
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic evaluation of phytochemicals in breast cancer remedy: current understanding and future perspectives
Muhammad Younasa,
Christophe Hanob,
Nathalie Giglioli-Guivarc'hc and
Bilal Haider Abbasi *abc
*abc
aDepartment of Biotechnology, Quaid-i-Azam University, Islamabad-45320, Pakistan. E-mail: bhabbasi@qau.edu.pk; Fax: +92-51-90644121; Tel: +92-51-90644121 Tel: +33-767-97-0619
bLaboratoire de Biologie des Ligneux et des Grandes Cultures (LBLGC), Plant Lignans Team, UPRES EA 1207, Université d'Orléans, F 28000 Chartres, France
cEA2106 Biomolecules et Biotechnologies Vegetales, Universite Francois-Rabelais de Tours, Tours, France
First published on 22nd August 2018
Abstract
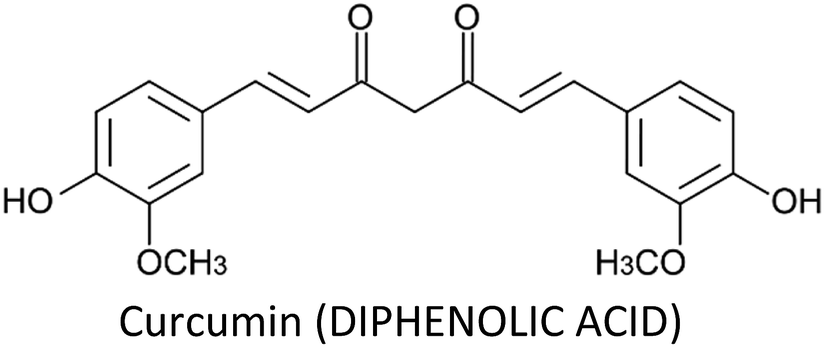
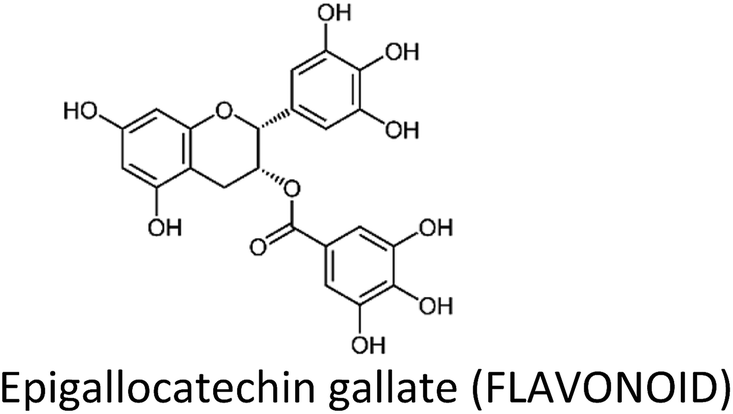
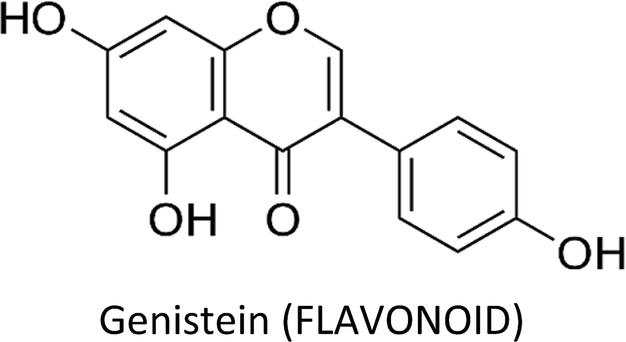
Breast cancer is one of the most commonly diagnosed cancers around the globe and accounts for a large proportion of fatalities in women. Despite the advancement in therapeutic and diagnostic procedures, breast cancer still represents a major challenge. Current anti-breast cancer approaches include surgical removal, radiotherapy, hormonal therapy and the use of various chemotherapeutic drugs. However, drug resistance, associated serious adverse effects, metastasis and recurrence complications still need to be resolved which demand safe and alternative strategies. In this scenario, phytochemicals have recently gained huge attention due to their safety profile and cost-effectiveness. These phytochemicals modulate various genes, gene products and signalling pathways, thereby inhibiting breast cancer cell proliferation, invasion, angiogenesis and metastasis and inducing apoptosis. Moreover, they also target breast cancer stem cells and overcome drug resistance problems in breast carcinomas. Phytochemicals as adjuvants with chemotherapeutic drugs have greatly enhanced their therapeutic efficacy. This review focuses on the recently recognized molecular mechanisms underlying breast cancer chemoprevention with the use of phytochemicals such as curcumin, resveratrol, silibinin, genistein, epigallocatechin gallate, secoisolariciresinol, thymoquinone, kaempferol, quercetin, parthenolide, sulforaphane, ginsenosides, naringenin, isoliquiritigenin, luteolin, benzyl isothiocyanate, α-mangostin, 3,3′-diindolylmethane, pterostilbene, vinca alkaloids and apigenin.
1 Introduction
Breast cancer is one of the most commonly diagnosed cancers around the world and accounts for a large proportion of mortalities in females. The American Cancer Society estimated that in 2017, there will be an expected 63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 410 new cases of in situ, 252710 cases of invasive and 40610 cases of breast cancer mortalities across women in the United States alone.1,2 On the basis of expression of the estrogen receptor (ER), breast cancer is categorized into ER-positive and ER-negative types. The human epidermal growth factor receptor 2 (HER2) and progesterone receptor (PR) further divide breast cancer into several molecular subtypes like HER2-positive, luminal A and luminal B. These types of breast carcinomas respond to aromatase inhibitors or hormonal therapy. However, another type of breast cancer, known as basal-like or triple negative breast cancer (TNBC), lacks these three receptors and thus does not respond to hormonal therapy.3–8 Current therapeutic interventions in breast cancer remedy include radiation, surgical exclusion and the use of various chemotherapeutic drugs such as paclitaxel, doxorubicin, cisplatin, docetaxel, carboplatin, epirubicin, bevacizumab and cyclophosphamide.9,10 However, the incidences of drug resistance and serious side-effects associated with these treatment methods have greatly reduced their therapeutic potential.11 These complications propel researchers to look into alternative and safer chemotherapeutic strategies.
410 new cases of in situ, 252710 cases of invasive and 40610 cases of breast cancer mortalities across women in the United States alone.1,2 On the basis of expression of the estrogen receptor (ER), breast cancer is categorized into ER-positive and ER-negative types. The human epidermal growth factor receptor 2 (HER2) and progesterone receptor (PR) further divide breast cancer into several molecular subtypes like HER2-positive, luminal A and luminal B. These types of breast carcinomas respond to aromatase inhibitors or hormonal therapy. However, another type of breast cancer, known as basal-like or triple negative breast cancer (TNBC), lacks these three receptors and thus does not respond to hormonal therapy.3–8 Current therapeutic interventions in breast cancer remedy include radiation, surgical exclusion and the use of various chemotherapeutic drugs such as paclitaxel, doxorubicin, cisplatin, docetaxel, carboplatin, epirubicin, bevacizumab and cyclophosphamide.9,10 However, the incidences of drug resistance and serious side-effects associated with these treatment methods have greatly reduced their therapeutic potential.11 These complications propel researchers to look into alternative and safer chemotherapeutic strategies.
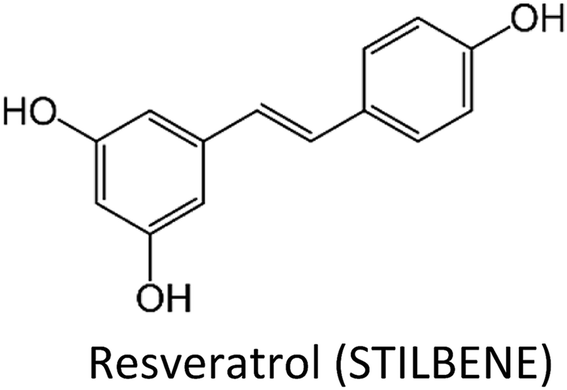
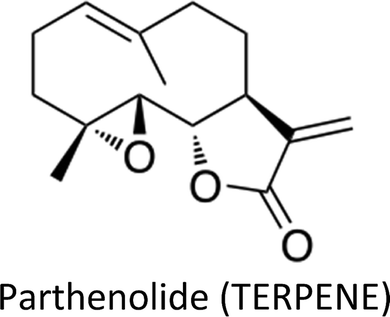
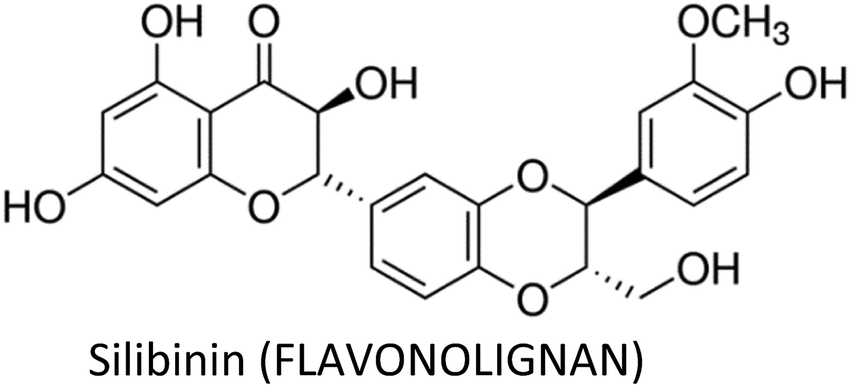
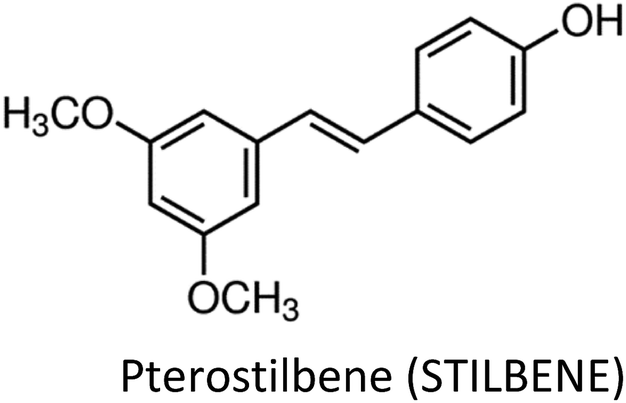
Human beings have always been suffered from infections by fungi, parasites, bacteria, viruses and many health disorders such as pain, inflammation, digestive complications and colds etc.12 Recent medicines, based on antibiotics and synthetic drugs, have come into practice during the previous 150 years. Before that, humans had to depend on drugs derived from plants, animals and fungi. The curing of health disorders and infections with herbal medicines includes active natural products, known as secondary metabolites or phytochemicals, which are found in almost all plant species.12 Secondary metabolites encompass a diverse group of organic compounds which have a vital role in plant defence systems and assist the interaction with the biotic environment. Many secondary metabolites like phenolics, alkaloids and terpenes are classified on the basis of their biosynthetic origin which possess different biological properties and are employed as pharmaceuticals, flavors, colors, agrochemicals, fragrances, food additives and biopesticides.13–15 Secondary metabolites specifically modulate a molecular target in humans or animals. Such targets often include neuroreceptors, ion pumps, ion channels and elements of the cytoskeleton or enzymes degrading neurotransmitters.12,16–20 An estimated more than 5000 distinct phytochemicals have been recognized in vegetables, fruits and grains, however, a large proportion is yet to be identified and understood before we can completely comprehend their health benefits in whole foods.21 Phytochemicals can be categorized into different classes such as phenolics, terpenes, organosulfur compounds and alkaloids (Fig. 1).393,394 The molecular structures of various phytochemicals involved in breast cancer chemoprevention with their major plant sources are given in Table 1.
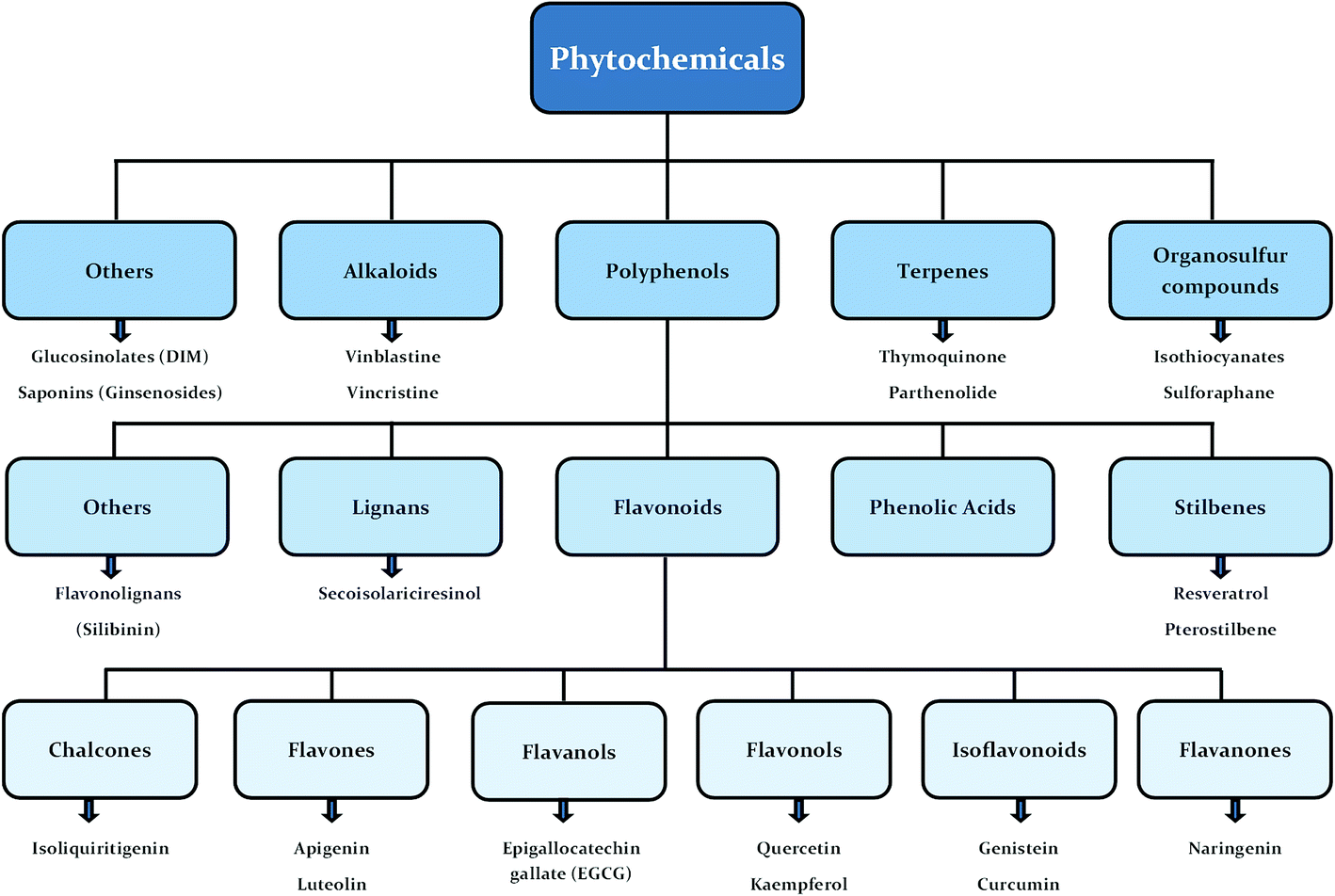 | ||
| Fig. 1 Classification of phytochemicals involved in breast cancer chemoprevention. | ||
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
Phytochemicals display diverse range of disease-preventing or protective effects. They have been used since ancient times to cope with various ailments including cancer, diabetes, cardiovascular diseases, inflammation, neurological disorders and skin diseases etc.22–28 Numerous epidemiological studies have predicted reduced cancer incidence with the use of phytochemicals.29–31 Phytochemicals such as curcumin,32 resveratrol,33,34 epigallocatechin gallate (EGCG),35,36 silibinin,37 benzyl isothiocyanate,38,39 genistein,40 kaempferol,41,42 thymoquinone,43 quercetin,44 parthenolide,45 sulforaphane,46 naringenin,47,327,330 isoliquiritigenin48,49 and ginsenosides50–52 have been shown to suppress breast carcinoma via modulation of various signalling transduction pathways, genes and gene products. These phytochemicals exert anti-breast cancer effects by inducing cellular apoptosis and reducing cell proliferation through modulation of various targets (Fig. 2 & 4). Additionally, phytochemicals inhibit the angiogenesis, metastasis and migratory behaviours in breast cancer cells (Fig. 3 & 4). Moreover, these compounds greatly enhanced the therapeutic efficacy of different anti-cancer drugs, overcame drugs resistance in breast cancer cells and also achieved sensitization to radiations.53–58 The compounds targeted breast cancer stem cells (bCSCs)/progenitor cells.59,60 CSCs are mainly involved in promotion of invasion, metastasis, abnormal proliferation, recurrence and drug resistance.61 The self-renewal capability of CSCs is related to the regulatory pathways of Notch, Wnt/β-catenin, hedgehog and P13K/Akt. These pathways might also contribute in the maintenance of CD44+/CD24−/low bCSCs stemness.59,62 Dandawate et al. (2016) reviewed the role of phytochemicals in targeting bCSCs.60 Abdal Dayem et al. (2016) reviewed the effect of polyphenols against breast cancer and CSCs (Fig. 5).395 Petric et al. (2015) reviewed some phytochemicals modulating signalling pathways in breast and hormone related cancers.63 Moreover, Siddiqui et al. (2015) have also put some light on therapeutic effect of phytochemicals in breast cancer.64 In this review article, we summarize various phytochemicals that are involved in breast cancer prevention, by putting in view the recently recognized molecular mechanisms, which may serve useful in future drugs development.
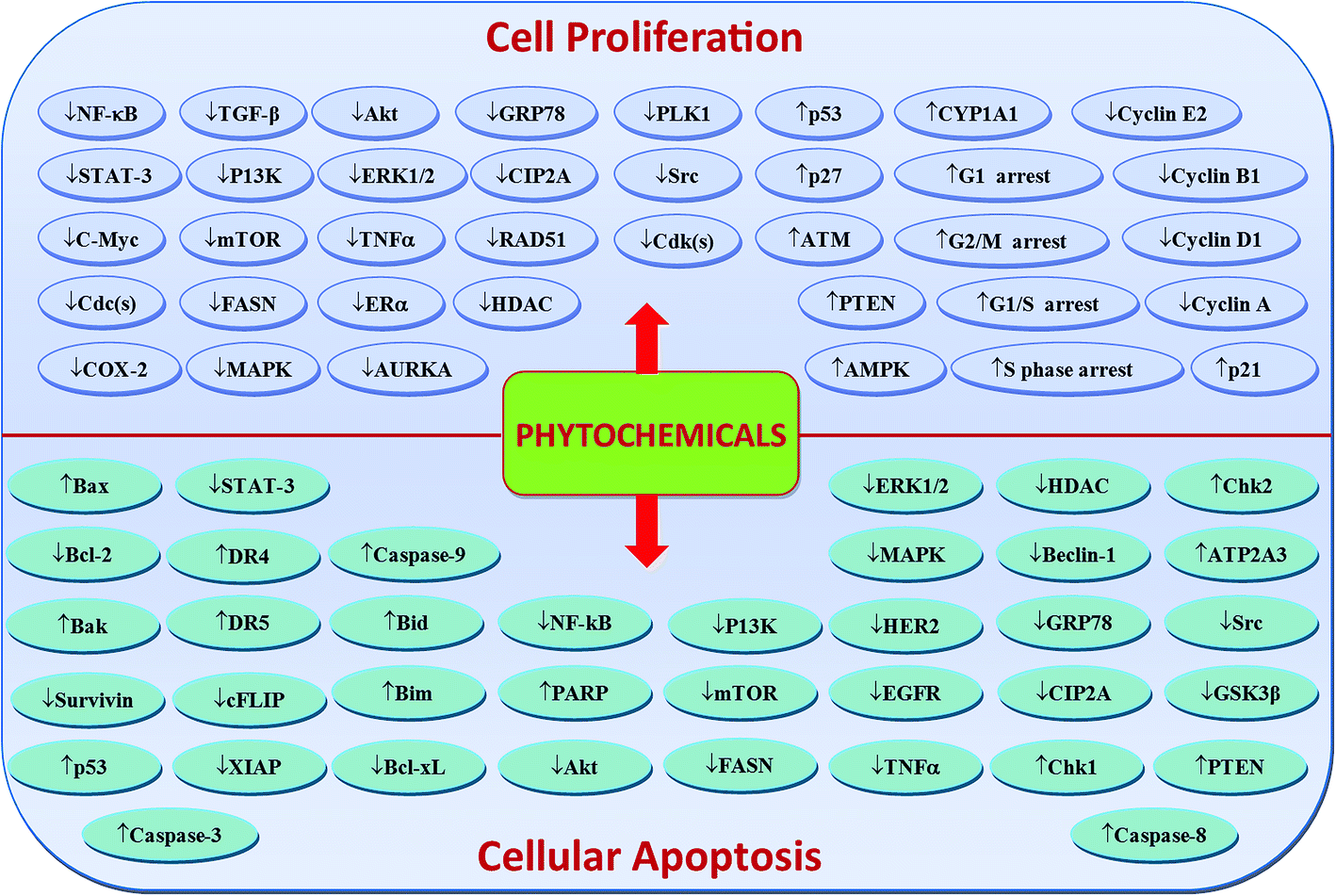 | ||
| Fig. 2 Phytochemicals-mediated regulation of molecular targets/signalling pathways involved in breast cancer cells proliferation and apoptosis. Downward arrows (↓) represent downregulation while upward arrows (↑) represent upregulation. | ||
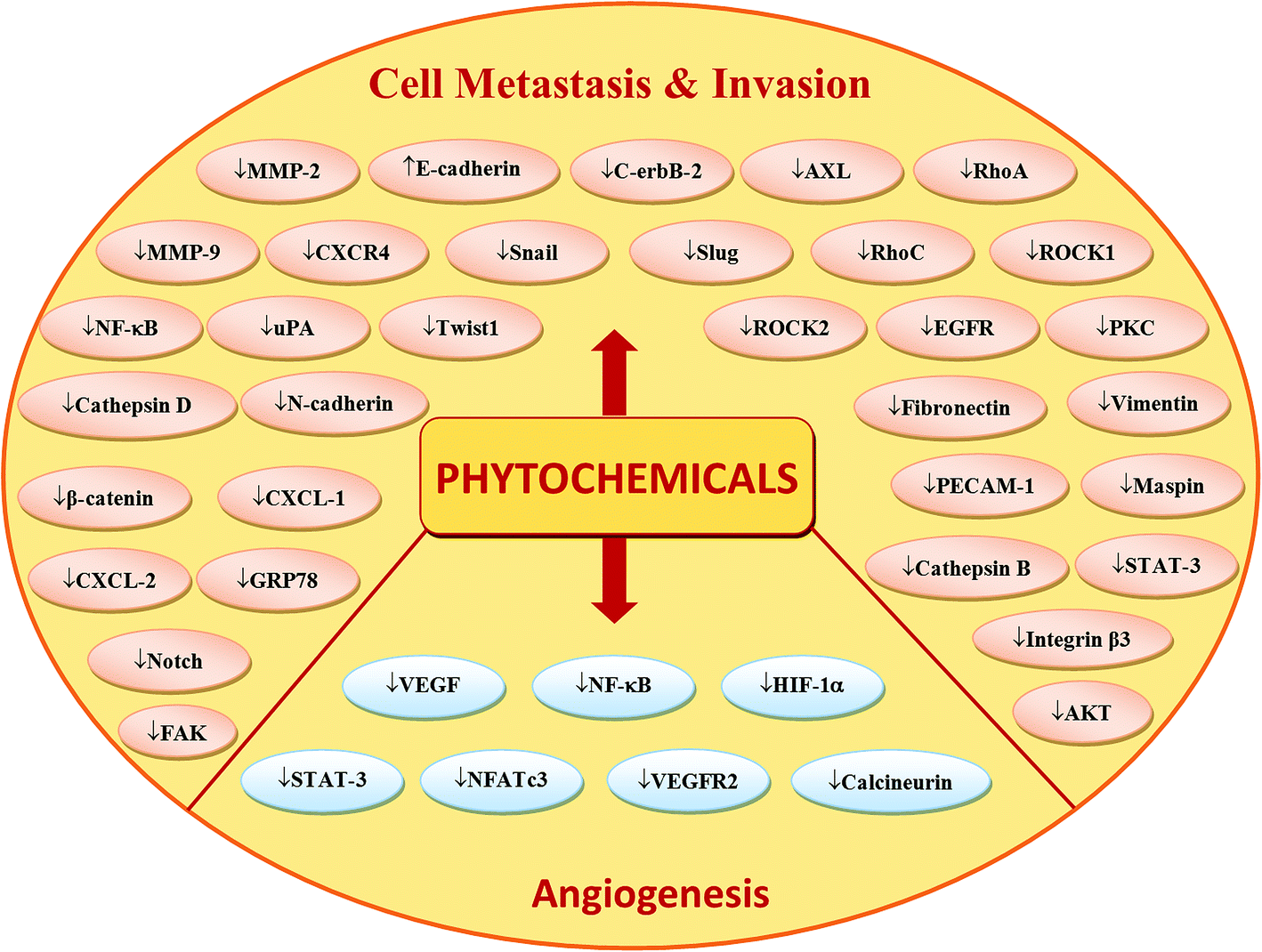 | ||
| Fig. 3 Phytochemicals-mediated regulation of molecular targets/signalling pathways involved in breast cancer cells invasion, metastasis and angiogenesis. Downward arrows (↓) represent downregulation while upward arrows (↑) represent upregulation. | ||
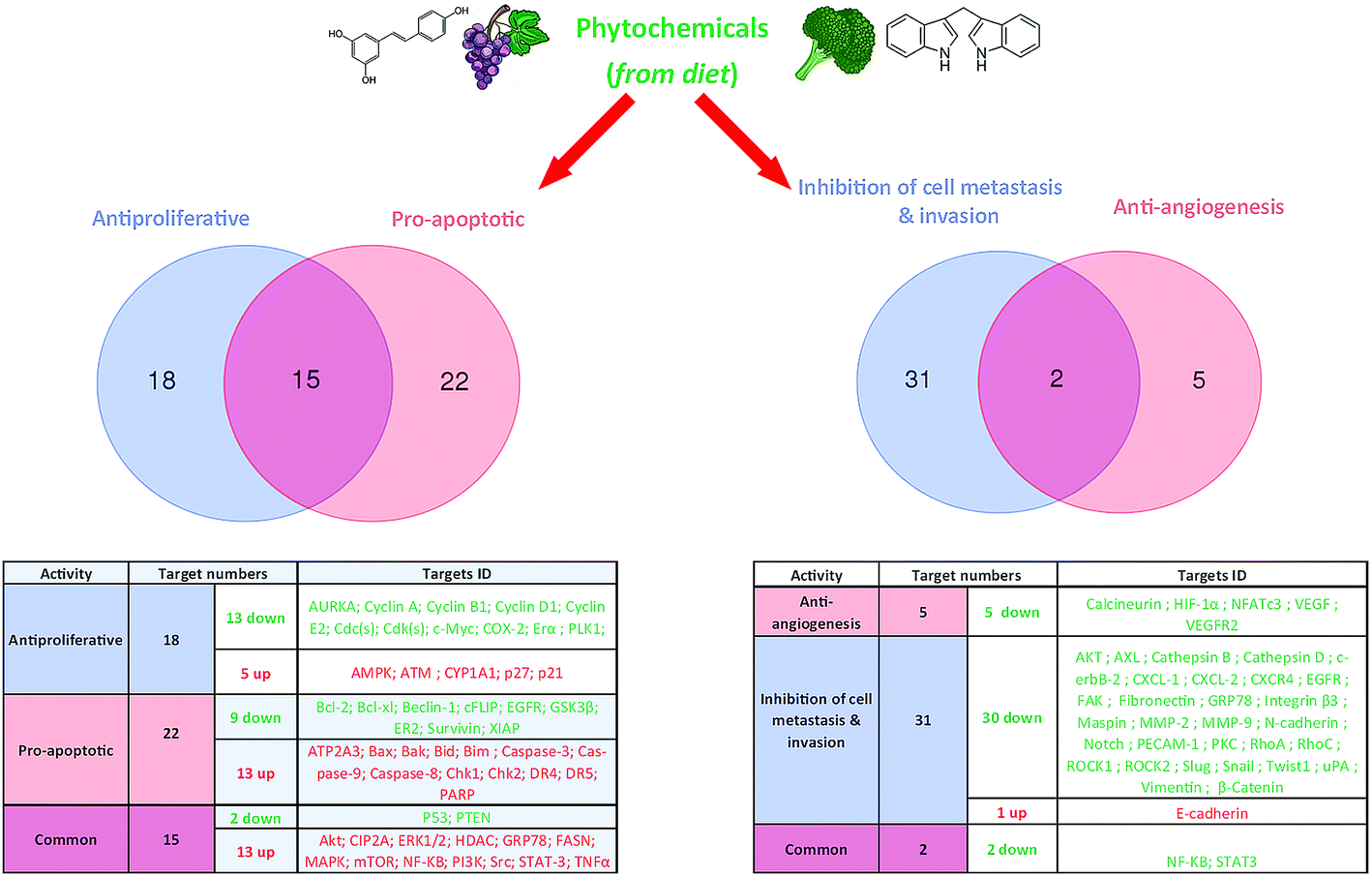 | ||
| Fig. 4 Possible number of key targets involved in breast cancer cells proliferation, apoptosis, angiogenesis, invasion and metastasis and their regulation by phytochemicals. | ||
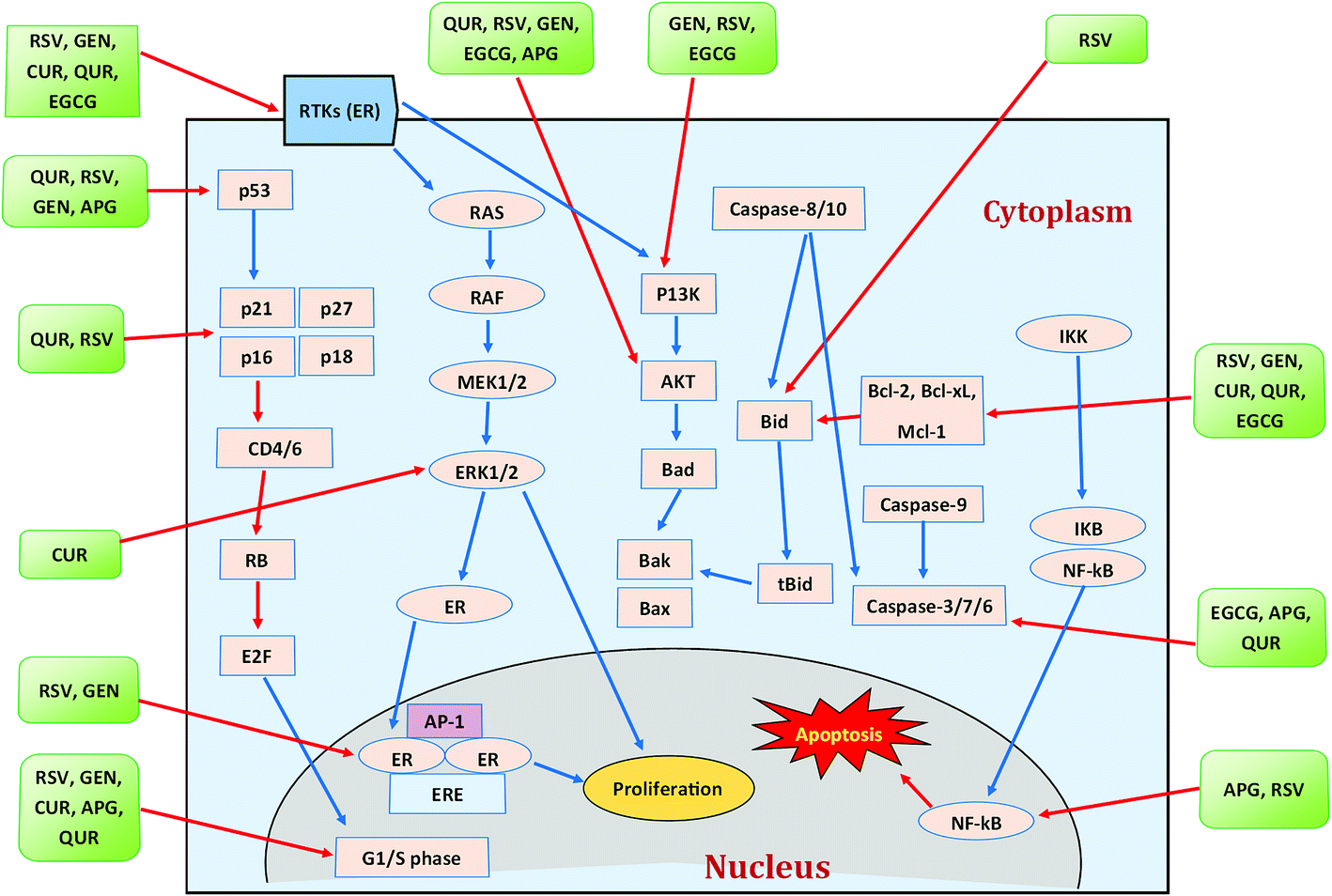 | ||
| Fig. 5 Polyphenols and their modes of action against breast cancer. Blue arrows indicate activation while red arrows indicate inhibition. CUR = curcumin, RSV = resveratrol, GEN = genistein, APG = apigenin, QUR = quercetin. Idea adopted from Abdal Dayem et al. (2016).395 | ||
2 Literature search
This review was conducted using various search engines including Google Scholar, PubMed, and Scopus by putting keywords such as breast cancer, chemoprevention, phytochemicals and individual names of respective phytochemicals plus breast cancer. All the research articles included in here are from the last half decade: 2012–2018.3 Phytochemicals involved in breast cancer chemoprevention
3.1. Curcumin
Curcumin—a yellow pigment found in the rhizome of turmeric—is one of the most extensively studied phytochemical in breast cancer therapy. It displayed a wide spectrum of activities such as anti-proliferative, apoptotic, anti-metastatic, anti-angiogenic, radio-protective, chemo-sensitization, induction of cell cycle arrest and inhibition of migration and invasion in breast cancer cells (Table 2). Curcumin can modulate the activities and functions of various gene products like enzymes, inflammatory cytokines, transcription factors and products related to cell proliferation and cell survival.65 Liu and Chen (2013) reviewed the effect of curcumin on breast cancer cells and showed that the compound modulate various molecular targets (Fig. 6).396| Phytochemical | Cell line/model | Proposed mechanism | Effect | Ref. |
|---|---|---|---|---|
| Curcumin | MCF-7 | ↓GSTP1 methylation | ↑Glutathione S-transferase Pi 1 | 72 |
| MDA-MB-361 | ↓Sp1 expression, ↓DLC1 methylation | ↓Growth, ↑re-expression of the tumor suppressor: deleted in liver cancer 1 | 73 | |
| MCF-7 | ↓miR-21, ↑PTEN, ↑Akt, ↑caspase-3, ↑caspase-9 | ↑Apoptosis | 66 | |
| MCF-7 & MCF-7/DPP | ↓CCAT1 expression, ↓PI3K, ↓p-Akt, ↓p-mTOR | ↑Autophagy, ↑sensitization to cisplatin | 80 | |
| MCF-10F, MDA-MB-231 & Tumor 2 | ↓β-Catenin, ↓N-cadherin, ↓E-cadherin, ↓slug, ↓Twist1, ↓AXL, ↓fibronectin, ↓vimentin | ↓Migration, ↓invasion | 88 | |
| MDA-MB-231 | ↑AMPK, ↓Akt | ↑Autophagy, ↓proliferation, ↓migration | 280 | |
| MDA-MB-231 | ↓Intracellular FAS | ↑Apoptosis | 69 | |
| MDA-MB-231 | ↓Slug/HK2 axis | ↑Apoptosis, ↓4-OHT resistance | 281 | |
| MCF-7, MCF10A, SUM149 | ↓SCD, ↓CD49f, ↓LDH1A3, ↓TP63, ↓PROM1 | ↓bCSCs self-renewal | 282 | |
| MCF-7 | ↓Cyclin B1, ↓Cdc2 | ↓Colonization | 283 | |
| MCF-7 | ↓RhoA, ↓ROCK1, ↓ROCK2, ↓MMP-2, ↓MMP-9 | ↓Invasion | 83 | |
| MDA-MB-231 | ↓Cyclin D1, ↓p65, ↓PECAM-1 | ↓Angiogenesis, ↓proliferation, ↑apoptosis | 84 | |
| MDA-MB-231 | ↑miR181b, ↓CXCL1, ↓CXCL2 | ↓Metastasis, ↓proliferation, ↓invasion, ↑apoptosis | 284 | |
| MDA-MB-231 & MCF-7 | ↓Bcl-2, ↑Bax | ↑Apoptosis | 68 | |
| MCF-7 & T47D | ↑E-Cadherin/β-catenin negative feedback loop | ↓Migration of bCSCs | 71 | |
| MCF-7 | ↑Nrf2 expression, ↓Fen1 expression | ↓Proliferation | 70 | |
| MCF-7 & MDA-MB-231 | ↓NF-κB-Snail signalling pathway, ↑E-cadherin, ↓vimentin | ↓Invasion | 285 | |
| MCF-7/LCC2 & MCF-7/LCC9 | ↓NF-κB, ↓Src, ↓FAK, ↓Akt, ↓mTOR, ↓EZH2, ↓Bcl-2, ↓Bcl-xL, ↓cyclin D1, ↓c-Myc, ↑ERK1/2 | ↓Proliferation, ↑apoptosis, ↓endocrine resistance, ↑sensitization to tamoxifen | 286 | |
| MCF-7 | ↓uPA expression, ↓NF-κB | ↓Adhesion, ↓invasion | 82 | |
| MCF-7, MCF-10A, MDA-MB-231, SK-BR-3h | ↓HER-2 oncoprotein, ↓MAPK, ↓p-Akt, ↓NF-κB | ↓Proliferation, ↓migration | 287 | |
| MCF-7 & MDA-MB-231 | ↓DNMT1, ↓RASSF1A methylation | ↑ RASSF1A | 74 | |
| MDA-MB-231 | ↓pERK1/2, ↓pEGFR | ↑Apoptosis, ↓proliferation | 288 | |
| MCF-7 | ↓MAPK, ↓PKC, ↓NF-κB | ↓Invasion | 289 | |
| EGCG | In vivo | ↓Ki-67 | ↓Proliferation | 290 |
| T-47D | ↓Telomerase, ↓PI3K/AKT | ↑Apoptosis | 92 | |
| MCF-7 | ↓Bcl-2, ↑p53 | ↑Apoptosis, ↓proliferation | 93 | |
| MDA-MB-231 | ↓β-Catenin signalling pathway | ↓Proliferation | 94 | |
| T-47D | ↓ERα protein levels | ↓Proliferation | 291 | |
| MCF-7/TAM | ↓Nrf2 expression | ↓Tamoxifen resistance | 100 | |
| MCF-7/DOX | ↓MMP-2, ↓MMP-9 activity | ↓Doxorubicin resistance | 101 | |
| MDA-MB-231 & MDA-MB-436 | ↓ER-α36, ↓MAPK/ERK, ↓EGFR, ↓PI3K/AKT | ↓Proliferation, ↓bCSCs growth | 98 | |
| MCF-7, T-47D & SK-BR-3 | ↓Hsp90/PR-B/HDAC interactions, ↑p38/CK2, ↓ERα | ↓Proliferation, ↓colonization | 96 | |
| MCF-7 & MDA-MB-231 | ↓H3K9/18 acetylation, ↓EZH2, ↓class I HDAC, ↓TIMP-3, ↓H3K2 trimethylation | ↓Progression & invasion | 292 | |
| SUM-149 & SUM-190 | ↓ALDH+ cells, ↓VEGF-D, ↓cyclin D1, ↓RhoC, ↓FN1, ↓E-cadherin, ↓Bcl-XL, ↓VIM, ↑c-PARP, ↑cleaved caspase-3 | ↓Proliferation, ↓migration, ↓invasion, ↑apoptosis, ↓lymphangiogenesis, ↓tumorsphere formation | 293 | |
| Hs578T | ↓VEGF | ↓Proliferation, ↓migration & invasion | 294 | |
| MCF-7 | ↓VEGF, ↓HIF-1α | ↓Proliferation | 295 | |
| 4T1 & RAW264.7 | ↑miR-16, ↓IKKα, ↓CSF-1, ↑IL-6, ↓CCL-2, ↑TGF-β, ↑TNF-α | ↓Tumor growth, ↓TAM infiltration, ↓M2 polarization | 296 | |
| MCF-7 & MDA-MB-231 | ↑p21WAF1, ↓DNMT1, ↓HDAC1, ↓MeCP2, ↓RARβ2, ↓cyclin D2 methylation, ↓TMS1 methylation, ↓MGMT methylation | ↓Cell viability | 297 | |
| Genistein | Sprague-Dawley rats | ↓UPR, ↓GRP78, ↓IRE1α, ↓Beclin-1, ↓ATF4, ↓TGFβ, ↓Foxρ3, ↑CD8a | ↓Tamoxifen resistance, ↓recurrence | 110 |
| MCF-7-C3 & T-47D | ↓CIP2A, ↓E2F1 | ↑Apoptosis, ↓proliferation | 106 | |
| MCF-7 | ↑miR-23b | ↑Cytotoxicity | 111 | |
| MDA-MB-231 & MCF-7 | ↓DNMTI, ↑ATM, ↑PTEN, ↑APC, ↑SERPINB5 | ↓Cell viability | 108 | |
| MCF-7/Adr | ↓ HER2/neu | ↑Apoptosis, ↓doxorubicin resistance | 298 | |
| MCF-7 & 3T3-L1 | ↓ERα, ↓cyclin D1, ↓Bcl-2, ↑Bax | ↓Proliferation, ↑apoptosis | 103 | |
| MCF-7 | ↓Hedgehog, ↓Gli1 | ↓bCSCs | 113 | |
| MDA-MB-231 & MCF-7 | ↑ATM, ↑Chk2, ↑Cdc25C, ↑Cdc2, ↑Bax, ↑p53, ↓Bcl-2, ↓Rad51 | ↑Radiosensitivity, ↑apoptosis | 299 | |
| MCF-7, SK-BR-3 & ZR-75-1 | ↓ERα, ↓c-erbB-2 expression | ↓Proliferation | 105 | |
| MDA-MB-231 | ↓pERK1/2, ↑Bax/Bcl-2 ratio | ↑Apoptosis, ↓proliferation | 300 | |
| MDA-MB-231 | ↓NF-κB, ↓cyclin B1, ↓Bcl-2, ↓Bcl-xL | ↓Proliferation, ↑apoptosis | 301 | |
| MCF-7 & UACC-3199 | ↓DNMT-1, ↓cyclin D1, ↑p53, ↑BRCA-1, ↓CpG methylation, ↑CYP1A1 | ↓Proliferation | 40 | |
| Resveratrol | MDA-MB-231 & MCF-7 | ↑ATP2A3, ↓Bcl-2, ↓Ki67, ↑Bcl-2L11 (BIM) | ↑Apoptosis, ↓proliferation | 114 |
| T47-D | ↓p53, ↓ERα | ↓Proliferation | 302 | |
| MDA-MB-231 | ↓AURKA, ↓PLK1, ↓cyclin D1, ↓cyclin B1 | ↓Cell cycle progression, ↑apoptosis, ↓viability | 117 | |
| MDA-MB-231 & MDA-MB-468 | ↓YAP, ↓RhoA | ↓Invasion | 116 | |
| MDA-MB-231 & MCF-7 | ↓ XIAP, ↓Bcl-2, ↑CASP-8, ↑CASP-9, modulation of miR-125b-5p, miR-409-3p, miR-200c-3, miR-542-3p & miR-122-5p | ↑Apoptosis | 128 | |
| MCF-7/DOX | ↓MDR-1, ↓P-glycoprotein | ↓Doxorubicin resistance, ↓proliferation | 124 | |
| MCF-7, SUM159 | ↓ Wnt, ↓β-catenin | ↓bCSCs proliferation, ↑autophagy, ↓mammospheres | 303 | |
| SKBR-3 | ↓FASN, ↓HER2, ↑PEA3, ↓p-Akt, ↑PTEN, ↓PI3K, ↓Akt, ↓mTOR | ↓Proliferation, ↑apoptosis | 115 | |
| MCF-7 | ↓PFK activity, ↓glucose consumption, ↓ATP content | ↓Cell viability | 304 | |
| MCF-7/TR | ↓TGF-β, ↓Smad cascade, ↓EMT | ↓Tamoxifen resistance, ↑apoptosis | 126 | |
| MCF-7 | ↓HSP27 | ↑Sensitization to doxorubicin | 125 | |
| MCF-7 & MDA-MB-231 | ↑p53, ↓procaspase 8, ↑CASP-7, ↑CASP-9, ↑p-Chk2, ↓cyclin A, ↓Thr160-phosphorylated CDK2, ↓CDK7, ↑cell cycle arrest | ↑Sensitization to melphalan | 127 | |
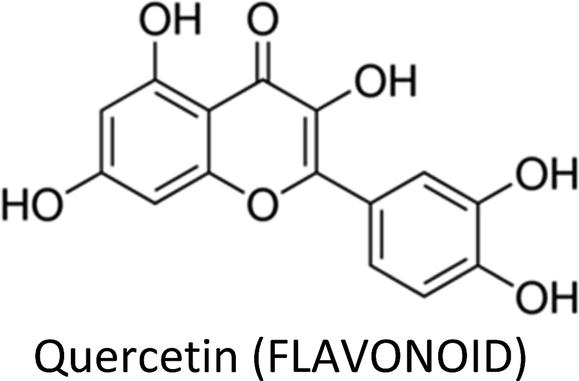
| Quercetin | MDA-MB-231 & MDA-MB-157 | ↓FASN, ↓β-catenin, ↓Bcl-2, ↑caspase-3 | ↑Apoptosis, ↓proliferation | 44 |
| MCF-7 | ↓Bcl-2, ↑Bax | ↑Apoptosis, ↑necroptosis | 130 | |
| MCF-7 | ↓VEGF, ↓VEGFR2, ↓NFATc3, ↓calcineurin pathway | ↓Angiogenesis | 137 | |
| MCF-7Ca/TAM-R | ↓Her-2, ↑ERα | ↓Proliferation, ↑apoptosis, ↓tamoxifen resistance | 134 | |
| MDA-MB-231 & MDA-MB-468 | ↑E-Cadherin, ↓vimentin, ↓c-Myc, ↓cyclin D1 | ↓Metastasis, ↓proliferation | 136 | |
| MCF-7 | ↓Twist, ↓p38MAPK, ↓cyclin D1, ↓p21 | ↑Apoptosis | 131 | |
| MCF-7 | ↓Proteasome, ↑CASP-3, ↑CASP-7 | ↓Proliferation, ↑apoptosis | 129 | |
| MCF-7 & MDA-MB-231 | ↑miR-146a, ↓EGFR, ↑Bax, ↑CASP-3 | ↓Proliferation, ↓invasion, ↑apoptosis | 138 | |
| MCF-7 | ↓Survivin | ↓Proliferation | 132 | |
| MCF-7/TR | ↓Cyclin E2 | ↑Sensitization to tamoxifen | 305 | |
| MCF-7 | ↓Bcl-2, ↑Bax | ↓Proliferation, ↑apoptosis | 306 | |
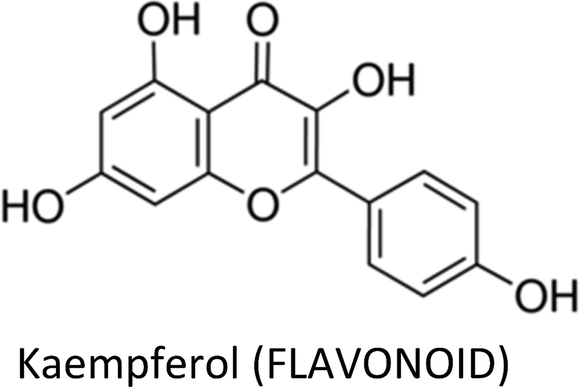
| Kaempferol | MCF-7 | ↓pIRS-1, ↓pAkt, ↓pMEK1/2, pERK1/2, ↓cyclin D1, ↓cyclin E, ↑p21, ↓cathepsin D | ↓Proliferation, ↓metastasis, ↑apoptosis | 243 |
| MCF-7 | ↓Bcl-2, ↑Bax, ↑PARP cleavage | ↑Apoptosis | 242 | |
| MCF-7 | ↓Cathepsin B, ↓cathepsin D, ↓N-cadherin, ↓snail, ↓slug, ↑E-cadherin | ↓Proliferation, ↓migration, ↓invasion, ↓metastasis | 307 | |
| MDA-MB-231 & MDA-MB-453 | ↓RhoA, ↓Rac1 | ↓Migration, ↓invasion | 42 | |
| MCF-7 | ↑E-cadherin, ↓MMP-9, ↓MMP-2, ↓cathepsin B, ↓cathepsin D, ↓N-cadherin, ↓snail, ↓slug | ↓Metastasis | 244 | |
| MCF-7 | ↓GLUT1, ↓MCT1 | ↓Proliferation, ↑cytotoxicity | 241 | |
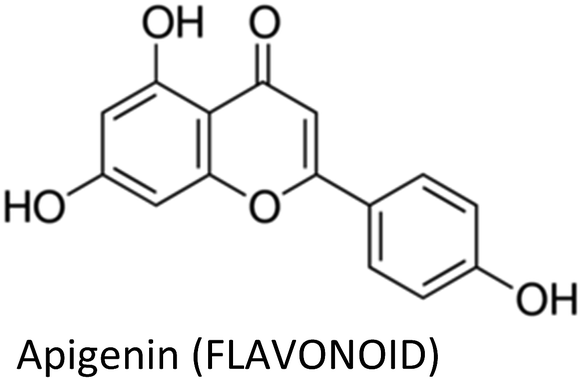
| Apigenin | MDA-MB-231 | ↓Cyclin A, ↓cyclin B, ↓CDK1, ↑p21WAF1/CIP1 | ↓Proliferation | 176 |
| MCF-7/ADR | ↓MDR1, ↓P-gp, ↓p-STAT3, ↓VEGF, ↓MMP-9 | ↑Apoptosis, ↓colonization, ↓adriamycin resistance | 173 | |
| MDA-MB-231 | ↓TNFα, ↓CCL2, ↓GMCSF, ↓IL-1α, ↓IL-6, ↓IKBKe | ↓Pro-inflammatory cytokines | 163 | |
| MDA-MB-231 & ZR75.1 | ↓CXCL10, ↓IL-6, ↓IL-1α, ↓IRAK1, ↓IRAK4, ↓NF-κB, ↓p38-MAPK, ↓IP10 | ↓Impact of senescent cells on breast cancer cells, ↓fibroblast proliferation | 308 | |
| BT-474 | ↓p-JAK1, ↓p-JAK2, ↓p-STAT3, ↓VEGF, ↑c-CASP-8, ↑c-CASP-3 | ↑Apoptosis, ↓proliferation, ↓colonization | 174 | |
| MDA-MB-468 | ↓Akt | ↓Proliferation | 177 | |
| MDA-MB-231 & T47D | ↑CASP-3, ↑c-PARP, ↑Bax, ↓Bcl-2, ↑LC3-II | ↑Apoptosis, ↓proliferation, ↓colonization, ↑autophagy | 309 | |
| MCF-7/HER2 & MCF-7 vec | ↑c-CASP-8, ↓p-HER-2, ↑p53, ↑p21, ↓p-JAK1, ↓p-STAT3, ↓NF-κB, ↓p-IκBα | ↑Apoptosis, ↓proliferation | 175 | |
| Silibinin | MCF-7 & T47D | ↓miR-21, ↓Bcl-2 | ↑Apoptosis | 149 |
| MCF-7 | ↓Maspin, ↓ERα | ↓Proliferation | 310 | |
| T47D | ↓hTERT, ↓cyclin D1 | ↓Proliferation | 147 | |
| MCF-7 | ↓Bcl-2, ↑p53, ↑Bax, ↑BRCA1, ↑ATM | ↑Apoptosis, ↓proliferation | 145 | |
| MCF-7 & T47D | ↑PTEN, ↑p21, ↓Bcl-2, ↑p27 | ↓Proliferation, ↑apoptosis, ↑necrosis | 311 | |
| MCF-7 | ↓miR-21, ↓miR-155, ↑CASP-9, ↑BID | ↑Apoptosis, ↓proliferation | 148 | |
| MCF-7 | ↓ERα, ↓Akt, ↓mTOR, ↓ERK, ↑CASP-6, ↑p53, ↓APAF-1, ↓p62, ↑Bax, ↓Bcl-2, ↑LC3-I to LC3-II | ↑Apoptosis, ↑autophagy | 151 | |
| MCF-7 | ↑Atg12-Atg5, ↑LC3-1 to LC3II, ↑Beclin-1, ↓Bcl-2, ↑BNIP3, ↑ROS | ↑Autophagy | 150 | |
| MCF-7 | ↑p53, ↑p21, ↑BRCA1, ↑Bak, ↑ATM, ↓Bcl-xl | ↑Apoptosis, ↓proliferation | 143 | |
| MCF-7 & MDA-MB-231 | ↓ERK, ↓Akt, ↓Notch-1 | ↑Apoptosis | 312 | |
| SKBR3 | ↓NF-κB | ↑Apoptosis, ↓proliferation | 142 | |
| MDA-MB-468 | ↓EGFR, ↓VEGF, ↓COX-2, ↓MMP-9 | ↓Metastasis, ↓infiltration, ↓tumor volume | 144 | |
| MCF-7 | ↓MMP-9, ↓MEK, ↓ERK | ↓Migration | 141 | |
| MDA-MB-231 & T47D | ↓Wnt, ↓β-catenin, ↓LRP6 | ↓Proliferation | 313 | |
| Pterostilbene | MDA-MB-468 | ↑ERK1/2, ↓cyclin D1, ↑p21, ↓Akt, ↓mTOR, ↑Bax | ↑Apoptosis, ↓proliferation | 189 |
| MCF, MDA-MB-231 & Hs578t | ↑E-cadherin, ↓vimentin, ↓snail, ↓slug, ↓ZEB1, ↑miR-205, ↓Src/Fak | ↓Metastasis | 191 | |
| MCF-7 | ↓CD44, ↑β-catenin, ↓hedgehog, ↓Akt, ↓GSK3β signalling, ↓cyclin D1, ↓c-Myc | ↓bCSCs, ↓mammospheres | 195 | |
| MCF-7 & MDA-MB-231 | ↓NFκB, ↓vimentin, ↓Twist1, ↑E-cadherin | ↓bCSCs, ↓metastasis | 196 | |
| MDA-MB-231 | ↓MMP-2, ↓MMP-9, ↓cortactin, ↓c-Src kinase, ↓MT1-MMP | ↓Metastasis | 314 | |
| Sulforaphane | SUM-149 & SUM-159 | ↓NF-κB p65 subunit, ↓p52 | ↓bCSCs, ↓mammospheres, ↓proliferation | 204 |
| MCF-7, MDA-MB-231 & SK-BR-3 | ↓DNMT1, ↓DNMT3B, ↑p21, ↑p27, ↓miR92b, ↓miR-23b, ↓miR-381, ↓miR-382, ↓Akt, ↓AMPK, ↓ATP | ↑Cellular senescence, ↑apoptosis, ↑autophagy | 210 | |
| MCF-7 & MDA-MB-231 | ↓Akt, ↓NF-κB, ↓Bcl-2 | ↑Apoptosis, ↑sensitization to paclitaxel | 205 | |
| MCF10DCIS.com | ↓TNF-α, ↓MMP-2, ↓MMP-9, ↓MMP-13 | ↓Migration, ↓invasion | 206 | |
| MCF-7 | ↓Bcl-2, ↓COX-2 | ↑Apoptosis, ↓proliferation | 315 | |
| MCF-7 | ↓MMP-9, ↓NF-κB | ↓Invasion | 207 | |
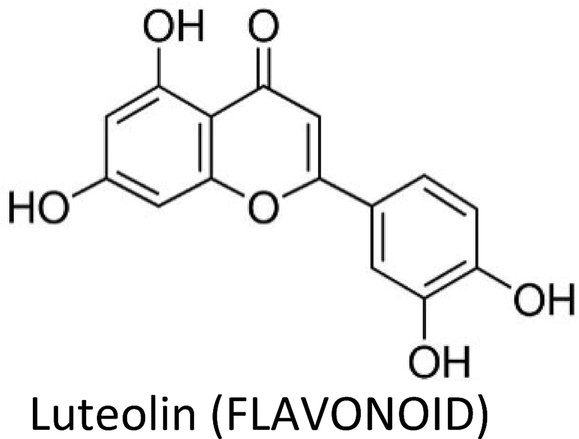
| Luteolin | MDA-MB-231 (4175) LM2 | ↓VEGF | ↓Angiogenesis, ↓lung metastasis | 274 |
| MCF-7 & MDA-MB-231 | ↓p-EGFR, ↓p-STAT3, ↓p-AKT, ↓p-ERK1/2 | ↓Proliferation | 316 | |
| BT-474 & T47D | ↓VEGF, ↓CD44, ↓ALDH | ↓Angiogenesis, ↓proliferation, ↑apoptosis | 317 | |
| MDA-MB-231, BT5-49 & female nude mice | ↓Vimentin, ↓slug, ↓β-catenin | ↓Lung metastasis | 275 | |
| MDA-MB-231 | ↓Notch signalling, ↓VEGF, ↓cyclin D1, ↓MMP-2, ↓MMP-9, ↓Hes-1 | ↓Migration, ↓angiogenesis, ↓cell survival | 318 | |
| MCF-7 (TAM-R) | ↓Cyclin E2 | ↓Tamoxifen resistance | 278 | |
| MDA-MB-231 | ↓AKT, ↓cyclin A, ↓PLK1, ↓CDC2, ↓CDK2,↓ cyclin B1, ↓Bcl-xL, ↑p21, ↑Bax, ↓EGFR, ↓MAPK | ↑Apoptosis, ↓proliferation | 319 | |
| MDA-MB-231, MCF-7 & SK-BR-3 | ↑ERK, ↑p38, ↑CASP-3, ↑c-PARP | ↑Apoptosis | 320 | |
| MCF-7 | ↓IGF-1, ↓Akt, ↓ERα | ↑Apoptosis, ↓proliferation | 321 | |
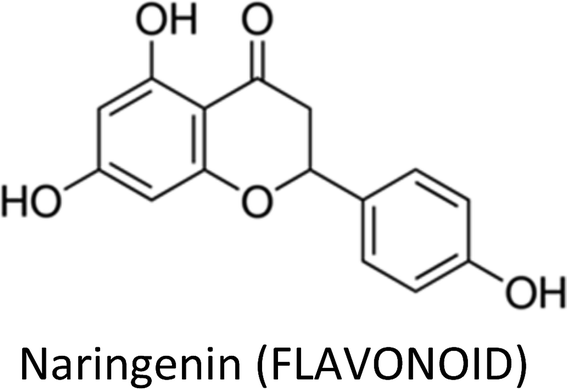
| Naringenin | MCF-7 (Tam-R) | ↓MAPK, CASP-7 | ↑Apoptosis, ↓proliferation | 322 |
| 4T1 | ↓TGF-β1, ↓PKC | ↓Pulmonary metastasis | 323 | |
| MDA-MB-231 | ↓Integrin β3, ↓MMP-2, ↓MMP-9 | ↓Migration & invasion | 324 | |
| HTB26 & HTB132 | ↓Bcl-2, ↓Cdk4, ↓Cdk6, Cdk7, ↓c-IAP-2, ↓x-IAP, ↑Bak, ↑Bax, ↑AIF, ↑CASP-3, ↑CASP-7, ↑CASP-8, ↑CASP-9, ↑p18, ↑p19, ↑p21, ↓pAkt, ↓P13K, ↓NFκB p65, ↓pIκBa | ↑Apoptosis, ↓proliferation, ↑chemo-sensitization | 325 | |
| E0771 | ↑AMPK, ↓Bcl-2, ↓cyclin D1 | ↑Apoptosis, ↓proliferation | 326 | |
| MDA-MB-231 | ↓Survivin, ↑p21, β-catenin | ↓Proliferation, ↑apoptosis | 339 | |
| MCF-7 | ↓P13K, ↓MAPK, ↓ERK1/2, ↓AKT, ↑CASP-7, ↑CASP-9 | ↑Apoptosis, ↓proliferation | 328 | |
| MCF-7, T47D & MDA-MB-231 | ↑CASP-3, ↓AKT, ↑p38 | ↑Apoptosis, ↓proliferation | 329 | |
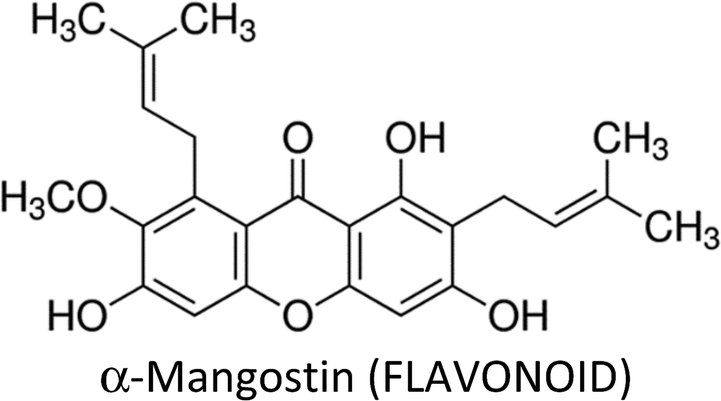
| α-Mangostin | T47D, MDA-MB-468, SKBR3 &AU565 | ↓Bcl-2, ↓Mcl-1, ↓P13K, ↓ERK1/2, ↓ERα, ↓HER2, ↓Akt, ↓ERK1/2, ↑p-p38, ↑p-JNK1/2, ↓MAPK | ↑Apoptosis, ↓proliferation, ↓colonization | 251 |
| MCF-7 & MDA-MB-231 | ↑p53, ↑Bax, ↑PARP cleavage, ↓Bcl-2, ↓Bid, ↓pS2, ↓ERα, ↑CASP-7, ↑CASP-8, ↑CASP-9 | ↑Apoptosis, ↓proliferation | 254 | |
| MCF-7 & MDA-MB-231 | ↓FASN, ↓FAK, ↓pAKT, ↑Bcl-2, ↓Bax, ↑p-ERK1/2 | ↑Apoptosis | 253 | |
| MDA-MB-231 | ↑CASP-3 | ↑Apoptosis, ↓proliferation | 252 | |
| MDA-MB-231 | ↑ p21cip1, ↑CASP-3, ↑CASP-8, ↑CASP-9, ↑CHEK2, ↓CDKs, ↓PCNA, ↓cdc(s) | ↑Apoptosis | 255 | |
| Thymoquinone | MCF-7, T47D & EMT6/p | ↓VEGF, ↑IFN-γ, ↑IL-4 | ↓Angiogenesis, ↑apoptosis, ↓proliferation | 118 |
| MCF-7 | ↓hsa04310 (Wnt), ↑hsa04115 (p53), ↓hsa04151 (P13K/AKT), ↓hsa04010 (MAPK) | ↑Apoptosis | 153 | |
| MCF-7 | ↑p53 | ↑Apoptosis | 331 | |
| EMT6/p | ↓VEGF, ↑IFN-γ, ↓IL-4, ↓AST, ↓ALT | ↑Apoptosis, ↓proliferation, ↑necrosis, ↓angiogenesis | 158 | |
| MCF-7 | ↑PTPRR, ↓MAPK, ↓p38-MAPK, ↑TGF-β, ↓TP53, ↓Bcl-2, ↓CARD16, ↓EGF-EGFR, ↓GPCR | ↑Apoptosis | 152 | |
| MCF-7 | ↑Bax, ↑p21, ↑Maspin, ↓Bcl-2, ↓HDAC | ↑Apoptosis, ↓proliferation, ↓migration | 155 | |
| 4T1, PMEF & Balb/c mice | ↑BRCA1, ↑p21, ↑HIC1, ↑CASP-3, ↑CASP-7, ↑CASP-12, ↑PARP, ↓p65, ↓p-Akt1 | ↑Apoptosis, ↓proliferation, ↓migration | 157 | |
| MCF7 & MDA-MB-231 | ↑TGF-β, ↑E-cadherin, ↑cytokeratin 19, ↓MMP-2, ↓MMP-9, ↓integrin αV, ↓snail, ↓Twist, ↓Smad2, ↓NF-κB | ↑Apoptosis, ↓proliferation, ↓migration, ↓invasion, ↓colonization, ↑sensitization to radiation | 154 | |
| MCF-7, T47D, MDA-MB-231 & MDA-MB-468 | ↓Cyclin D1, ↓cyclin E, ↓p27, ↓survivin, ↓Bcl-xl, ↓Bcl-2, ↑Bax, ↑PARP, ↑procaspase-3, ↑Cyt c, ↓Akt, ↑PTEN, ↓PDK1 | ↑Apoptosis, ↓proliferation, ↓viability | 332 | |
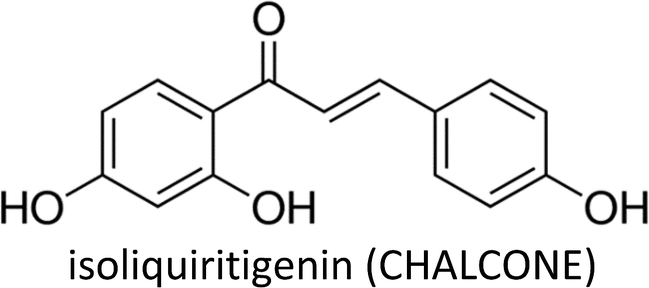
| Isoliquiritigenin | MCF-7, MDA-MB-231, 4T1, BT474, MCF-10A | ↓miR-374a, ↑PTEN, ↓Akt, ↑Bax, ↓Bcl-2, ↑c-CASP-9, ↑Cyt C, ↑MMP-7, ↓p-GSK3β, ↓β-catenin | ↑Apoptosis, ↓proliferation, ↓lung metastasis, ↓migration, ↓invasion | 181 |
| MDA-MB-231 & Hs-578T | ↓miR-21, ↓STAT3 | ↓Invasion | 184 | |
| MDA-MB-231 & Hs-578T | ↑RECK, ↓miR-21 | ↓Invasion | 185 | |
| MCF-7 & MDA-MB-231, MMTV-PyMT mice | ↓DNMT1, ↓β-catenin, ↑WIF1, ↓Wnt, ↓β-catenin, ↓Cyclin D1, ↓Survivin, ↓C-Myc, Oct-4 | ↓Proliferation, ↓lung metastasis, ↓initiation & progression, ↓bCSCs & its self-renewal | 187 | |
| BT549 & MDA-MB-231 | ↓COX-2, ↓CYP 4A, ↓PGE2, ↓PLA2, ↑cleaved caspase-3 & −9 | ↓Metastasis, ↑apoptosis, ↑anoikis | 333 | |
| BT549, MCF-7 & MDA-MB-231 | ↓β-catenin, ↓ABCG2, ↓GRP78, ↑proteasome degradation pathway, ↓CD44+CD24−/low, ↓Survivin, ↓Cyclin D1, ↓Oct-4, ↓c-Myc, ↓GSK-3β, | ↓Proliferation, ↓colonization, ↑apoptosis, ↓bCSCs self-renewal & differentiation, ↑chemosensitization | 188 | |
| MCF-7, MCF-7/ADR & MCF-10A | ↓miR-25, ↓ABCG2, ↑ULK1, ↑LC3-II | ↓proliferation, ↑chemosensitization, ↓colonization | 186 | |
| MDA-MB-231 | ↓VEGF, ↓HIF-1α, ↓MMP-2, ↓MMP-9, ↓p38, ↓Akt, ↓NF-κB, ↓P13K | ↓Migration, ↓proliferation, ↓angiogenesis | 48 | |
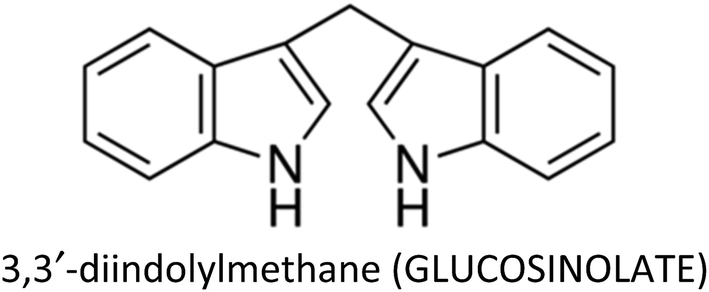
| 3,3′-Diindolylmethane | MCF-7 | ↓EMT, ↓CXCR4, ↓N-cadherin, ↑E-cadherin, ↓snail, ↓slug, ↓cathepsin B, ↓cathepsin D, ↓MMP-2, ↓MMP-9 | ↓Metastasis, ↓proliferation | 264 |
| MCF-7 & T47D | ↑p21 | ↓Proliferation | 334 | |
| MDA-MB-231 | ↓Akt | ↓Proliferation | 266 | |
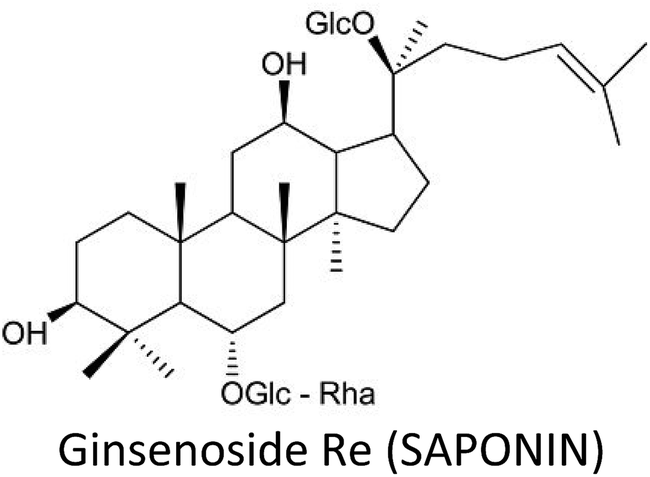
| Ginsenosides | MCF-7 | ↓CASP1, ↓INSL5, ↓OR52A1, ↑CLINT1, ↑ST3GAL4, ↑C1orf198 | ↓Proliferation, ↑apoptosis | 52 |
| MDA-MB-453, MDA-MB-231 & BT-549 | ↓NF-κB, ↓Bcl-2, ↑Bax, ↑CASP-3 | ↑Chemosensitization, ↑apoptosis, ↓proliferation | 215 | |
| T47D & BT-474 | ↑AMPK | ↓Proliferation | 218 | |
| MCF-7 | ↓MMP-2, ↓MMP-9, ↓mTOR, ↓Akt, ↓p62, ↓JNK, ↓P13K, ↓VEGFA, ↓VEGFB, ↓VEGFC, ↓Beclin-1, ↑LC3-II | ↓Angiogenesis, ↓invasion, ↑autophagy | 217 | |
| 4T1 | ↓miR-18a, ↓Smad2 | ↓Metastasis | 335 | |
| MCF-7 & MDA-MB-453 | ↑p21WAF1/cip1, ↑p53, ↑p15INK4B, ↓CDK4, ↓cyclin E2, ↓Cyclin D1, ↑CASP-6, ↑CASP-7, ↑CASP-8, ↑CASP-9, ↑p38 MAPKs, ↑Bax, ↑DR4, ↑DR5, ↓survivin, ↑PARP | ↓Proliferation, ↑apoptosis | 219 | |
| MDA-MB-231 | ↓NF-κB, ↓Akt, ↓ERK | ↓Proliferation, ↑apoptosis | 216 | |
| Benzyl isothiocyanate | MDA-MB-231, MCF-7, MDA-MB-468, T47D, HBL-100, Hs578t, BT474 | ↑ERK, ↑p53, ↑LKB1, ↑p73, ↓Ki-67, ↓survivin, ↓XIAP, ↑p-ERK, ↑CREB phosphorylation, ↓PRAS40 phosphorylation | ↓Proliferation, ↑apoptosis, ↓colonization | 39 |
| SUM159, MCF-7 & MDA-MB-231 | ↑KLF4 | ↓bCSCs | 336 | |
| SUM159, MDA-MB-231, MCF-7, & MDA-MB-361 | ↓BMI-1, ↓ALDH1, ↑Notch-4 | ↓bCSCs, ↑apoptosis, ↓migration | 162 | |
| MDA-MB-231, MCF-7 & SUM159 | ↓Ron, ↓sfRon, ↑SOX-2, ↑Nanog, ↑Oct-4, ↓ALDH1 | ↓bCSCs, ↓mammospheres | 161 | |
| MDA-MB-231, MCF-7 & SUM159 | ↑Notch2 | ↓Migration | 337 | |
| MCF-7, MDA-MB-231, MDA-MB-468, MCF-10A | ↑FOXO1, ↓p62, ↓mTOR, ↑LC3-II | ↑Autophagy, ↓viability | 338 | |
| MDA-MB-231 | ↓uPA, ↑PAI-1, ↓NF-κB, ↓p-Akt, ↓c-Met phosphorylation | ↓Migration & invasion | 160 | |
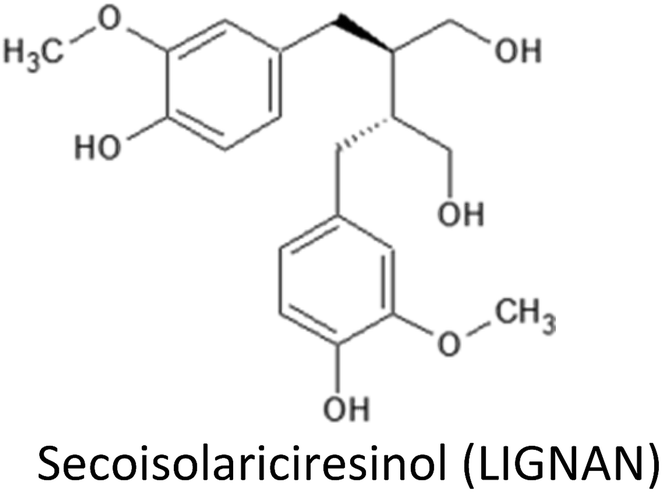
| Secoisolariciresinol | MCF-7 | ↑Erk1/2, ↑PI3K/Akt expression | Modulate estradiol effect (possibly ↓cell proliferation) | 221 |
| MCF-7 | ↓ERα and PR expression, ↑estradiol secretion | Modulate estradiol effect (possibly ↓cell proliferation) | 222 | |
| MCF-7 | Opposite action of ENL and END on telomerase activity | Possible opposite effect on cell division | 223 | |
| MDA-MB-231 | ↓uPA-induced plasmin activation ↓MMP-2 and MMP-9 (ECM-mediated remodelling) | ↓Proliferation, ↓migration, ↓metastasis | 225 | |
| MDA-MB-231 | ↓Ki-67, PCNA & FoxM1 gene expression ↓cyclin A2, B1, B2 & E1 gene expression ↓phosphorylation of FAK/paxillin pathway | ↓Proliferation | 224 | |
| SKBR3, MDA-MB-231 | ↑Cytotoxic effects of chemotherapeutic agents ↓FASN expression and activity | Improve efficiency of chemotherapeutic agents | 220 | |
| MCF7, MDA-MB-231 | ↑Cytotoxic effects of chemotherapeutic agents ↓GSTP1 expression and activity | ↓Cell growth, ↑apoptosis improve efficiency of chemotherapeutic agents | 232 | |
| Animal models (ACI rats) | ↓Ki-67 gene expression ↓SHBG & IGFBP-3 serum levels, ↑ESR2 (ERβ) | ↓Tumor progression and burden ↓cell proliferation & disphasia | 226 |
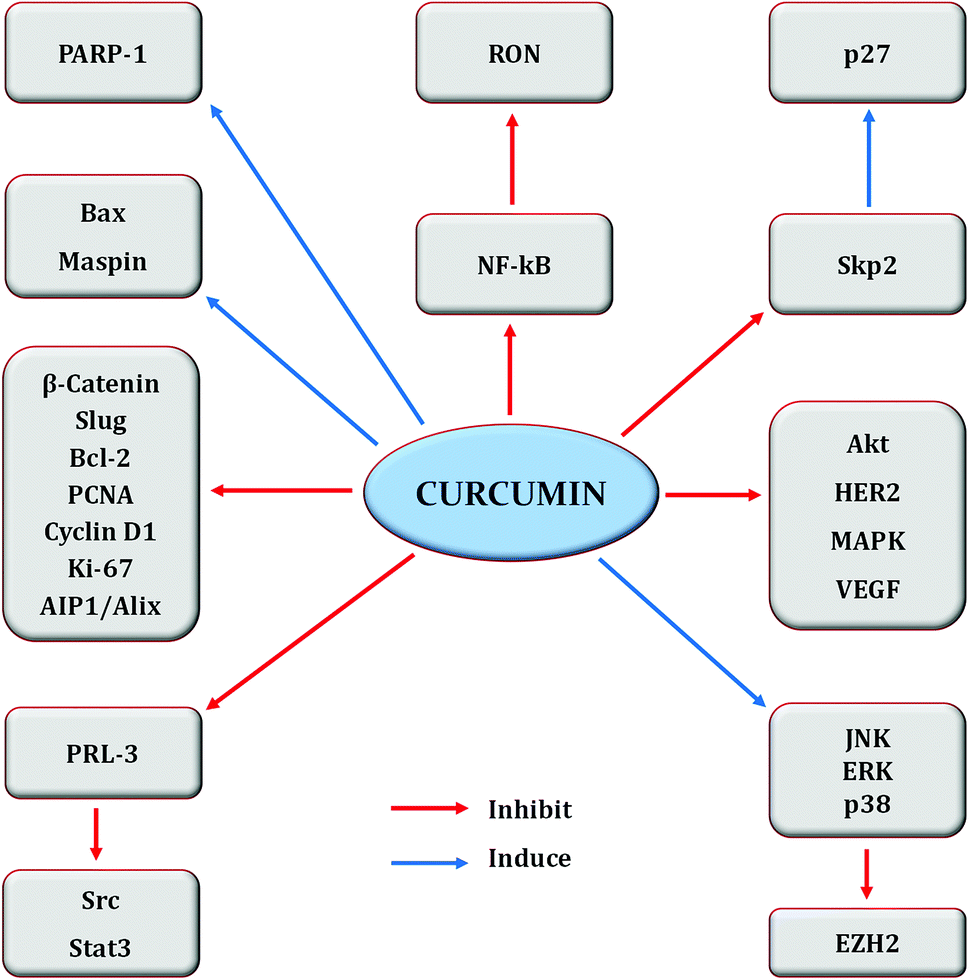 | ||
| Fig. 6 Regulation of molecular targets by curcumin in breast cancer cells. Idea adopted from Liu and Chen (2013).396 | ||
Recently, curcumin was shown to induce apoptosis in MCF-7 cells through increased caspase-3 and caspase-9 expressions. Additionally, this compound suppressed miR-21 expression via up-regulation of PTEN/Akt signalling.66 Curcumin also reduced the proliferation of MCF-7 cells by decreasing nitric oxide and ROS levels. Synergistic anti-proliferative effect was observed when ECM proteins: fibronectin or collagen were used along with curcumin.67 Moreover, the compound upregulated Bax and downregulated Bcl-2 expressions in MDA-MB-231 cells, leading to growth inhibition and apoptosis induction.68 Fatty acid synthase inhibition by curcumin also resulted in breast cancer cells apoptosis.69 Curcumin downregulated flap endonuclease 1 (DNA repair enzyme) expression via Nrf2 stimulation thereby reduced the proliferation of MCF-7 cells.70
Similarly, curcumin inhibited the nuclear translocation of β-catenin, hence impeded the trans-activation of slug and subsequent restoration of E-cadherin which led to increased formation of E-cadherin/β-catenin complex and more β-catenin in cytosol to finally suppress bCSCs migration and EMT.71 Recently, hypomethylation reactivation of tumor suppressors by curcumin has also been presented. The compound reversed the hypermethylation status of Glutathione S-Transferase Pi 1 (GSTP1) gene in MCF-7 cells, leading to its reactivation.72 Likewise, it also activated Deleted in Liver Cancer 1 (DLC1) promoter by decreasing its methylation level via down-regulation of Sp1 transcription factor to constrain the expression of DNA methyltransferase 1 (DNMT1) in MDA-MB-361 cells.73 Curcumin was also shown to decrease the methylation status of tumor suppressor termed as Ras-association domain family protein 1A (RASSF1A), thereby activating it. This was achieved through interference of curcumin with DNMT1.74
Co-treatment of curcumin with other drugs/phytochemicals have greatly enhanced their therapeutic efficacy in various malignancies, including breast cancer. Recently, the combined effect of curcumin and metformin (oral anti-diabetic drug) was explored in breast cancer cell lines which markedly suppressed VEGF expression, activated Th2 immune response and induced Trp53-independent apoptosis.75 Similarly, arabinogalactan—a polysaccharide obtained from larch wood—in combination with curcumin induced in vivo and in vitro apoptosis in breast cancer cells by increasing reactive oxygen species (ROS), cleaved-caspase-3 and Bax/Bcl-2 ratio. Reduced glutathione, p53 overexpression and a change in mitochondrial membrane were also observed.76,77 Curcumin synergistically enhanced the effects of retinoic acid (RA) and IFN-β on breast cancer cells by upregulating GRIM-19 (cell death regulatory gene) via STAT3-dependent and- independent pathways.78 RA induction of apoptosis via CRABPII/RAR signalling pathway has been reported and curcumin was recently shown to upregulate CRABPII and RAR thus overcoming RA-resistance in TNBC cell lines.79 Similarly, curcumin sensitized MDR breast cancer cells to cisplatin via downregulation of CCAT1 and inactivation of P13K/Akt/mTOR pathway.80 Moreover, curcumin chemosensitized breast cancer cells to 5-fluorocuracil (drug which exerts its action by inhibiting thymidylate synthase (TS)) through TS-dependent downregulation of NF-κB.81
Curcumin inhibited the invasion and adhesion of MCF-7 cells by reducing the expression of uPA via activation of NF-κB.82 Furthermore, the compound suppressed lysophosphatidic acid-induced invasion and metastasis in MCF-7 cells by interfering with RhoA/ROCK/MMPs pathway.83 Similarly, inhibition of angiogenesis and tumor growth through downregulation of NF-κB and its related gene products like PECAM-1, p65 and cyclin D1 by curcumin in MDA-MB-231 cells has also been reported.84 Curcumin upregulated p16INK4A and several other tumor suppressors, inhibited JAK2 and STAT3 pathways, leading to decreased α-smooth muscle actin and invasion/migration abilities of breast cancer-associated fibroblasts. Curcumin also suppressed lamin B1 and triggered p16INK4A-dependent senescence in these fibroblasts.85 Matrix metalloproteinases (MMPs) possess a significant part in remodelling of the extracellular matrix. Tissue inhibitor of metalloproteinases (TIMPs) regulate the activities of MMPs. Curcumin regulated cell metastasis by up-regulating expression of TIMP1/4 genes and inhibiting MMP-2 and MMP-9 in breast cancer cells.86 Mo et al. (2012) revealed that curcumin inhibit invasion of MDA-MB-231 cells via reduction of p-ERK and Smad2 signalling.87 The compound also affected genes related to Epithelial–Mesenchymal Transition (EMT), leading to reduced expression of β-catenin, N-cadherin, E-cadherin, Twist1, slug, AXL, fibronectin and vimentin, subsequently inhibiting migration and invasion in breast cancer cells.88 Other pathways and genes modulated by curcumin are presented in Table 2.
3.2. Epigallocatechin gallate (EGCG)
EGCG—a polyphenol and predominant catechin in green tea (Camellia sinensis)—has long been studied for its health benefits, including cancer chemoprevention. Sinha et al. (2017) have recently reviewed the role of green tea and its constituents in breast cancer chemoprevention. EGCG induced anti-proliferative, anti-metastatic, apoptotic, anti-angiogenic, anti-genotoxic and epigenetic effects by targeting several pathways and genes.36 There is also evidence that topical EGCG prevented radiation-induced dermatitis in breast cancer patients, thus making it a radio-protective agent.89–91EGCG induced apoptosis through downregulation of telomerase and P13K/AKT and increased Bax/Bcl-2 ratio and p53 expression in T47D cells. The expression of hTERT gene was decreased while that of CASP3, CASP9 and PTEN was increased.92 Huang et al. (2017) also observed an increase in p53 and decrease in Bcl-2 in MCF-7 cells.93 Moreover, EGCG suppressed the growth of MDA-MB-231 cells through downregulation of β-catenin, cyclin D1 and pAkt.94 In tumor microenvironment, tumor-associated macrophages (TAM) carry a significant role. EGCG reduced TAM infiltration and M2 macrophage polarization by upregulating miR-16, thereby decreased tumor growth in murine breast cancer model.95
EGCG suppressed heat shock protein 90 (Hsp90) and promoted the translocation of progesterone receptor B (PR-B) into the nucleus, consequently resulting in downregulation of ERα.96 Physiological concentrations of EGCG inhibited the growth of ERα-positive MCF-7 cells through reduction in ERα and insulin-like growth factor (IGF) binding protein 2 and increased p53 and p21 (tumour suppressors).97 Furthermore, the compound downregulated expression of ER-α36 and thus suppressed the growth of ER-negative breast cancer stem/progenitor cells.98
EGCG sensitized MCF-7 cells to 5-fluorouracil by regulating the expression of Bcl-xL.99 The compound also achieved sensitization to tamoxifen by reducing the expression of Nrf2 (transcription factor) in tamoxifen-resistant MCF-7 cells.100 Similarly, a reduction in MMP-2 and MMP-9 in doxorubicin resistant MCF-7 cells has also been observed.101
3.3. Genistein
Genistein—a phytoestrogen in soybeans—primarily exerts its function by interacting with estrogen receptors (ERα & ERβ).102 Varinska et al. (2015) reviewed the molecular targets regulated by genistein in cancer cells, with special preference to angiogenesis (Fig. 7).397 Genistein repressed the differentiation and proliferation of 3T3-L1 and MCF-7 cells by regulating the expression of ERα.103 It also reactivated ERα expression by remodelling its promoter chromatin structure and inhibited the growth of ERα-negative breast cancer cells. This epigenetic restoration of ERα further enhanced the therapeutic efficacy of tamoxifen.104 Additionally, the compound reduced the expression of ERα and c-erB-2, consequently inhibiting the proliferation of ERα/c-erB-2-positive breast cancer cells.105 Genistein also induced apoptosis in MCF-7 and T47D cells via downregulation of CIP2A (cancerous inhibitor of PP2A).106 A phosphoproteomic level study using TNBC cells revealed that several biological processes are regulated by genistein during cell cycle such as kinetochore formation, cohesion complex cleavage and DNA replication. Moreover, DNA damage response involving BRCA1 complex and ATR can also be activated by genistein.107 | ||
| Fig. 7 Molecular targets regulated by genistein in cancer cells. Downward arrows (↓) represent decrease of activity/expression/secretion while upward arrows (↑) represent increase of activity/expression/secretion. Idea adopted from Varinska et al. (2015).397 | ||
Genistein resulted in reactivation of several tumor suppressor genes such as adenomatous polyposis coli (APC), mammary serpin peptidase inhibitor (SERPINB5), ataxia telangiectasia mutated (ATM) and PTEN in breast cancer cell lines by decreasing their methylation status. This was accomplished through reduced expression of DNMT1 by genistein.108 Moreover, it also suppressed cyclin D1 and DNMT-1 in MCF-7 cells, leading to BRCA1 CpG demethylation and consequent reactivation of BRCA1.40 Prepubertal exposure to Bisphenol A (BPA) changes the signalling pathways which may add to carcinogenesis, as revealed by DNA methylation studies. Conversely, prepubertal exposure to genistein or genistein + BPA resulted in hypomethylation of several genes in rat mammary tissues out of which HPSE and RPS9 genes were linked with enhanced long term survival, thus emphasizing cancer preventive properties of genistein.109 In another study, lifetime genistein intake improved response of mammary tumors to tamoxifen resistance in Sprague-Dawley rats and also reduced the risk of recurrence. Genistein administration downregulated the unfolded protein response (UPR) and genes linked to autophagy such as ATF4, GRP78, Beclin-1 and IRE1α, as well as, immunosuppression-linked genes like Foxp3 and TGFβ and upregulated CD8a (cytotoxic T-cell marker) in tumors. This demonstrates that the enhanced response to endocrine therapy was pre-programmed early in life.110
Avci et al. (2015) found that genistein suppress the growth of MCF-7 cells via upregulation of miR-23b.111 Furthermore, it inhibited NF-κB via Notch-1 signalling and reduced the growth of TNBC cells.111 Long-term low-dose genistein administration sensitized inflammatory breast cancer cell lines to radiation and reduced mammosphere formation and the growth of stem cell populations.112 Downregulation of Hedgehog–Gli1 signalling by genistein is also linked with reduced bCSCs.113
3.4. Resveratrol
Resveratrol (RSV)—a phytoestrogen mainly produced by grapes, berries and peanuts in response to stress conditions—has also been explored well for its anti-malignant properties. Sinha et al. (2016) comprehensively reviewed the effect of RSV on breast cancer cells.398 RSV regulate various molecular targets underlying breast cancer cells proliferation, apoptosis, EMT/metastasis, as well as, epigenetic responses and sensitization to chemotherapy (Fig. 8).398 Recently, RSV was found to induce the expression of ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 3 (ATP2A3)—gene encoding enzyme which maintains homeostasis of intracellular Ca2+ by pumping it into endoplasmic reticulum (ER)—in breast cancer cells, thereby induced apoptosis, reduced cell viability and caused alteration in cytosolic levels of Ca2+ along with its releasing capacity by ER.114 RSV induced apoptosis in human epidermal growth factor receptor 2 (HER2)-positive SKBR-3 cells via suppression of fatty acid synthase and HER2 genes. The compound also up-regulated PTEN expression and down-regulated Akt phosphorylation (pAkt), thereby alleviated P13K/Akt/mTOR pathway.115 RSV prevented breast cancer cell invasion by directly inactivating RhoA which in turn phosphorylated YAP via Lats1 activation, leading to reduced expression of YAP gene.116 Moreover, it downregulated the polo-like kinase-1 and aurora kinase A (cell cycle regulatory proteins), cyclin D1 and cyclin B1, thereby suppressed cell cycle in breast cancer cells.117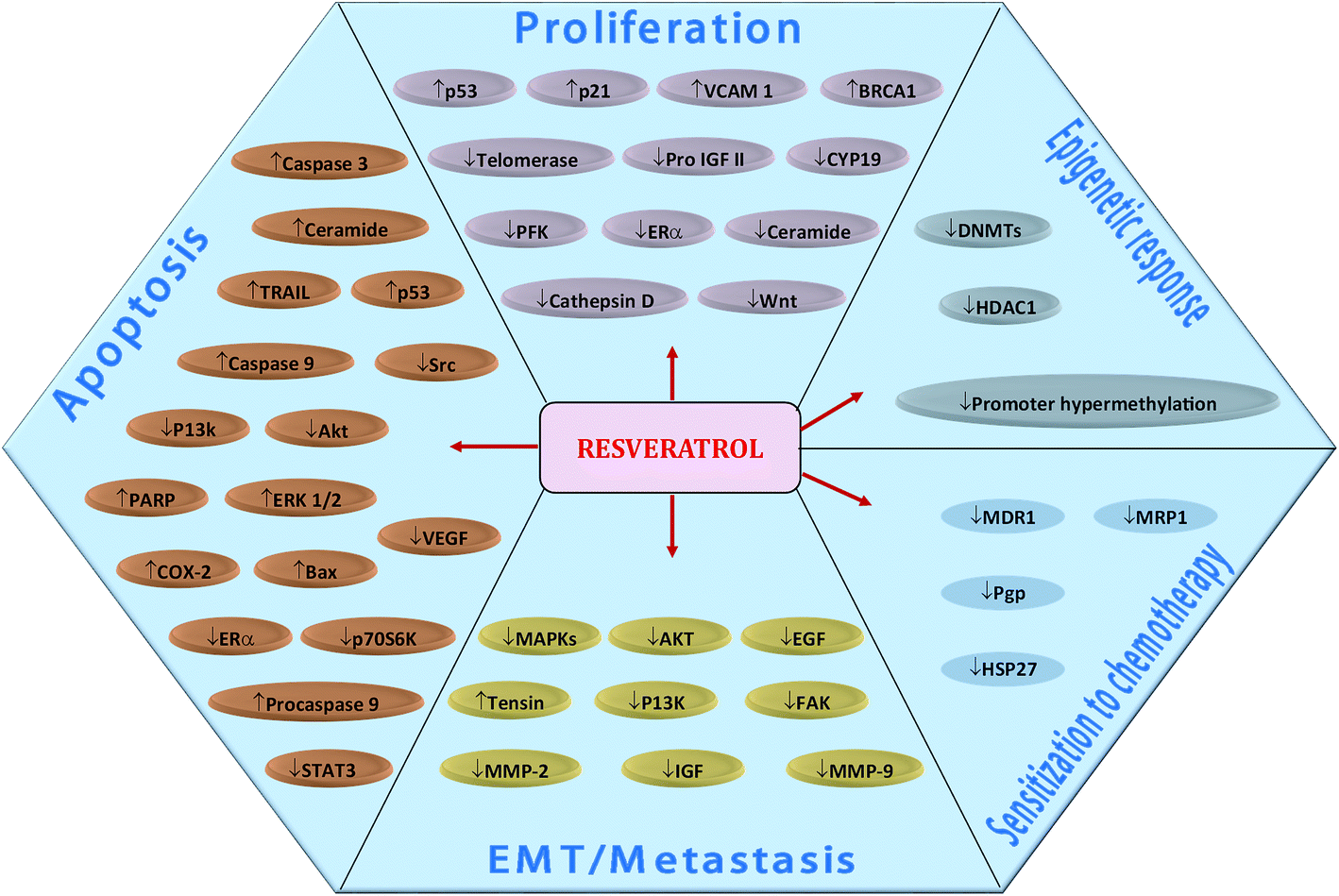 | ||
| Fig. 8 Resveratrol-mediated regulation of molecular targets underlying breast cancer cells proliferation, apoptosis, EMT/metastasis, epigenetic responses and sensitization to chemotherapy. Downward arrows (↓) represent downregulation while upward arrows (↑) represent upregulation. Idea adopted from Sinha et al. (2016).398 | ||
Combination of RSV with other compounds greatly improved its efficacy and bioavailability. Co-treatment of thymoquinone and RSV increased apoptosis, induced geographic necrosis, reduced VEGF and increased serum levels of IFN-γ in breast cancer cell lines.118 Likewise, combination with salinomycin markedly enhanced the anti-cancer properties of RSV in breast cancer cells via downregulation of canonical Wnt signalling proteins and vimentin.119 Similarly, RSV and pterostilbene reactivated the expression of ERα in ERα-negative breast cancer cell lines by increasing active chromatin markers such as acetyl-H3lysine9 and acetyl-H3/4 in ERα promoter region. Reduction in the activity of DNMT and level of 5-methylcytosine along with alteration in histone acetyl transferase and HDAC were also noted.120 Enhanced apoptosis of MCF-7 cells occurred when Sorafenib (drug inhibitor of angiogenesis and tyrosine kinase) was used in combination with RSV. The treatment induced the expression of p53, enhanced intracellular ROS generation and Bax/Bcl-2 expressions while reduced mitochondrial membrane potential. Moreover, downregulation of cyclin B1 and cyclin D1 also occurred while cleaved caspase-3, -9, cleaved poly(ADP-ribose) polymerase (PARP) and apaf-1 were found to be upregulated.121 RSV and rapamycin prevented mTORC1 (mechanistic target of rapamycin), autophagy and Akt activation, consequently instigating apoptosis in breast cancer cells.122 Co-treatment with melatonin inhibited aromatase, thus making them suitable for hormonal treatment of breast cancer.123 RSV also sensitized resistant MCF-7/DOX cells to doxorubicin via downregulation of MDR1.124 Furthermore, the compound enhanced the cellular accumulation of doxorubicin by reducing ABC transporter genes, MRP1 and MDR1 in resistant breast cancer cells.54 In another study, down-regulation of HSP27 by RSV has been linked with sensitization to doxorubicin in resistant MCF-7 cells.125 Similarly, RSV chemosensitized resistant MCF-7 cells to tamoxifen by modulating Smad phosphorylation and endogenous TGF-β production,126 and to melphalan by arresting cell cycle at S phase.127
Resveratrol modulation of microRNAs has also been observed. Venkatadri et al. (2016) demonstrated that the compound modulate crucial tumor-suppressive microRNAs such as miR-122-5p, miR-125b-5p, miR-409-3p, miR-542-3p and miR-200c-3p in breast cancer cells. These microRNAs regulate key cell cycle and anti-apoptotic proteins like X-linked inhibitor of apoptosis protein, Bcl-2 and CDKs.128
3.5. Quercetin
Quercetin—a well-researched and potential chemopreventive agent ubiquitous in plant foods—suppressed the growth of T47D cells by inhibiting the gene expression and secretion of leptin, thus making leptin a novel target in breast cancer therapy.129 The compound increased Bax and decreased Bcl-2 proteins expressions in MCF-7 cells. In the presence of Nec-1 (necroptosis inhibitor), the expression of Bax and apoptotic index decreased and an increase in proliferation and viability of MCF-7 cells occurred, suggesting that the death pathway mainly involve necroptosis.130 Additionally, quercetin induced apoptosis in TNBC cells by reducing the expressions of β-catenin and fatty acid synthase (FASN).44 Similarly, the compound displayed apoptotic activity in MCF-7 cells by inhibiting Twist through p38MAPK pathway.131 Likewise, anti-proliferative and apoptotic activity in MCF-7 cells via reduced survivin expression and Go/G1-phase arrest was also reported.132Combination therapy of quercetin and ascorbic acid (vitamin C) along with therapeutic drugs such as paclitaxel or doxorubicin markedly enhanced their anti-cancer properties in breast cancer cells by significant reduction in S and Go/G1 phases.133 Quercetin downregulated Her-2 and upregulated ERα, thereby reversed tamoxifen resistance in MCF-7 cells.134 Pentagalloylglucose (5GG), which shows structural resemblance to (−)-EGCG, in combination with quercetin persuaded S-phase arrest and caused apoptosis in MDA-MB-231 cells via decreased expression of S-phase kinase, while G2/M-phase arrest and apoptosis in AU565 cells occurred through reduced expression of Her-2.135
Quercetin revealed anti-metastatic property in TNBC cells by modulating EMT markers. It increased E-cadherin and decreased vimentin, leading to mesenchymal-to-epithelial transition, associated with modulation of cyclin D1 and c-Myc (β-catenin target genes) and a change in β-catenin nuclear localization.136 The calcineurin/NFAT pathway play a key role in angiogenesis which was inhibited by quercetin in breast cancer xenograft via downregulation of VEGF, VEGFR2 and NFATc3.137 Quercetin up-regulated miR-146a and prevented proliferation in breast cancer cells. The compound also inhibited invasion via EGFR suppression and induced mitochondrial-mediated apoptosis.138
3.6. Silibinin
Silibinin—a main active constituent of silymarin complex extracted from Silybum marianum—has a potent hepatoprotective activity.139 The compound also displayed anticancer properties and prevented breast cancer cells metastasis through inhibition of CXCR4 (chemokine receptor type 4).140 Moreover, it suppressed the migration of MCF-7 cells and markedly reduced MMP-9 expression via inhibition of p-ERK and p-MEK.141 It also inhibited proliferation and induced apoptosis in ERα-negative SKBR3 cells via suppression of NF-κB.142 Silibinin decreased p53, p21, Bcl-xL, Bak and BRCA1 in MCF-7 cells, thereby induced apoptosis and prevented proliferation.143 Furthermore, it conveyed anti-tumorigenic property in TNBC xenograft model by inhibiting the phosphorylation of EGFR and subsequently suppressing VEGF, MMP-9 and COX-2.144Co-treatment of silibinin with cisplatin or paclitaxel increased early apoptosis by decreasing Bcl-2 and increasing mRNA levels of Bax, ATM, BRCA1 and p53 in MCF-7 cells.145 Silibinin also sensitized resistant breast cancer cells to paclitaxel and doxorubicin through inhibition of the key oncogenic pathways encompassing ERK, AKT and STAT3 in doxorubicin-resistant MDA-MB-435 cells and paclitaxel-resistant MCF-7 cells at 400 μM concentration.146 Similarly, silibinin and chrysin synergistically inhibited proliferation of T47D cells via downregulation of cyclin D1 and hTERT.147
Silibinin reduced proliferation of MCF-7 cells via down-regulation of miR-155 and miR-21 and induced apoptosis through intrinsic and extrinsic pathways by upregulating their apoptotic targets such as BID and CASP-9.148 However, Jahanafrooz et al. (2017) presented that down-regulation of miR-21 upon silibinin treatment has minimal effect on its anti-tumorigenic property in breast cancer cells and that other pathways are responsible for its anti-apoptotic affect.149
Silibinin exerted autophagic cell death in MCF-7 cells via ROS-dependent mitochondrial dysfunction (ΔΨm) and a reduction in ATP levels involving BNIP3 (Bcl-2 interacting protein 3, a member of pro-death Bcl-2 protein family).150 However, according to Zheng et al. (2015), the anti-apoptotic and autophagy induction properties of silibinin in ERα-positive MCF-7 cells are due to down-regulation of ERα expression and subsequent inhibition of ERK and mTOR signalling pathways.151
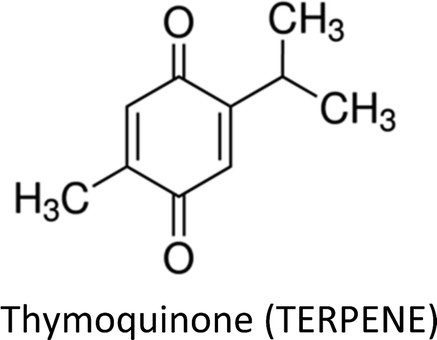
3.7. Thymoquinone
Thymoquinone (TQ)—an anti-inflammatory, antioxidant, anti-cancerous and cytotoxic constituent in black seed oil—has also been investigated in breast cancer therapy. The compound led to differential expression of apoptosis related genes in ER-positive (MCF-7) cells. Up-regulation of PTPRR gene resulted in inhibition of p38-MAPK and MAPK pathways. Inhibition of p38-MAPK triggered down-regulation of Bcl-2 and up-regulation of TP53 which indicates that intrinsic apoptotic pathway is involved. Moreover, participation of caspase in apoptosis was confirmed through downregulation of CARD16 gene.152 Recently, the effect of TQ on miRNA profile and molecular mechanism using MCF-7 cells was highlighted and it was found that hsa04310 (Wnt), hsa04115 (p53), hsa04151 (P13K/AKT) and hsa04010 (MAPK) are the key targets of the compound.153 TQ pre-sensitization restored cytokeratin 19 and E-cadherin (epithelial markers), as well as, MMP-2, MMP-9, integrin-αV (mesenchymal markers) and TGF-β in radiation-induced metastatic progression of breast cancer.154 TQ also exerted anticancer action in breast cancer cells by attenuating the activity of global histone deacetylase (HDAC). The compound increased Bax, decreased Bcl-2, reactivated HDAC target genes (maspin and p21) and induced G2/M phase arrest.155TQ and tamoxifen synergistically induced apoptosis and decreased cell viability in both estrogen positive (MCF-7) and estrogen negative (MDA-MB-231) cell lines.156 The compound sensitized breast cancer cells to paclitaxel through multiple cascades involving extrinsic apoptosis, p53 signalling and tumor suppressor genes. Moreover, TQ regulated apoptosis inducible genes through death receptors and upregulated tumor suppressor genes including BRCA1, p21 and Hic1. Additionally, high doses of TQ downregulated pro-apoptotic factors like caspases and upregulated growth factors such as EGF and VEGF.157 Likewise, co-treatment of thymoquinone and melatonin greatly decreased tumor size via induction of apoptosis, activation of Th1 immune response and inhibition of angiogenesis.158
3.8. Benzyl isothiocyanate
Benzyl isothiocyanate (BITC)—an isothiocyanate from cruciferous vegetables—exhibit anti-carcinogenic effect. BITC induced apoptosis in breast cancer cells by altering mitochondrial dynamics. The compound greatly reduced levels of Drp1, p-S616, Fis1 (fission proteins) and Mfn1, Mf2 (fusion proteins) in breast cancer cells, suggesting that it negatively target the fission and fusion machinery of mitochondrial dynamics. Furthermore, it was shown that multi-domain pro-apoptotic proteins Bak and Bax regulated the BITC-mediated inhibition of mitochondrial fusion.159 BITC triggered p53-signalling network and repressed p53-mutant cells growth. The compound induced expression of p73 in these mutant cells, interrupting the interaction of mutant-p53 and p73 and subsequently emancipating p73 from sequestration and permitting it to be active transcriptionally. In molecular mechanisms, liver kinase B1 (LKB1), a tumour suppressor, was found to be a key node underlying the anti-cancer role of the compound. The p73 and p53 transcriptionally upregulated LKB1 in a mutant- and wild-type-p53 breast cancer cells, respectively. Furthermore, it was elucidated that LKB1 tethers with p73 and p53 in a feed-forward mechanism for recruitment into p53-responsive gene promoters.39 BITC also suppressed the migration and invasion of MDA-MB-231 cells through decreased uPA activity and reduction of p-Akt.160Truncated Recepteur d'Origine Nantais (sfRON) was also found as a novel mechanistic target for induction of apoptosis in breast cancer cells. BITC induced apoptosis in MDA-MB-361 and MCF-7 cells by regulating sfRON, whereby overexpression of sfRON in these cells intensified apoptosis, independent of JNK or MAPK hyperphosphorylation. Moreover, activation of Bak and Bax in sfRON overexpressing cells following BITC treatment were greatly enhanced while G2/M phase arrest and ROS generation were attenuated.159 However, according to Kim et al. (2013), full-length RON and sfRON overexpression offered protection against inhibition of bCSCs by BITC in MCF-7 cells and down-regulation of RON and its truncated form are associated with breast cancer stem cells inhibition by BITC.161 BITC inhibited bCSCs via downregulation of oncogene BMI-1 (Polycomb complex protein) and activation of Notch-4 in in vivo and in vitro breast cancer cells.162
3.9. Apigenin
Apigenin—a well-known anti-inflammatory flavonoid in parsley and variety of other plants—suppressed TNFα-mediated release of chemokines including CCL2, IL-1α, IL-6, and granulocyte macrophage colony-stimulating factor in TNBC cells. These chemokines direct the inward migration/infiltration of tumor-associated neutrophils, T-regulatory cells, tumor-associated macrophages, T-helper IL-17-producing cells, myeloid-derived suppressor cells, cancer-associated fibroblasts and metastasis-associated macrophages, which collectively enables tumor growth, immune evasion, metastasis and angiogenesis. The inhibition of CCL2 by apigenin occurred through suppression of IKBKe signalling.163–172 Apigenin overcame drug resistance and decreased colonization and cell growth in adriamycin-resistant MCF-7 cells via downregulation of MDR1 and P-glycoprotein, as well as, suppression of STAT3 signalling and its nuclear translocation. The compound also diminished the secretion of MMP-9 and VEGF (STAT3 target genes) in these cells.173Moreover, apigenin inhibited the proliferation and clonogenic survival of HER2-expressing BT-474 cells via caspase-dependent extrinsic apoptosis by upregulating cleaved caspase-3, cleaved caspase-8 and PARP cleavage. The compound decreased phospho-STAT3, phospho-JAK1 and phospho-JAK2 and prevented CoCl2-induced VEGF secretion and STAT3 nuclear translocation in HER2-expressing breast cancer cells.174 The same authors also mentioned before that apigenin induce apoptosis in HER2-overexpressing MCF-7 cells via extrinsic pathway, inhibit NF-κB and STAT3 and induce p53.175
Mechanism behind induction of cell cycle arrest by apigenin in MDA-MB-231 cells was also pointed out and it was shown that the compound suppress cyclin dependent kinase-1 (CDK1), cyclin A and cyclin B which are vital for G2-to-M-phase transition in cell cycle. Furthermore, apigenin increased p21WAF1/CIP1 and its interaction with proliferating cell nuclear antigen, thus obstructing cell cycle progression. Moreover, inhibition of histone deacetylase activity and an increase in acetylated histone H3 was also observed.176 Harrison et al. (2014) reported that apigenin reduce the proliferation of MDA-MB-468 cells by arresting cell cycle at G2/M phase and enhancing ROS production. Additionally, the compound reduced p-Akt.177
3.10. Isoliquiritigenin
Isoliquiritigenin (ISL)—a flavonoid mostly found in the root of liquorice—exhibits anti-cancer properties at multistage carcinogenesis processes such as cell cycle arrest, proliferation suppression, apoptosis induction, metastasis obstruction and angiogenesis inhibition in different kinds of cancer.178–183Peng et al. (2017) proposed that ISL induces apoptosis and prevent metastasis in breast cancer cells via downregulation of miR-374a. Decreased expression of miR-374a led to increased PTEN expression which prevented abnormal Akt signalling.181 ISL transcriptionally downregulated the expression of primary and mature miR-21 and decreased STAT3 signalling activity in breast cancer cells.184 The compound also suppressed breast cancer cells invasion by upregulating RECK (tumor suppressor gene) and downregulating miR-21.185 Moreover, ISL caused cell cycle arrest, chemosensitized and induced autophagy in MCF-7/ADR cells and also stimulated the degradation of ABCG2 through autophagy-lysosome pathway. Autophagy induction was associated with inhibition of miR-25 which led to increased expression of ULK1 (a kinase involved in autophagy).186 ISL treatment inhibited NF-κB, P13K/Akt and p38, ensuing decreased expressions of MMP-2, MMP-9, VEGF and HIF-1α which reduced the migration of breast cancer cells.48
ISL encouraged the demethylation of WIF1 promoter by docking into the catalytic domain of DNMT1, thereby increasing the gene expression of WIF1 and subsequently prevented mammary carcinogenesis by inhibiting bCSCs.187 The compound also chemosensitized bCSCs by inhibiting β-catenin/ABCG2 signalling via docking into ATP domain of GRP78 (binding immunoglobulin protein), resulting in inhibition of its ATPase activity and subsequent dissociation from β-catenin.188
3.11. Pterostilbene
Pterostilbene—resveratrol analogue extracted from blueberries—markedly inhibited the growth and induced apoptosis in TNBC cells through G0/G1 phase arrest. The compound maintained the activation of ERK1/2, associated with decreased cyclin D1 and increased p21 expressions. Moreover, it also repressed p-AKT and mTOR and a subsequently increased Bax protein without affecting Bcl-xL.189 Pterostilbene enhanced TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in TNBC cells via ROS-mediated activation of p38/C/EBP-homologous protein pathways which subsequently induced the expression of death receptors DR4 and DR5. The compound also reduced the expression of decoy receptor 1 and decoy receptor 2 and suppressed the anti-apoptotic proteins Bcl-2, Bcl-xL, XIAP, c-FLIPS/L and survivin, as well as, increased Bax and Bid cleavage in these cells.190Pterostilbene inhibited metastasis of TNBC cells through induction of miR-205 and negative modulation of EMT markers including vimentin, ZEB1, slug and snail and upregulation of E-cadherin. Downregulation of miR-205 caused decreased expression of Src—a nonreceptor tyrosine kinase whose activation and overexpression leads to different types of cancers, involving invasive breast cancer.191–194
Interestingly, pterostilbene and 6-shogaol inhibited bCSCs via reduced expression of CD44+ surface antigen and stimulated phosphorylation of β-catenin through hedgehog/Akt/GSK3β signalling inhibition thus downregulating downstream cyclin D1 and c-Myc.195 The occurrence of M2-polarized tumour associated macrophages (TAMs) enhanced metastatic abilities and CD44+/CD− CSCs in breast cancer cells. M2 TAMs-associated properties were suppressed by pterostilbene via inhibition of NF-κB, E-cadherin, vimentin and Twist1.196
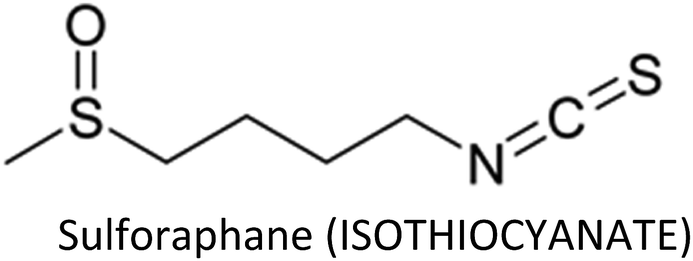
3.12. Sulforaphane
Sulforaphane (SFN)—an antioxidative and anti-inflammatory compound found in cruciferous vegetables—modulates various cellular targets that are involved in cancer development.197–203 SFN stimulated the anti-cancer activity of taxane (docetaxel or paclitaxil) in TNBC cells by preventing CSCs via reduced translocation of NF-κB p65 and decreased p52 expression. The compound also restored taxane-induced ALDH+ cell enrichment and markedly suppressed mammospheres.204 Additionally, co-treatment of SFN and paclitaxel enhanced paclitaxel-induced apoptosis in breast cancer cells through upregulation of cytochrome c, caspase-3, -8 and -9 and downregulation of NF-κB, Bcl-2 and p-Akt serine/threonine kinase.205 SFN prevented TNF-α mediated migration and invasion in breast cancer cells and suppressed MMP-2, -9 and -13 at both transcriptional and translational levels.206 Lee et al. (2012) reported before that SFN suppress MMP-9 expression and invasion in MCF-7 cells via inhibition of NF-κB.207Atwell et al. (2015) reviewed that most of the chemopreventive actions of SFN in breast and prostate cancers are due to modification of epigenetic mechanisms.208 Co-treatment of sulforaphane and withaferin A synergistically induced apoptosis and reduced cell viability and epigenetic processes in breast cancer cell lines through upregulation of Bax and downregulation of histone deacetylase 1 (HDAC1) and Bcl-2.209 On the other hand, SFN-induced senescence and cell cycle arrest in breast cancer cells are arbitrated by epigenetic alterations including decreased DNMT1 and DNMT3B expressions, global DNA hypo-methylation and variations in microRNA profile. The compound reduced methylation of N6-methyladenosine RNA (epigenetic regulation at RNA level) and exerted cytostatic action via induction of nitro-oxidative stress and downregulation of Akt signalling.210
3.13. Ginsenosides
Ginsenosides—major pharmacologically active saponins in ginseng root—are well-known for their promising restorative and healing abilities. Ginsenosides include Rg3, Rb1, Rb2, Rh2, Rh3 (protopanaxadiols) and Rh1, Rg1, Rg2 (protopanaxatriols).211–213Ginsenoside Rh2 provoked epigenetic methylation alteration in genes that are involved in immunity and tumorigenesis, thus increased immunogenicity and inhibited the growth of MCF-7 cells. Hyper-methylated genes like INSL5, OR52A1 and CASP1 experienced downregulation, while hypo-methylated genes including C1orf198, ST3GAL4 and CLINT1 displayed upregulation. Moreover, LINE1 (a global methylation marker) also displayed hypomethylation at specific CpGs.52 Rh2 also reversed resistance to docetaxel or adriamycin in resistant MCF-7 cells by differential microRNA expressions including miR-34a, miR-222 and miR-29a.214
Ginsenoside Rg3 enhanced cytotoxic effect of paclitaxel in TNBC cells via inhibition of NF-κB and Bcl-2 and upregulation of Bax and caspase-3.215 Kim et al. (2014) also reported that inhibition of NF-κB via inactivation of Akt and ERK is responsible for Rg3-induced apoptosis in breast cancer cells.216 Rg3 combined with recombinant human endostar suppressed the growth of breast cancer, inhibited cell invasion and angiogenesis and increased autophagy via decreased mRNA contents of MMP-2, MMP-9, VEGFA, VEGFB, VEGFC, p62, Beclin-1, P13K, mTOR, Akt and JNK.217
Similarly, ginsenoside Rg5 suppressed the proliferation of breast carcinoma via enhanced activation of AMPK and subsequent reduction in S6 and p70S6K activation.218 The compound induced apoptosis via regulation of Bax, cytochrome c and PARP and promoted cell cycle arrest at G0/G1 phase through downregulation of CDK4, cyclin E2 and cyclin D1 and upregulation of p21WAF1/CIP1, p53 and p15INK4B in breast cancer cell lines.219
3.14. Secoisolariciresinol and derived enterolignans
Secoisolariciresinol (SECO) is the most widespread phytoestrogenic lignan (diphenolic compound), abundantly found in flaxseed. Following their ingestion, plant phytoestrogenic lignans are converted into enterolignans (aka mammalian lignans): enterodiol (END) and enterolactone (ENL) that can bind to ER and are therefore considered as selective estrogen receptor modulator (SERM). During the last two decades, evidences suggest that lignan consumption and more precisely the serum lignan concentration are linked with a reduction of breast cancer incidence,220 but the last half decade was very informative concerning the possible underlying molecular mechanism.Examination of the estrogenic activity of ENL using DNA microarray based gene expression profiling in MCF-7 cells revealed that both estradiol and ENL initiated the same estrogen signalling but their signals are then differentially and directionally modulated later in the pathway, resulting in the differences at cell function levels.221 Particularly, ENL have been shown to activate the ERK1/2 and PI3K/Akt pathways.221 ENL have significantly decreased both ERα and PR (progesterone receptor) expression and increased estradiol secretion in MCF-7 cells. Different dose–responses were observed between ENL and END, with END acting at highest concentration level.222 In the same way ENL and END produced opposite action on telomerase activity since ENL, but not END, have been shown to decrease telomerase activity in MCF-7 cells.223
Antiproliferative action of lignans was also observed in MDA-MB-231 breast cancer cells and has been attributed to the gene expression downregulation of Ki-67, PCNA and FoxM1.224 It also exerts a control on cell cycle by lowering gene expression of cyclin E1, A2, B1 and B2 and interfering with the cytoskeleton by downregulating phosphorylation of FAK/paxillin pathway, thus suppressing cell migration and invasion.224 ENL have been shown to suppress proliferation, migration and metastasis using MDA-MB-231 breast cancer cells through the reduction of uPA-induced plasmin activation and the matrix metalloproteases MMP-2 and MMP-9 mediated by ECM remodelling.225
A reduced tumor progression and burden was observed in a model deriving from ACI rats either with flaxseed or with SECO supplemented groups in both xerograph and carcinogen animal models of human breast cancer.226 Hypothesized mechanisms include reduction in estrogen related signalling, anti-angiogenic and antioxidant activity at higher doses. Decrease in serum levels of follicular phase SHBG and IGFBP-3 were noted in supplemented animals, whereas, ESR2 (ERβ) gene expression increased.226 Authors suggested that ESR2 can oppose or reverse the proliferation signalling in mammary epithelial tissue. SECO may activate this pathway, suggesting that increased ERβ signalling could be one mechanism behind the decrease of mammary proliferation and dysplasia observed with SECO administration. Together a decrease in the cell proliferation marker Ki-67 gene expression was also observed for flax and SECO supplemented animals. SECO also did not promote ovarian dysplasia in these models.226
High lignan exposure have been associated with reduced mortality in breast cancer patients. For examples recent studies have found high ENL blood concentrations or high dietary lignans intake to be associated with a better prognosis among post-menopausal women.227,228 In a population of 2182 breast cancer patients, ENL was found to be associated with reduced all-cause mortality and breast cancer-specific mortality. This association was restricted to early stage breast cancer and to patients with normal BMI.227 However, no association between ENL and HER2 status and Ki-67 gene expression were observed229 which is in good agreement with a smaller study (n = 24) that also reported no effect on Ki-67 gene expression together with no impact on both ERβ and CASPs expressions.230 On the contrary, a previously reported by Flower et al. (2005), a meta-analysis of 10 clinical studies evidenced both the antiproliferative and antiangiogenic activities for flax lignans.231 Interestingly, these effects appeared to be correlated with BMI, with significant actions restricted to normal BMI (<25), but without any significant correlation with the menopausal or the ER+/ER− status of the patients. These observational data suggested associations between flax intake and decreased risk of primary breast cancer, lower mortality among breast cancer patients and better mental health.231 A large pre-diagnostic plasma ENL levels and breast cancer prognosis among Danish post-menopausal women study evidence interesting results in crude models higher ENL levels were associated with lower risk of breast cancer-specific mortality but after adjustment for lifestyle factors this association was only borderline significant (P = 0.0501). However for women who never smoked or used hormones a more significant lower risk of mortality was noted and certainly deserve further investigations.229
Flaxseed lignan SECO and ENL were also reported to enhance the cytotoxic effect of classic chemotherapeutic agents (docetaxel, doxorubicin and carboplatin) in the metastatic breast cancer cell lines SKBR3 and MDA-MB-231, suggesting possible future direction in improving chemotherapeutic efficacy in breast cancer by adjuvant therapy with flaxseed lignans.220 Combination of dietary supplement along with the reduction in dosage of conventional breast cancer chemotherapeutic agent (doxorubicin) may retain its benefits while minimizing the cytotoxic side effects and thus may enhance its therapeutic efficiency.232 Interaction of SECO with gluthathione-S-transferase PI-1 (GSTP1) gene has also been observed. Inhibition of GSTP1 expression at both gene and protein level by SECO was confirmed in MCF-7 and MDA-MB-231 breast cancer cells. SECO was also shown to inhibit cell growth and increase cellular apoptosis by up-regulating pro-apoptotic Casp-3 and Bax expression and down-regulating anti-apoptotic Bcl-xl and Bcl-2 expression.232
3.15. Vinca alkaloids
Vinca alkaloids, such as vincristine and vinblastine, are anti-microtubule and antimitotic agents which are obtained from Catharanthus roseus.399,400 Combination of vincristine and Thymus caramanicus extract significantly induced cell death in MCF-7 breast cancer cells by increasing protein expression of cleaved caspase 3 and decreasing cyclin D1.233 Similar synergistic effect of vincristine and Sutureja khuzestanica extract has also been observed for MCF-7 cells.234 On the other hand, vincristine combined with suspension culture has been used for the isolation of cancer stem cells from MDA-MB-231 cell lines.235 Recently, dasatinib plus vincristine liposomes were successfully employed for the treatment of triple negative breast cancer using MDA-MB-231 cells and its xenografts in nude mice. The liposomes induced apoptosis via activation of caspase-3, -8 and -9, increased ROS generation and Bax expression while decreasing Mcl-1. Furthermore, deletion of vasculogenic mimicry channels also occurred.236Vinblastine is extensively used for the treatment of breast cancer and Kaposi's sarcoma.238,401 Vinblastine not only inhibits the tumor growth but also malignant angiogenesis and can specifically bind to tubulin, thereby restricting its polymerization and subsequent microtubules association.237 Recently, it was shown that vinblastine induced apoptosis is MCF-7 cells by interfering with microtubules. The compound increased PARP cleavage, however, it was demonstrated that colchicine was more effective than vinblastine in inducing apoptosis in docetaxel resistant breast cancer (MCF-7TXT) cells.238 Interestingly, it was observed that enhanced sensitization of MDA-MB-231 cells to vinblastine or actinomycin D can be achieved by disrupting the function of Golgi apparatus inside these cells, thus resulting in increased apoptosis and decreased cell migration and proliferation.239 Likewise, co-treatment of cells with vinblastine or actinomycin D and golgicide A or brefeldin A (disrupting agents of ADP-ribosylation factor 1 (ARF1)—a protein required for homeostasis of Golgi complex) caused the similar effects via reducing the levels of either phospho-AKT or phospho-ERK1/2.239 Furthermore, combined treatment of MDA-MB-231 cells with vinblastine and docetaxel decreased survivin expression and induced apoptosis. A synergistic effect was observed when deguelin (a survivin inhibitor) was used along with vinblastine and docetaxel, suggesting that survivin downregulation may be useful in increasing the therapeutic efficacy of these chemotherapeutics.240
3.16. Miscellaneous phytochemicals
4 Critical bottlenecks in clinical translation
Although previous observations have directed various fruitful pre-clinical studies, still inadequate amount of clinical trials have been made to completely depict the distinct impact that each phytochemical may exert on cancer prevention. Several of these failures have been linked to the distribution of compounds, variable bioavailability and optimum mixtures of phytochemicals.340 Bioavailability is the rate and degree to which an active moiety/ingredient is absorbed from a drug and becomes presented at the action site.341 When describing bioavailability, the mode of application of active ingredient also needs to be taken into consideration. In case of oral administration, both the digestive and metabolic processing must be taken into account while in case of intravenous application only the capacity to metabolize the nutrient by host organism must be considered.342–344 A major hurdle in phytochemical research is the absence of consensus about the required optimum dose of several of these agents to be employed in trials. Phytochemicals exhibit their optimal cancer preventive property at relevant doses.340Several obstacles limit the clinical efficacy of curcumin such as poor bioavailabilty, quick metabolism and slow aqueous solubility.345,346 To improve the potential clinical efficacy of curcumin in high-risk populations, present research is mainly focusing on enhancing bioavailability to overcome both rapid compound metabolism and variability of absorption. Several efforts combining glucuronidation inhibitors (piperine, for instance) with curcumin to restrict intestinal and hepatic metabolism have revealed promising results.347 Other recent attempts have focused on using nanoparticles, curcumin analogs or delivery via phospholipid or liposomal complexing.348,349 A phase-I dose-escalation trial revealed that participants getting single dose of liposomal curcumin had a dose-dependent enhancement in plasma level of curcumin, as well as, its active metabolite (tetrahydrocurcumin) without any clinical side effects. However, at higher doses of curcumin (120 mg m−2), changes in morphology of RBC were seen, signifying a dose restricting sign of toxicity.350
Despite the promising anti-proliferative effects of resveratrol in vitro and in animal models, its translation to clinics also face several challenges. One obstacle is limited bioavailability as resveratrol is metabolically excluded from the body very fast, thereby creating difficulty in maintaining a therapeutically effective level in bloodstream.351 Recently, several efforts have been made in combining other natural substances with resveratrol to enhance its overall therapeutic value, especially cancer prevention.351 Pharmacokinetic studies of resveratrol showed extensive and rapid metabolism to resveratrol-3-O-sulfate, resveratrol-3′-O-glucuronide and resveratrol-4′-O-glucuronide following oral administration at different doses, which doesn't allow for sufficient anticancer property of resveratrol.352,353 Thus, efforts are underway to somehow sustain its metabolism in order to achieve better tissue exposure in the body. As resveratrol is rapidly metabolized into its metabolites, therefore, it is unclear whether metabolites may have different biological activity than free resveratrol or not. Resveratrol-3-O-sulfate has also been found to be a chemopreventive agent.354 Another effort is to find the optimal route and dose of resveratrol.355
The main constituent of green tea i.e. EGCG, though extensively supported by results from cell culture, epidemiological, clinical and animal studies, however, there are several hurdles like bioavailability, stability and metabolic transformations under physiological circumstances.356 Several studies have displayed conflicting results concerning the cancer risk decreasing properties of green tea in different populations. This incompatibility in results may be because of variable tea preparations, variable bioavailability of compounds across populations and unknown concentration of antioxidants.340
Quercetin is also considered as a potent chemopreventive agent in various cancers, including breast cancer but its application in clinics is limited due to poor bioavailability, instability, low solubility and poor permeability.357 Several approaches have been developed to improve its bioavailability such as using nanoparticles, micelles, liposomes or inclusion complexes. Enhanced bioavailability will enable to bring this agent in forefront in disease therapeutics in the near future.357 Table 3 further depicts the bottlenecks involved in the clinical translation of various phytochemicals involved in breast cancer remedy.
| Phytochemical | Bottlenecks in clinical translation | Ref. |
|---|---|---|
| Curcumin | Poor bioavailability, quick metabolism, slow aqueous solubility | 345 and 346 |
| Resveratrol | Poor bioavailability, quick metabolism, lack of proper dose | 351 |
| EGCG | Poor bioavailability, low stability, metabolic transformation under physiological circumstances | 356 |
| Quercetin | Poor bioavailability, instability, low permeability, low solubility | 357 |
| Apigenin | Poor bioavailability, poor aqueous solubility, low lipid solubility | 358 and 359 |
| Kaempferol | Low bioavailability, poor solubility | 360 |
| Genistein | Poor water solubility, low serum level after oral delivery (bioavailability), bitter taste | 361 and 362 |
| Silibinin | Poor bioavailability, poor water and lipid solubility | 363 and 364 |
| Parthenolide | Poor bioavailability & solubility in blood plasma, off-target effects, increased hydrophobicity | 258 |
| Sulforaphane | Instability, poor gastrointestinal absorption, hydrophobicity, poor bioavailability | 365 and 366 |
| 3,3′-Diindolylmethane | Poor absoption, poor biodistribution | 367 |
| Thymoquinone | Poor aqueous solubility, poor bioavailability, high lipophilicity | 368 and 369 |
| Naringenin | Poor bioavailability, instability, low aqueous solubility, low permeability, extensive first pass metabolism | 370 and 371 |
| Isoliquiritigenin | Poor bioavailability, poor solubility, low targeting ability, high effective dose | 187, 372 and 373 |
| Ginsenosides | Poor oral absorption, poor bioavailability | 374 |
| Secoisolariciresinol | Poor bioavailability | 375 |
| Luteolin | Poor water solubility, poor bioavailability | 376 |
5 Conclusions and future perspective
Breast cancer is challenging and account for maximum deaths across females in both developed and developing countries. As revealed by different epidemiological studies, there exists an inverse correlation between the use of phytochemicals and breast cancer incidence. Phytochemicals such as curcumin, resveratrol, silibinin, EGCG, thymoquinone, genistein, luteolin, α-mangostin, luteolin, kaempferol, quercetin, parthenolide, apigenin, pterostilbene, isoliquiritigenin, DIM, ginsenosides and isothiocyanates possess great potential towards breast cancer chemoprevention. These phytochemicals display anti-breast cancer activities through various mechanisms such as inhibition of proliferation, angiogenesis, migration, invasion, metastasis, as well as, induction of cell cycle arrest and apoptosis via modulation of several genes, gene products and signalling pathways. Furthermore, they have the potential to target breast cancer stem cells and overcome drugs resistance, along with sensitization to radiation and radio-protection. In recent years, much attention has been paid to natural remedies which have the capability to reduce adverse effects associated with currently available marketed drugs. Several synergistic approaches of phytochemicals with conventional chemotherapeutic drugs such as paclitaxel, doxorubicin, tamoxifen and adriamycin etc. have been addressed in various breast cancer cell lines for enhanced chemoprevention and sensitization.High costs and adverse reactions associated with the use of conventional drugs and irradiations reduced their therapeutic efficacy. In this regard, phytochemicals provide a safer and cost-effective approach. Most phytochemicals are readily available in vegetables, grains and fruits and their steady consumption would be effective to prevent breast cancer. It has been estimated that proper lifestyle adjustment could reduce cancer incidence by as much as two-thirds. Therefore, awareness should be raised among the general public about the proper and stable consumption of phytochemicals. Although, our understanding about multistage carcinogenesis has advanced, still the mechanism of action of most phytochemicals lacks adequate knowledge. The anticancer effects that most phytochemicals employ are probably the sum of several distinct mechanisms. Furthermore, deregulation or disruption of the intracellular signalling cascades sometime subject the cells to malignant transformation, so it is vital to recognize molecules in the signalling cascades that can be affected by specific phytochemicals in order to assess properly their underlying molecular mechanisms. Several phytochemicals have displayed non-toxic anti-tumor effects in xenograft model of nude mice, hence suggesting that they are effective in vivo.
A detailed insight about bioavailability and proper concentration of the required compounds are necessary, since most of them exerted dose-dependent inhibitory effects in breast cancer cells. Additionally, low concentration of some phytochemicals has shown to induce proliferation and activation of pro-survival autophagy in breast cancer, which further facilitate tumorigenesis. Likewise, large doses of phytochemicals resulted in development of additional diseases. For example, it has been shown that high doses of phytochemicals lead to chromosomal translocation, gastrointestinal disturbances and sexual dysfunction.377–379 Moreover, after absorption, phytochemicals are transformed to other conjugated forms, which may reduce their bioavailability. Thus, some alternations are required to enhance bioavailability of these phytochemicals, such as use of delivery systems like encapsulation, emulsion and other nanomedicine strategies. Pharmaceutical companies do not take interest in manufacturing phytochemical-based drugs due to intellectual property rights, which further creates hurdles. Some phytochemicals are produced by wild plants in relatively smaller proportions which make them difficult to be collected in large quantities, in which cases elicitation strategies and other plant tissue culture techniques provide a valuable insights for efficient accumulation of cancer related marker compounds, as well as, conservation of the respective plant species.
Strategies to decrease the side effects of chemotherapy are necessary, in which case, nanotechnology provides a useful opportunity in breast cancer therapy. The toxicity of conventional chemotherapeutic agents can be greatly reduced by administering them in nanoparticles and their combination with phytochemicals. Furthermore, new approach is required to enhance the intracellular stability, constant release and bioavailability of phytochemicals.380 Recently, it was reported that combination nanoparticles of paclitaxel and EGCG inhibited NF-κB activation, increased apoptosis, downregulated the genes involved in angiogenesis, cell survival and metastasis in MDA-MB-231 cell lines. Targeting the nanoparticles with anti-EGFR antibodies greatly enhanced these effects. Moreover, the EGCG-containing nanoparticles also overcame multidrug resistance in MDA-MB-231 cells via downregulation of P-glycoprotein.380 In another study, the intracellular concentration, stability and sustained release of EGCG was enhanced when loaded into chitosan-coated liposomes. The combined treatment significantly increased apoptosis and decreased cell viability.381 Similarly, quercetin encapsulation in MPEG-PLA (methoxypolyethylene glycol-polylactic acid) is useful in defeating its hydrophobicity. Quercetin nanoparticles induced apoptosis in MDA-MB-231 cells with sustained release for a duration of 10 days.382 In another report, quercetin-loaded mesoporous silica nanoparticles with folic acid tag ensured increased bioavailability and targeted delivery in breast cancer cells. The nanostructure caused apoptosis and cell cycle arrest in breast cancer cells via regulation of Bax and Akt signalling.383 Curcumin exerts numerous anticancer effects, however, it is faced with several limitations such as low bioavailability, quick metabolism, quick degradation and low aqueous solubility. Nanotechnology is revealing promising results in improving the bioavailability of hydrophobic agents such as curcumin. It was observed that curcumin-loaded PLGA-PEG greatly improved the cytotoxic effects in MCF-7 cell lines as compared to pure curcumin.384 Recently, curcumin-loaded folate-modified chitosan nanoparticles having targeted ability were developed which displayed good potential for breast cancer therapy. It was observed that reducing the pH of the release medium increased the release rate of curcumin from nanoparticles, showing pH responsive capacity of modified nanoparticles.385 Curcumin-loaded and calcium-doped dendritic mesoporous silica nanoparticles adapted with folic acid was also designed for effective treatment of breast cancer. This nanoformulation showed remarkable dispersal in aqueous solution, displayed pH responsive curcumin release and efficiently targeted the MCF-7 cells. The combination resulted in enhanced apoptosis, inhibition of proliferation, cell cycle arrest at G2/M phase, increased ROS generation and reduced mitochondrial membrane potential, in contrast to free curcumin.386 Resveratrol is also a promising candidate in breast cancer therapy, however, poor bioavailability and stability limit its clinical applications. Resveratrol-capped gold nanoparticles were synthesized previously, which repressed the TPA-induced invasion and migration abilities of breast cancer cells. The treatment greatly inhibited COX-2, ERK, MMP-9, NF-kB, P13K/Akt and AP-1.387 Resveratrol co-encapsulation with paclitaxel in a PEGylated liposome was successfully applied to drug resistant MCF-7/Adr cells which generated potent cytotoxicity. Moreover, the composite liposome also increased the bioavailability of the drug.388 In a recent study, resveratrol-load solid lipid nanoparticles were targeted on MDA-MB-231 cells which promoted Bax/Bcl-2 ratio and reduced the expression of c-Myc and cyclin D1, thereby greatly inhibiting proliferation, migration and invasion, as compared to free resveratrol.389 Thymoquinone-encapsulated nanoparticles were made by using hydrophilic and biodegradable polymers like PEG and PVP to overcome its poor aqueous solubility, minimal systemic bioavailability and light and thermal sensitivity. The resultant nanoparticles was more effective in killing cancer cells and less toxic to normal cells. Moreover, it displayed more potent anti-migratory activity on cancer cells.390 In another study, PLGA nanoparticles loaded with thymoquinone and paclitaxel were synthesized which showed improved anticancer activity in MCF-7 cells. This nanoformulation also aided in decreasing the toxic effects of paclitaxel by depressing its effective dosage.391 Similarly, nanostructured lipid carrier loading thymoquinone also improved the cytotoxicity and bioavailability of thymoquinone in MCF-7 and MDA-MB-231 cells.392 These results demonstrate that nanoformulations based on phytochemicals provide a useful strategy to enhance their cytotoxicity, bioavailability, stability and sustained release, as well as, to overcome drugs resistance and other harmful side effects associated with conventional chemotherapeutic agents.
Conflicts of interest
All the authors declare no conflicts of interest, financial or otherwise, associated with this publication.References
- C. E. DeSantis, J. Ma, A. Goding Sauer, L. A. Newman and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 439–448 CrossRef PubMed.
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 87–108 CrossRef PubMed.
- C. M. Perou and A.-L. Børresen-Dale, Cold Spring Harbor Perspect. Biol., 2011, 3, a003293 CrossRef PubMed.
- J. S. Reis-Filho and L. Pusztai, Lancet, 2011, 378, 1812–1823 CrossRef.
- T. Sørlie, C. M. Perou, R. Tibshirani, T. Aas, S. Geisler, H. Johnsen, T. Hastie, M. B. Eisen, M. Van De Rijn and S. S. Jeffrey, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10869–10874 CrossRef PubMed.
- C. Sotiriou, S.-Y. Neo, L. M. McShane, E. L. Korn, P. M. Long, A. Jazaeri, P. Martiat, S. B. Fox, A. L. Harris and E. T. Liu, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10393–10398 CrossRef PubMed.
- C. A. Hudis and L. Gianni, Oncologist, 2011, 16, 1–11 CrossRef PubMed.
- K. Reddy, Curr. Oncol., 2011, 18, e173 Search PubMed.
- S. J. Isakoff, Cancer J., 2010, 16, 53 CrossRef PubMed.
- S. Moulder and G. Hortobagyi, Clin. Pharmacol. Ther., 2008, 83, 26–36 CrossRef PubMed.
- M. Cazzaniga and B. Bonanni, BioMed Res. Int., 2012, 2012, 985620 Search PubMed.
- M. Wink, Medicines, 2015, 2, 251–286 CrossRef PubMed.
- R. Verpoorte, A. Contin and J. Memelink, Phytochem. Rev., 2002, 1, 13–25 CrossRef.
- H. N. Murthy, E.-J. Lee and K.-Y. Paek, Plant Cell, Tissue Organ Cult., 2014, 118, 1–16 CrossRef.
- M. Wink, Theor. Appl. Genet., 1988, 75, 225–233 CrossRef.
- M. Wink, T. Schmeller and B. Latz-Brüning, J. Chem. Ecol., 1998, 24, 1881–1937 CrossRef.
- M. Wink and B.-E. Van Wyk, Mind-altering and poisonous plants of the world, Timber Press Portland, OR, 2008 Search PubMed.
- M. Wink and O. Schimmer, Functions and biotechnology of plant secondary metabolites, 2010, vol. 39, pp. 21–161 Search PubMed.
- M. Wink, The Alkaloids: Chemistry and Biology, 2007, vol. 64, pp. 1–47 Search PubMed.
- M. Wink, in Studies in natural products chemistry, Elsevier, 2000, vol. 21, pp. 3–122 Search PubMed.
- R. H. Liu, Am. J. Clin. Nutr., 2003, 78, 517S–520S CrossRef PubMed.
- E. K. Arendt and E. Zannini, Cereal grains for the food and beverage industries, Elsevier, 2013 Search PubMed.
- Z.-h. Zhou, J. Yang and A.-N. Kong, Curr. Pharmacol. Rep., 2017, 3, 77–91 CrossRef.
- M. Govindappa, J. Diabetes Metab., 2015, 6, 1000565 Search PubMed.
- B. Pagliaro, C. Santolamazza, F. Simonelli and S. Rubattu, BioMed Res. Int., 2015, 2015, 918069 Search PubMed.
- M. A. Islam, F. Alam, M. Solayman, M. I. Khalil, M. A. Kamal and S. H. Gan, Oxidative medicine and cellular longevity, 2016, vol. 2016 Search PubMed.
- A. Sunkaria, A. Yadav, S. K. Sharma and R. Sandhir, Neuroprotective Effects of Phytochemicals in Neurological Disorders, 2017, pp. 515–559 Search PubMed.
- A. M. Abbasi, M. Khan, M. Ahmad, M. Zafar, S. Jahan and S. Sultana, J. Ethnopharmacol., 2010, 128, 322–335 CrossRef PubMed.
- M. S. Farvid, W. Y. Chen, K. B. Michels, E. Cho, W. C. Willett and A. H. Eliassen, BMJ, 2016, 353, i2343 CrossRef PubMed.
- P. E. Miller and D. C. Snyder, Nutr. Clin. Pract., 2012, 27, 599–612 CrossRef PubMed.
- M. K. Kim, J. H. Kim, S. J. Nam, S. Ryu and G. Kong, Nutr. Cancer, 2008, 60, 568–576 CrossRef PubMed.
- C. D. Mock, B. C. Jordan and C. Selvam, RSC Adv., 2015, 5, 75575–75588 RSC.
- R. Venkatadri, C. Wright, V. Kaushik, Y. Kulkarni, J. S. Yakisich, A. K. V. Iyer and N. Azad, Cancer Res., 2015, 3801 Search PubMed.
- L. Le Corre, N. Chalabi, L. Delort, Y. J. Bignon and D. Bernard-Gallon, Mol. Nutr. Food Res., 2005, 49, 462–471 CrossRef PubMed.
- E. C. Stuart, M. J. Scandlyn and R. J. Rosengren, Life Sci., 2006, 79, 2329–2336 CrossRef PubMed.
- D. Sinha, J. Biswas, S. M. Nabavi and A. Bishayee, Semin. Cancer Biol., 2017, 46, 33–54 CrossRef PubMed.
- M. Dastpeyman, N. Motamed, K. Azadmanesh, E. Mostafavi, V. Kia, A. Jahanian-Najafabadi and M. A. Shokrgozar, Med. Oncol., 2012, 29, 2512–2518 CrossRef PubMed.
- D. Xiao, V. Vogel and S. V. Singh, Mol. Cancer Ther., 2006, 5, 2931–2945 CrossRef PubMed.
- B. Xie, A. Nagalingam, P. Kuppusamy, N. Muniraj, P. Langford, B. Győrffy, N. K. Saxena and D. Sharma, Sci. Rep., 2017, 7, 40070 CrossRef PubMed.
- D. F. Romagnolo, M. G. Donovan, A. J. Papoutsis, T. C. Doetschman and O. I. Selmin, Curr. Dev. Nutr., 2017, 1, e000562 CrossRef PubMed.
- E. J. Choi and W. S. Ahn, Nutr. Res. Pract., 2008, 2, 322–325 CrossRef PubMed.
- S. Li, T. Yan, R. Deng, X. Jiang, H. Xiong, Y. Wang, Q. Yu, X. Wang, C. Chen and Y. Zhu, OncoTargets Ther., 2017, 10, 4809 CrossRef PubMed.
- M. Barkat, H. Abul, J. Ahmad, M. Khan, S. Beg and F. Ahmad, Curr. Drug Targets, 2018, 19, 70–80 Search PubMed.
- A. S. Sultan, M. I. Khalil, B. M. Sami, A. F. Alkhuriji and O. Sadek, Int. J. Clin. Exp. Pathol., 2017, 10, 156–172 Search PubMed.
- D. Carlisi, G. Buttitta, R. Di Fiore, C. Scerri, R. Drago-Ferrante, R. Vento and G. Tesoriere, Cell Death Dis., 2016, 7, e2194 CrossRef PubMed.
- A. Pledgie-Tracy, M. D. Sobolewski and N. E. Davidson, Mol. Cancer Ther., 2007, 6, 1013–1021 CrossRef PubMed.
- A. W. Harmon and Y. M. Patel, Breast Cancer Res. Treat., 2004, 85, 103–110 CrossRef PubMed.
- K.-L. Wang, S.-M. Hsia, C.-J. Chan, F.-Y. Chang, C.-Y. Huang, D.-T. Bau and P. S. Wang, Expert Opin. Ther. Targets, 2013, 17, 337–349 CrossRef PubMed.
- M. Maggiolini, G. Statti, A. Vivacqua, S. Gabriele, V. Rago, M. Loizzo, F. Menichini and S. Amdò, J. Steroid Biochem. Mol. Biol., 2002, 82, 315–322 CrossRef PubMed.
- Y. Cui, X.-O. Shu, Y.-T. Gao, H. Cai, M.-H. Tao and W. Zheng, Am. J. Epidemiol., 2006, 163, 645–653 CrossRef PubMed.
- H. R. Shin, J. Y. Kim, T. K. Yun, G. Morgan and H. Vainio, Cancer Causes Control, 2000, 11, 565–576 CrossRef PubMed.
- H. Lee, S. Lee, D. Jeong and S. J. Kim, J. Ginseng Res., 2017 DOI:10.1016/j.jgr.2017.05.003.
- M. Labbozzetta, M. Notarbartolo, P. Poma, A. Maurici, L. Inguglia, P. Marchetti, M. Rizzi, R. Baruchello, D. Simoni and N. D'Alessandro, Ann. N. Y. Acad. Sci., 2009, 1155, 278–283 CrossRef PubMed.
- T. H. Kim, Y. J. Shin, A. J. Won, B. M. Lee, W. S. Choi, J. H. Jung, H. Y. Chung and H. S. Kim, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 615–625 CrossRef PubMed.
- N. S. Alamolhodaei, A. M. Tsatsakis, M. Ramezani, A. W. Hayes and G. Karimi, Food Chem. Toxicol., 2017, 103, 223–232 CrossRef PubMed.
- T. Nabekura, Toxins, 2010, 2, 1207–1224 CrossRef PubMed.
- J. F. Weiss and M. R. Landauer, Toxicology, 2003, 189, 1–20 CrossRef PubMed.
- F. H. Sarkar and Y. Li, Cancer Res., 2006, 66, 3347–3350 CrossRef PubMed.
- B. T. Kawasaki, E. M. Hurt, T. Mistree and W. L. Farrar, Mol. Interventions, 2008, 8, 174 CrossRef PubMed.
- P. R. Dandawate, D. Subramaniam, R. A. Jensen and S. Anant, Semin. Cancer Biol., 2016, 40, 192–208 CrossRef PubMed.
- S.-Y. Yi, Y.-B. Hao, K.-J. Nan and T.-L. Fan, Cancer Treat. Rev., 2013, 39, 290–296 CrossRef PubMed.
- R. Pardal, M. F. Clarke and S. J. Morrison, Nat. Rev. Cancer, 2003, 3, 895–902 CrossRef PubMed.
- R. C. Petric, C. Braicu, L. Raduly, O. Zanoaga, N. Dragos, P. Monroig, D. Dumitrascu and I. Berindan-Neagoe, OncoTargets Ther., 2015, 8, 2053 CrossRef PubMed.
- J. Akhtar Siddiqui, A. Singh, M. Chagtoo, N. Singh, M. Madhav Godbole and B. Chakravarti, Curr. Cancer Drug Targets, 2015, 15, 116–135 CrossRef.
- B. J. Cridge, L. Larsen and R. J. Rosengren, Oncology Discovery, 2013, 1, 6 CrossRef.
- X. Wang, Y. Hang, J. Liu, Y. Hou, N. Wang and M. Wang, Oncol. Lett., 2017, 13, 4825–4831 CrossRef PubMed.
- B. L. Regassa and A. Vaidya, Breast Cancer Manage., 2016, 5, 93–106 CrossRef.
- Z.-D. Lv, X.-P. Liu, W.-J. Zhao, Q. Dong, F.-N. Li, H.-B. Wang and B. Kong, Int. J. Clin. Exp. Pathol., 2014, 7, 2818 Search PubMed.
- H. Fan, Y. Liang, B. Jiang, X. Li, H. Xun, J. Sun, W. He, H. T. Lau and X. Ma, Oncol. Rep., 2016, 35, 2651–2656 CrossRef PubMed.
- B. Chen, Y. Zhang, Y. Wang, J. Rao, X. Jiang and Z. Xu, J. Steroid Biochem. Mol. Biol., 2014, 143, 11–18 CrossRef PubMed.
- S. Mukherjee, M. Mazumdar, S. Chakraborty, A. Manna, S. Saha, P. Khan, P. Bhattacharjee, D. Guha, A. Adhikary and S. Mukhjerjee, Stem Cell Res. Ther., 2014, 5, 116 CrossRef PubMed.
- U. Kumar, U. Sharma and G. Rathi, Tumor Biol., 2017, 39 DOI:10.1177/1010428317692258.
- Y. Liu, J. Zhou, Y. Hu, J. Wang and C. Yuan, Mol. Cell. Biochem., 2017, 425, 47–58 CrossRef PubMed.
- L. Du, Z. Xie, L.-c. Wu, M. Chiu, J. Lin, K. K. Chan, S. Liu and Z. Liu, Nutr. Cancer, 2012, 64, 1228–1235 CrossRef PubMed.
- R. R. Falah, W. H. Talib and S. J. Shbailat, Ther. Adv. Med. Oncol., 2017, 9, 235–252 CrossRef PubMed.
- H. Moghtaderi, H. Sepehri and F. Attari, Biomed. Pharmacother., 2017, 88, 582–594 CrossRef PubMed.
- P. Odonmažig, A. Ebringerová, E. Machová and J. Alföldi, Carbohydr. Res., 1994, 252, 317–324 CrossRef.
- M. Ren, Y. Wang, X. Wu, S. Ge and B. Wang, J. Drug Targeting, 2017, 25, 247–254 CrossRef PubMed.
- P. Thulasiraman, G. Garriga, V. Danthuluri, D. J. McAndrews and I. Q. Mohiuddin, Oncol. Rep., 2017, 37, 2007–2015 CrossRef PubMed.
- L. Xiao-ai, W. Bei, X. Xiao-hong, P. Lei, W. Bin, D. Xiao-xue, Z. Chen-hui and D. Qi-wei, RSC Adv., 2017, 7, 33572–33579 RSC.
- B. Vinod, J. Antony, H. Nair, V. Puliyappadamba, M. Saikia, S. S. Narayanan, A. Bevin and R. J. Anto, Cell Death Dis., 2013, 4, e505 CrossRef PubMed.
- H. Zong, F. Wang, Q.-x. Fan and L.-x. Wang, Mol. Biol. Rep., 2012, 39, 4803–4808 CrossRef PubMed.
- K. Sun, X. Duan, H. Cai, X. Liu, Y. Yang, M. Li, X. Zhang and J. Wang, Clin. Exp. Med., 2016, 16, 37–47 CrossRef PubMed.
- S. Bimonte, A. Barbieri, G. Palma, D. Rea, A. Luciano, M. D'Aiuto, C. Arra and F. Izzo, BioMed Res. Int., 2015, 2015, 878134 Search PubMed.
- S.-F. Hendrayani, H. H. Al-Khalaf and A. Aboussekhra, Neoplasia, 2013, 15, 631IN611–640IN611 CrossRef.
- Z. K. Hassan and M. H. Daghestani, Asian Pac. J. Cancer Prev., 2012, 13, 3259–3264 CrossRef PubMed.
- N. Mo, Z.-Q. Li, J. Li and Y.-D. Cao, Asian Pac. J. Cancer Prev., 2012, 13, 5709–5714 CrossRef PubMed.
- M. Gallardo and G. M. Calaf, Int. J. Oncol., 2016, 49, 1019–1027 CrossRef PubMed.
- W. Zhu, L. Jia, G. Chen, H. Zhao, X. Sun, X. Meng, X. Zhao, L. Xing, J. Yu and M. Zheng, Oncotarget, 2016, 7, 48607 Search PubMed.
- M. Bonucci, M. Fioranelli, M. G. Roccia and T. Lotti, Dermatol. Ther., 2017, 30, e12493 CrossRef PubMed.
- H. Zhao, W. Zhu, L. Jia, X. Sun, G. Chen, X. Zhao, X. Li, X. Meng, L. Kong and L. Xing, Br. J. Radiol., 2015, 89, 20150665 CrossRef PubMed.
- M. Moradzadeh, A. Hosseini, S. Erfanian and H. Rezaei, Pharmacol. Rep., 2017, 69, 924–928 CrossRef PubMed.
- C.-Y. Huang, Z. Han, X. Li, H.-H. Xie and S.-S. Zhu, Oncol. Lett., 2017, 14, 3623–3627 CrossRef PubMed.
- O. Y. Hong, E. M. Noh, H. Y. Jang, Y. R. Lee, B. K. Lee, S. H. Jung, J. S. Kim and H. J. Youn, Oncol. Lett., 2017, 14, 441–446 CrossRef PubMed.
- R. León-Cachón, J. Ascacio-Martínez and H. A. Barrera-Saldaña, Rev. Invest. Clin., 2012, 64, 364–376 Search PubMed.
- F. De Amicis, A. Russo, P. Avena, M. Santoro, A. Vivacqua, D. Bonofiglio, L. Mauro, S. Aquila, D. Tramontano and S. A. Fuqua, Mol. Nutr. Food Res., 2013, 57, 840–853 CrossRef PubMed.
- L. Zeng, J. M. Holly and C. M. Perks, Front. Endocrinol., 2014, 5, 61 Search PubMed.
- X. Pan, B. Zhao, Z. Song, S. Han and M. Wang, J. Pharmacol. Sci., 2016, 130, 85–93 CrossRef PubMed.
- S. Sun, Y. Dai, Z. Lu, M. Li, Z. Zhai, X. Ren and D. Li, Int. J. Clin. Exp. Pathol., 2016, 9, 4251–4259 Search PubMed.
- M. A. Esmaeili, Journal of Chemical Biology, 2016, 9, 41–52 CrossRef PubMed.
- A. Nowakowska and J. Tarasiuk, Acta Biochim. Pol., 2016, 63, 571–575 CrossRef PubMed.
- D. G. Pons, M. Nadal-Serrano, M. Torrens-Mas, J. Oliver and P. Roca, J. Cell. Biochem., 2016, 117, 218–229 CrossRef PubMed.
- E. J. Choi, J. Y. Jung and G.-H. Kim, Exp. Ther. Med., 2014, 8, 454–458 CrossRef PubMed.
- Y. Li, S. M. Meeran, S. N. Patel, H. Chen, T. M. Hardy and T. O. Tollefsbol, Mol. Cancer, 2013, 12, 9 CrossRef PubMed.
- E. J. Choi and G.-H. Kim, Mol. Med. Rep., 2013, 7, 781–784 CrossRef PubMed.
- Q. Zhao, M. Zhao, A. B. Parris, Y. Xing and X. Yang, Int. J. Oncol., 2016, 49, 1203–1210 CrossRef PubMed.
- Y. Fang, Q. Zhang, X. Wang, X. Yang, X. Wang, Z. Huang, Y. Jiao and J. Wang, Int. J. Oncol., 2016, 48, 1016–1028 CrossRef PubMed.
- Q. Xie, Q. Bai, L. Y. Zou, Q. Y. Zhang, Y. Zhou, H. Chang, L. Yi, J. D. Zhu and M. T. Mi, Genes, Chromosomes Cancer, 2014, 53, 422–431 CrossRef PubMed.
- R. R. Jadhav, J. Santucci-Pereira, Y. V. Wang, J. Liu, T. D. Nguyen, J. Wang, S. Jenkins, J. Russo, T. H.-M. Huang and V. X. Jin, Genes, 2017, 8, 144 CrossRef PubMed.
- X. Zhang, K. L. Cook, A. Warri, I. M. Cruz, M. Rosim, J. Riskin, W. Helferich, D. Doerge, R. Clarke and L. Hilakivi-Clarke, Clin. Cancer Res., 2017, 23, 814–824 CrossRef PubMed.
- C. B. Avci, S. Y. Susluer, H. O. Caglar, T. Balci, D. Aygunes, Y. Dodurga and C. Gunduz, Contemp. Oncol., 2015, 19, 32 Search PubMed.
- J. Sims-Mourtada, L. Opdenaker, J. Davis and C. Wu, Cancer Stud. Mol. Med., 2015, 2, 60–65 CrossRef.
- P. Fan, S. Fan, H. Wang, J. Mao, Y. Shi, M. M. Ibrahim, W. Ma, X. Yu, Z. Hou and B. Wang, Stem Cell Res. Ther., 2013, 4, 146 CrossRef PubMed.
- E. Izquierdo-Torres, G. Rodríguez, I. Meneses-Morales and A. Zarain-Herzberg, Mol. Carcinog., 2017, 56, 1703–1711 CrossRef PubMed.
- A. Khan, A. N. Aljarbou, Y. H. Aldebasi, S. M. Faisal and M. A. Khan, Cancer Epidemiol., 2014, 38, 765–772 CrossRef PubMed.
- Y. N. Kim, S. R. Choe, K. H. Cho, D. Y. Cho, J. Kang, C. G. Park and H. Y. Lee, Exp. Mol. Med., 2017, 49, e296 CrossRef PubMed.
- R. Medina-Aguilar, L. A. Marchat, E. Arechaga Ocampo, P. Gariglio, J. García Mena, N. Villegas Sepúlveda, M. Martínez Castillo and C. López-Camarillo, Oncol. Rep., 2016, 35, 3696–3704 CrossRef PubMed.
- O. H. Alobaedi, W. H. Talib and I. A. Basheti, Asian Pac. J. Trop. Med., 2017, 10, 400–408 CrossRef PubMed.
- R. Venkatadri, A. K. V. Iyer, V. Kaushik and N. Azad, Pharmacol. Rep., 2017, 69, 788–797 CrossRef PubMed.
- R. Kala and T. O. Tollefsbol, PLoS One, 2016, 11, e0155057 CrossRef PubMed.
- A. Mondal and L. L. Bennett, Biomed. Pharmacother., 2016, 84, 1906–1914 CrossRef PubMed.
- A. Alayev, S. M. Berger, M. Y. Kramer, N. S. Schwartz and M. K. Holz, J. Cell. Biochem., 2015, 116, 450–457 CrossRef PubMed.
- S. Chottanapund, M. Van Duursen, P. Navasumrit, P. Hunsonti, S. Timtavorn, M. Ruchirawat and M. Van den Berg, Toxicol. in Vitro, 2014, 28, 1215–1221 CrossRef PubMed.
- F. Huang, X.-N. Wu, J. Chen, W.-X. Wang and Z. F. Lu, Exp. Ther. Med., 2014, 7, 1611–1616 CrossRef PubMed.
- J. Díaz-Chávez, M. A. Fonseca-Sánchez, E. Arechaga-Ocampo, A. Flores-Pérez, Y. Palacios-Rodríguez, G. Domínguez-Gómez, L. A. Marchat, L. Fuentes-Mera, G. Mendoza-Hernández and P. Gariglio, PLoS One, 2013, 8, e64378 CrossRef PubMed.
- X.-P. Shi, S. Miao, Y. Wu, W. Zhang, X.-F. Zhang, H.-Z. Ma, H.-L. Xin, J. Feng, A.-D. Wen and Y. Li, Int. J. Mol. Sci., 2013, 14, 15655–15668 CrossRef PubMed.
- F. Casanova, J. Quarti, D. C. F. da Costa, C. A. Ramos, J. L. da Silva and E. Fialho, J. Cell. Biochem., 2012, 113, 2586–2596 CrossRef PubMed.
- R. Venkatadri, T. Muni, A. Iyer, J. Yakisich and N. Azad, Cell Death Dis., 2016, 7, e2104 CrossRef PubMed.
- D. Pradhan, R. K. Pradhan, G. Tripathy and S. Pradhan, J. Young Pharm., 2015, 7, 225 CrossRef.
- L. Khorsandi, M. Orazizadeh, F. Niazvand, M. Abbaspour, E. Mansouri and A. Khodadadi, Bratisl. Lek. Listy, 2017, 118, 123–128 Search PubMed.
- S. Ranganathan, D. Halagowder and N. D. Sivasithambaram, PLoS One, 2015, 10, e0141370 CrossRef PubMed.
- X.-H. Deng, H.-Y. Song, Y.-F. Zhou, G.-Y. Yuan and F.-J. Zheng, Exp. Ther. Med., 2013, 6, 1155–1158 CrossRef PubMed.
- F. Ramezani, N. Samadi and Z. Mostafavi-Pour, Nutr. Cancer, 2017, 69, 881–891 CrossRef PubMed.
- H. Wang, L. Tao, K. Qi, H. Zhang, D. Feng, W. Wei, H. Kong, T. Chen and Q. Lin, J. BUON, 2015, 20, 707–713 Search PubMed.
- C. Huang, S.-Y. Lee, C.-L. Lin, T.-H. Tu, L. H. Chen, Y. J. Chen and H.-C. Huang, J. Agric. Food Chem., 2013, 61, 6430–6445 CrossRef PubMed.
- A. Srinivasan, C. Thangavel, Y. Liu, S. Shoyele, R. B. Den, P. Selvakumar and A. Lakshmikuttyamma, Mol. Carcinog., 2016, 55, 743–756 CrossRef PubMed.
- X. Zhao, Q. Wang, S. Yang, C. Chen, X. Li, J. Liu, Z. Zou and D. Cai, Eur. J. Pharmacol., 2016, 781, 60–68 CrossRef PubMed.
- S.-f. Tao, H.-f. He and Q. Chen, Mol. Cell. Biochem., 2015, 402, 93–100 CrossRef PubMed.
- M. Younas, S. Drouet, M. Nadeem, N. Giglioli-Guivarc'h, C. Hano and B. H. Abbasi, J. Photochem. Photobiol., B, 2018, 184, 61–70 CrossRef PubMed.
- Y. Wang, W.-C. Liang, W.-L. Pan, W.-K. Law, J.-S. Hu, D. T.-M. Ip, M. M.-Y. Waye, T.-B. Ng and D. C.-C. Wan, Phytomedicine, 2014, 21, 1310–1317 CrossRef PubMed.
- S.-J. Oh, S. P. Jung, J. Han, S. Kim, J. S. Kim, S. J. Nam, J. E. Lee and J.-H. Kim, Oncol. Rep., 2013, 29, 1343–1348 CrossRef PubMed.
- K. Alimoghaddam and A. Ghavamzadeh, Arch. Iran. Med., 2014, 17, 366 Search PubMed.
- M. B. Pirouzpanah, M. Sabzichi, S. Pirouzpanah, H. Chavoshi and N. Samadi, Asian Pac. J. Cancer Prev., 2015, 16, 2087–2092 CrossRef PubMed.
- W. H. Kil, S. M. Kim, J. E. Lee, K. S. Park and S. J. Nam, Ann. Surg. Treat. Res., 2014, 87, 167–173 CrossRef PubMed.
- H. Chavoshi, V. Vahedian, S. Saghaei, M. B. Pirouzpanah, M. Raeisi and N. Samadi, Asian Pac. J. Cancer Prev., 2017, 18, 2243 Search PubMed.
- O. Molavi, F. Narimani, F. Asiaee, S. Sharifi, V. Tarhriz, A. Shayanfar, M. Hejazi and R. Lai, Pharm. Biol., 2017, 55, 729–739 CrossRef PubMed.
- Z. J. Maasomi, Y. P. Soltanahmadi, M. Dadashpour, S. Alipour, S. Abolhasani and N. Zarghami, Asian Pac. J. Cancer Prev., 2017, 18, 1283 Search PubMed.
- M. M. Zadeh, N. Motamed, N. Ranji, M. Majidi and F. Falahi, J. Breast Cancer, 2016, 19, 45–52 CrossRef PubMed.
- Z. Jahanafrooz, N. Motamed and B. Bakhshandeh, Cytotechnology, 2017, 1–14 Search PubMed.
- K. Jiang, W. Wang, X. Jin, Z. Wang, Z. Ji and G. Meng, Oncol. Rep., 2015, 33, 2711–2718 CrossRef PubMed.
- N. Zheng, P. Zhang, H. Huang, W. Liu, T. Hayashi, L. Zang, Y. Zhang, L. Liu, M. Xia and S.-i. Tashiro, J. Pharmacol. Sci., 2015, 128, 97–107 CrossRef PubMed.
- M. Marjaneh, M. Narazah and H. Shahrul, Apoptosis, 2015, 1, 10001014 Search PubMed.
- B. Saraçligil, B. Öztürk, Ş. B. Bozkurt and Y. Kahveci, Int. J. Pharm. Sci. Res., 2017, 8, 2849–2852 Search PubMed.
- S. Rajput, B. Kumar, P. Banik, S. Parida and M. Mandal, J. Cell. Physiol., 2015, 230, 620–629 CrossRef PubMed.
- S. Parbin, A. Shilpi, S. Kar, N. Pradhan, D. Sengupta, M. Deb, S. K. Rath and S. K. Patra, Mol. BioSyst., 2016, 12, 48–58 RSC.
- S. Ganji-Harsini, M. Khazaei, Z. Rashidi and A. Ghanbari, Cell, 2016, 18, 245 Search PubMed.
- Ç. Şakalar, K. İzgi, B. İskender, S. Sezen, H. Aksu, M. Çakır, B. Kurt, A. Turan and H. Canatan, Tumor Biol., 2016, 37, 4467–4477 CrossRef PubMed.
- L. H. Odeh, W. H. Talib and I. A. Basheti, J. Cancer Res. Ther., 2018, 14, 324 CrossRef PubMed.
- A. Sehrawat, C. S. Croix, C. J. Baty, S. Watkins, D. Tailor, R. P. Singh and S. V. Singh, Mitochondrion, 2016, 30, 67–77 CrossRef PubMed.
- E. J. Kim, S. J. Eom, J. E. Hong, J.-Y. Lee, M.-S. Choi and J. H. Y. Park, Mol. Cell. Biochem., 2012, 359, 431–440 CrossRef PubMed.
- S.-H. Kim, A. Sehrawat and S. V. Singh, Cancer Prev. Res., 2013, 6, 782–790 CrossRef PubMed.
- S.-H. Kim and S. V. Singh, Breast Cancer Res. Treat., 2015, 149, 681–692 CrossRef PubMed.
- D. Bauer, N. Redmon, E. Mazzio and K. F. Soliman, PLoS One, 2017, 12, e0175558 CrossRef PubMed.
- G. Soria, M. Ofri-Shahak, I. Haas, N. Yaal-Hahoshen, L. Leider-Trejo, T. Leibovich-Rivkin, P. Weitzenfeld, T. Meshel, E. Shabtai and M. Gutman, BMC Cancer, 2011, 11, 130 CrossRef PubMed.
- A. Papi, G. Storci, T. Guarnieri, S. De Carolis, S. Bertoni, N. Avenia, A. Sanguinetti, A. Sidoni, D. Santini and C. Ceccarelli, PLoS One, 2013, 8, e54968 CrossRef PubMed.
- A. Ben-Baruch, Cancer Microenviron., 2012, 5, 151–164 CrossRef PubMed.
- J. L. Steiner and E. A. Murphy, Int. J. Biol. Markers, 2012, 27, e179–185 CrossRef PubMed.
- B. Kindlund, Å. Sjöling, C. Yakkala, J. Adamsson, A. Janzon, L.-E. Hansson, M. Hermansson, P. Janson, O. Winqvist and S. B. Lundin, Gastric Cancer, 2017, 20, 116–125 CrossRef PubMed.
- S. Tabariès, V. Ouellet, B. E. Hsu, M. G. Annis, A. A. Rose, L. Meunier, E. Carmona, C. E. Tam, A.-M. Mes-Masson and P. M. Siegel, Breast Cancer Res., 2015, 17, 45 CrossRef PubMed.
- P. Mishra, D. Banerjee and A. Ben-Baruch, J. Leukocyte Biol., 2011, 89, 31–39 CrossRef PubMed.
- S. A. Ibrahim, G. K. Katara, A. Kulshrestha, M. K. Jaiswal, M. A. Amin and K. D. Beaman, Oncotarget, 2015, 6, 33033 CrossRef PubMed.
- C. N. Vrakas, R. M. O'Sullivan, S. E. Evans, D. A. Ingram, C. B. Jones, T. Phuong and R. A. Kurt, Immunol. Invest., 2015, 44, 174–188 CrossRef PubMed.
- H. S. Seo, J. M. Ku, H. S. Choi, J. K. Woo, B. H. Lee, D. S. Kim, H. J. Song, B. H. Jang, Y. C. Shin and S. G. Ko, Oncol. Rep., 2017, 38, 715–724 CrossRef PubMed.
- H.-S. Seo, J. K. Jo, J. M. Ku, H.-S. Choi, Y. K. Choi, J.-K. Woo, S.-y. Kang, K. min Lee, K. W. Nam and N. Park, Biosci. Rep., 2015, 35, e00276 CrossRef PubMed.
- H.-S. Seo, H.-S. Choi, S.-R. Kim, Y. K. Choi, S.-M. Woo, I. Shin, J.-K. Woo, S.-Y. Park, Y. C. Shin and S.-K. Ko, Mol. Cell. Biochem., 2012, 366, 319–334 CrossRef PubMed.
- T. H. Tseng, M. H. Chien, W. L. Lin, Y. C. Wen, J. M. Chow, C. K. Chen, T. C. Kuo and W. J. Lee, Environ. Toxicol., 2017, 32, 434–444 CrossRef PubMed.
- M. E. Harrison, M. R. P. Coombs, L. M. Delaney and D. W. Hoskin, Exp. Mol. Pathol., 2014, 97, 211–217 CrossRef PubMed.
- G. Chen, L. Zhu, Y. Liu, Q. Zhou, H. Chen and J. Yang, Phytother. Res., 2009, 23, 498–506 CrossRef PubMed.
- G. Chen, X. Hu, W. Zhang, N. Xu, F.-Q. Wang, J. Jia, W.-F. Zhang, Z.-J. Sun and Y.-F. Zhao, Apoptosis, 2012, 17, 90–101 CrossRef PubMed.
- F. Peng, Q. Du, C. Peng, N. Wang, H. Tang, X. Xie, J. Shen and J. Chen, Phytother. Res., 2015, 29, 969–977 CrossRef PubMed.
- F. Peng, H. Tang, P. Liu, J. Shen, X. Guan, X. Xie, J. Gao, L. Xiong, L. Jia and J. Chen, Sci. Rep., 2017, 7, 9022 CrossRef PubMed.
- L. Si, X. Yang, X. Yan, Y. Wang and Q. Zheng, Oncol. Lett., 2017, 14, 241–249 CrossRef PubMed.
- S. K. Lee, K.-K. Park, K. R. Kim, H.-J. Kim and W.-Y. Chung, Journal of Cancer Prevention, 2015, 20, 281 CrossRef PubMed.
- S. Ning, X. Ma, D. Zhu, Z. Shen, J. Liu, Y. Liu, J. Chen and Z. Li, RSC Adv., 2017, 7, 18085–18092 RSC.
- S. Ning, J. Mu, Z. Shen, D. Zhu, F. Jiang, X. Wang, Y. Li and Z. Li, RSC Adv., 2016, 6, 24719–24727 RSC.
- Z. Wang, N. Wang, P. Liu, Q. Chen, H. Situ, T. Xie, J. Zhang, C. Peng, Y. Lin and J. Chen, Oncotarget, 2014, 5, 7013 Search PubMed.
- N. Wang, Z. Wang, Y. Wang, X. Xie, J. Shen, C. Peng, J. You, F. Peng, H. Tang and X. Guan, Oncotarget, 2015, 6, 9854 Search PubMed.
- N. Wang, Z. Wang, C. Peng, J. You, J. Shen, S. Han and J. Chen, Carcinogenesis, 2014, 35, 2544–2554 CrossRef PubMed.
- R. Wakimoto, M. Ono, M. Takeshima, T. Higuchi and S. Nakano, Anticancer Res., 2017, 37, 6153–6159 Search PubMed.
- C.-M. Hung, L.-C. Liu, C.-T. Ho, Y.-C. Lin and T.-D. Way, J. Agric. Food Chem., 2017, 65, 11179–11191 CrossRef PubMed.
- C.-M. Su, W.-H. Lee, A. T. Wu, Y.-K. Lin, L.-S. Wang, C.-H. Wu and C.-T. Yeh, J. Nutr. Biochem., 2015, 26, 675–685 CrossRef PubMed.
- M. Guarino, J. Cell. Physiol., 2010, 223, 14–26 Search PubMed.
- A. Varkaris, A. D. Katsiampoura, J. C. Araujo, G. E. Gallick and P. G. Corn, Cancer Metastasis Rev., 2014, 33, 595–606 CrossRef PubMed.
- L. C. Kim, L. Song and E. B. Haura, Nat. Rev. Clin. Oncol., 2009, 6, 587–595 CrossRef PubMed.
- C.-H. Wu, B.-H. Hong, C.-T. Ho and G.-C. Yen, J. Agric. Food Chem., 2015, 63, 2432–2441 CrossRef PubMed.
- K. K. Mak, A. T. Wu, W. H. Lee, T. C. Chang, J. F. Chiou, L. S. Wang, C. H. Wu, C. Y. F. Huang, Y. S. Shieh and T. Y. Chao, Mol. Nutr. Food Res., 2013, 57, 1123–1134 CrossRef PubMed.
- L. Kong, B. Liu, C. Zhang, B. Wang, H. Wang, X. Song, Y. Yang, X. Ren, L. Yin and H. Kong, Neurochem. Int., 2016, 100, 52–61 CrossRef PubMed.
- M. Lenzi, C. Fimognari and P. Hrelia, in Advances in Nutrition and Cancer, Springer, 2014, pp. 207–223 Search PubMed.
- S. M. Tortorella, S. G. Royce, P. V. Licciardi and T. C. Karagiannis, Antioxid. Redox Signaling, 2015, 22, 1382–1424 CrossRef PubMed.
- J. D. Clarke, R. H. Dashwood and E. Ho, Cancer Lett., 2008, 269, 291–304 CrossRef PubMed.
- E. Heiss, C. Herhaus, K. Klimo, H. Bartsch and C. Gerhäuser, J. Biol. Chem., 2001, 276, 32008–32015 CrossRef PubMed.
- Y. W. An, K. A. Jhang, S.-Y. Woo, J. L. Kang and Y. H. Chong, Neurobiol. Aging, 2016, 38, 1–10 CrossRef PubMed.
- H. Tilg, Clin. Phytosci., 2015, 1, 10 CrossRef.
- J. P. Burnett, G. Lim, Y. Li, R. B. Shah, R. Lim, H. J. Paholak, S. P. McDermott, L. Sun, Y. Tsume and S. Bai, Cancer Lett., 2017, 394, 52–64 CrossRef PubMed.
- S. H. Kim, H. J. Park and D. O. Moon, Oncol. Lett., 2017, 13, 4427–4432 CrossRef PubMed.
- C. Bao, J. Ko, H.-C. Park, M. C. Kim, J. Kim, J.-H. Auh and H. J. Lee, Food Sci. Biotechnol., 2015, 24, 347–351 CrossRef.
- Y.-R. Lee, E.-M. Noh, J.-H. Han, J.-M. Kim, B.-M. Hwang, B.-S. Kim, S.-H. Lee, S. H. Jung, E. Y. Chung and J.-S. Kim, BMB Rep., 2013, 46, 201 CrossRef PubMed.
- L. L. Atwell, L. M. Beaver, J. Shannon, D. E. Williams, R. H. Dashwood and E. Ho, Curr. Pharmacol. Rep., 2015, 1, 102–111 CrossRef PubMed.
- K. J. Royston, N. Udayakumar, K. Lewis and T. O. Tollefsbol, Int. J. Mol. Sci., 2017, 18, 1092 CrossRef PubMed.
- A. Lewinska, J. Adamczyk-Grochala, A. Deregowska and M. Wnuk, Theranostics, 2017, 7, 3461 CrossRef PubMed.
- H. Yu, C. Zhang, M. Lu, F. Sun, Y. Fu and F. Jin, Chem. Pharm. Bull., 2007, 55, 231–235 CrossRef PubMed.
- J.-M. Lu, Q. Yao and C. Chen, Curr. Vasc. Pharmacol., 2009, 7, 293–302 CrossRef PubMed.
- J. Y. Park, H. You, D. Lee, W. Huh, G. S. Hwang, K. T. No, K. H. Kim, J. Ham, N. Yamabe and Y. Kim, Bull. Korean Chem. Soc., 2016, 37, 52–55 CrossRef.
- X. Wen, H.-D. Zhang, L. Zhao, Y.-F. Yao, J.-H. Zhao and J.-H. Tang, Asian Pac. J. Cancer Prev., 2015, 16, 1105–1109 CrossRef PubMed.
- Z. Yuan, H. Jiang, X. Zhu, X. Liu and J. Li, Biomed. Pharmacother., 2017, 89, 227–232 CrossRef PubMed.
- B.-M. Kim, D.-H. Kim, J.-H. Park, Y.-J. Surh and H.-K. Na, Journal of Cancer Prevention, 2014, 19, 23 CrossRef PubMed.
- Y. Zhang, Q.-Z. Liu, S.-P. Xing and J.-L. Zhang, Asian Pac. J. Trop. Med., 2016, 9, 180–183 CrossRef PubMed.
- Y. Zou and P. Liu, Int. J. Clin. Exp. Med., 2016, 9, 17664–17669 Search PubMed.
- S.-J. Kim and A. K. Kim, J. Ginseng Res., 2015, 39, 125–134 CrossRef PubMed.
- Y. Di, F. De Silva, E. S. Krol and J. Alcorn, Nutr. Cancer, 2018, 1–10 Search PubMed.
- Y. Zhu, K. Kawaguchi and R. Kiyama, PLoS One, 2017, 12, e0171390 CrossRef PubMed.
- L. Schröder, D. U. Richter, B. Piechulla, M. Chrobak, C. Kuhn, S. Schulze, S. Abarzua, U. Jeschke and T. Weissenbacher, Nutrients, 2016, 8, 616 CrossRef PubMed.
- D. Ilbeigi, M. Nourbakhsh, S. Khaghani, N. Einollahi, N. Kheiripour, Z. Gholinejad, M. Alaee and M. Saberian, Cell, 2017, 19, 37 Search PubMed.
- X.-Y. Xiong, X.-J. Hu, Y. Li and C.-M. Liu, Nutr. Cancer, 2015, 67, 1326–1334 CrossRef PubMed.
- A. V. Mali, A. A. Joshi, M. V. Hegde and S. S. Kadam, Asian Pac. J. Cancer Prev., 2017, 18, 905 Search PubMed.
- D. M. Delman, C. J. Fabian, B. F. Kimler, H. Yeh and B. K. Petroff, Nutr. Cancer, 2015, 67, 857–864 CrossRef PubMed.
- P. Seibold, A. Vrieling, T. S. Johnson, K. Buck, S. Behrens, R. Kaaks, J. Linseisen, N. Obi, J. Heinz and D. Flesch-Janys, Int. J. Cancer, 2014, 135, 923–933 CrossRef PubMed.
- C. Kyrø, L. Hansen, K. Frederiksen, N. P. Nørskov, K. E. B. Knudsen, A. K. Eriksen, M. Holm, A. Tjønneland and A. Olsen, Clin. Nutr., 2017 DOI:10.1016/j.clnu.2017.10.023.
- S. Jaskulski, A. Y. Jung, A. Rudolph, T. Johnson, K. Thöne, E. Herpel, P. Sinn and J. Chang-Claude, Mol. Nutr. Food Res., 2017, 61, 1700449 CrossRef PubMed.
- S. E. McCann, S. B. Edge, D. G. Hicks, L. U. Thompson, C. D. Morrison, G. Fetterly, C. Andrews, K. Clark, J. Wilton and S. Kulkarni, Nutr. Cancer, 2014, 66, 566–575 CrossRef PubMed.
- G. Flower, H. Fritz, L. G. Balneaves, S. Verma, B. Skidmore, R. Fernandes, D. Kennedy, K. Cooley, R. Wong and S. Sagar, Integr. Cancer Ther., 2014, 13, 181–192 CrossRef PubMed.
- S. Dutta, P. S. Kharkar, N. U. Sahu and A. Khanna, Life Sci., 2017, 185, 73–84 CrossRef PubMed.
- S. Esmaeili-Mahani, F. Falahi and M. M. Yaghoobi, J. Evidence-Based Complementary Altern. Med., 2014, 2014, 893247 Search PubMed.
- S. Esmaeili-Mahani, M. R. Samandari-Bahraseman and M. M. Yaghoobi, Iran. J. Pharm. Res., 2018, 17, 343 Search PubMed.
- T. Ghanbari, M. Azadbakht, A. Vesi-Raygani and M. Khazaei, Int. J. Morphol., 2016, 34, 1197–1202 CrossRef.
- F. Zeng, R.-J. Ju, L. Liu, H.-J. Xie, L.-M. Mu, Y. Zhao, Y. Yan, Y.-J. Hu, J.-S. Wu and W.-L. Lu, Oncotarget, 2015, 6, 36625 Search PubMed.
- C.-T. Lee, Y.-W. Huang, C.-H. Yang and K.-S. Huang, Curr. Top. Med. Chem., 2015, 15, 1491–1500 CrossRef PubMed.
- R. C. Wang, X. Chen, A. M. Parissenti, A. A. Joy, J. Tuszynski, D. N. Brindley and Z. Wang, PLoS One, 2017, 12, e0182400 CrossRef PubMed.
- C. Luchsinger, M. Aguilar, P. V. Burgos, P. Ehrenfeld and G. A. Mardones, PLoS One, 2018, 13, e0195401 CrossRef PubMed.
- P. Ghanbari, M. Mohseni, M. Tabasinezhad, B. Yousefi, A. A. Saei, S. Sharifi, M. R. Rashidi and N. Samadi, Appl. Biochem. Biotechnol., 2014, 174, 667–681 CrossRef PubMed.
- C. Azevedo, A. Correia-Branco, J. R. Araújo, J. T. Guimarães, E. Keating and F. Martel, Nutr. Cancer, 2015, 67, 504–513 CrossRef PubMed.
- X. Yi, J. Zuo, C. Tan, S. Xian, C. Luo, S. Chen, L. Yu and Y. Luo, Afr. J. Tradit., Complementary Altern. Med., 2016, 13, 210–215 CrossRef PubMed.
- S.-H. Kim, K.-A. Hwang and K.-C. Choi, J. Nutr. Biochem., 2016, 28, 70–82 CrossRef PubMed.
- G.-A. Lee, K.-C. Choi and K.-A. Hwang, Environ. Toxicol. Pharmacol., 2017, 49, 48–57 CrossRef PubMed.
- K. Matsumoto, Y. Akao, H. Yi, K. Ohguchi, T. Ito, T. Tanaka, E. Kobayashi, M. Iinuma and Y. Nozawa, Bioorg. Med. Chem., 2004, 12, 5799–5806 CrossRef PubMed.
- K. Matsumoto, Y. Akao, K. Ohguchi, T. Ito, T. Tanaka, M. Iinuma and Y. Nozawa, Bioorg. Med. Chem., 2005, 13, 6064–6069 CrossRef PubMed.
- Y. Akao, Y. Nakagawa and Y. Nozawa, Int. J. Mol. Sci., 2008, 9, 355–370 CrossRef PubMed.
- P. Moongkarndi, N. Kosem, S. Kaslungka, O. Luanratana, N. Pongpan and N. Neungton, J. Ethnopharmacol., 2004, 90, 161–166 CrossRef PubMed.
- C.-H. Lee, T.-H. Ying, H.-L. Chiou, S.-C. Hsieh, S.-H. Wen, R.-H. Chou and Y.-H. Hsieh, Oncotarget, 2017, 8, 47425 Search PubMed.
- M.-A. Shibata, M. Iinuma, J. Morimoto, H. Kurose, K. Akamatsu, Y. Okuno, Y. Akao and Y. Otsuki, BMC Med., 2011, 9, 69 CrossRef PubMed.
- S. Kritsanawong, S. Innajak, M. Imoto and R. Watanapokasin, Int. J. Oncol., 2016, 48, 2155–2165 CrossRef PubMed.
- N. Atluri, V. Thirumalanadhuni and U. M. devi Palempalli, Int. J. Phytomed., 2014, 6, 354–359 Search PubMed.
- P. Li, W. Tian and X. Ma, Mol. Cancer, 2014, 13, 138 CrossRef PubMed.
- Y.-S. Won, J.-H. Lee, S.-J. Kwon, J.-Y. Kim, K.-H. Park, M.-K. Lee and K.-I. Seo, Food Chem. Toxicol., 2014, 66, 158–165 CrossRef PubMed.
- H. Kurose, M.-A. Shibata, M. Iinuma and Y. Otsuki, BioMed Res. Int., 2012, 2012, 672428 Search PubMed.
- S. P. Hehner, T. G. Hofmann, W. Dröge and M. L. Schmitz, J. Immunol., 1999, 163, 5617–5623 Search PubMed.
- V. B. Mathema, Y.-S. Koh, B. C. Thakuri and M. Sillanpää, Inflammation, 2012, 35, 560–565 CrossRef PubMed.
- A. Ghantous, A. Sinjab, Z. Herceg and N. Darwiche, Drug discovery today, 2013, 18, 894–905 CrossRef PubMed.
- A. Pareek, M. Suthar, G. S. Rathore and V. Bansal, Pharmacogn. Rev., 2011, 5, 103 CrossRef PubMed.
- C. Lu, W. Wang, Y. Jia, X. Liu, Z. Tong and B. Li, J. Cell. Biochem., 2014, 115, 1458–1466 CrossRef PubMed.
- A. D'Anneo, D. Carlisi, S. Emanuele, G. Buttitta, R. Di Fiore, R. Vento, G. Tesoriere and M. Lauricella, Int. J. Oncol., 2013, 43, 1895–1900 CrossRef PubMed.
- A. D'anneo, D. Carlisi, M. Lauricella, R. Puleio, R. Martinez, S. Di Bella, P. Di Marco, S. Emanuele, R. Di Fiore and A. Guercio, Cell Death Dis., 2013, 4, e891 CrossRef PubMed.
- D. Carlisi, M. Lauricella, A. D'Anneo, G. Buttitta, S. Emanuele, R. Di Fiore, R. Martinez, C. Rolfo, R. Vento and G. Tesoriere, J. Cell. Physiol., 2015, 230, 1276–1289 CrossRef PubMed.
- G.-A. Lee, K.-A. Hwang and K.-C. Choi, Food Chem. Toxicol., 2017, 109, 284–295 CrossRef PubMed.
- W. Wang, M. Lv, Y. Wang and J. Zhang, Pharm. Biol., 2016, 54, 3164–3168 CrossRef PubMed.
- H. L. Nicastro, G. L. Firestone and L. F. Bjeldanes, J. Nutr. Biochem., 2013, 24, 1882–1888 CrossRef PubMed.
- A. Ahmad, S. Ali, A. Ahmed, A. S. Ali, A. Raz, W. A. Sakr and K. W. Rahman, PLoS One, 2013, 8, e54657 CrossRef PubMed.
- E. R. Kasala, L. N. Bodduluru, C. C. Barua and R. Gogoi, Biomed. Pharmacother., 2016, 82, 568–577 CrossRef PubMed.
- J. Xiong, K. Wang, C. Yuan, R. Xing, J. Ni, G. Hu, F. Chen and X. Wang, Int. J. Mol. Med., 2017, 39, 113–125 CrossRef PubMed.
- G. Seelinger, I. Merfort and C. M. Schempp, Planta Med., 2008, 74, 1667–1677 CrossRef PubMed.
- L. Ziyan, Z. Yongmei, Z. Nan, T. Ning and L. Baolin, Planta Med., 2007, 73, 221–226 CrossRef PubMed.
- M. Kimata, N. Inagaki and H. Nagai, Planta Med., 2000, 66, 25–29 CrossRef PubMed.
- N. E. Wilsher, R. R. Arroo, M. T. Matsoukas, A. M. Tsatsakis, D. A. Spandidos and V. P. Androutsopoulos, Food Chem. Toxicol., 2017, 110, 383–394 CrossRef PubMed.
- M. T. Cook, Y. Liang, C. Besch-Williford and S. M. Hyder, Breast Cancer: Targets Ther., 2017, 9, 9 Search PubMed.
- D. Lin, G. Kuang, J. Wan, X. Zhang, H. Li, X. Gong and H. Li, Oncol. Rep., 2017, 37, 895–902 CrossRef PubMed.
- L. Zhang, F. Yang, L. Huang, A. Liu and J. Zhang, Biomed. Res., 2017, 28, 4902–4907 Search PubMed.
- Y. W. Jeon, Y. E. Ahn, W. S. Chung, H. J. Choi and Y. J. Suh, Tumor Biol., 2015, 36, 6349–6359 CrossRef PubMed.
- S.-H. Tu, C.-T. Ho, M.-F. Liu, C.-S. Huang, H.-W. Chang, C.-H. Chang, C.-H. Wu and Y.-S. Ho, Food Chem., 2013, 141, 1553–1561 CrossRef PubMed.
- Y. Sato, N. Sasaki, M. Saito, N. Endo, F. Kugawa and A. Ueno, Biol. Pharm. Bull., 2015, 38, 703–709 CrossRef PubMed.
- F. Guan, Y. Ding, Y. Zhang, Y. Zhou, M. Li and C. Wang, PLoS One, 2016, 11, e0146553 CrossRef PubMed.
- C. Geng, J. Li, F. Ding, G. Wu, Q. Yang, Y. Sun, Z. Zhang, T. Dong and X. Tian, Biochem. Biophys. Res. Commun., 2016, 473, 147–153 CrossRef PubMed.
- J. A. Colacino, S. P. McDermott, M. A. Sartor, M. S. Wicha and L. S. Rozek, Breast Cancer Res. Treat., 2016, 158, 29–41 CrossRef PubMed.
- Ö. Berrak, Y. Akkoç, E. D. Arısan, A. Çoker-Gürkan, P. Obakan-Yerlikaya and N. Palavan-Ünsal, Biomed. Pharmacother., 2016, 77, 150–160 CrossRef PubMed.
- E. Kronski, M. E. Fiori, O. Barbieri, S. Astigiano, V. Mirisola, P. H. Killian, A. Bruno, A. Pagani, F. Rovera and U. Pfeffer, Mol. Oncol., 2014, 8, 581–595 CrossRef PubMed.
- T. Huang, Z. Chen and L. Fang, Oncol. Rep., 2013, 29, 117–124 CrossRef PubMed.
- M. Jiang, O. Huang, X. Zhang, Z. Xie, A. Shen, H. Liu, M. Geng and K. Shen, Molecules, 2013, 18, 701–720 CrossRef PubMed.
- H.-W. Lai, S.-Y. Chien, S.-J. Kuo, L.-M. Tseng, H.-Y. Lin, C.-W. Chi and D.-R. Chen, J. Evidence-Based Complementary Altern. Med., 2012, 2012, 486568 Search PubMed.
- X.-D. Sun, X.-E. Liu and D.-S. Huang, Mol. Med. Rep., 2012, 6, 1267–1270 CrossRef PubMed.
- J.-M. Kim, E.-M. Noh, K.-B. Kwon, J.-S. Kim, Y.-O. You, J.-K. Hwang, B.-M. Hwang, B.-S. Kim, S.-H. Lee and S. J. Lee, Phytomedicine, 2012, 19, 1085–1092 CrossRef PubMed.
- M. Lazzeroni, A. Guerrieri-Gonzaga, S. Gandini, H. Johansson, D. Serrano, M. Cazzaniga, V. Aristarco, D. Macis, S. Mora and P. Caldarella, Cancer Prev. Res., 2017, 10, 363–370 CrossRef PubMed.
- K. Hallman, K. Aleck, M. Quigley, B. Dwyer, V. Lloyd, M. Szmyd and S. Dinda, Breast Cancer: Targets Ther., 2017, 9, 365 Search PubMed.
- G. Deb, V. S. Thakur, A. M. Limaye and S. Gupta, Mol. Carcinog., 2015, 54, 485–499 CrossRef PubMed.
- N. D. Mineva, K. E. Paulson, S. P. Naber, A. S. Yee and G. E. Sonenshein, PLoS One, 2013, 8, e73464 CrossRef PubMed.
- C. Braicu, C. D. Gherman, A. Irimie and I. Berindan-Neagoe, J. Nanosci. Nanotechnol., 2013, 13, 632–637 CrossRef PubMed.
- H. Luo, M. Xu, W. Zhong, Z. Cui, F. Liu, K. Zhou and X. Li, J. BUON, 2014, 19, 435–439 Search PubMed.
- J.-Y. Jang, J.-K. Lee, Y.-K. Jeon and C.-W. Kim, BMC Cancer, 2013, 13, 421 CrossRef PubMed.
- S. Mirza, G. Sharma, R. Parshad, S. D. Gupta, P. Pandya and R. Ralhan, J. Breast Cancer, 2013, 16, 23–31 CrossRef PubMed.
- J.-P. Xue, G. Wang, Z.-B. Zhao, Q. Wang and Y. Shi, Oncol. Rep., 2014, 32, 1647–1653 CrossRef PubMed.
- X. Liu, C. Sun, X. Jin, P. Li, F. Ye, T. Zhao, L. Gong and Q. Li, Molecules, 2013, 18, 13200–13217 CrossRef PubMed.
- T. T. Rajah, K. J. Peine, N. Du, C. A. Serret and N. R. Drews, Anticancer Res., 2012, 32, 1181–1191 Search PubMed.
- H. Pan, W. Zhou, W. He, X. Liu, Q. Ding, L. Ling, X. Zha and S. Wang, Int. J. Mol. Med., 2012, 30, 337–343 CrossRef PubMed.
- J. Saluzzo, K. M. Hallman, K. Aleck, B. Dwyer, M. Quigley, V. Mladenovik, A. E. Siebert and S. Dinda, Genes Cancer, 2016, 7, 414 Search PubMed.
- Y. Fu, H. Chang, X. Peng, Q. Bai, L. Yi, Y. Zhou, J. Zhu and M. Mi, PLoS One, 2014, 9, e102535 CrossRef PubMed.
- L. S. Gomez, P. Zancan, M. C. Marcondes, L. Ramos-Santos, J. R. Meyer-Fernandes, M. Sola-Penna and D. Da Silva, Biochimie, 2013, 95, 1336–1343 CrossRef PubMed.
- S.-H. Tu, K.-C. Yang, C.-T. Ho, C.-S. Huang, C.-S. Chen, H.-W. Chang, C.-H. Chang, C.-H. Wu and Y.-S. Ho, Journal of Cancer Research and Practice, 2013, 29, 167–181 Search PubMed.
- J. Duo, G.-G. Ying, G.-W. Wang and L. Zhang, Mol. Med. Rep., 2012, 5, 1453–1456 Search PubMed.
- G. Lee, K.-A. Hwang and K.-C. Choi, Environ. Toxicol. Pharmacol., 2017, 49, 48–57 CrossRef PubMed.
- K. M. Perrott, C. D. Wiley, P.-Y. Desprez and J. Campisi, GeroScience, 2017, 39, 161–173 CrossRef PubMed.
- X. Cao, B. Liu, W. Cao, W. Zhang, F. Zhang, H. Zhao, R. Meng, L. Zhang, R. Niu and X. Hao, Chin. J. Cancer Res., 2013, 25, 212 Search PubMed.
- M. Karimi, H. Babaahmadi-Rezaei, G. Mohammadzadeh and M.-A. Ghaffari, Iran. J. Pathol., 2017, 12, 135–143 Search PubMed.
- Z. Jahanafrooz, N. Motameh and B. Bakhshandeh, Asian Pac. J. Cancer Prev., 2016, 17, 2661–2665 Search PubMed.
- T. H. Kim, J. S. Woo, Y. K. Kim and K. H. Kim, J. Pharmacol. Exp. Ther., 2014, 349, 268–278 CrossRef PubMed.
- W. Lu, C. Lin, T. D. King, H. Chen, R. C. Reynolds and Y. Li, Cell. Signalling, 2012, 24, 2291–2296 CrossRef PubMed.
- B. H. Hong, C. H. Wu, C. T. Yeh and G. C. Yen, Mol. Nutr. Food Res., 2013, 57, 886–895 CrossRef PubMed.
- A. Hussain, M. Javeria, A. P. Sathyen, B. Salema, N. Qurrat El-Ain, H. Geetganga, J. Elham, M. Ali Khan and C. Sharma, Asian Pac. J. Cancer Prev., 2013, 14, 5855–5860 CrossRef PubMed.
- J. Sui, K. Xie and M. Xie, Shengli Xuebao, 2016, 68, 27–34 Search PubMed.
- M. T. Cook, Y. Liang, C. Besch-Williford, S. Goyette, B. Mafuvadze and S. M. Hyder, SpringerPlus, 2015, 4, 444 CrossRef PubMed.
- D.-W. Sun, H.-D. Zhang, L. Mao, C.-F. Mao, W. Chen, M. Cui, R. Ma, H.-X. Cao, C.-W. Jing and Z. Wang, Cell. Physiol. Biochem., 2015, 37, 1693–1711 CrossRef PubMed.
- E.-J. Lee, S.-Y. Oh and M.-K. Sung, Food Chem. Toxicol., 2012, 50, 4136–4143 CrossRef PubMed.
- M. J. Kim, J. S. Woo, C. H. Kwon, J. H. Kim, Y. K. Kim and K. H. Kim, Cell Biol. Int., 2012, 36, 339–344 CrossRef PubMed.
- L.-M. Wang, K.-P. Xie, H.-N. Huo, F. Shang, W. Zou and M.-J. Xie, Asian Pac. J. Cancer Prev., 2012, 13, 1431–1437 CrossRef PubMed.
- L. Eanes and Y. M. Patel, Biochimie Open, 2016, 3, 64–71 CrossRef PubMed.
- F. Zhang, W. Dong, W. Zeng, L. Zhang, C. Zhang, Y. Qiu, L. Wang, X. Yin, C. Zhang and W. Liang, Breast Cancer Res., 2016, 18, 38 CrossRef PubMed.
- Y. Sun and J. Gu, Zhongguo Zhongyao Zazhi, 2015, 40, 1144–1150 Search PubMed.
- M. S. I. Abaza, K. Y. Orabi, E. Al-Quattan and J. Raja'a, Cancer Cell Int., 2015, 15, 46 CrossRef PubMed.
- J.-Y. Ke, Y.-H. Hsiao, S. R. Straka, L. D. Yee and M. A. Belury, Cancer Res., 2015, 903 Search PubMed.
- J. Helle, K. Kräker, M. I. Bader, A. M. Keiler, O. Zierau, G. Vollmer, J. Welsh and G. Kretzschmar, Mol. Cell. Endocrinol., 2014, 392, 125–135 CrossRef PubMed.
- T. Hatkevich, J. Ramos, I. Santos-Sanchez and Y. M. Patel, Exp. Cell Res., 2014, 327, 331–339 CrossRef PubMed.
- S. Kim and T. I. Park, Int. J. Clin. Exp. Med., 2013, 6, 890 Search PubMed.
- P. Bulzomi, A. Bolli, P. Galluzzo, F. Acconcia, P. Ascenzi and M. Marino, IUBMB Life, 2012, 64, 690–696 CrossRef PubMed.
- M. N. Dastjerdi, E. M. Mehdiabady, F. G. Iranpour and H. Bahramian, Int. J. Prev. Med., 2016, 7, 66 CrossRef PubMed.
- S. Rajput, B. P. Kumar, K. K. Dey, I. Pal, A. Parekh and M. Mandal, Life Sci., 2013, 93, 783–790 CrossRef PubMed.
- H. Zheng, Y. Li, Y. Wang, H. Zhao, J. Zhang, H. Chai, T. Tang, J. Yue, A. M. Guo and J. Yang, Toxicol. Appl. Pharmacol., 2014, 280, 10–20 CrossRef PubMed.
- M. Marques, L. Laflamme, I. Benassou, C. Cissokho, B. Guillemette and L. Gaudreau, BMC Cancer, 2014, 14, 524 CrossRef PubMed.
- P. Wang, X. Du, M. Xiong, J. Cui, Q. Yang, W. Wang, Y. Chen and T. Zhang, Sci. Rep., 2016, 6, 33709 CrossRef PubMed.
- S.-H. Kim, Y. Wan and S. V. Singh, Cancer Res., 2017, 5265 Search PubMed.
- S.-H. Kim, A. Sehrawat and S. V. Singh, Breast Cancer Res. Treat., 2012, 134, 1067–1079 CrossRef PubMed.
- D. Xiao, A. Bommareddy, S.-H. Kim, A. Sehrawat, E.-R. Hahm and S. V. Singh, PLoS One, 2012, 7, e32597 CrossRef PubMed.
- H. Li, B. Yang, J. Huang, T. Xiang, X. Yin, J. Wan, F. Luo, L. Zhang, H. Li and G. Ren, Toxicol. Lett., 2013, 220, 219–228 CrossRef PubMed.
- R. Kotecha, A. Takami and J. L. Espinoza, Oncotarget, 2016, 7, 52517 CrossRef PubMed.
- S. W. Hoag and A. S. Hussain, J. Nutr., 2001, 131, 1389S–1391S CrossRef PubMed.
- M.-M. Mocanu, P. Nagy and J. Szöllősi, Molecules, 2015, 20, 22578–22620 CrossRef PubMed.
- V. S. Srinivasan, J. Nutr., 2001, 131, 1349S–1350S CrossRef PubMed.
- R. P. Heaney, J. Nutr., 2001, 131, 1344S–1348S CrossRef PubMed.
- P. Anand, A. B. Kunnumakkara, R. A. Newman and B. B. Aggarwal, Mol. Pharmaceutics, 2007, 4, 807–818 CrossRef PubMed.
- A. Shehzad, F. Wahid and Y. S. Lee, Arch. Pharm., 2010, 343, 489–499 CrossRef PubMed.
- G. Shoba, D. Joy, T. Joseph, M. M. R. Rajendran and P. Srinivas, Planta Med., 1998, 64, 353–356 CrossRef PubMed.
- Q. Huang, H. Yu and Q. Ru, J. Food Sci., 2010, 75, R50–R57 CrossRef PubMed.
- S. Prasad, A. K. Tyagi and B. B. Aggarwal, Cancer Res. Treat., 2014, 46, 2 CrossRef PubMed.
- A. Storka, B. Vcelar, U. Klickovic, G. Gouya, S. Weisshaar, S. Aschauer, G. Bolger, L. Helson and M. Woltz, Int. J. Clin. Pharmacol. Ther., 2015, 53, 54–65 CrossRef PubMed.
- C. K. Singh, M. A. Ndiaye and N. Ahmad, Biochim. Biophys. Acta, Mol. Basis Dis., 2015, 1852, 1178–1185 CrossRef PubMed.
- D. J. Boocock, G. E. Faust, K. R. Patel, A. M. Schinas, V. A. Brown, M. P. Ducharme, T. D. Booth, J. A. Crowell, M. Perloff and A. J. Gescher, Cancer Epidemiol. Biomark. Prev., 2007, 16, 1246–1252 CrossRef PubMed.
- L. Almeida, M. Vaz-da-Silva, A. Falcão, E. Soares, R. Costa, A. I. Loureiro, C. Fernandes-Lopes, J. F. Rocha, T. Nunes and L. Wright, Mol. Nutr. Food Res., 2009, 53, S7–S15 CrossRef PubMed.
- J. Hoshino, E.-J. Park, T. P. Kondratyuk, L. Marler, J. M. Pezzuto, R. B. Van Breemen, S. Mo, Y. Li and M. Cushman, J. Med. Chem., 2010, 53, 5033–5043 CrossRef PubMed.
- P. R. Van Ginkel, D. Sareen, L. Subramanian, Q. Walker, S. R. Darjatmoko, M. J. Lindstrom, A. Kulkarni, D. M. Albert and A. S. Polans, Clin. Cancer Res., 2007, 13, 5162–5169 CrossRef PubMed.
- C. Huo, S. Wan, W. Lam, L. Li, Z. Wang, K. Landis-Piwowar, D. Chen, Q. Dou and T. Chan, Inflammopharmacology, 2008, 16, 248–252 CrossRef PubMed.
- X. Cai, Z. Fang, J. Dou, A. Yu and G. Zhai, Curr. Med. Chem., 2013, 20, 2572–2582 CrossRef PubMed.
- J. Zhang, D. Liu, Y. Huang, Y. Gao and S. Qian, Int. J. Pharm., 2012, 436, 311–317 CrossRef PubMed.
- S.-m. Ding, Z.-h. Zhang, J. Song, X.-d. Cheng, J. Jiang and X.-b. Jia, Int. J. Nanomed., 2014, 9, 2327 CrossRef PubMed.
- K. Zhang, L. Gu, J. Chen, Y. Zhang, Y. Jiang, L. Zhao, K. Bi and X. Chen, J. Pharm. Biomed. Anal., 2015, 114, 168–175 CrossRef PubMed.
- H.-Y. Si, D.-P. Li, T.-M. Wang, H.-L. Zhang, F.-Y. Ren, Z.-G. Xu and Y.-Y. Zhao, J. Nanosci. Nanotechnol., 2010, 10, 2325–2331 CrossRef PubMed.
- R. Cohen, B. Schwartz, I. Peri and E. Shimoni, J. Agric. Food Chem., 2011, 59, 7932–7938 CrossRef PubMed.
- N. Dixit, S. Baboota, K. Kohli, S. Ahmad and J. Ali, Indian J. Pharmacol., 2007, 39, 172 CrossRef.
- Y. Song, J. Zhuang, J. Guo, Y. Xiao and Q. Ping, Pharmazie, 2008, 63, 35–42 Search PubMed.
- C. A. Houghton, R. G. Fassett and J. S. Coombes, Nutr. Rev., 2013, 71, 709–726 CrossRef PubMed.
- H. Danafar, A. Sharafi, H. Kheiri Manjili and S. Andalib, Pharm. Dev. Technol., 2017, 22, 642–651 CrossRef PubMed.
- M. Paltsev, V. Kiselev, E. Muyzhnek, V. Drukh, I. Kuznetsov and O. Pchelintseva, EPMA J., 2013, 4, 25 CrossRef PubMed.
- M. Elmowafy, A. Samy, M. A. Raslan, A. Salama, R. A. Said, A. E. Abdelaziz, W. El-Eraky, S. El Awdan and T. Viitala, AAPS PharmSciTech, 2016, 17, 663–672 CrossRef PubMed.
- F. Odeh, S. I. Ismail, R. Abu-Dahab, I. S. Mahmoud and A. Al Bawab, Drug Delivery, 2012, 19, 371–377 CrossRef PubMed.
- F.-L. Yen, T.-H. Wu, L.-T. Lin, T.-M. Cham and C.-C. Lin, Pharm. Res., 2009, 26, 893–902 CrossRef PubMed.
- D. V. Ratnam, D. Ankola, V. Bhardwaj, D. K. Sahana and M. R. Kumar, J. Controlled Release, 2006, 113, 189–207 CrossRef PubMed.
- M. Cuendet, J. Guo, Y. Luo, S. Chen, C. P. Oteham, R. C. Moon, R. B. Van Breemen, L. E. Marler and J. M. Pezzuto, Cancer Prev. Res., 2010, 1940–6207 Search PubMed.
- F. Gao, J. Zhang, C. Fu, X. Xie, F. Peng, J. You, H. Tang, Z. Wang, P. Li and J. Chen, Int. J. Nanomed., 2017, 12, 4147 CrossRef PubMed.
- Z. Yang, S. Gao, J. Wang, T. Yin, Y. Teng, B. Wu, M. You, Z. Jiang and M. Hu, Drug Metab. Dispos., 2011, 111, 040006 Search PubMed.
- A. Gupta, L. D. Kagliwal and R. S. Singhal, in Advances in food and nutrition research, Elsevier, 2013, vol. 69, pp. 183–217 Search PubMed.
- H. Dang, M. H. W. Meng, H. Zhao, J. Iqbal, R. Dai, Y. Deng and F. Lv, J. Nanopart. Res., 2014, 16, 2347 CrossRef.
- G. Galati and P. J. O'brien, Free Radicals Biol. Med., 2004, 37, 287–303 CrossRef PubMed.
- R. Strick, P. L. Strissel, S. Borgers, S. L. Smith and J. D. Rowley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 4790–4795 CrossRef PubMed.
- H. L. Bonkovsky, Ann. Intern. Med., 2006, 144, 68 CrossRef PubMed.
- S. Narayanan, U. Mony, D. K. Vijaykumar, M. Koyakutty, B. Paul-Prasanth and D. Menon, Nanomedicine, 2015, 11, 1399–1406 CrossRef PubMed.
- R. C. C. de Pace, X. Liu, M. Sun, S. Nie, J. Zhang, Q. Cai, W. Gao, X. Pan, Z. Fan and S. Wang, J. Liposome Res., 2013, 23, 187–196 CrossRef PubMed.
- G. Sharma, J. Park, A. R. Sharma, J.-S. Jung, H. Kim, C. Chakraborty, D.-K. Song, S.-S. Lee and J.-S. Nam, Pharm. Res., 2015, 32, 723–735 CrossRef PubMed.
- A. Sarkar, S. Ghosh, S. Chowdhury, B. Pandey and P. C. Sil, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 2065–2075 CrossRef PubMed.
- F. S. Tabatabaei Mirakabad, A. Akbarzadeh, M. Milani, N. Zarghami, M. Taheri-Anganeh, V. Zeighamian, F. Badrzadeh and M. Rahmati-Yamchi, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 423–430 CrossRef PubMed.
- S. Esfandiarpour-Boroujeni, S. Bagheri-Khoulenjani, H. Mirzadeh and S. Amanpour, Carbohydr. Polym., 2017, 168, 14–21 CrossRef PubMed.
- J. Wang, Y. Wang, Q. Liu, L. Yang, R. Zhu, C. Yu and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 26511–26523 CrossRef PubMed.
- S. Y. Park, S. Y. Chae, J. O. Park, K. J. Lee and G. Park, Oncol. Rep., 2016, 35, 3248–3256 CrossRef PubMed.
- J. Meng, F. Guo, H. Xu, W. Liang, C. Wang and X.-D. Yang, Sci. Rep., 2016, 6, 22390 CrossRef PubMed.
- W. Wang, L. Zhang, T. Chen, W. Guo, X. Bao, D. Wang, B. Ren, H. Wang, Y. Li and Y. Wang, Molecules, 2017, 22, 1814 CrossRef PubMed.
- S. Bhattacharya, M. Ahir, P. Patra, S. Mukherjee, S. Ghosh, M. Mazumdar, S. Chattopadhyay, T. Das, D. Chattopadhyay and A. Adhikary, Biomaterials, 2015, 51, 91–107 CrossRef PubMed.
- P. Soni, J. Kaur and K. Tikoo, J. Nanopart. Res., 2015, 17, 18 CrossRef.
- W. K. Ng, L. Saiful Yazan, L. H. Yap, W. N. Hafiza, W. A. Ghani, C. W. How and R. Abdullah, BioMed Res. Int., 2015, 2015, 263131 Search PubMed.
- Y. Bellik, L. Boukraâ, H. A. Alzahrani, B. A. Bakhotmah, F. Abdellah, S. M. Hammoudi and M. Iguer-Ouada, Molecules, 2012, 18, 322–353 CrossRef PubMed.
- A. A. Ramos, C. F. Lima and C. Pereira-Wilson, in Selected Topics in DNA Repair, InTech, 2011 Search PubMed.
- A. Abdal Dayem, H. Y. Choi, G.-M. Yang, K. Kim, S. K. Saha and S.-G. Cho, Nutrients, 2016, 8, 581 CrossRef PubMed.
- D. Liu and Z. Chen, J. Breast Cancer, 2013, 16, 133–137 CrossRef PubMed.
- L. Varinska, P. Gal, G. Mojzisova, L. Mirossay and J. Mojzis, Int. J. Mol. Sci., 2015, 16, 11728–11749 CrossRef PubMed.
- D. Sinha, N. Sarkar, J. Biswas and A. Bishayee, Semin. Cancer Biol., 2016, 40, 209–232 CrossRef PubMed.
- R. H. Himes, Pharmacol. Ther., 1991, 51, 257–267 CrossRef PubMed.
- M. Moudi, R. Go, C. Y. S. Yien and M. Nazre, Int. J. Prev. Med., 2013, 4, 1231 Search PubMed.
- C. Vassallo, A. Carugno, F. Derlino, O. Ciocca, V. Brazzelli and G. Borroni, Cutis, 2015, 95, E28–E34 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |