
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion influences on reactivity and NMR spectroscopic features of NHC precursors†
Han Vinh Huynh *,
Truc Tien Lam and
Huyen T. T. Luong
*,
Truc Tien Lam and
Huyen T. T. Luong
Department of Chemistry, National University of Singapore, 3 Science Drive 3, 117543 Singapore, Republic of Singapore. E-mail: chmhhv@nus.edu.sg; Fax: +65 6779 1691; Tel: +65 6516 2670
First published on 12th October 2018
Abstract
A series of 16 benzimidazolium salts of the type iPr2-bimyH+X− with various anions X were synthesized and characterized by various spectroscopic and spectrometric methods. Significant anion and solvent effects on the chemical shifts of the C2–H protons were found, which allows for a ranking of the anions in terms of their hydrogen-bond acceptor properties. Stronger acceptors could increase the acidity of their respective salts leading to a faster H/D exchange. Similar but less pronounced anion influences were detected for the 13CC2 NMR resonances, while 1JC2–H coupling constants appear to be anion and solvent independent.
Introduction
N-Heterocyclic carbenes (NHCs) and their complexes are nowadays ubiquitous in the areas of catalysis and organometallic chemistry.1–5 Most conveniently, they are accessed by deprotonation of azolium salts differing in backbones and substituents. The structural diversity allows certain control over the stereoelectronic properties of the respective NHCs. Thus, detailed knowledge of the properties and preparative routes to these most common precursors is imperative for NHC chemistry.6 One general synthetic pathway involves double N-alkylation of azoles using electrophiles such as alkyl halides and oxonium salts. In such SN2 reactions, azolium salts are routinely obtained which contain either different halides (e.g. Cl−, Br−, I−) or weakly coordinating species (e.g. BF4−, PF6−) as counteranions. Many of such compounds are also ionic liquids, and it is well known that anions influence the physical properties of the salts, e.g. melting point and solubility. Moreover, the influence of substituents and the heterocyclic backbone on the acidities of azolium salts have also been established.7,8 Systematic studies detailing the comparison between different anions of a given cation and their influences on reactivity and spectroscopic characteristics are surprisingly rare. A recent study suggests that anion effects are negligible in dimethylsulfoxide8 or in water.9 However, most NHC chemistry is conducted in organic solvents, and we hypothesize that counteranions could affect the acidity of an azolium cation. For example, we have observed that direct metalation of azolium salts with weakly coordinating anions (e.g. BF4−, PF6−) is difficult even under harsh conditions, but with addition of halides the reaction proceeds at ambient temperature.10,11 Such anion effects have also been observed in catalysis where the active metal–NHC species is prepared in situ from azolium precursors. Again, halide salts were much more effective than the respective tetrafluoroborates, which could be due to their increased acidity facilitating NHC complex formation.12,13 As part of our broader research program on NHCs and in contribution to a more detailed understanding of NHC precursors and ionic liquids in general, we herein report a detailed spectroscopic profiling on such anion influences in selected common organic solvents.Results and discussion
Syntheses and characterizations of iPr2-bimyH+X− salts
The proposed study on anion influences in NHC precursors calls for the preparation of various azolium salts that contain the same heterocyclic cation, but differ in the counteranion. The question thus arises, which heterocyclic system would be best for our purpose. Among the types of classical azolium salts depicted in Chart 1, we decided to explore the N,N-dialkylbenzimidazolium system III, because (i) their preparation is easy, and (ii) they contain only one acidic proton in the five-membered ring system, which should simplify the study.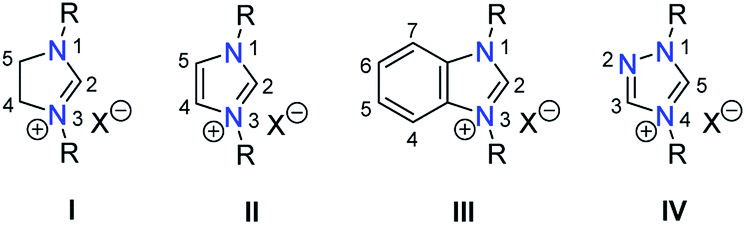 | ||
| Chart 1 Classical azolium salts for the most common NHCs and their numbering scheme. | ||
In particular, 1,3-diisopropylbenzimidazolium bromide (A)14 is regularly used in our laboratories to prepare NHC complexes for donor strength determination using the HEP system.15–18 Using this selected salt as the starting point a series of anion exchange reactions were conducted, which led to 16 new benzimidazolium salts as summarized in Scheme 1.
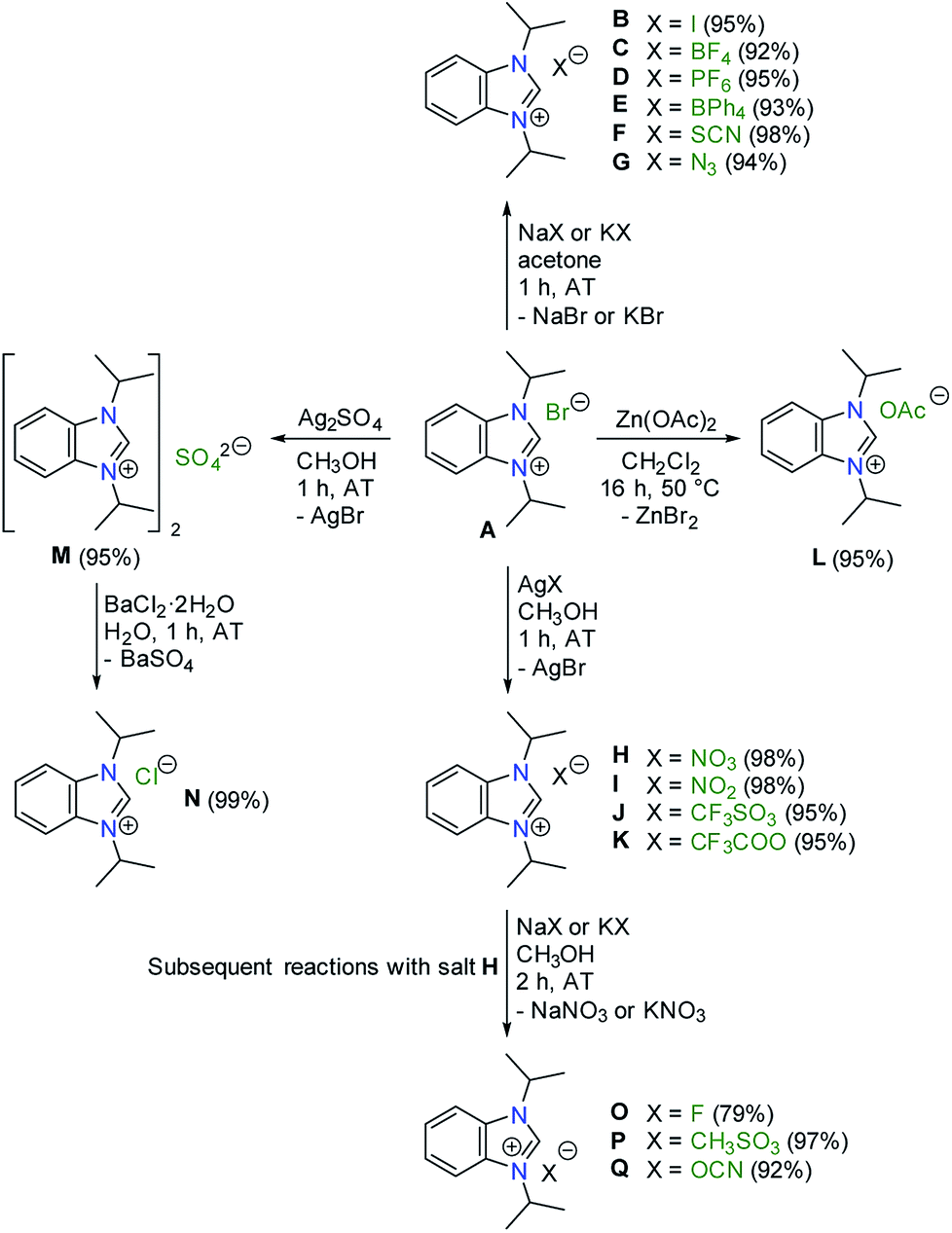 | ||
| Scheme 1 Preparative routes to benzimidazolium salts with diverse counteranions. | ||
The preparation of salts B–G was most straightforward, and simply involved exposure of compound A to a slight excess of sodium or potassium salts containing the desired anion at ambient temperature (AT). In general, the comparatively big azolium cations favour larger anions (e.g. PF6−), while small alkali metals form more stable salts with small anions (e.g. halides) due to better interionic interactions (e.g. lattice energy). The precipitation of sodium or potassium bromide from acetone drives these reactions forward. However, the same approach could not be applied for the synthesis of H–K, where silver salts had to be employed instead. The more favourable formation of silver bromide drove anion exchange to completion. More difficult was the synthesis of the acetate L. Reaction of salt A with sodium acetate did not occur, while its treatment with silver(I) acetate led to in situ deprotonation to afford a silver(I)–NHC complex instead. Eventually, salt L could be prepared by prolonged heating species A with zinc(II) acetate in dichloromethane under reflux.
The replacement of bromide with lighter halides is difficult (vide supra), and often require ion-exchange resins. However, such anion exchange can also be achieved by exploiting the solubilities of common salts. This approach was demonstrated in the 2-step preparation of the chloride N. First, the bromide in salt A was precipitated as silver bromide using silver(I) sulfate to afford the bis(benzimidazolium) sulfate M. Subsequently, the sulfate anion can be easily precipitated as barium(II) sulfate using an aqueous solution of barium(II) chloride to give salt N as a hygroscopic solid. The very favourable formation of insoluble barium(II) sulfate drives this reaction.
Finally, the salts O–Q were prepared in a similar manner by reaction of the nitrate H with sodium or potassium salts containing the desired anions.
All salts B–Q were all obtained as white to off-white powders and characterized by NMR spectroscopy. Their formations were confirmed by the significant shifts of the C2–H signals compared to the starting material A. In addition, new NMR signals arising from the newly introduced anions were found for C, D, E, F, J, K, L, P and Q. Apart from the C2–H signals, there were little changes of other 1H NMR signals of the iPr2-bimyH+ cations in these salts. Fig. 1 depicts a representative comparison of the 1H NMR spectra between the bromide salt A and the tetrafluoroborate salt C in the range from 12–4 ppm.
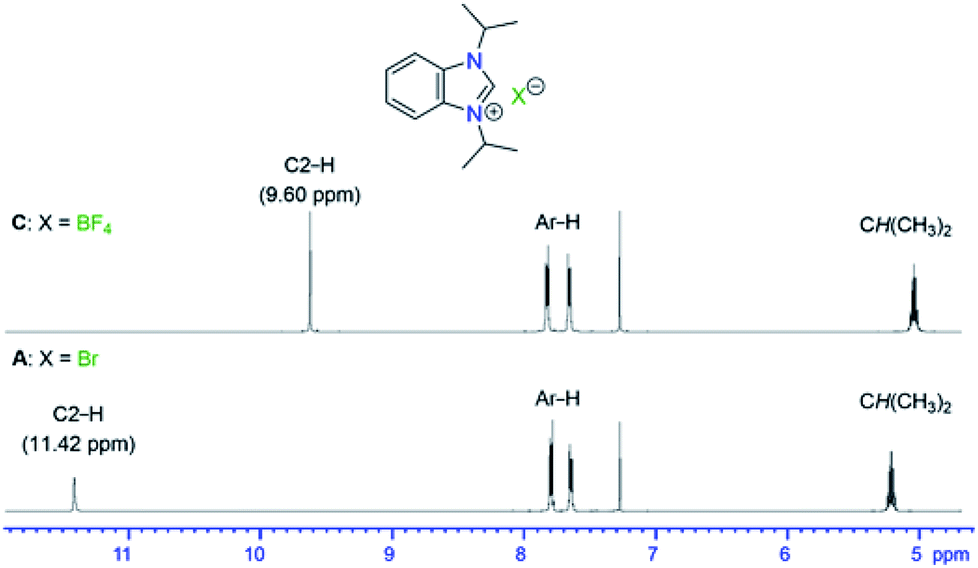 | ||
| Fig. 1 1H NMR spectroscopic comparison of BF4− vs. Br− salts in the range from 12–4 ppm. | ||
Moreover, ESI mass spectrometry was also employed as a viable method, especially for salts with heavier anions, to determine the compositions of the newly synthesized salts. Apparently, all new salts show the same base peak at m/z = 203 for the [iPr2-bimyH]+ cation in the positive-ion mode. Peaks attributed to the anions could also be observed in the case of B (X = I; m/z = 127), D (X = PF6; m/z = 145), E (X = BPh4; m/z = 319) and J (X = CF3SO3; m/z = 149) in the negative-ion mode. Furthermore, peaks assignable to the [2M − X]+ fragments were also observed for the three salts bearing comparatively big anions, i.e. D (X = PF6; m/z = 551), E (X = BPh4; m/z = 725) and J (X = CF3SO3; m/z = 555) pointing to their greater tendencies to form ion pairs in the gas phase.
In addition, single crystals of E, F and N·H2O suitable for X-ray diffraction were obtained by slow evaporation of their solutions in dichloromethane/hexane (E, F) and dichloromethane/ethyl acetate (N·H2O). Fig. 2 depicts the molecular structures of these three salts. As anticipated, the bond lengths and angles within the iPr2-bimyH+ cations of all three salts are typical and do not vary much from those of their starting material A.14 For salt E, no apparent cation–anion interactions can be found in the solid state molecular structure. Salt F, however, shows short contacts of 2.695(2) Å between the nitrogen of the anion with the C2–H proton of the cation and a C–H⋯NCS angle of 107.4(1)°. Despite several attempts, it was not possible to crystallize salt N without water, since it is very hygroscopic. Instead, we obtained crystals of the solvate N·H2O. Despite the water molecule, hydrogen bonding of the chloride to the C2–H proton with a separation of 2.4982(7) Å and a C–H⋯Cl angle of 155.0(1)° is apparent. In addition, the chloride engages in two hydrogen bonds with water with distances of 2.43(4) and 2.49(4) Å and angles of 167(3) and 172(4)°, respectively. The latter leads to a chain-like arrangement of the salts in solid state.
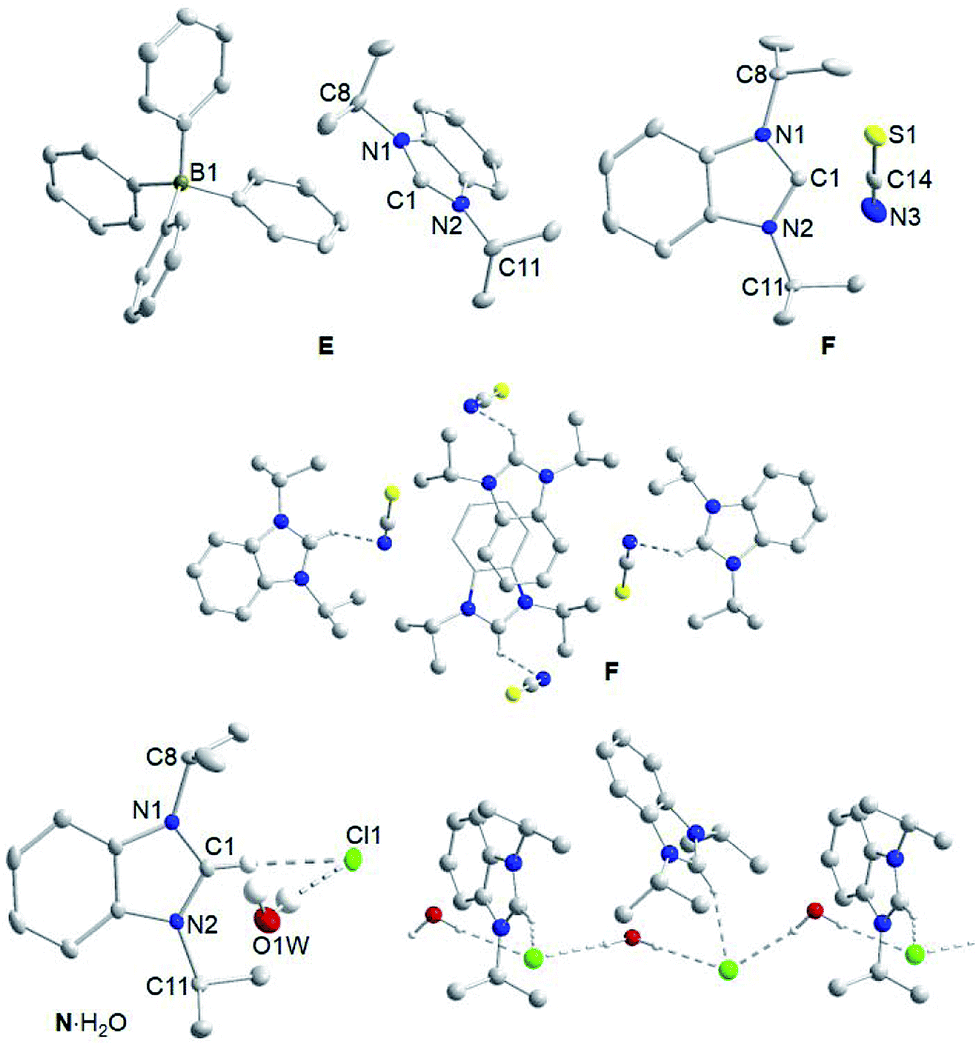 | ||
| Fig. 2 Molecular structures of E, F and N·H2O showing 50% probability ellipsoids; hydrogen atoms are omitted for clarity with exception of those engaged in hydrogen bonding. Selected bond lengths [Å] and bond angles [deg]: E, C1–N1 1.328(3), C1–N2 1.326(3), N1–C8 1.485(3), N2–C11 1.490(2); N1–C1–N2 110.8(2). F, C1–N1 1.335(2), C1–N2 1.331(2), N1–C8 1.485(2), N2–C11 1.485(2), S1–C14 1.646(2), N3–C14 1.154(3); N1–C1–N2 110.6(2), S1–C14–N3 179.0(2). N·H2O, C1–N1 1.329(3), C1–N2 1.336(2), N1–C8 1.488(3), N2–C11 1.488(3); N1–C1–N2 110.5(2). | ||
Anion influence on C2–H NMR chemical shifts and IR bands of iPr2-bimyH+X− salts
Hydrogen bonding has been recognized as a main interaction between the cations and anions of azolium salts leading to close arrangement between the counteranions and the most acidic C2–H proton.19–22However, this type of interaction can be affected by many factors, such as functional groups attached to the cation as well as solvents and concentrations used for measuring. In order to investigate only anion influences, all salts in our study contain the same iPr2-bimyH+ cation, which bears innocent and inert N-isopropyl substituents. Since azolium salts are notoriously hygroscopic, they were dried under vacuum for a prolonged time (1 day) to remove as much water as possible. The very hygroscopic salt N was dried under vacuum at 120 °C for 5 hours. In some cases, complete removal of crystal water was not possible, and the effect of trace water was investigated separately with the parent salt A (vide infra). The dried salts were initially measured in CDCl3 at room temperature at a concentration of 83 μmol mL−1, in order to eliminate any other effects that could affect the cation–anion interaction. Table 1 summarizes the 1HC2–H as well as 13CC2 NMR resonances for salts A–Q, with the exception of M, which bears the less comparable dianionic sulfate anion. The IR data for the C2–H vibration has also been included for comparison. The 1HC2–H NMR signals of the 16 salts cover chemical shifts from 8.99–11.68 ppm, which supposedly reflects a diverse strength of hydrogen bonding of the anions.
| Salt | X | 1HC2–Ha [ppm] | 13CC2a [ppm] | ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) C2–Hb [cm−1] C2–Hb [cm−1] |
|---|---|---|---|---|
| a Measured in CDCl3 at concentrations of 83 μmol mL−1 and internally referenced to the residual protio-solvent signal at 7.26 and 77.7 ppm relative to TMS, respectively.b Data was collected by ATR methodology with solid sample. | ||||
| N | Cl | 11.68 | 142.1 | 2983 |
| A | Br | 11.42 | 141.3 | 2983 |
| O | F | 11.10 | 142.0 | 2982 |
| B | I | 10.92 | 140.4 | 2985 |
| G | N3 | 10.89 | 141.1 | 2979 |
| I | NO2 | 10.84 | 140.3 | 2981 |
| Q | OCN | 10.71 | 141.2 | 2984 |
| H | NO3 | 10.70 | 139.7 | 2988 |
| P | CH3SO3 | 10.67 | 141.7 | 2989 |
| F | SCN | 10.50 | 140.5 | 2980 |
| L | CH3CO2 | 9.98 | 139.8 | 2982 |
| J | CF3SO3 | 9.78 | 138.5 | 2982 |
| K | CF3CO2 | 9.71 | 138.7 | 2983 |
| C | BF4 | 9.60 | 139.2 | 2995 |
| D | PF6 | 9.17 | 138.5 | 2996 |
| E | BPh4 | 8.99 | 139.6 | 2985 |
We anticipate that a stronger hydrogen bond acceptor would also polarize the C2–H bond more, thus slightly increasing the acidity of the salt. As expected, halide anions with four lone pairs of electrons are the best hydrogen bonding acceptors that lead to the most downfield shifts of the 1HC2–H signal The gradual downfield shifts from 10.92 (I), 11.42 (Br) to 11.68 ppm (Cl) reflects the increasing electronegativity of the halogens in the order of I < Br < Cl.
However, the fluoride salt O does not follow this trend with an intermediate 1HC2–H NMR resonance of 11.10 ppm. In fact, the relative position of the fluoride among the halides is not clear and difficult to determine.23,24 Intuitively, the highly electronegative and small-sized fluoride is thought to be a superior hydrogen bond acceptor. Instead, this study places its hydrogen bonding capability with the iPr2-bimyH+ donor in between that of the bromide and iodide anions. Notably, it has been reported that intramolecular hydrogen bonding of 2-halophenols decrease in the order F ≈ Cl > Br > I in the vapour phase, while the order in solution is Cl > Br > F ≈ I.24,25 The latter is in agreement with our results. It has been suggested that the highly electronegative fluorine holds onto its electron lone pairs so tightly that charge transfer from the non-bonding orbital of fluorine (nF) to the anti-bonding orbital of the C2–H interaction 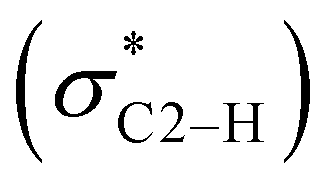 cannot occur properly at a certain distance.26 Therefore, the ability to form hydrogen bonding of fluoride anion could be reduced compared to its intuitively expected value.
cannot occur properly at a certain distance.26 Therefore, the ability to form hydrogen bonding of fluoride anion could be reduced compared to its intuitively expected value.
The substitution of other groups with fluorine can affect the hydrogen-bonding capability of an anion as well. For example, the acetate anion of L and methanesulfonate anion of P are stronger hydrogen bonding acceptors compared to their fluorine-substituted analogues K and J, respectively. The negative inductive (−I) effects exerted by the three fluorine atoms withdraw electrons and decrease the hydrogen bonding capability of the anions as a whole.
It is also our interest to analyse the different effects of linear triatomic anions, such as SCN−, OCN− and N3−. The influence of these pseudohalides was anticipated to be very similar, yet they can be differentiated by the 1HC2–H NMR chemical shifts of their salts. For ambivalent species, it has been noted that nitrogen are superior acceptors than oxygen atoms.27 Sulfur is expected to be a poorer acceptor than oxygen due to its larger size, which weakens interactions with the much smaller hydrogen atoms (cf. Fig. 2).
All three anions can be represented by simple resonance structures (taught in classes), which are depicted in Fig. 3 along with the calculated Mulliken charges. The more favourable resonance form of the N3− anion contains two negative charges at two terminal nitrogen atoms, which should result in greater ability to form hydrogen bonds. Salt G indeed shows the most downfield 1HC2–H NMR signal of 10.89 ppm in comparison to its counterparts. The preferred resonance structure for OCN− anion has the negative charge on the oxygen atom with three lone pairs of electrons, which is reflected in the Mulliken charge as well. For the SCN− anion, the favourable resonance form places the negative charge on the nitrogen atom with only two lone pairs (cf. Fig. 2). Therefore, the former is expected to be a slightly better hydrogen bond acceptor. Notably, the 1HC2–H NMR resonances of 10.71 (OCN−) and 10.50 ppm (SCN−) recorded for salts Q and F, respectively, are in line with this argument. Nevertheless, it could be more accurate to compare the calculated Mulliken charges at nitrogen, since nitrogen was reported to be a more dominant acceptor than oxygen or sulfur. The Mulliken charges at nitrogen decrease in the order N3− > OCN− > SCN−, which could explain the upfield shifts of the C2–H protons of the respective salts in the same order.
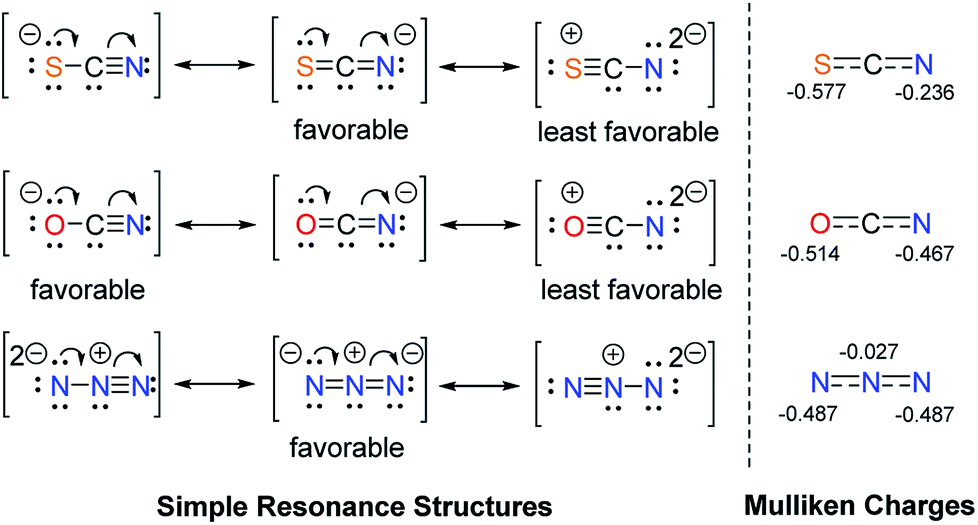 | ||
| Fig. 3 Resonance structures and calculated Mulliken charges of the pseudohalides SCN−, OCN− and N3−. | ||
Similar considerations can be used to rationalize differences between nitrate salt H (10.70 ppm) and the nitrite salt I (10.84 ppm). On average, each of the three oxygen atoms in the ‘delocalized’ NO3− anion retains two thirds of the negative charge, which can be used to interact with the benzimidazolium C2–H proton (Fig. 4). However, there is also a positive charge at nitrogen in all resonance forms, which is reflected in the Mulliken charge.
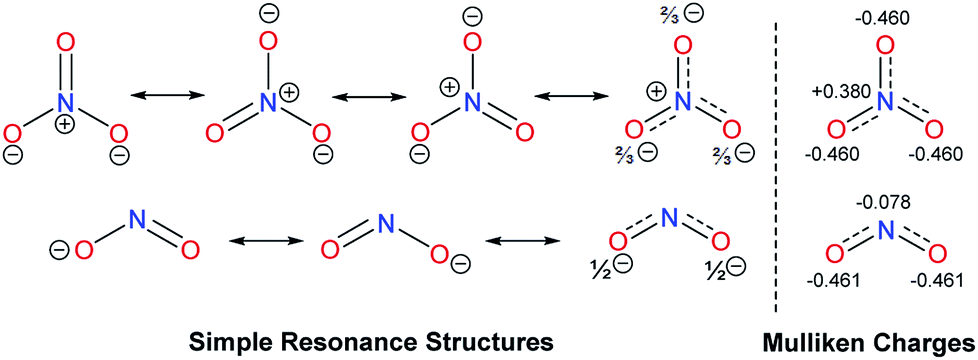 | ||
| Fig. 4 Resonance structures and calculated Mulliken charges of NO3− and NO2− anions. | ||
The two oxygen atoms of the NO2− anion, on the other hand, have one-half of the negative charge for such an interaction. Here, there is no positive charge at the central nitrogen. The calculation even indicates a small negative Mulliken charge at nitrogen, while the charge at oxygen is essentially identical to that of the NO3− anion. Therefore, NO2− anion could be considered as a better hydrogen bonding acceptor than NO3− anion, which is supported by 1H NMR spectroscopy.
Finally, the salts C, D, J and K containing the weakly coordinating anions BF4−, PF6−, CF3SO3 and CF3CO2−, respectively, show the most upfield C2–H 1H NMR signals. This observation lends support to the notion that they can hardly form strong hydrogen bonds. The BPh4− ion in salt E represents a special case, where the downfield shift cannot be used to estimate hydrogen bonding. Here, it has been reported that stronger hydrogen bonding leads to an upfield shift instead. This is so, since stronger interactions would place the C2–H proton closer to the shielding area of the phenyl substituents of the borate anion.28 Overall, the 1H NMR resonances of C2–H proton in most iPr2-bimyH+X− salts can be used to rank the effects of monoanions on the 1H NMR spectroscopic scale (Fig. 5).
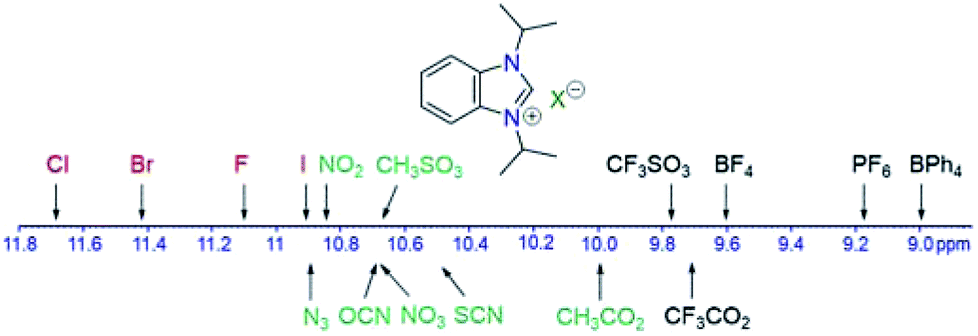 | ||
| Fig. 5 1HC2–H NMR chemical shifts of benzimidazolium salts with different anions. | ||
As mentioned above, many azolium salts are hygroscopic, and it can be rather difficult to remove all traces of water from the crystal lattice. Thus, we studied the effects of water on the 1HC2–H NMR chemical shifts of salt A as a representative. Fig. 6 depicts the 1H NMR spectroscopic changes of the bromide salt A in CDCl3 upon addition of 1–3 equivalents of water. In the presence of one equivalent of water (b), a slightly more upfield NMR signal for the C2–H proton can be noted compared to the water-free sample (a), while all other signals remain essentially unchanged. Addition of another equivalent induces a smaller upfield change, while addition of a third equivalent appears to have little further effect on the C2–H signal. The presence of water reduces interaction of the bromide with the azolium cation. Instead, more favourable and competing bromide–water interactions kick in. As expected, a new signal due to water starts to appear. Interestingly, a second water signal surfaces in spectrum (c), which becomes very apparent in spectrum (d). This observation indicates the presence of unsymmetrical water molecules. It appears that with excess of water only one of the two water hydrogen atoms can efficiently engage in hydrogen bonding giving rise to two well separated signals. Thus, water competes efficiently with the azolium cation for good hydrogen-bond acceptors leading to an upfield shift of the C2–H signal.
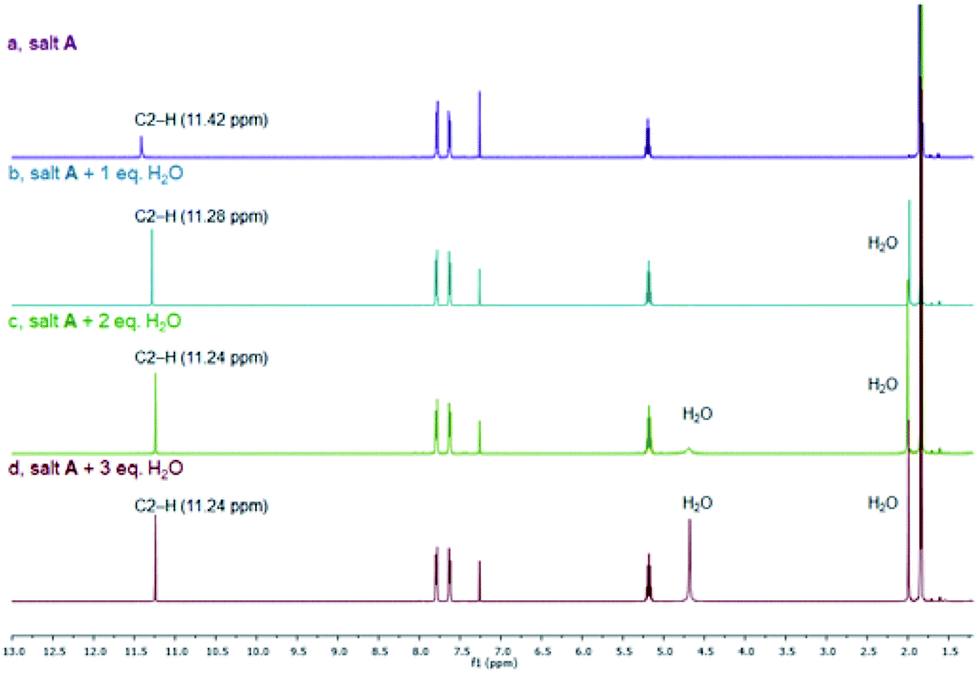 | ||
| Fig. 6 Changes of the 1H NMR spectrum of salt A in CDCl3 upon addition of 1–3 equivalents of water. | ||
According to the 13C NMR data listed in Table 1, the influence of the anions on the C2 carbon signals is less pronounced. This is not entirely surprising since the C2-carbon atom is overall further away and “indirectly” affected by the anion. Nevertheless, salts with strong and moderate hydrogen bond acceptors gave more downfield 13CC2 NMR signals ranging from 140.3–142.1 ppm, while those with weaker ones resonate at more upfield regions from 138.5–139.2 ppm. Again, the BPh4− ion is a special case, since the chemical shift of 139.6 ppm for its salt suggests a stronger acceptor compared to 1H NMR data.
In addition to NMR studies, we also have also determined the C2–H stretches of the salts by IR spectroscopy. In general, a smaller wavenumber indicates a weaker C–H bond. However, comparison of the relevant data is less conclusive, and a clear trend is absent. IR spectroscopy cannot reveal smaller differences between halides and pseudohalides due to its insufficient resolution.29 However, one piece of information that can be extracted from the data is that halide salts have notably smaller wavenumbers than their BF4− or PF6− counterparts, which is within expectations. Notably, the IR data for the BPh4− salt appears to be in line with its 13C NMR data.
Finally, four salts A–C and N with common anions (Cl, Br, I and BF4) in NHC chemistry were chosen to study the conductivity of their equimolar solutions in chloroform. Strong ion pairs are expected to have lower conductivity, while solvated ions would give larger values. The chloride (N) and bromide (A) salts showed the lowest conductivity of 1.8 and 2.5 μS cm−1, respectively, which could reflect their tendency to form ion pairs via hydrogen bonds. The salts with the larger iodide and tetrafluoroborate anions show conductivities of 5.1 and 4.1 μS cm−1, respectively. It appears that larger anions can interact less efficiently with the small C2–H hydrogen leading to a larger separation of cation and anion in solution. Nevertheless, this study only provides a qualitative argument.
Solvent and anion influence on chemical shifts and 1JC–H coupling constants
Next, solvent effects on the 1H and 13C NMR signals of the C2–H group were studied. Equimolar solutions of nine selected salts A–F, H, I and N in CD3CN and DMSO-d6 as two common polar deuterated solvents were subjected to NMR spectroscopy and the results compared to the data obtained in CDCl3. The respective protio-solvents have polarity indexes of 4.1 (chloroform), 5.8 (acetonitrile) and 7.2 (dimethylsulfoxide), respectively.Table 2 summarizes the 1H NMR resonances in decreasing order (CDCl3) observed in different solvents. It becomes apparent, that the chemical shift differences among the salts are most pronounced in CDCl3 (Δδ 2.69 ppm) as the least polar solvent. With increasing solvent polarity, the resonances of the four salts become more and more similar (CD3CN: Δδ 1.50 ppm). The smallest differences are observed in DMSO-d6 (Δδ 0.3 ppm), which can engage best in hydrogen bonds. As such, the interactions with each individual anion becomes less significant, and very similar chemical shifts are thus obtained. In CDCl3 as the worst hydrogen bond acceptor, the anion influences are most pronounced. Another striking observation is that the chemical shifts for salts C, D and E with weakly coordinating anions are more downfield in DMSO-d6 than in CDCl3. Apparently, these shifts are due to hydrogen bonds with the DMSO solvent molecules. This implies that DMSO is a better hydrogen bond acceptor than the BF4−, PF6− and BPh4− anions, but significantly weaker than the iodide. Consequently, such anion effects cannot be studied in water, which is best known for hydrogen bonds (cf. Fig. 2).
| Salt | X | CDCl3 | CD3CN | DMSO-d6 |
|---|---|---|---|---|
| a Measured in at concentrations of 83 μmol mL−1 and internally referenced to the residual protio-solvent signal at 7.26 (CDCl3), 1.94 (CD3CN) and 2.50 ppm (DMSO-d6) relative to TMS, respectively. | ||||
| N | Cl | 11.68 ppm | 10.38 ppm | 10.04 ppm |
| A | Br | 11.42 ppm | 9.96 ppm | 9.91 ppm |
| B | I | 10.92 ppm | 9.35 ppm | 9.79 ppm |
| I | NO2 | 10.84 ppm | 9.39 ppm | 9.79 ppm |
| H | NO3 | 10.70 ppm | 9.34 ppm | 9.77 ppm |
| F | SCN | 10.50 ppm | 9.16 ppm | 9.76 ppm |
| C | BF4 | 9.60 ppm | 9.10 ppm | 9.75 ppm |
| D | PF6 | 9.17 ppm | 8.95 ppm | 9.74 ppm |
| E | BPh4 | 8.99 ppm | 8.88 ppm | 9.74 ppm |
A similar, but less pronounced trend, likely due to the same reasons, is also noted for 13CC2 NMR signals, which are summarized in Table 3. Again, it is notable that the chemical shift differences become smaller and smaller with increasing polarity of the solvent, i.e. CDCl3: Δδ 3.6 ppm; CD3CN: Δδ 2.2 ppm; DMSO-d6: 0.3 ppm.
| Salt | X | CDCl3 | CD3CN | DMSO-d6 |
|---|---|---|---|---|
| a Measured in at concentrations of 83 μmol mL−1 and internally referenced to the solvent signal at 77.7 (CDCl3), 118.3 (CD3CN) and 39.5 ppm (DMSO-d6) relative to TMS, respectively. | ||||
| N | Cl | 142.1 ppm | 140.4 ppm | 139.3 ppm |
| A | Br | 141.3 ppm | 139.6 ppm | 139.2 ppm |
| F | SCN | 140.5 ppm | 138.7 ppm | 139.0 ppm |
| B | I | 140.4 ppm | 138.9 ppm | 139.0 ppm |
| I | NO2 | 140.3 ppm | 139.3 ppm | 139.0 ppm |
| H | NO3 | 139.7 ppm | 139.2 ppm | 139.0 ppm |
| E | BPh4 | 139.6 ppm | 138.2 ppm | 138.9 ppm |
| C | BF4 | 139.2 ppm | 138.6 ppm | 139.0 ppm |
| D | PF6 | 138.5 ppm | 138.3 ppm | 139.0 ppm |
Recently, 1JC–H coupling constants in azolium salts have been used to estimate the donating ability of the respective NHC.30 However, it was noted that conclusions made did not consider neither anion nor solvent influences on the 1JC–H values.29 In order to address this issue, we decided compare the 1JC2–H of the four salts A–D in the aforementioned solvents. The data summarized in Table 4 indicate that anion or solvent influences on the 1JC2–H are surprisingly small or even negligible. Salts with different anions show the same coupling constants in the same solvent. When measured in different solvents only marginal changes of ∼1 Hz were observed. Nevertheless, it is still advisable only to compare salts with the same counteranion, where the data was obtained in the same solvent to avoid any unexpected interferences that could lead to misinterpretations in terms of donating ability of the respective NHC.
| Salt | X | CDCl3 | CD3CN | DMSO-d6 |
|---|---|---|---|---|
| a Measured in at concentrations of 83 μmol mL−1 and internally referenced to the solvent signal at 77.7 (CDCl3), 118.3 (CD3CN) and 39.5 ppm (DMSO-d6) relative to TMS, respectively. | ||||
| A | Br | 217.6 Hz | 216.3 Hz | 217.6 Hz |
| B | I | 217.6 Hz | 216.3 Hz | 217.6 Hz |
| C | BF4 | 217.6 Hz | 216.3 Hz | 217.6 Hz |
| D | PF6 | 217.6 Hz | 216.3 Hz | 217.6 Hz |
H/D exchange reactions
Increased hydrogen-bond acceptor properties of an anion could also better polarize the C2–H bond and therefore increase its acidity, which may be related to the ease of C2–H/D exchange. To test this notion, the ease of C2–H/D exchange of the three salts containing Br− (A), I− (B) and BF4− (C) anions was studied in CDCl3 at ambient temperature with the addition of one equivalent of CD3OD as an deuterium source. Salt A exchanges rapidly to give >30% conversion in seconds, after which the rate drops significantly (Fig. 7). After 18 hours, 68% H/D exchange was noted for the bromide salt A. The iodide salt B shows an extremely slow H/D exchange leading to only 11% conversion, while almost no H/D exchange was observed for salt C containing BF4− as the weakest hydrogen-bond acceptor. The very different reaction profiles support an anion influence on the acidity of azolium salts.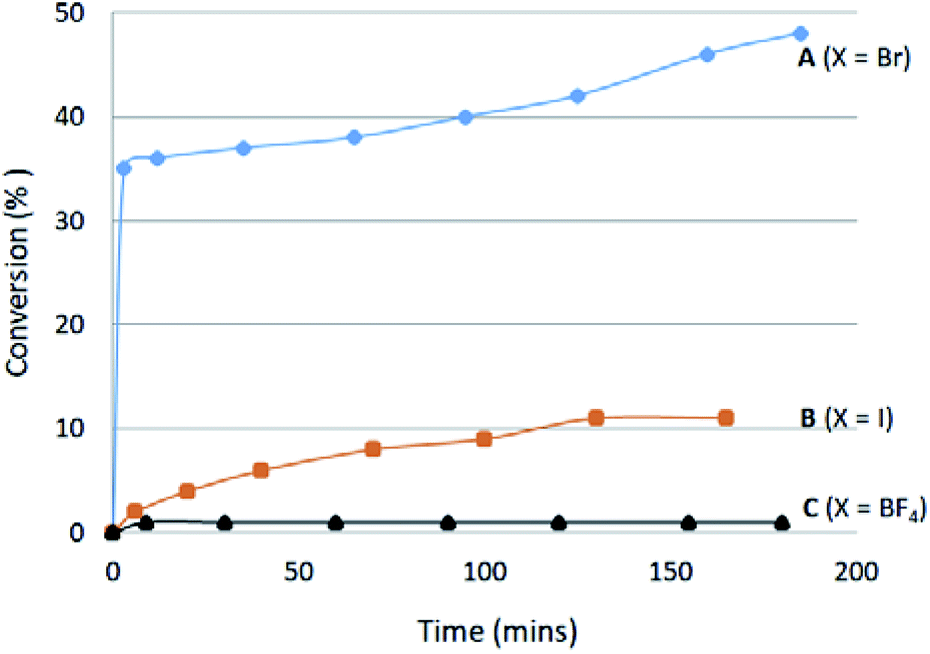 | ||
| Fig. 7 Time-dependant H/D exchange of salts A–C. | ||
Salt N was expected to undergo H/D exchange with ease. However, it is too hygroscopic to be obtained as a completely water-free salt. Attempts to study the exchange using N·H2O led to an unexpected outcome. Here the CD3OD exchanges rapidly with the initial crystal water instead of the less acidic C2–H proton of the benzimidazolium cation.
Conclusions
We have studied the influences of various anions on the NMR spectroscopic profiles of azolium salts. It was found that the acidic C2–H proton of 1,3-diisopropylbenzimidazolium salts is most sensitive to the nature of the counteranion. The chemical shifts of the C2–H protons allows for a ranking of the anions in terms of their hydrogen-bond acceptor properties, whereby better acceptors affect a more downfield resonance, which could be related to a faster H/D exchange. Moreover, it was observed that the C2–H chemical shift differences become significantly smaller with increasing polarity of the solvent, which competes for hydrogen bonding and thus diminishes the effects of the anions. This study also reveals that the 1JC2–H coupling constants of the azolium salts are almost anion and solvent independent. Since such salts are ubiquitously used as ionic liquids and NHC precursors for organocatalysis and organometallic chemistry, we believe that a better understanding of their anion influences will aid in the studies of their potential applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National University of Singapore and the Singapore Ministry of Education (WBS R-143-000-669-112). Technical support from staff at the Chemical, Molecular and Materials Analysis Centre (CMMAC) of our department is appreciated.Notes and references
- H. V. Huynh, The Organometallic Chemistry of N-heterocyclic Carbenes, John Wiley & Sons, Ltd, Chichester, UK, 2017 Search PubMed.
- F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS PubMed.
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed.
- T. L. Amyes, S. T. Diver, J. P. Richard, F. M. Rivas and K. Toth, J. Am. Chem. Soc., 2004, 126, 4366–4374 CrossRef CAS PubMed.
- Y. Chu, H. Deng and J. P. Cheng, J. Org. Chem., 2007, 72, 7790–7793 CrossRef CAS PubMed.
- E. M. Higgins, J. A. Sherwood, A. G. Lindsay, J. Armstrong, R. S. Massey, R. W. Alder and A. C. O'Donoghue, Chem. Commun., 2011, 47, 1559–1561 RSC.
- J. C. Bernhammer and H. V. Huynh, Organometallics, 2012, 31, 5121–5130 CrossRef CAS.
- H. Sivaram, J. Tan and H. V. Huynh, Organometallics, 2012, 31, 5875–5883 CrossRef CAS.
- W. Wu, Q. Teng, Y.-Y. Chua, H. V. Huynh and H. A. Duong, Organometallics, 2017, 36, 2293–2297 CrossRef CAS.
- Q. Teng, W. Wu, H. A. Duong and H. V. Huynh, Chem. Commun., 2018, 54, 6044–6047 RSC.
- H. V. Huynh, Y. Han, J. H. H. Ho and G. K. Tan, Organometallics, 2006, 25, 3267–3274 CrossRef CAS.
- H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395–5404 CrossRef CAS.
- S. Guo, H. Sivaram, D. Yuan and H. V. Huynh, Organometallics, 2013, 32, 3685–3696 CrossRef CAS.
- Q. Teng and H. V. Huynh, Inorg. Chem., 2014, 53, 10964–10973 CrossRef CAS PubMed.
- Q. Teng and H. V. Huynh, Dalton Trans., 2017, 46, 614–627 RSC.
- A. G. Avent, P. A. Chaloner, M. P. Day, K. R. Seddon and T. Welton, J. Chem. Soc., Dalton Trans., 1994, 3405 RSC.
- J. A. Cowan, J. A. C. Clyburne, M. G. Davidson, R. L. W. Harris, J. A. K. Howard, P. Küpper, M. A. Leech and S. P. Richards, Angew. Chem., Int. Ed., 2002, 41, 1432–1434 CrossRef CAS PubMed.
- N. Kuhn and A. Alsheikh, Coord. Chem. Rev., 2005, 249, 829–857 CrossRef CAS.
- P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef.
- G. A. Jeffrey and W. Saenger, Hydrog. Bond. Biol. Struct., 1994, pp. 161–163 Search PubMed.
- J.-M. Dumas, M. Gomel and M. Guerin, Halides, Pseudo-Halides and Azides Part 2, 1983, pp. 985–1020 Search PubMed.
- B. E. Smart, in Organoflourine Chemistry, 1994, pp. 57–88 Search PubMed.
- P. N. Edwards, in Organofluorine Chemistry, 1994, pp. 515–520 Search PubMed.
- H.-J. Böhm, S. Brode, U. Hesse and G. Klebe, Chem.–Eur. J., 1996, 2, 1509–1513 CrossRef.
- J. Van den Broeke, M. Stam, M. Lutz, H. Kooijman, A. L. Spek, B. J. Deelman and G. Van Koten, Eur. J. Inorg. Chem., 2003, 2798–2811 CrossRef CAS.
- H. V. Huynh, Chem. Rev., 2018 DOI:10.1021/acs.chemrev.8b00067.
- K. Verlinden, H. Buhl, W. Frank and C. Ganter, Eur. J. Inorg. Chem., 2015, 2416–2425 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, selected crystallographic data. CCDC 1853200, 1853201 and 1867791. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra05839c |
| This journal is © The Royal Society of Chemistry 2018 |