 CrossRef PubMed.
- H. Yuan and Z. He, Nanoscale, 2015, 7, 7022–7029 RSC.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-h. Ahn, P. Kim, J.-y. Choi and B. H. Hong, Nature, 2008, 457, 706–710 CrossRef PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–389 CrossRef PubMed.
- K. S. Novoselov, V. I. Fal, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef PubMed.
- Y. Zhang, G. Mo, X. Li, W. Zhang, J. Zhang, J. Ye, X. Huang and C. Yu, J. Power Sources, 2011, 196, 5402–5407 CrossRef.
- J. Liu, Y. Qiao, C. X. Guo, S. Lim, H. Song and C. M. Li, Bioresour. Technol., 2012, 114, 275–280 CrossRef PubMed.
- X. Cao, Z. Yin and H. Zhang, Energy Environ. Sci., 2014, 7, 1850–1865 RSC.
- Y.-C. Yong, X.-C. Dong, M. B. Chan-Park, H. Song and P. Chen, ACS Nano, 2012, 6, 2394–2400 CrossRef PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef PubMed.
- R. Atlas, Handbook of Microbiological Media, 4th edn, 2010, 978-1-4398-0408-7, Ebook-PDF Search PubMed.
- M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef PubMed.
- M. H. Zwietering, I. l. Jongenburger, F. M. Rombouts and K. van't Riet, Appl. Environ. Microbiol., 1990, 56, 1875–1881 Search PubMed.
- D. Verotta, J. Haagensen, A. M. Spormann and K. Yang, Comput. Math. Methods Med., 2017, 2017 Search PubMed.
- J. M. Dubois, G. Ouanounou and B. Rouzaire-Dubois, Prog. Biophys. Mol. Biol., 2009, 99, 87–93 CrossRef PubMed.
- X. Zhang, Z. Sui, B. Xu, S. Yue, Y. Luo, W. Zhan and B. Liu, J. Mater. Chem., 2011, 21, 6494–6497 RSC.
- D. L. Cologgi, S. Lampa-pastirk, A. M. Speers, S. D. Kelly and G. Reguera, Proc. Natl. Acad. Sci., 2011, 108, 15248–15252 CrossRef PubMed.
- G. Reguera, K. P. Nevin, J. S. Nicoll, S. F. Covalla, T. L. Woodard and D. R. Lovley, Appl. Environ. Microbiol., 2006, 72, 7345–7348 CrossRef PubMed.
- G. Reguera, R. B. Pollina, J. S. Nicoll and D. R. Lovley, J. Bacteriol., 2007, 189, 2125–2127 CrossRef PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef PubMed.
- L. Chen, Y. Tang, K. Wang, C. Liu and S. Luo, Electrochem. Commun., 2011, 13, 133–137 CrossRef.
- Y. C. Yong, Y. Y. Yu, X. Zhang and H. Song, Angew. Chem., Int. Ed., 2014, 53, 4480–4483 CrossRef PubMed.
- V. Ragoonanan, J. Malsam, D. R. Bond and A. Aksan, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 2283–2290 CrossRef PubMed.
- H. Muhamadali, Y. Xu, D. I. Ellis, J. W. Allwood, N. J. Rattray, E. Correa, H. Alrabiah, J. R. Lloyd and R. Goodacre, Appl. Environ. Microbiol., 2015, 81, 3288–3298 CrossRef PubMed.
- J. A. Cornejo, C. Lopez, S. Babanova, C. Santoro, K. Artyushkova, L. Ista, A. J. Schuler and P. Atanassov, J. Electrochem. Soc., 2015, 162, 597–603 CrossRef.
- C. I. Torres, A. K. Marcus and B. E. Rittmann, Biotechnol. Bioeng., 2008, 100, 872–881 CrossRef PubMed.
- M. R. Karim, K. Hatakeyama, T. Matsui, H. Takehira, T. Taniguchi, M. Koinuma, Y. Matsumoto, T. Akutagawa, T. Nakamura, S.-i. Noro, T. Yamada, H. Kitagawa and S. Hayami, J. Am. Chem. Soc., 2013, 135, 8097–8100 CrossRef PubMed.
- M. E. Hernandez and D. K. Newman, Cell. Mol. Life Sci., 2001, 58, 1562–1571 CrossRef PubMed.
- G. Reguera, K. D. Mccarthy, T. Mehta, J. S. Nicoll, M. T. Tuominen and D. R. Lovley, Nature, 2005, 435, 1098–1102 CrossRef PubMed.
- J. A. Gralnick and D. K. Newman, Mol. Microbiol., 2007, 65, 1–11 CrossRef PubMed.
- E. Marsili, D. B. Baron, I. D. Shikhare, D. Coursolle, J. a. Gralnick and D. R. Bond, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3968–3973 CrossRef PubMed.
- E. Marsili, J. B. Rollefson, D. B. Baron, R. M. Hozalski and D. R. Bond, Appl. Environ. Microbiol., 2008, 74, 7329–7337 CrossRef PubMed.
- K. P. Katuri, P. Kavanagh, S. Rengaraj and D. Leech, Chem. Commun., 2010, 46, 4758–4760 RSC.
- H. Richter, K. P. Nevin, H. Jia, D. a. Lowy, D. R. Lovley and L. M. Tender, Energy Environ. Sci., 2009, 2, 506 RSC.
- P. S. Jana, K. Katuri, P. Kavanagh, A. Kumar and D. Leech, Phys. Chem. Chem. Phys., 2014, 16, 9039–9046 RSC.
- Y.-Y. Yu, D.-D. Zhai, R.-W. Si, J.-Z. Sun, X. L. Yong and Y.-C. Yong, Int. J. Mol. Sci., 2017, 18, 90 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |